

Abstract
General interest in the biological functions of IFN type I in Mycobacterium tuberculosis (Mtb) infection increased after the recent identification of a distinct IFN gene expression signature in tuberculosis (TB) patients. Here, we demonstrate that TB-susceptible mice lacking the receptor for IFN I (IFNAR1) were protected from death upon aerogenic infection with Mtb. Using this experimental model to mimic primary progressive pulmonary TB, we dissected the immune processes affected by IFN I. IFNAR1 signaling did not affect T-cell responses, but markedly altered migration of inflammatory monocytes and neutrophils to the lung. This process was orchestrated by IFNAR1 expressed on both immune and tissue-resident radioresistant cells. IFNAR1-driven TB susceptibility was initiated by augmented Mtb replication and in situ death events, along with CXCL5/CXCL1-driven accumulation of neutrophils in alveoli, followed by the discrete compartmentalization of Mtb in lung phagocytes. Early depletion of neutrophils rescued TB-susceptible mice to levels observed in mice lacking IFNAR1. We conclude that IFN I alters early innate events at the site of Mtb invasion leading to fatal immunopathology. These data furnish a mechanistic explanation for the detrimental role of IFN I in pulmonary TB and form a basis for understanding the complex roles of IFN I in chronic inflammation.
Keywords: Alveolar macrophages, Inflammation, Interferon type I, Mycobacterium tuberculosis, Neutrophils
Introduction
Tuberculosis (TB) is a pulmonary disease caused by Mycobacterium tuberculosis (Mtb), which afflicts millions of people worldwide 1. The failure to eradicate TB by available measures calls for novel intervention strategies based on deeper insights into pathophysiological processes underlying active disease. A unique IFN signature in blood leukocytes has recently been described in active TB 2–4 raising questions about the role of IFN I in TB pathogenesis and whether IFN I modulation can be harnessed for immune intervention.
Type I IFN performs multiple functions in immunity and inflammation 5. Consistent with this multifunctionality is the ubiquitous expression of its receptor (receptor for IFN I, IFNAR1) and its propensity to induce complex autocrine and paracrine signaling cascades 6. IFN I has well-assigned protective functions in viral infections. In bacterial infections, these effects depend on the microbial habitat. Generally, IFN I is considered detrimental in diseases caused by intracellular bacteria 7, including TB. Experimental studies in TB-resistant mice report heightened IFN I induction by clinical isolates of Mtb with increased virulence 8,9 and fatal TB progression upon exogenous delivery of IFN I inducers 10. However, Mtb infection of mice deleted of IFNAR1 resulted in inconclusive results 11–13, likely as a consequence of different infection routes with impact on tissue-dependent disease processes.
Besides the IFN I and II gene expression signatures in blood leukocytes revealed in TB, cross-regulation of IFN I and IFN II was recently described in leprosy, another mycobacterial disease affecting skin, mucosa, and the peripheral nervous system 14. IFN interdependence was identified in skin specimens and found to imprint polarization of tuberculoid or lepromatous lesions. Mtb primarily affects the respiratory parenchyma, an environment with distinct immune regulation. In resistant mice, inhibition by IFN I of IL-1 cognates in isolated immune cells 15, and IFN-driven recruitment of Mtb-permissive mononuclear cells during pulmonary TB, were observed 10,16. Yet, the precise biological functions of IFN I at the site of infection in progressive TB remain elusive. We embarked on dissecting the cellular and molecular effects of IFN I in pulmonary TB.
We found that the lethally TB-susceptible phenotype of 129S2 mice, which resembles primary progressive TB, can be rescued by IFNAR1 deletion. Susceptibility correlated with exacerbated inflammation, which was ameliorated in the absence of IFN I signaling. Early during infection, IFN I signaling fostered Mtb replication and triggered accelerated dissemination of tubercle bacilli to recruited phagocyte populations while diminishing alveolar macrophage (AM) lung content. Functional IFNAR1 on both hematopoietic and tissue-resident cells was critical for fatal inflammation. This process was initiated by heightened cell death events within AM populations, elevated chemokine release, and enhanced recruitment of detrimental neutrophils into the airways before onset of adaptive immunity. These novel insights into TB pathogenesis provide guidelines for future immune intervention strategies for progressive TB.
Results
Signaling via IFNAR1 is detrimental in TB and promotes inflammation
We investigated downstream effects of IFNAR1 signaling on TB in highly susceptible 129S2 mice (Fig.1A and B; Supporting Information Fig. 1A and B) and their counterparts with deleted IFNAR1 (Ifnar1−/−). Mice were infected with a low dose of Mtb (∼200 CFUs) via the aerogenic route. The 129S2 mice gradually lost weight and most of them died within 40 days postinfection (p.i.), whereas Ifnar1−/− animals survived acute and chronic TB (Fig.1A; Supporting Information Fig. 1C). The absence of IFN I signaling ameliorated lung inflammation, as monitored by lung/body weight ratio (Supporting Information Fig. 1D) and improved protection against Mtb compared to WT 129S2 controls (129S2 WT; Fig.1B and C). This was accompanied by milder tissue pathology, namely smaller inflammatory foci in the lung parenchyma, observed in Ifnar1−/− mice compared to that observed in 129S2 WT controls at 21 days p.i. (Fig.1D). As the disease progressed, these differences became more prominent (Supporting Information Fig. 1E). Large necrotic lesions developed in 129S2 WT mice by 25 days p.i., but not in Ifnar1−/− animals. The milder pathology in Ifnar1−/− animals was accompanied by a lower, albeit insignificantly reduced, serum lactate dehydrogenase concentration, a systemic indicator of tissue destruction at 21 days p.i. (Supporting Information Fig. 1F). Stronger iNOS expression in WT lung tissue (Fig.1E) suggests that improved control of Mtb replication in the absence of IFN I signaling was independent of local NO production, a well-known antimycobacterial effector molecule in mice. In a complementary approach, the TB-resistant C57BL/6 mice and their Ifnar1−/− counterparts were infected with a higher but still sublethal inoculum of Mtb (∼500 CFUs). Ifnar1−/− mice were protected from TB at day 40 p.i., as revealed by significantly reduced Mtb growth in lungs (Supporting Information Fig. 2A) and they controlled acute and chronic infection when exposed to low-dose Mtb (Supporting Information Fig. 2B). Together, our data demonstrate that IFNAR1 signaling is detrimental in murine TB, independent of the genetic background of the mice. Moreover, it appears that IFN I dictated susceptibility to TB in 129S2 mice.
Figure 1.

Signaling through IFNAR1 is detrimental in TB. (A) Survival of WT (129S2) and Ifnar1−/− mice (npooled = 18–21) after aerosol infection with virulent Mycobacterium tuberculosis (Mtb) H37Rv (∼200 CFUs), Kaplan–Meier curves. Data were pooled from three independent experiments. (B) Bacterial burdens in lungs of Mtb-infected WT (129S2) and Ifnar1−/− mice were determined at day 21 by colony counts and shown as median ± IQR of n = 25–29 pooled from five independent experiments (infection dose ∼200 CFUs). Mann–Whitney test for statistical analysis. (C) Acid-fast staining of Mtb in lungs of infected mice was performed (scale bar: 10 μm). (D) Histopathology of lungs with Giemsa staining of paraffin-embedded tissue (scale bar, 100 μm). (C and D) Images are representative of two independent experiments. Lung morphometry data, including the number of granulomas per lung section, area of diseased lung and granuloma size, were also determined by microscopy from these experiments (right, npooled = 9–10). (E) Immunostaining for iNOS (scale bar, 100 μm) was also performed on WT and Ifnar1−/− mice after Mtb infection. Images are representative of two independent experiments. The fluorescent signal (relative mean fluorescence intensity, MFI) was quantified from these experiments, for each granuloma observed in one tissue section per mouse (right, npooled = 6). (D and E) Data are shown as median ± IQR of the number of samples indicated. *p < 0.05, ****p < 0.0001.
The absence of wasting disease in Ifnar1−/− mice led us to investigate the inflammation status in the affected lungs. The phosphorylation status of IκB-α, a regulator of NF-κB activity, was markedly reduced in absence of IFNAR1 (Fig.2A). Lung concentrations of key inflammatory mediators, IL-1β, IL-1α, TNF-β, and IL-6, were significantly reduced in homogenates of Ifnar1−/− mice compared to WT mice, and were similar or even lower as compared to resistant C57BL/6 mice (Fig.2B). Chemokines, such as CXCL1, CCL2, and CCL4 showed a similar pattern (Fig.2B). We conclude that signaling through IFN I promotes secretion of key inflammatory cytokines, which accelerate progressive lung pathology.
Figure 2.
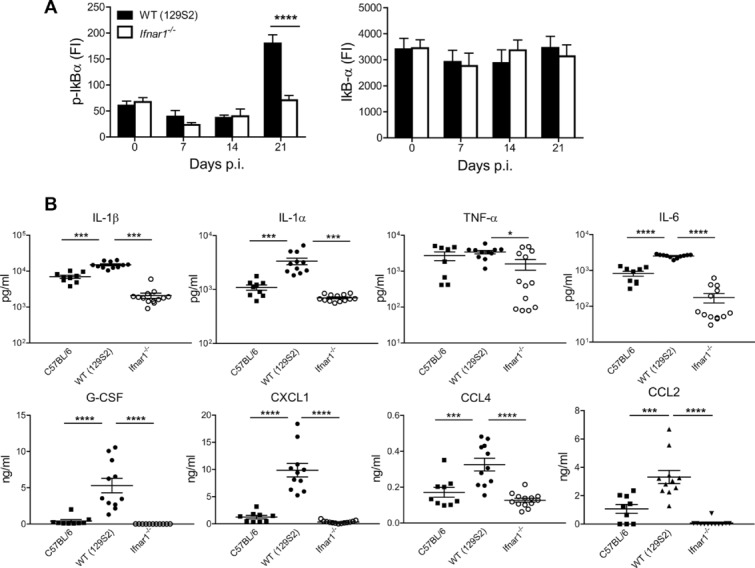
Type I IFN controls inflammation during TB. (A) IκB-α phosphorylation in lung tissue from WT (129S2) and Ifnar1−/− mice at various time points following aerosol infection with Mtb. Two-way ANOVA with Bonferroni post test. Data are shown as mean + SEM of n = 10–13 pooled from two independent experiments. (B) Cytokine and chemokine levels in lung homogenates collected at 21 days p.i. from WT (129S2), Ifnar1−/− and WT (C57BL/6) mice were determined by mutiplex immunoassay and shown as mean ± SEM of n = 9–13 pooled from two independent experiments. Two-way ANOVA with Bonferroni post test. *p < 0.05, ***p < 0.001, ****p < 0.0001.
IFN I signaling promotes pulmonary recruitment of inflammatory monocytes and neutrophils
IFN-γ produced by type 1 Th (Th1) cells is essential for TB control 17 and IFN I influences Th1 responses 5. To determine whether absence of IFNAR1 impacts on T-cell responses during TB, we isolated leukocyte populations from infected lungs and analyzed them by flow cytometry (see Supporting Information Fig. 3A for gating strategy). The frequencies and numbers of Mtb (purified protein derivative) specific IFN-γ-producing T-cell populations and Foxp3+ regulatory T lymphocytes were comparable between Ifnar1−/− and controls (Supporting Information Fig. 3A and B). These results argue against profound effects of IFN I signaling on T-cell responses in TB. Intact IFNAR1 signaling resulted in increased innate immune inflammation (Fig.2), but left adaptive immunity apparently unaffected (Supporting Information Fig. 3). Consequently, we analyzed the kinetics of innate myeloid cells in lungs. We enumerated various myeloid populations in lung tissue at 14 and 21 days p.i. using flow cytometry (see Supporting Information Fig. 4A for gating strategies). Significantly more inflammatory macrophages (iMs) and PMNs were counted in WT mice by day 21 p.i. (Fig.3A). This observation was verified by immunohistochemical analysis of myeloperoxidase (MPO)+ cells in the lung (Fig.3B). Intriguingly, Ifnar1−/− mice showed higher frequencies of AMs both at 14 and 21 days p.i. (Fig.3A). The frequency of AMs did not differ in naïve animals, and was only apparent following Mtb infection, ruling out developmental defects in AM differentiation (Supporting Information Fig. 4B). These results substantiate that IFNAR1 promotes the recruitment of inflammatory myeloid cells to the lung during Mtb infection and regulates resident AM populations, without apparent effects on T cells.
Figure 3.

Type I IFN augments PMN and iM recruitment into the lung. (A) WT (129S2) and Ifnar1−/− mice were infected with Mtb and innate pulmonary leukocyte populations were analyzed by flow cytometry at indicated time points. The numbers and frequencies of alveolar macrophages (AMs), lung macrophages (LMs), neutrophils (PMNs), and inflammatory macrophages (iMs) among lung leukocytes are shown as mean ± SEM of n = 8–10 pooled from two independent experiments, along with representative dot plots. Student's ttest for statistical analysis. (B) Immunostaining for MPO in lung tissue at 21 days p.i. (scale bar upper panel 100 μm, lower panel 10 μm). Images are representative of two independent experiments. The fluorescent signal MFI was quantified from these experiments, for each granuloma observed in one tissue section per mouse (right, npooled = 6). **p < 0.01, ***p < 0.001, ****p < 0.0001.
IFN I signaling in both hematopoietic and lung-resident cells controls TB inflammation
To define cell types affected by IFNAR1 deficiency, we created BM chimeras (WT→Ifnar1−/−, WT→WT, Ifnar1−/−→WT, Ifnar1−/−→ Ifnar1−/−) and infected them with Mtb. Transplantation of WT BM cells into Ifnar1−/− mice (WT→KO), and of Ifnar1−/− BM cells into WT mice (KO→WT), resulted in comparable TB susceptibility (Fig.4A). Mice from both groups succumbed between 28 and 150 days p.i., revealing comparable intermediate resistance and suggesting that IFN I signaling in both hematopoietic and radioresistant cells was important for TB control (Fig.4A). Proinflammatory cytokines IL-1β, IL-1α, and IL-6, as well as the chemokines CXCL1 and CXCL2, were most abundant in WT mice and their concentrations were decreased upon transfer of Ifnar1−/− BM cells (Fig.4B). A similar trend was indicated by MPO staining of the lung to visualize lung recruitment of PMNs and iMs (Fig.4C). Thus, IFNAR1 expression by both radiosensitive hematopoietic and radioresistant tissue cells was important for cytokine/chemokine release and leukocyte recruitment during acute TB.
Figure 4.
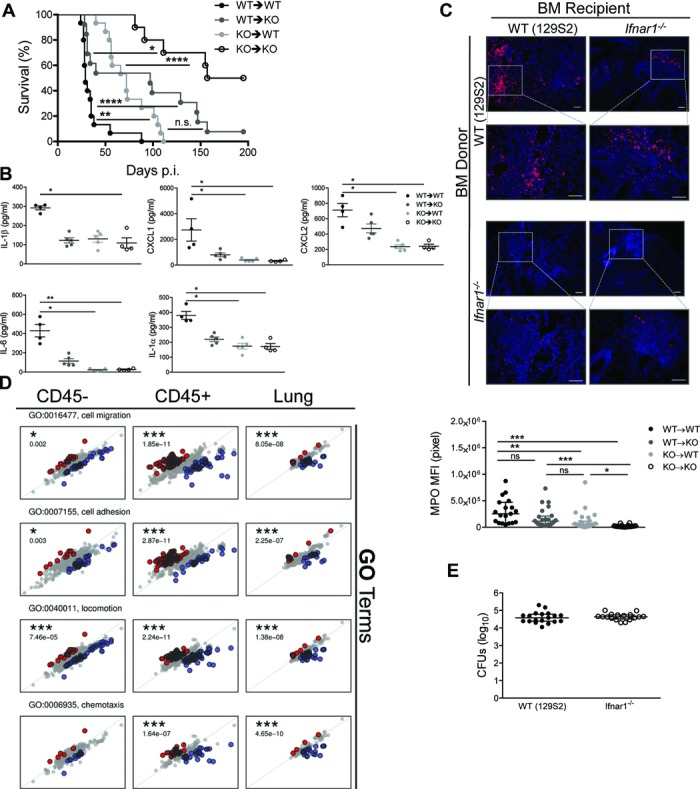
IFN I signaling in hematopoietic and tissue-resident cells modulates inflammation during TB. (A) Survival of chimeric mice (npooled = 10–15) after aerosol infection with Mtb (∼200 CFUs), Kaplan–Meier curves. Data shown are pooled from two independent experiments. (B) Cytokine and chemokine levels in lung homogenates collected at 21 days p.i. from chimeric mice were measured by multiplex immunoassay and shown as mean ± SEM of n = 4–5 from a single experiment. Two-way ANOVA with Bonferroni post test. (C) Immunostaining for MPO (scale bar, 100 μm). Images representative of one experiment performed with five samples, which was used to quantify fluorescent signals (MFI, bottom). Data are shown as median ± IQR of all granulomas detected per tissue section. One-way ANOVA with Dunn's post test. (D) Gene ontology (GO) enrichment analysis. Plots show log fold changes in gene expression between naive mice for the WT (horizontal axis) and the Ifnar1−/− strains (vertical axis) at day 14 p.i.; each point corresponds to one gene from the given GO category. Three plot columns correspond to the samples tested: RNA isolated from CD45− cells, CD45+ cells, and whole lung. Genes depicted in color show significant interactions between infection and mouse strain. Red denotes genes for which the log fold change is higher in the Ifnar1−/− than in WT, and blue marks genes for which the log fold change is higher in WT than in Ifnar1−/−. p-values correspond to the significance of GO enrichment. (E) Bacterial burdens in the lungs of infected WT (129S2) and Ifnar1−/− mice were determined on day 14 and shown as median ± IQR of n = 17–19 pooled from three independent experiments. *p < 0.05, **p < 0.01, ****p < 0.0001.
To elucidate how IFNAR1 modulates inflammation, we determined global gene expression profiles of total lung tissue as well as of selected hematopoietic and parenchyma cells during TB (Fig.4D). Lung cells were MACS-sorted based on the leukocyte marker CD45. At day 14, when lung CFU burdens were similar in infected groups (Fig.4E), we performed analysis of differentially expressed genes and used the web-based resource Gene Ontology Enrichment Analysis and Visualization tool (Gorilla) 18 for enrichment of defined gene ontology terms. Gene ontology categories related to chemotaxis, locomotion, migration and adhesion were highly enriched in total lung tissue as well as in both CD45+ cells (hematopoietic) and CD45− cells (parenchyma) of WT animals (Fig.4D). A compatible pathway enrichment was defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) database (Supporting Information Fig. 4C). Altogether, our results emphasize roles for both hematopoietic and radioresistant resident lung cells in IFNAR1-driven inflammation in TB.
IFNAR1 controls early CXC–chemokine release, in situ cell death and PMN recruitment to lung alveoli
To further investigate the effect of IFNAR1 on chemotaxis, locomotion and migration we evaluated the propensity of myeloid cells to secrete CXC and CC chemokines following exposure to Mtb. The absence of IFNAR1 in BM-derived macrophages (BMDMs) markedly affected production of CCL2 and CCL5, but left CXCL1, CXCL2, CCL3, and CCL4 unaffected (Fig.5A). The CXCL2 and CCL3 production by Mtb-infected PMNs did not differ (Fig.5B). We subsequently determined the abundance of chemokines in the bronchoalveolar fluid (BALF) early during infection. BALF from WT mice contained more CXCL1 and CXCL5 compared to Ifnar1−/− samples (Fig.5C), whereas CXCL2, CCL2, CCL3, and CCL5 were below the detection limit in both groups. Frequencies of PMNs were decreased in BALF of Ifnar1−/− mice (Fig.5D). In TB-resistant mice (C57BL/6), we did not observe early IFNAR1-dependent recruitment of PMNs into the alveoli, even when mice were infected with sublethal infection doses (Supporting Information Fig. 4D). CXCL5 is produced exclusively by the respiratory epithelium 19, while CXCL1 can also be produced by myeloid cells 20. The stimuli for inducing CXCL5/CXCL1 release by pneumocytes could be of bacterial or host origin. Concentrations of TNF-α and IL-1β, known regulators of CXC chemokine secretion by pneumocytes, in BALF were reduced and did not differ at 14 days p.i. (data not shown). However, we observed significantly higher cell death rates, in BALF of WT mice, exclusively at 14 days p.i. (Fig.5E; Supporting Information Fig. 4E). We subsequently determined the frequencies of annexinV+7AAD+ cells within BALF to identify the myeloid cell population(s) affected by cell death (see Supporting Information Fig. 5A for gating strategy). In agreement with the nucleosome measurements, we observed higher frequencies of dead leukocytes among WT cells (Fig.5F). These observations were recapitulated by AMs, the major myeloid population within airways at this time point of infection. Although higher numbers of PMNs were recruited to the bronchoalveolar space of WT mice, their death rates were below those observed for Ifnar1−/− cells (Fig.5E). Endogenous ligands liberated by dying cells are potent activators of CXC chemokines 21 and probably contributed to PMN recruitment in the presence of IFN I during TB. Thus, our data suggest that heightened local cell death concurrent with IFN I stimulation of nonhematopoietic cells induced PMN recruitment into the alveoli, promptly after bacterial invasion. At later time points, recruitment of iMs and PMNs was perpetuated by IFNAR1-dependent release of CCL2/CCL5 or CXCL2/CCL3 from macrophages or PMNs, respectively.
Figure 5.
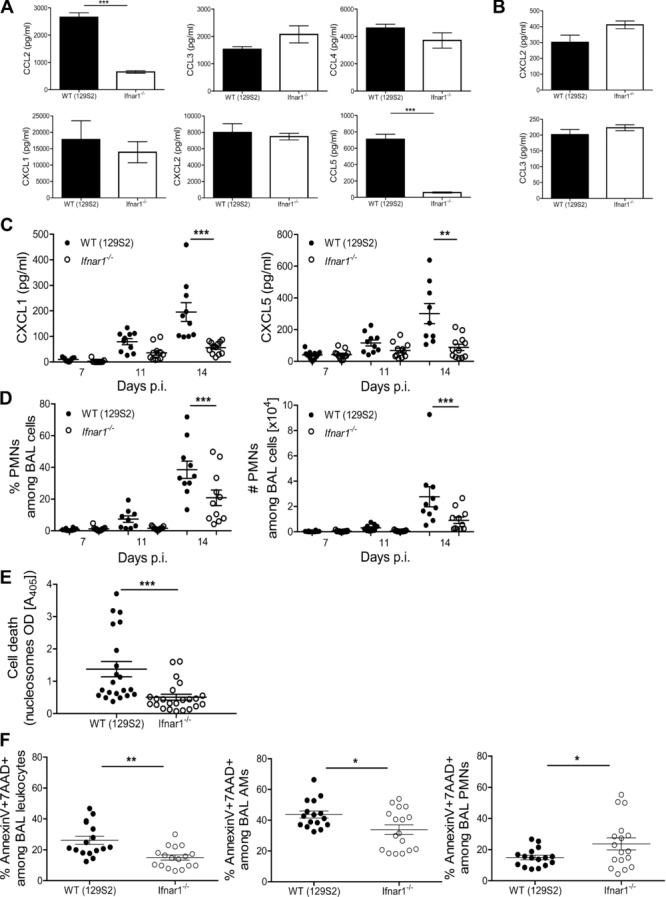
IFN I signaling modulates chemokine release, cell death, and early neutrophil dynamics in TB. (A) Cytokine levels in supernatants from BMDMs infected with Mtb at MOI 5 and collected at 24 h p.i. were measured by ELISA and shown as mean ± SEM of three replicates from a single experiment representative of three performed. Two-way ANOVA with Bonferroni post test. (B) Chemokine abundance in supernatants from PMNs infected with Mtb at MOI 10 and collected at 20 h p.i. was determined by ELISA and shown as mean ± SEM of four replicates from one experiment representative of three independent experiments performed. (C) The CXCL1 and CXCL5 concentrations in BALF from WT (129S2) and Ifnar1−/− mice were measured by ELISA at indicated time points p.i. Data are shown as mean ± SEM of n = 9–12 samples pooled from two independent experiments. Two-way ANOVA with Bonferroni post test. (D) PMN frequencies and numbers in BAL were measured by flow cytometry. Data are shown as mean ± SEM of n = 9–12 pooled from two independent experiments. (E) Levels of nucleosomes in BALF obtained at 14 days p.i. were determined by photometric enzyme immunoassay. Data are shown as mean ± SEM of 21–23 samples pooled from four independent experiments. (F) Frequencies of annexinV+7AAD+ leukocytes, AMs, and PMNs in BAL obtained at 14 days p.i. were measured by flow cytometry. Data are shown as mean ± SEM of n = 17 samples pooled from three independent experiments. (D–F) Student's t-test for analysis, *p < 0.05, **p < 0.01, ***p < 0.001.
IFNAR1 affects early Mtb compartmentalization among lung phagocytes
Reduced numbers of AMs due to higher death events in WT animals concomitant with more abundant chemokines and local PMN recruitment prompted us to investigate whether signaling via type I IFN offered a replication advantage for Mtb inside cells lining the alveoli. Despite similar bacterial loads in lung homogenates, we detected higher bacterial burdens in BAL cells collected from WT animals (Fig.6A). To identify myeloid cells that could serve as a replicative niche for Mtb in the presence of IFNAR1, we infected BMDMs, BM dendritic cells and ex vivo-isolated lung macrophages (LMs). In BM-derived resting and IFN-γ-activated cells, the growth kinetics of Mtb were similar between WT and Ifnar1−/− cells, while lung-purified macrophages revealed minor differences (Supporting Information Fig. 6A). In a subsequent approach, we investigated bacterial growth in AMs. Numbers of Mtb were elevated at 48 h p.i. in ex vivo-isolated WT AMs (Fig.6B), despite similar phagocytic capacities at 4 h p.i. (Supporting Information Fig. 6B). To further assess the preferential Mtb growth in presence of IFNAR1 in vivo, we infected mice with GFP-expressing Mtb H37Rv and determined by flow cytometry the frequencies and numbers of GFP+ myeloid cells within alveoli (see Supporting Information Fig. 5B for gating strategy). At 14 days p.i., overall bacterial loads in lung parenchyma were similar (Fig.4E), but proportions of AMs-containing Mtb were higher in WT mice (Fig.6C). Percentages of infected PMNs were similar between mouse strains, suggesting that the absence of IFNAR1 did not affect bacterial uptake by these cells. The propensity of isolated BMs and spleen PMNs to phagocytose mycobacteria in vitro was indeed similar in WT and Ifnar1−/− cells (Supporting Information Fig. 7). However, numbers of infected PMNs were significantly elevated in WT mice (Fig.6D). In a complementary approach, we enumerated acid-fast bacilli (AFB) in BAL cells. Congruent with FACS data, we detected more abundant bacteria in WT cells (Fig.6E), no striking differences in the numbers of AFB per phagocyte between mouse strains (Fig.6F) and a discrete compartmentalization of AFB in BAL phagocytes (Fig.6G). The higher numbers of Mtb-containing WT PMNs was a consequence of a combination of increased Mtb replication in WT AMs, increased in situ cell death and more abundant CXCR2 ligands at the site of bacterial invasion. We conclude that Mtb spreads more severely from AMs to lung-infiltrating myeloid phagocytes in WT mice and preadaptive immune events within alveoli are controlled by IFNAR1. The pathogenic role of PMNs recruited early upon infection was underlined by the profound consequences of PMN depletion on TB susceptibility. Mice receiving Abs against Ly6G for 1 week, starting day 7 p.i., survived in a similar manner as Ifnar1−/− mice (Fig.6H). We conclude that IFNAR1 impacts on Mtb replication in myeloid cells, induces distinct compartmentalization of Mtb among phagocytes in the early phase of infection, and fosters accumulation of PMNs with immunopathologic functions.
Figure 6.
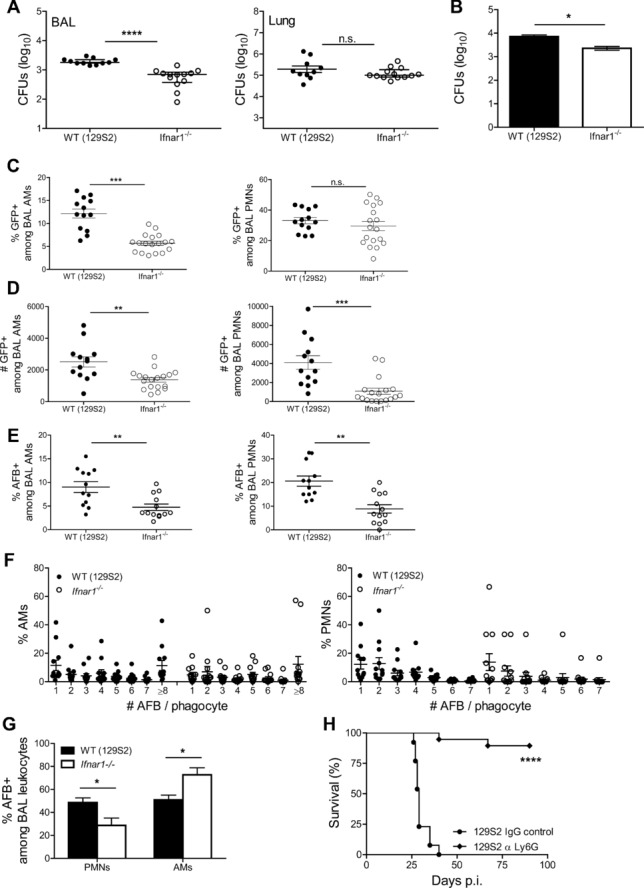
IFN I signaling drives Mtb spreading in lung phagocytes. (A) Bacterial burdens in BAL cells (left) and lung tissue devoid of BAL cells (right) of infected WT (129S2) and Ifnar1−/− mice at 14 days p.i. were measured by colony counts and are shown as median ± IQR of n = 11–13 samples pooled from two experiments. Mann–Whitney test for statistical analysis. (B) AMs isolated from the airways of WT and Ifnar1−/− mice were infected with Mtb at MOI 10 and bacterial replication was determined at 48 h p.i. by colony counts. Data are shown as mean ± SEM of four replicates from one experiment representative of two independent experiments performed, each with four to seven replicates. Mann–Whitney test for statistical analysis. (C) Frequencies and (D) numbers of AMs and PMNs positive for GFP-Mtb in BAL obtained from WT (129S2) and Ifnar1−/− mice at day 14 p.i. were determined by flow cytometry and shown as mean ± SEM of n = 13–18 samples pooled from two independent experiments. (E) Frequencies of AMs (left) and PMNs (right) positive for AFB in BAL obtained from WT (129S2) and Ifnar1−/− mice at day 14 p.i. were determined by microscopy. (F) Frequencies of BAL phagocytes with various numbers of AFB+ bacilli per cell were determined by microscopy. (G) Frequencies of AFB+ BAL cells were determined by microscopy. (E–G) Data are shown as mean ± SEM of n = 12–13 pooled from three independent experiments. (C–E) Student's t-test for analysis, (G) two-way ANOVA with Bonferroni post test. (H) Survival of WT (129S2) mice after aerosol infection with virulent Mtb H37Rv (infection dose ∼200 CFUs) and treatment with mAb against Ly6G or isotype IgG control, Kaplan–Meier curves. Data represent n = 13–19 mice pooled from two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
The role of IFN I in TB remains a matter of controversy. Here, we demonstrate that IFN I fosters lung inflammation in primary progressive TB, and establish a direct link between IFNAR1 signaling and disease immunopathology. These effects were most obvious in highly TB-susceptible 129S2 mice. Shortly after infection, IFN I promoted intracellular Mtb replication, diminished AM numbers, and caused a distinct compartmentalization of Mtb within myeloid cells. By controlling in situ cell death and release of proinflammatory cytokines and chemokines by lung parenchyma cells, IFNAR1 induced early PMN accumulation in alveoli and, later on, recruitment of both iMs and PMNs to the Mtb-infected lung. IFN I drove exacerbated tissue damage and promoted Mtb dissemination, thereby benefiting the pathogen.
To contest the role of IFN I in TB, we harnessed lethally TB-susceptible mice. The 129 mouse genotype family comprises various substrains 22 with varying TB susceptibility 23. For our investigations, we selected the substrain 129S2 (129SvPas), which has been used for generation of the Ifnar1−/− mice 24,25. Single-nucleotide polymorphism analysis confirmed the congenic status of the control strain (data not shown). IFN I was a critical component of the high TB susceptibility of the 129S2 mice because IFNAR1 deletion was essential and sufficient for conversion of a susceptible to a resistant phenotype. Deletion of IFNAR1 further heightened TB resistance in C57BL/6 mice, though less dramatically. Our data emphasize a crucial role of IFN I in hyperinflammation during TB and demonstrate an early detrimental effect of IFNAR1 in TB susceptibility. The 129S2 mice could serve as model for primary progressive TB pathogenesis in susceptible human hosts.
Exacerbated inflammation and lethality of Mtb-infected 129S2 mice was independent from IFN I effects on adaptive Th1 responses. Our finding agrees with previous observations that Poly I:C-induced IFN I worsens TB without affecting T cells 10. We also did not detect impaired CD8 T-cell functions, consistent with previous observations in aged Mtb-infected 129 mice 13. Our data exclude aberrant adaptive T-cell immunity, including Treg responses, as a cause of excessive inflammation in this model. This conclusion is also aligned with the marginal effects on T cells reported in Mtb-infected Ifnar1−/− mutants with resistant C57BL/6 background 16. Rather, we discovered that IFN I regulates myeloid cell recruitment into the Mtb-infected lung, with iM and PMN infiltrations accompanying TB disease progression. Treatment with Poly I:C, an IFNAR1-independent activator of IFN I production, has been shown to drive exclusive iM accumulation in the lung parenchyma in a CCL2/CCR2-dependent manner 10. In TB-resistant C57BL/6 mice, IFNAR1 regulates recruitment and survival of CD11c−CD11b+Gr-1lo/neg monocytes 16. It appears that exogenous stimuli for IFN I and Mtb infection trigger different pathways, which depend of host genotype. Profound variability has been observed in IFNAR1-driven tissue recruitment of myeloid cells in different diseases, implying that pathogen-specific signatures shape downstream innate immune responses. In postinfluenza streptococcal pneumonia 26 and polymicrobial sepsis 27, IFN I constrains access of PMNs to tissue sites through interference with leukotriene pathways and modulation of CXCL1/2 release. In contrast, infection with Chlamydia muridarum 28, Candida albicans 29, and prolonged sterile inflammation 30 cause IFN I-dependent influx of PMNs and iMs leading to exacerbated inflammation, similar to TB described here. Recruitment of inflammatory myeloid cells is orchestrated by CCL2, CCL7, CCL12, CXCL1, and CXCL2 under these conditions. IFNAR1-competent macrophages infected with Mtb released abundant CCL2 and deletion of IFNAR1 did not alter CXC chemokine release by myeloid cells in vitro. The early source of PMN chemoattractants in airways was lung-resident cells, as indicated by the abundance of CXCL5 in BALF. Propensity of pneumocytes to release CXCL5 is controlled by recognition of bacterial signatures in TB 31. Mtb sensing by pneumocytes is likely accelerated when AMs undergo cell death, as noticed for 129S2 WT mice. Later during Mtb infection, increasing numbers of PMNs secreting CXCL2 and CCL3 likely created a positive feedback, promoting additional IFNAR-dependent leukocyte recruitment to the lung. Thus, CXC chemokine-releasing tissue-resident cells along with hematopoietic cells, which release Mtb upon death, are the IFNAR1-dependent drivers of early leukocyte recruitment in the TB-susceptible hosts. Despite reduced bacterial loads at 6 weeks p.i., we did not observe early PMN accumulation in the airways of resistant WT (C57BL/6) mice compared to their Ifnar1−/− counterparts, as noticed for 129 mice. These results suggest that kinetics of the immune response differs between strains, and careful selection of the animal model is called for when addressing TB pathogenesis.
Intriguingly, despite similar overall lung bacterial burdens early during infection, we detected a unique Mtb compartmentalization in airway myeloid cells in TB susceptible mice, which was triggered by augmented Mtb replication concurrent with cell death of AMs. How exactly IFN I offers a replicative advantage to Mtb at the cellular level and what the precise cell death processes affected by IFN I are, remain to be elucidated. Interestingly, IFNAR1-driven necroptosis, a highly inflammatory cell death, was reported for the intracellular pathogen Salmonella enterica 32, and recently described in the zebrafish model of TB 33. In our TB-susceptible mouse model, higher frequencies of Mtb+ AMs and abundant cell death correlated with increased levels of CXC chemokines. The accelerated accumulation of IFNAR1-competent PMNs into the alveoli during early TB, synchronous with limited frequencies of annexinV+7AAD+ PMNs, suggests that IFN I drives recruitment of PMNs to the site of infection via chemokines, rather than causing heightened PMN cell death. Yet a role for cell death in limiting PMN numbers in absence of IFNAR1 cannot be excluded. Although IFN I did not alter the intrinsic phagocytic capacity of PMNs, we detected higher numbers of Mtb+ PMNs in BALF from WT mice early during infection suggesting that distinct compartmentalization was due to heightened Mtb availability at the time of accelerated PMN migration to the port of Mtb entry. This early surge in bacterial uptake was critical for TB disease outcome as determined by disease amelioration in TB-susceptible 129S2 mice following PMN depletion. The deleterious role of PMNs as disease dissemination elements in TB is supported by their capacity to release cytokines and chemokines, notably TNF-α 34, CCL3, and CXCL2 35, which impact on accumulation of Mtb-permissive phagocytes. In addition, PMNs could migrate and directly promote spread of mycobacteria 36.
Only a minor proportion of the 5–10% of Mtb-infected individuals will develop active TB during lifetime. In the vast majority, the pathogen is successfully controlled over long periods of time. We provide evidence that IFN I drives primary TB progression and propose that IFNAR1 profoundly impacts on the outcome of postprimary TB, with excessive IFN I secretion aggravating PMN-driven disease (re)activation. Our findings contribute to a better understanding of the biological relevance of IFN I signaling in lung immune and nonimmune cells during mycobacterial insult. Due to the severe inflammatory responses caused by aberrant IFN I signaling, we assume that Mtb takes advantage of IFN signaling pathways to intensify inflammation to its own benefit. This notion is supported by TB reactivation observed following antiviral IFN I therapy 37–39. The apparent beneficial effects of IFN I therapy in adjunct to conventional TB chemotherapy 40–43 may be due to resuscitation of dormant Mtb, as only actively replicating, not dormant Mtb is susceptible to TB drug treatment.
In conclusion, our data emphasize a central role of IFN I in pulmonary TB, with detrimental consequences of aberrant IFN I production for the host. IFN I effects emerge from both hematopoietic and tissue-resident cell populations suggesting a more productive crosstalk between these cell types than previously anticipated. The consequences of these events at the site of the infection are early mycobacterial dissemination and accumulation of harmful inflammatory PMNs. Increased understanding of IFN I-driven pathology can promote rational design of immunomodulatory interventions in TB.
Materials and methods
Mice and Mtb infection
Mice, 129S2 (129SvPas), Ifnar1−/− in 129S2 background 24, and Ifnar1−/− 44 in C57BL/6 background were bred and kept under specific pathogen-free conditions at the Max Planck Institute for Infection Biology in Berlin, Germany. C57BL/6 animals were purchased from Charles Rivers Laboratories. Mice were 10–12 weeks of age at the beginning of the experiments. Aerosol infection with Mtb strain H37Rv and enumeration of bacteria in lung tissue were performed as previously described 34. To generate GFP-expressing Mtb, competent H37Rv was transformed with the plasmid pGFM-11 by electroporation and selected on kanamycin 45. Chimeric mice were obtained by reconstituting lethally irradiated mice with BM cells. Mtb infection was performed 8–10 weeks following transplantation. PMNs were depleted by administration of 250 μg mAb Ly6G (clone 1A8, BioXCell) i.p. for 1 week at 2-day intervals, starting 7 days p.i. All experiments were performed in accordance with German Animal Protection Law, in Berlin, by the State Office of Health for Social Affairs in Berlin (Landesamt für Gesundheit and Soziales, LAGeSo; approvals G0310/06, G0221/11, T0223/11, and T0087/13).
Lung cell isolation and flow cytometry
Single-cell lung suspensions were prepared as previously described 34. Innate immune cells were classified based on surface expression of the following markers: AMs: CD11chiCD11b−, PMNs: Ly6GhiCD11bhi, LMs: CD11c−Ly6G−F4/80+CD11b+, iMs: CD11c−Ly6G−F4/80+CD11b+Ly6Chi. The following mAbs were used for these investigations: anti-Ly6G (clone 1A8; BD), anti-Ly6C (clone AL-21; BD), anti-CD11b (clone M1/70; BD), anti-F4/80 (clone BM8; ebiosciences), and anti-CD11c (clone HL3; ATCC).
For intracellular cytokine staining (ICS), 2–4 × 106 lung cells were cultured for 6 h in medium containing Brefeldin A, either alone or supplemented with purified protein derivative (SSI). Cells were phenotyped according to their CD4 and CD8 surface expression (anti-CD4, clone RM4–5; BD), (anti-CD8, clone 53–6.7, BD) and staining intracellularly with anti-IFN-γ (clone XMG1.2, BD) and anti-TNF-α (clone XT-22, ATCC) Abs. Stained cells were acquired using a BD FACS Canto II instrument. Data analysis was carried out using BD FACSDiva and FACS Analyzer software.
Histology and immunohistochemistry
Lung lobes were processed as previously described 34 and pathology was evaluated following Giemsa staining. Acid-fast (Ellis) staining was performed to visualize Mtb in situ 46 and Abs against MPO (DAKO) and iNOS (NeoMarkers) were used to identify PMNs and iNOS, respectively, in lung specimens. Lung morphometry and immunofluorescence analysis was performed on tissue obtained from two independent experiments with at least five animals per group. For lung lesion scoring, images were collected with the Leica DMLB microscope. Numbers of granulomas per tissue section were manually enumerated, granuloma size and area of damaged lung were quantified using the software ProgRes CT5 (Jenoptik). For immunofluorescence investigations, images were captured using the Leica DRMB microscope equipment with the ProgResC12 (Jenoptik) camera and quantification was performed using the Volocity software.
Bronchoalveolar lavage
Airways were instilled with PBS-containing protease inhibitors (Roche). The BAL fluid was used for cytokine/chemokine quantification. BAL cells were phenotyped using the same criteria as described above. For microscopy studies, BAL cells on glass coverslips were stained for Mtb (TB-Fluor, Merck) and nuclei (Hoechst, Invitrogen). A minimum of 400 cells were evaluated for each specimen.
Primary cell cultures and innate immunity assays
BMDMs and BM dendritic cells were obtained and stimulated as described 34. LMs were enriched from lung cell suspensions by adherence. AMs were obtained from BALF. PMNs were purified from BM and spleen with Ly6G magnetic beads (Miltenyi Biotech). Cells were infected with Mtb H37Rv as previously reported 34. Mtb replication in infected cells was estimated by [3H]uracil uptake or CFU counts. Abundance of chemokines and cytokines in cell-free supernatants was measured at 24 h p.i. using bead-based assays (Bio-Rad) and commercial ELISA (R&D). Phagocytosis of GFP-BCG by purified PMNs was estimated at 4 h p.i. by flow cytometry.
Multiplex cytokine and IκB-α phosphorylation assays in lung homogenates
Lung tissue was dispersed in PBS 0.05% v/v Tween 20 containing protease inhibitors (Roche) or BioPlex lysis buffer for phosphorylation studies. Measurements were performed using multiplex immunoassay kits (Bio-Rad). Samples were acquired on a Bio-Rad instrument.
RNA extraction from lung tissue and lung cells
Lung lobes were homogenized in Trizol (Invitrogen) and RNA extraction was carried out according to manufacturer's instructions. RNA amounts were estimated with a NanoDropND-100 spectrometer (NanoDrop Technologies) and RNA integrity was confirmed using the 2100 Bioanalyzer (Agilent Technologies). Selection of lung CD45+ cells was performed with magnetic beads (Miltenyi Biotech). CD45+ and CD45− cell fractions were processed for RNA extraction using Trizol reagent.
Gene expression array analysis
Dual-color hybridization microarrays were performed for gene expression experiments (Agilent Technologies). Data were analyzed using the Rosetta Resolver platform Build 7.2.2 SP1.31 (Rosetta Biosoftware). Microarray data were submitted to NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and can be accessed under GSE44848.
Microarray analysis
Limma version 3.12.1 47 was used. Normalization was done using normexp background correction, loess within-array normalization, and quantile between-array normalization. The differences in the reaction of the mouse strains used in response to infection were estimated by testing for significant interaction between infection and strain. Calculated p-values were corrected for multiple testing using Benjamini and Hochberg correction 48 and the resulting q-values were used throughout the subsequent analyses.
Functional analysis
To test for enrichment in sets of differentially expressed genes, we applied the Gorilla algorithm in a single-list mode sorted by absolute log fold change 18, as well as by comparing differentially expressed sets of genes defined by abs (log fold change) >0.5 and q < 0.005. Pathway analysis was done using the SPIA package for R 49, which allows testing both, the pathway enrichment and pathway perturbation.
Statistical analysis
PRISM Graphpad software was applied for statistical analyses. Bacterial titers were analyzed by the Mann–Whitney test. Concentrations of cytokines, chemokines, and phosphoproteins were compared using one-way or two-way ANOVA followed by Bonferroni's post test. Differences in lung cell populations were assessed using Student's t-test. p values < 0.05 were considered statistically significant.
Acknowledgments
The authors thank Mary Louise Grossman for help preparing, and Natalie Nieuwenhuizen for critically reading, the manuscript. We thank C. Köberle for developing software for analysis of flow cytometry data.
This work was supported by the European Union's Seventh Framework Programme (EU FP7) projects “NEWTBVAC” (Health-F3–2009–241745), “Phagosys” (Health-F3–2008–223451), and “TB-VIR” (Health-F3–2008–200973).
Glossary
- AFB
acid-fast bacilli
- AM
alveolar macrophage
- BALF
bronchoalveolar fluid
- BMDM
BM-derived macrophage
- IFNAR1
receptor for IFN I
- iMs
inflammatory macrophages
- LM
lung macrophage
- MPO
myeloperoxidase
- Mtb
Mycobacterium tuberculosis
- p.i.
postinfection
- TB
tuberculosis
Conflict of interest
The authors declare no financial or commercial conflict of interest.
References
- WHO. Global Tuberculosis Report 2013. Geneva: WHO Press; 2013. [Google Scholar]
- Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, Walzl G, et al. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun. 2011;12:15–22. doi: 10.1038/gene.2010.51. [DOI] [PubMed] [Google Scholar]
- Ottenhoff TH, Dass RH, Yang N, Zhang MM, Wong HE, Sahiratmadja E, Khor CC, et al. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS One. 2012;7:e45839. doi: 10.1371/journal.pone.0045839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Type I interferon: friend or foe? J. Exp. Med. 2010;207:2053–2063. doi: 10.1084/jem.20101664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE, III, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha /beta. Proc. Natl. Acad. Sci. USA. 2001;98:5752–5757. doi: 10.1073/pnas.091096998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, Barry C, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon. Cytokine Res. 2005;25:694–701. doi: 10.1089/jir.2005.25.694. [DOI] [PubMed] [Google Scholar]
- Antonelli LR, Gigliotti RA, Goncalves R, Roffe E, Cheever AW, Bafica A, Salazar AM, et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Invest. 2010;120:1674–1682. doi: 10.1172/JCI40817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AM, Pearl JE, Brooks JV, Ehlers S, Orme IM. Expression of the nitric oxide synthase 2 gene is not essential for early control of Mycobacterium tuberculosis in the murine lung. Infect. Immun. 2000;68:6879–6882. doi: 10.1128/iai.68.12.6879-6882.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 2007;178:3143–3152. doi: 10.4049/jimmunol.178.5.3143. [DOI] [PubMed] [Google Scholar]
- Turner J, Orme IM. The expression of early resistance to an infection with Mycobacterium tuberculosis by old mice is dependent on IFN type II (IFN-gamma) but not IFN type I. Mech. Ageing Dev. 2004;125:1–9. doi: 10.1016/j.mad.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. 2013;339:1448–1453. doi: 10.1126/science.1233665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. 2011;35:1023–1034. doi: 10.1016/j.immuni.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvignes L, Wolf AJ, Ernst JD. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J. Immunol. 2012;188:6205–6215. doi: 10.4049/jimmunol.1200255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JL, Chan J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyaseelan S, Manzer R, Young SK, Yamamoto M, Akira S, Mason RJ, Worthen GS. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am. J. Respir. Cell Mol. Biol. 2005;32:531–539. doi: 10.1165/rcmb.2005-0063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balamayooran G, Batra S, Fessler MB, Happel KI, Jeyaseelan S. Mechanisms of neutrophil accumulation in the lungs against bacteria. Am. J. Respir. Cell Mol. Biol. 2010;43:5–16. doi: 10.1165/rcmb.2009-0047TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Wei F, Tewary P, Howard OM, Oppenheim JJ. Alarmin-induced cell migration. Eur. J. Immunol. 2013;43:1412–1418. doi: 10.1002/eji.201243138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ. Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat. Genet. 1997;16:19–27. doi: 10.1038/ng0597-19. [DOI] [PubMed] [Google Scholar]
- Medina E, North RJ. Resistance ranking of some common inbred mouse strains to Mycobacterium tuberculosis and relationship to major histocompatibility complex haplotype and Nramp1 genotype. Immunology. 1998;93:270–274. doi: 10.1046/j.1365-2567.1998.00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, Diamond MS, et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005;6:722–729. doi: 10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 2009;119:1910–1920. doi: 10.1172/JCI35412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weighardt H, Kaiser-Moore S, Schlautkotter S, Rossmann-Bloeck T, Schleicher U, Bogdan C, Holzmann B. Type I IFN modulates host defense and late hyperinflammation in septic peritonitis. J. Immunol. 2006;177:5623–5630. doi: 10.4049/jimmunol.177.8.5623. [DOI] [PubMed] [Google Scholar]
- Qiu H, Fan Y, Joyee AG, Wang S, Han X, Bai H, Jiao L, et al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J. Immunol. 2008;181:2092–2102. doi: 10.4049/jimmunol.181.3.2092. [DOI] [PubMed] [Google Scholar]
- Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M, Muller M, et al. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog. 2012;8:e1002811. doi: 10.1371/journal.ppat.1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PY, Li Y, Kumagai Y, Xu Y, Weinstein JS, Kellner ES, Nacionales DC, et al. Type I interferon modulates monocyte recruitment and maturation in chronic inflammation. Am. J. Pathol. 2009;175:2023–2033. doi: 10.2353/ajpath.2009.090328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J, III, Fae KC, Arrey F, et al. CXCL5/LIX-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J. Clin. Invest. 2014;124:1268–1282. doi: 10.1172/JCI72030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 2012;13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153:521–534. doi: 10.1016/j.cell.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, Hanke K, et al. The adaptor molecule CARD9 is essential for tuberculosis control. J. Exp. Med. 2010;207:777–792. doi: 10.1084/jem.20090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorhoi A, Iannaccone M, Farinacci M, Fae KC, Schreiber J, Moura-Alves P, Nouailles G, et al. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J. Clin. Invest. 2013;123:4836–4848. doi: 10.1172/JCI67604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, Fiette L, et al. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. 2005;106:1843–1850. doi: 10.1182/blood-2005-03-1281. [DOI] [PubMed] [Google Scholar]
- Telesca C, Angelico M, Piccolo P, Nosotti L, Morrone A, Longhi C, Carbone M, et al. Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J. Infect. 2007;54:e223–e226. doi: 10.1016/j.jinf.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Babudieri S, Soddu A, Murino M, Molicotti P, Muredda AA, Madeddu G, Fois AG, et al. Tuberculosis screening before anti-hepatitis C virus therapy in prisons. Emerg. Infect. Dis. 2012;18:689–691. doi: 10.3201/eid1804.111016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkahla N, Kchir H, Maamouri N, Ouerghi H, Hariz FB, Chouaib S, Chaabouni H, et al. Reactivation of tuberculosis during dual therapy with pegylated interferon and ribavirin for chronic hepatitis C. Rev. Med. Interne. 2010;31:e1–e3. doi: 10.1016/j.revmed.2009.11.017. [DOI] [PubMed] [Google Scholar]
- Giosue S, Casarini M, Alemanno L, Galluccio G, Mattia P, Pedicelli G, Rebek L, et al. Effects of aerosolized interferon-alpha in patients with pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 1998;158:1156–1162. doi: 10.1164/ajrccm.158.4.9803065. [DOI] [PubMed] [Google Scholar]
- Palmero D, Eiguchi K, Rendo P, Castro ZL, Abbate E, Gonzalez Montaner LJ. Phase II trial of recombinant interferon-alpha2b in patients with advanced intractable multidrug-resistant pulmonary tuberculosis: long-term follow-up. Int. J. Tuberc. Lung Dis. 1999;3:214–218. [PubMed] [Google Scholar]
- Zhukova EM, Krasnov VA, Petrenko TI, Romanov VV. Interferon alpha-2b in the complex therapy of patients with pulmonary tuberculosis concurrent with bronchoobstructive syndrome. Probl. Tuberk. Bolezn. Legk. 2009;12:58–61. [PubMed] [Google Scholar]
- Tsai MC, Lin MC, Hung CH. Successful antiviral and antituberculosis treatment with pegylated interferon-alfa and ribavirin in a chronic hepatitis C patient with pulmonary tuberculosis. J. Formos. Med. Assoc. 2009;108:746–750. doi: 10.1016/S0929-6646(09)60400-9. [DOI] [PubMed] [Google Scholar]
- Fejer G, Drechsel L, Liese J, Schleicher U, Ruzsics Z, Imelli N, Greber UF, et al. Key role of splenic myeloid DCs in the IFN-alphabeta response to adenoviruses in vivo. PLoS Pathog. 2008;4:e1000208. doi: 10.1371/journal.ppat.1000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer L, Baulard A, Estaquier J, Poulain-Godefroy O, Locht C. Green fluorescent protein as a new expression marker in mycobacteria. Mol. Microbiol. 1995;17:913–922. doi: 10.1111/j.1365-2958.1995.mmi_17050913.x. [DOI] [PubMed] [Google Scholar]
- Dorhoi A, Nouailles G, Jorg S, Hagens K, Heinemann E, Pradl L, Oberbeck-Muller D, et al. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur. J. Immunol. 2012;42:374–384. doi: 10.1002/eji.201141548. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and computational biology solutions using R and bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. 1995;57:289–300. [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, Kim CJ, et al. A novel signaling pathway impact analysis. Bioinformatics. 2009;25:75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.