

Abstract
The NMR spectrum of a mixture of small molecules is a fingerprint of all of its components. Herein, we present an NMR fingerprint method that takes advantage of the fact that fractions contain simplified NMR profiles, with minimal signal overlap, to allow the identification of unique spectral patterns. The approach is exemplified in the identification of a novel natural product, iotrochotazine A (1), sourced from an Australian marine sponge Iotrochota sp. Compound 1 was used as a chemical probe in a phenotypic assay panel based on human olfactory neurosphere-derived cells (hONS) from idiopathic Parkinson’s disease patients. Compound 1 at 1 μm was not cytotoxic but specifically affected the morphology and cellular distribution of lysosomes and early endosomes.
Keywords: natural products, NMR spectroscopy, NMR fingerprints, Parkinson’s disease, total synthesis
Natural products, having undergone millions of years of evolutionary optimization, can be thought of as pre-validated starting points for exploring target interactions. The basis for the wide-ranging biological activity of natural products can be explained by the concept of protein fold topology, which describes the similarity of the biosynthetic imprint, the molecular recognition between substrate and biosynthetic enzyme, and the natural-product binding motif, the molecular recognition between a ligand and its therapeutic target.[1] Having been assembled by biosynthetic enzymes for the purpose of interacting with a biological target, natural products have been, and will continue to be, an important source of new drugs and drug leads.
We have previously reported a strategy to prepare a library of drug-like natural products.[2] This was achieved by analyzing all known compounds that comply with Lipinski’s “rule of five” in the Dictionary of Natural Products, and then targeting species known to contain those metabolites. This study was successful in producing a library in which 85 % of the isolated compounds were Lipinski-compliant and therefore drug-like, however the library was biased towards reported compounds, and contained only a limited number of novel structures (around 1 %). We therefore sought to develop a methodology to yield drug-like natural products while maintaining high diversity and novelty. Recently, we reported on the generation of lead-like enhanced (LLE) extracts and fractions with a method that allowed for the retention of lead- and drug-like constituents by selecting favorable physicochemical properties such as log P<5.[3] While this process achieves an ability to examine the complete diversity of the drug-like natural product metabolome, it does not intrinsically provide any structural information about the class of the small molecules comprising the fraction library. Herein, we detail a metabolic fingerprint procedure, based on the 1H NMR spectroscopic analysis of the LLE fraction library, and demonstrate the effectiveness of the NMR-guided approach towards revealing the unique components of the drug-like natural product metabolome. The applicability of the technique is exemplified through the identification of a novel natural product iotrochotazine A (1), its rapid total synthesis, and the interrogation of the biological activity of the compound using a cell model of Parkinson’s disease (PD).
 |
The NMR fingerprinting method was modelled to take advantage of the LLE library generation conditions,[3] where 300 mg of freeze-dried biota was extracted in organic solvents, selected for lead and drug-like characteristics on a solid phase extraction (SPE) cartridge, and then fractionated in 50 mg dry-weight equivalent aliquots on a reverse-phase HPLC column. The LLE fractions were then analyzed by high-field NMR spectroscopy. We found that with the analysis of fractions containing only a few small molecules each, rather than crude extracts containing hundreds of compounds, the complexity of spectra and signal overlap was kept to a minimum (see Supporting Information Figures S2 and S3). In contrast to the traditional NMR dereplication where pure or semi-purified compounds are analyzed for the presence of structural fragments that can be searched against a library of known natural products,[4] the NMR fingerprint approach presented herein identifies unique spectral patterns and aims to search for novelty against a library of fractions, rather than published natural products. Thus, the methodology enables a non-targeted interrogation of the drug-like natural product metabolome and has the potential to simplify and accelerate the current efforts in the isolation and identification of new natural products.
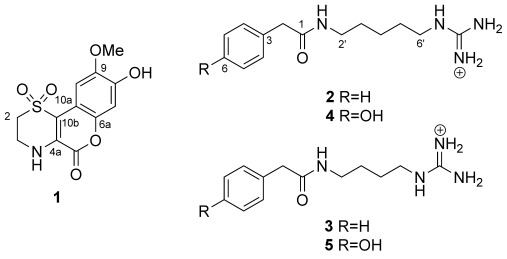
To exemplify our approach, a small subset of twenty sponges from the order Poecilosclerida sourced from the in-house Nature Bank biota repository were screened for the presence of unique 1H NMR spectral patterns. Figure 1 outlines the NMR-guided workflow; in a spectral library of 220 sponge-sourced fractions, five fractions showed a fingerprint that was identified as unique and not present in any other fractions. Resonances associated with this fingerprint were two multiplets at δH=3.55 and 3.80 ppm which were present in two fractions and multiplet resonance signals in the spectral region δH=1.10 to 1.50 ppm which were present in five fractions in the dataset. All five unique fractions were sourced from the same sponge extract, a Great Barrier Reef collection of the Iotrochota sp. sponge (for complete NMR fingerprint, see Supporting Information). The Iotrochota sp. sponge specimen was then subjected to NMR-guided large-scale natural-product isolation work to yield five new natural products, namely iotrochotazine A (1) and iotrochotadines A–D (2–5; for structural elucidation of iotrochotadines A–D, see Supporting Information). Compound 1, which contains a 1,1-dioxo-1,4-thiazine ring fused to a coumarin backbone, represents a novel structural class of marine alkaloid.
Figure 1.
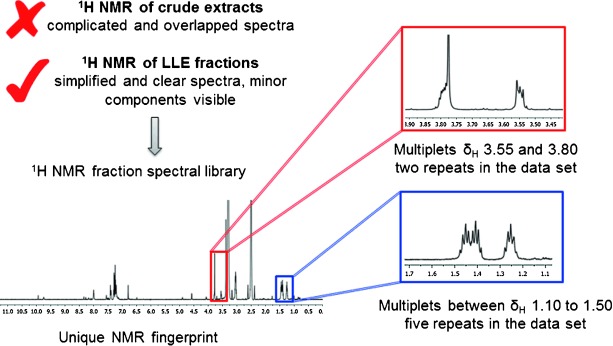
Summary of the 1H NMR-guided workflow. The 1H NMR fingerprints of the LLE fraction showed simplified, non-overlapping spectra which allowed the minor components of the metabolome to be detected. A dataset of 220 fractions was manually examined for the presence of unusual, non-repeating NMR resonance signals. Five fractions were identified as unique based on the spectral regions highlighted in red and blue rectangles.
Iotrochotazine A (1) was isolated as optically inactive yellow oil with a molecular formula of C12H11NO6S established by HR-ESI-FT-MS, which required ten double bond equivalents. The 1H NMR spectrum of 1 in [D6]DMSO revealed seven resonances: two exchangeable (δH=9.74 and 7.56 ppm) and five non-exchangeable signals composed of two sp2 singlets from methine protons (δH=7.42 and 6.79 ppm), two coupled sp3 multiplets from methylene protons (δH=3.80 and 3.55 ppm) and a singlet from a methoxy group (δH=3.77 ppm). The 13C NMR spectrum contained twelve signals, five of which were from protonated carbon atoms: two sp2 methine carbon atoms (δC=105.0 and 103.6 ppm), two sp3 methylene carbon atoms (δC=48.4 and 38.8 ppm) and a resonance from a methoxy group (δC=56.0 ppm), in addition to seven resonances from quaternary carbon atoms: four oxygen-bearing sp2 carbon atoms (δC=156.8, 145.9, 145.2, and 140.7 ppm) and three other sp2 carbon resonances (δC=129.9, 112.1, and 105.9 ppm). Interpretation of the 2D NMR spectroscopy data allowed for the identification of a 1,3-disubstituted ethyl amine spin system and a 1,2,4,5-tetrasubstituted benzene ring. The -NHCH2CH2- fragment was further elaborated into a 1,1-dioxo-1,4-thiazine ring upon comparison of the characteristic 1H and 13C NMR shifts of the two methylene resonances for C-2 and C-3 (δH=3.55, m, δC=48.4 and δH=3.80, m, δC=38.8 ppm, respectively), with similar natural products containing the same moiety.[5] 2D HMBC correlations placed the two singlet resonances for aromatic methine groups on the same ring in a para relationship to each other at C-7 (δH=6.79 ppm, δC=103.6 ppm) and C-10 (δH=7.42 ppm, δC=105.0 ppm), with the methoxy group at C-9 (δC=145.2 ppm), and a hydroxy substituent at C-8 (δC=145.9 ppm). The remaining atoms, according to the molecular formula and the double bond equivalents were assigned to a γ-lactone functionality. In an HMBC experiment optimized for 8.0 Hz, the singlet methine at C-10 showed two strong 3J correlations to C-10b (δC=112.1 ppm) and C-6a (δC=140.7 ppm), while H-7 showed a strong 3J correlation to C-10a (δC=105.9 ppm), establishing the benzopyran-2-one moiety in the molecule. Finally, on the 1,1-dioxo-1,4-thiazine ring, H-2 showed a correlation to C-10b, H-3 showed a strong 3J correlation to C-4a, and the exchangeable NH resonance showed a strong 3J correlation to the lactone carbonyl at C-5 (δ=156.8 ppm) as well as C-10b, securing the regiochemistry of the 1-dioxo-1,4-thiazine ring and completing the structure of 1.
Based on our previous work in the total synthesis of a dioxothiazine natural product,[6] we envisaged that 1 could be accessed by an oxidative condensation between hypotaurine and a 3-hydroxycoumarin (i.e. 6; Scheme 1), which could be produced from the Erlenmeyer condensation of the corresponding aldehyde (i.e. 7). In the event, 2,4-dihydroxy-5-methoxybenzaldehyde (7) was condensed with N-acetylglycine in the presence of sodium acetate and acetic anhydride to provide 3-acetamido-6-methoxy-7-acetoxycoumarin in moderate yield. Trituration of this material with ethanol was found to be crucial for the success of the subsequent hydrolysis (3 m HCl, AcOH) to yield 3,7-dihydroxy-6-methoxycoumarin (6). Upon treatment with hypotaurine under an atmosphere of oxygen, 6 underwent a one-pot enamine formation/intramolecular conjugate addition/oxidation sequence to produce iotrochotazine A (1). This reaction also proceeded under reflux in air, but was found to be more reproducible under an oxygen atmosphere.
Scheme 1.
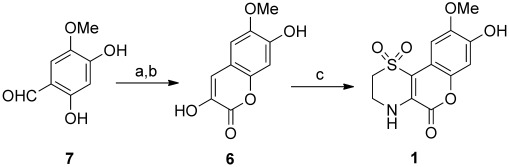
Synthesis of iotrochotazine A (1). a) N-acetylglycine, Ac2O, NaOAc, 140 °C, 3 h, 25 %; b) 3 m HCl, acetic acid, 130 °C, 16 h, 59 %; c) hypotaurine, O2, EtOH, CH3CN, H2O, 80 °C, 6 days, 70 %.
The biological activity of compound 1 was assessed in a multiparametric phenotypic assay on non-transformed human olfactory neurosphere-derived cells (hONS), which model functional aspects of Parkinson’s disease.[7] The hONS cells were cultured in 384-well plates to 1350 cells per well, and treated with 1 μm of compound 1 for 24 h. Phenotypic parameters were assessed by staining cells with fluorescent probes targeting various cellular organelles and compartments. These included the mitochondria, early endosomes, lysosomes, and cytoskeleton. Cellular processes such as autophagy and apoptosis were similarly assessed. In total, 38 phenotypic changes across the individual cell line were examined. Nuclear, cytoplasmic, mitochondrial, and autophagy markers were not altered indicating 1 μm iotrochotazine A is not cytotoxic (Figure S4 Supporting Information). However, a significant increase in the number of EEA1-associated early endosomes throughout the cytoplasm was evident with a corresponding decrease in signal intensity (Figure 2 B). A decrease in lysosomal staining was also detected (Figure 2 A) suggesting 1 may specifically effect vesicular trafficking in patient-derived hONS cells. When assayed on hONS cells sourced from two neurologically healthy individuals, iotrochotazine A up to a concentration of 10 μm did not show any significant effect on the lysosomal or endosomal markers. Given that mutations in the vesicular trafficking protein VPS35 can cause genetic PD[8] this natural product may be a useful tool to understand vesicular trafficking pathways in Parkinson’s disease.
Figure 2.
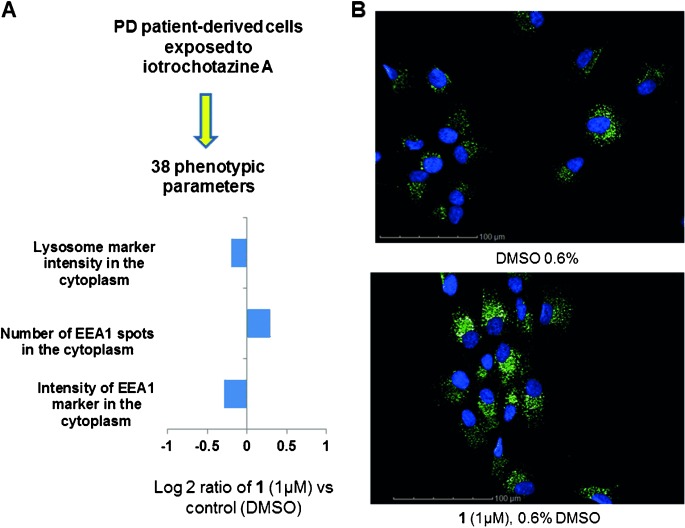
Immunofluorescence analysis of human olfactory neurosphere-derived cells from a Parkinson’s disease patient. A) In the presence of 1 μm of 1, high content image analysis revealed a decrease in the lysosomal staining, and an increase in the number of EEA1-associated early endosomes throughout the cytoplasm together with a decrease in signal intensity. The full description of all 38 cellular parameters and their response to the natural product 1 is given in Supporting Information Figure S4. B) Effect of 1 on the early endosome EEA-1 marker. Cell nuclei are stained with DAPI (blue), while the green stain depicts the anti-EEA1 antibody.
To validate our results as target-specific, compound 1 was assessed in a range of cytotoxic, anti-parasitic, and anti-microbial assays. Iotrochotazine A did not show any significant toxicity against transformed cells in the Developmental Therapeutics Programme’s 60-cell line screen, nor any inhibition of malarial, mycobacterial, Pseudomonas or MRSA growth. To investigate possible molecular pathways affected by compound 1 a panel of 48 kinases (ExpresS Diversity Kinase profile, Cerep), were assessed for inhibitory activity at a single concentration of 10 μm, with iotrochotazine A not showing any significant kinase inhibition.
In conclusion, we have developed a new method for the isolation and identification of novel, drug-like natural products through NMR-guided metabolic fingerprinting. Focusing on the fraction, rather than crude extracts, we were able to identify unique spectral patterns of the small molecules present and isolate four new natural products, namely iotrochotadines A–D (2–5), and a novel compound, iotrochotazine A (1). The total synthesis of iotrochotazine A through a one-pot enamine formation/intramolecular conjugate addition/oxidation sequence allowed for structural confirmation and biological investigations of this novel natural product. In a screen for biological function using a cell model of Parkinson’s disease,[7] iotrochotazine A had specific effects on the early endosome and lysosome markers, while not showing any cytotoxicity in a number of other mammalian, parasite, and bacterial cell-based assays. The natural product may be a useful tool to investigate the molecular mechanisms underlying Parkinson’s disease.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Kellenberger E, Hofmann A, Quinn RJ. Nat. Prod. Rep. 2011;28:1483–1492. doi: 10.1039/c1np00026h. [DOI] [PubMed] [Google Scholar]
- 2.Quinn RJ, Carroll AR, Pham NB, Baron P, Palframan ME, Suraweera L, Pierens GK, Muresan S. J. Nat. Prod. 2008;71:464–468. doi: 10.1021/np070526y. [DOI] [PubMed] [Google Scholar]
- 3.Camp D, Davis RA, Campitelli M, Ebdon J, Quinn RJ. J. Nat. Prod. 2012;75:72–81. doi: 10.1021/np200687v. [DOI] [PubMed] [Google Scholar]
- 4.Lang G, Mayhudin NA, Mitova MI, Sun L, van dSS, Blunt JW, Cole ALJ, Ellis G, Laatsch H, Munro MHG. J. Nat. Prod. 2008;71:1595–1599. doi: 10.1021/np8002222. [DOI] [PubMed] [Google Scholar]
- 5a.Morris BD, Prinsep MR. J. Org. Chem. 1998;63:9545–9547. [Google Scholar]
- 5b.Aiello A, Fattorusso E, Luciano P, Menna M, Esposito G, Iuvone T, Pala D. Eur. J. Org. Chem. 2003:898–900. [Google Scholar]
- 5c.Aiello A, Fattorusso E, Luciano P, Mangoni A, Menna M. Eur. J. Org. Chem. 2005:5024–5030. [Google Scholar]
- 5d.Pearce AN, Chia EW, Berridge MV, Clark GR, Harper JL, Larsen L, Maas EW, Page MJ, Perry NB, Webb VL, Copp BR. J. Nat. Prod. 2007;70:936–940. doi: 10.1021/np060626o. [DOI] [PubMed] [Google Scholar]
- 5e.Menna M, Aiello A, D’Aniello F, Imperatore C, Luciano P, Vitalone R, Irace C, Santamaria R. Eur. J. Org. Chem. 2013:3241–3246. [Google Scholar]
- 5f.Davis RA, Duffy S, Fletcher S, Avery VM, Quinn RJ. J. Org. Chem. 2013;78:9608–9613. doi: 10.1021/jo400988y. [DOI] [PubMed] [Google Scholar]
- 6.Pouwer RH, Deydier SM, Van LP, Schwartz BD, Franken NC, Davis RA, Coster MJ, Charman SA, Edstein MD, Skinner-Adams TS, Andrews KT, Jenkins ID, Quinn RJ. ACS Med. Chem. Lett. 2014;5:178–182. doi: 10.1021/ml400447v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a.Matigian N, Abrahamsen G, Sutharsan R, Cook AL, Vitale AM, Nouwens A, Bellette B, An J, Anderson M, Beckhouse AG, Bennebroek M, Cecil R, Chalk AM, Cochrane J, Fan Y, Feron F, McCurdy R, McGrath JJ, Murrell W, Perry C, Raju J, Ravishankar S, Silburn PA, Sutherland GT, Mahler S, Mellick GD, Wood SA, Sue CM, Wells CA, Mackay-Sim A. Dis. Models Mech. 2010;3:785–798. doi: 10.1242/dmm.005447. [DOI] [PubMed] [Google Scholar]
- 7b.Cook AL, Vitale AM, Ravishankar S, Matigian N, Sutherland GT, Shan J, Sutharsan R, Perry C, Silburn PA, Mellick GD, Whitelaw ML, Wells CA, Mackay-Sim A, Wood SA. PLoS One. 2011;6:e21907. doi: 10.1371/journal.pone.0021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S, Zhe Y, Wood SA, Mellick GD, Silburn PA, Collins BM, Bugarcic A, Teasdale RD. Traffic. 2014;15:230–244. doi: 10.1111/tra.12136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information