

Abstract
Two privileged drug scaffolds have been hybridized to create the novel heteromorphic nucleoside 5-(2-amino-3-cyano-5-oxo-5,6,7,8-tetrahydro-4H-chromen-4-yl)-1-(2-deoxypentofuranosyl)pyrimidine-2,4-(1H,3H)-dione (2). Compound 2 inhibited the replication of two orthopoxviruses, vaccinia virus (VV) (EC50 = 4.6 ± 2.0 μM), and cowpox virus (CV) (EC50 = 2.0 ± 0.3 μM). Compound 2 exhibited reduced activity against a thymidine kinase (TK) negative strain of CV, implying a requirement for 5′-monophosphorylation for antiorthopoxvirus activity. Compound 2 was efficiently phosphorylated by VV TK, establishing that VV TK is more promiscuous than previously believed.
Smallpox, although declared eradicated as a natural disease in 1983 by the World Health Organization, now stands as the most potentially devastating of all bioterrorist threats.1,2 It is presently the policy of the U.S. Government to provide two FDA-approved drugs for the treatment of smallpox and to have two others in the pipeline, ideally with different modes of action.3 One drug, cidofovir (Vistide), licensed to treat cytomegalovirus (CMV) retinitis in HIV-infected patients, is available through a special protocol (Investigational New Drug, IND) for emergency treatment of smallpox or vaccine reactions (http://www.bt.cdc.gov/agent/smallpox/vaccination/cidofovir.asp) if vaccinia immune globulin (VIG, in limited supply) is not effective.4,5 Progress has been made on development of oral dosage forms of cidofovir,6–10 but these are not yet available in the clinic. Some agents for the treatment of orthopoxvirus infections are in preclinical or clinical development. These include inhibitors of viral morphogenesis (TTP-6171)11 and viral release (ST-246)12 as well as cellular (i.e., Erb-1 kinase inhibitors, CI-1033)13,14 and tyrosine kinase inhibitors (Gleveec, STI-571).15 Nonetheless, there presently is no drug approved by the FDA to treat smallpox.
We have pursued a chemistry-driven strategy for the discovery of lead molecules with anti-orthopoxvirus activity.16,17 Our approach to new orthopoxvirus antivirals has been guided by the following considerations: (a) since the “privileged”18,19 structure of nucleosides has led to a variety of efficacious antiviral agents,20 the nucleoside scaffold is an excellent point of departure in the search for new antiviral drugs; (b) other privileged18,19 molecular scaffolds exist that have spawned a significant number of drugs and other biologically active agents, and these also can be used to discover molecular “masterkeys”;21 (c) 5-formyl-2′-deoxyuridine is a neglected but powerful synthon for the generation of novel nucleoside structures that can be employed in multicomponent reactions22–25 (MCR) to generate chemical diversity.
In this study, a modified benzofuran–nucleoside chimera was generated in a MCR originating with 5-formyl-2′-deoxyuridine.26–28 Benzofuran congeners form the nucleus of many biological active molecules.29–32 Singh et al.33 gained entry to these fused pyrans by reactions of 1,3-oxazinanes and oxazolidines with various carbon nucleophiles. We adapted this to the reaction of 5-formyl-2′-deoxyuridine with malononitrile and 1,3-cyclohexanedione to obtain a novel nucleoside. The synthesis was carried out using 5-formyl-2′-deoxyuridine26–28 in a multicomponent reaction with malononitrile and 1,3-cyclohexanedione in EtOH to give 5-(2-amino-3-cyano-5-oxo-5,6,7,8-tetrahydro-4H-chromen-4-yl)-1-(2-deoxypentofuranosyl)pyrimidine-2,4(1H,3H)-dione (2) (Scheme 1). Compound 2 was obtained as a 1:1 diastereomeric mixture arising from the generation of a chiral carbon at position 4 of the chromone ring.
Scheme 1.
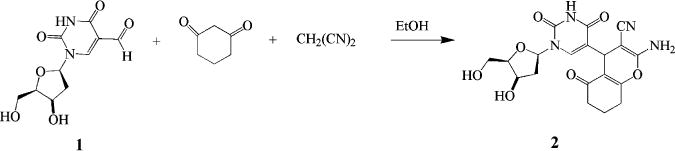
Synthesis of Compound 2
The antiviral activities of 2 (Table 1) were determined in human foreskin fibroblast cells, and the challenge orthopoxviruses were vaccinia virus (VV) or cowpox virus (CV). An initial evaluation was performed using the viral cytopathogenic effect as the endpoint. A second confirmatory assay involved plaque reduction. The concentration of agent that inhibited viral CPE or plaque formation by 50% was defined as the EC50. The effect of the potential antiviral agent on uninfected host cell viability was ascertained by Neutral Red uptake as a measure of cellular cytotoxicity. The concentration that reduced Neutral Red uptake by 50% was defined as the CC50. Compound 2 had no significant cytopathic effect on uninfected cells under these conditions (CC50 > 300 μM).
Table 1.
Antiorthopoxvirus Activitiesa
| compd | efficacy (EC50,c
μM)
|
toxicity (CC50,e
μM) Neutral Red uptake |
||||
|---|---|---|---|---|---|---|
| VVb CPE | VVb PR | CPVd TK+ lacZ | CPVb PR | CPVd TK− lacZ | ||
| cidofovir | 3.2 | 24 ± 12 | 3.3 ± 1.1 | 40 ± 6.1 | 5.2 ± 3.9 | >317 ± 0 |
| 2 | 0.6 | 4.6 ± 2.0 | 0.8 ± 0.1 | 2.0 ± 0.3 | 28 ± 2.7 | >300 ± 0 |
| 5-iodo-2′-deoxyuridine | 6.0 ± 0.2 | 0.4 ± 0.1 | 2.0 ± 0.2 | 27 ± 3 | >260a | |
Procedures adapted from Kern et al.34 Assays were performed according to the procedures described previously35–37 for activity against VV and CV and for cytotoxicity (Neutral Red uptake assay) in human foreskin fibroblast (HFF) cells. Briefly, to determine efficacy, initial cytopathogenic effect (CPE) assays were performed in 96-well plates seeded with HFF cells. Varying concentrations of drug were added to monolayers of HFF cells and challenged with VV or CV at 1000 PFU per well (incubation at 37 °C for 7 days). Confirmatory assays involving plaque reduction (PR) assays were performed using HFF cells seeded in six-well plates 2 days prior to use and infected with VV or CV by the addition of 20–30 PFU per well. Plates were incubated for 1 h. Various concentrations of drug were then added to triplicate wells, and plates were incubated at 37 °C for 3 days. Toxicity was evaluated using uninfected HFF cells seeded in 96-well plates incubated with various concentrations of drug for 7 days at 37 °C.
Virus used for challenge: VV (Copenhagen) or CV (Brighton).
Values are the mean (standard deviation of two or more assays.
CV strains δ crmA (TK+) and TK:GFP lacZ (TK−) were obtained from Pete Turner (University of Florida, Gainesville, FL) and were described previously.38 Values were obtained using a β-galactosidase assay to determine antiviral activity.
CC50: concentration that causes a cytotoxic effect (as ascertained by Neutral Red uptake) on 50% of uninfected cells.
Compound 2 was also evaluated against a thymidine kinase (TK) deficient strain (TK:GFP lacZ) of CV. CDV does not require phosphorylation to be active because it is a monophosphate analogue.4,5,39,40 Therefore, its activity is quite similar in TK+ and TK− virus strains. 5-Iodo-2′-deoxyuridine (idoxuridine) is known to be activated by the viral TK41 such that it is much less effective against TK− viruses.
The data of Table 1 clearly show that 2 is active only against the TK+ strain of CV, suggesting a specific 5′-monophosphorylation of this compound by the virus enzyme. That 2 indeed is a substrate for VV TK was confirmed by in vitro assays with recombinant VV TK. Under conditions wherein thymidine itself possessed a Km of 49 ± 7.6 μM and a Vmax of 289 ± 137 μmol min−1 mg−1, 2 was found to have a Km of 43 ± 1.4 μM and a Vmax of 77 ± 5 μmol min−1 mg−1. Thus, 2 is a good substrate and is efficiently phosphorylated by the enzyme.
These results have several important consequences for orthopoxvirus antiviral discovery and development. First, the requirement for the poxvirus TK for antiviral activity attests that 2 can be expected (as so far suggested by the cell culture studies of Table 1) to be of minimal toxicity to uninfected cells. Second, these foregoing data also imply that the orthopoxvirus TK (as embodied by the VV and CV genomes) may not exhibit the extremely limited substrate specificity characteristic of other type II highly discriminating TKs. VV TK originally was classified as a type II TK because of its substrate specificity, sequence homology to other type II kinases, and tetrameric configuration.42–46 To date, the only published recognized substrates for VV TK are thymidine, 2′-deoxyuridine, and 5-bromo-2′-deoxyuridine. The data reported here with 2 signify that, as for the herpes virus TKs, orthopoxvirus TKs are more promiscuous kinases than the cellular homologues, thereby providing fertile terrain for more diverse structure interrogation for candidate antiorthopoxvirus agents. Third, the unique structure of 2 suggests the possibility of a novel mode of action. Last, the recruitment of the versatile 5-formyl-2′-deoxyuridine and the adoption of the multicomponent reaction strategy provide access to an uncharted domain of structural diversity for exploration in antiviral drug discovery.
Supplementary Material
Acknowledgments
The authors acknowledge Contract US-AMRIID DAMD 17-03-C-0081 from the U.S. Army Medical Research Materiel Command and the State of Arizona Proposition 301 Funds for financial support, and Robert Smith and Shalisa Sanders for excellent technical assistance. The authors thank Dr. Ming Luo for the kind gift of VV TK. The in vitro evaluation for antiviral activity was supported by Public Health Service Contract No. NO1-AI-30049 (E.R.K.) from NIAID, NIH, Bethesda, MD.
Footnotes
Supporting Information Available: Synthesis, characterization, and HPLC purity of new compounds reported. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bray M. Viral Bioterrorism and Antiviral Countermeasures. In: Torrence PF, editor. Antiviral Drug Discovery for Emerging Diseases and Bioterrorism Threats. John Wiley & Sons; New York: 2005. p. 17. [Google Scholar]
- 2.Torrence PF. Introduction: Pestilence, Plague, Bioterrorism. In: Torrence PF, editor. Antiviral Drug Discovery for Emerging Diseases and Bioterrorism Threats. John Wiley & Sons; New York: 2005. p. 3. [Google Scholar]
- 3.Tseng CK. Overview of Antiviral Drug Discovery and Development. In: Torrence PF, editor. Antiviral Drug Discovery for Emerging Diseases and Bioterrorism Threats. John Wiley & Sons; New York: 2005. p. 31. [Google Scholar]
- 4.Kern ER. Discovery and Development of New Antivirals for Smallpox. In: Torrence PF, editor. Antiviral Drug Discovery for Emerging Diseases and Bioterrorism Threats. John Wiley & Sons; New York: 2005. p. 331. [Google Scholar]
- 5.De Clercq E. Antiviral Drug Targets and Strategies for Emerging Viral Disease and Bioterrorism Threats. In: Torrence PF, editor. Antiviral Drug Discovery for Emerging Diseases and Bioterrorism Threats. John Wiley & Sons; New York: 2005. p. 83. [Google Scholar]
- 6.Bradbury J. Orally available cidofovir derivative active against smallpox. Lancet. 2002;359(9311):1041. doi: 10.1016/S0140-6736(02)08115-1. [DOI] [PubMed] [Google Scholar]
- 7.Buller RM, Owens G, Schriewer J, Melman L, Beadle JR, Hostetler KY. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology. 2004;318(2):474. doi: 10.1016/j.virol.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 8.Ciesla SL, Trahan J, Wan WB, Beadle JR, Aldern KA, Painter GR, Hostetler KY. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Res. 2003;59(3):163. doi: 10.1016/s0166-3542(03)00110-4. [DOI] [PubMed] [Google Scholar]
- 9.Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY. Increased antiviral activity of 1-O-hexadecyloxypropyl-[2-(14)C]-cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol Pharmacol. 2003;63(3):678. doi: 10.1124/mol.63.3.678. [DOI] [PubMed] [Google Scholar]
- 10.Keith KA, Wan WB, Ciesla SL, Beadle JR, Hostetler KY, Kern ER. Inhibitory activity of alkoxyalkyl and alkyl esters of cidofovir and cyclic cidofovir against orthopoxvirus replication in vitro. Antimicrob Agents Chemother. 2004;48(5):1869. doi: 10.1128/AAC.48.5.1869-1871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byrd CM, Bolken TC, Mjalli AM, Arimilli MN, Andrews RC, Rothlein R, Andrea T, Rao M, Owens KL, Hruby DE. New class of orthopoxvirus antiviral drugs that block viral maturation. J Virol. 2004;78(22):12147. doi: 10.1128/JVI.78.22.12147-12156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RL, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus challenge. J Virol. 2005;79(20):13139. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H, Kim SK, Kim M, Reche PA, Morehead TJ, Damon IK, Welsh RM, Reinherz EL. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J Clin Invest. 2005;115(2):379. doi: 10.1172/JCI23220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fauci AS, Challberg MD. Host-based antipoxvirus therapeutic strategies: turning the tables. J Clin Invest. 2005;115(2):231. doi: 10.1172/JCI24270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reeves PM, Bommarius B, Lebeis S, McNulty S, Christensen J, Swimm A, Chahroudi A, Chavan R, Feinberg MB, Veach D, Bornmann W, Sherman M, Kalman D. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat Med. 2005;11(7):731. doi: 10.1038/nm1265. [DOI] [PubMed] [Google Scholar]
- 16.Fan X, Zhang X, Zhou L, Keith KA, Kern ER, Torrence PF. A Pyrimidine–pyrazolone nucleoside chimera with potent in vitro anti-orthopoxvirus activity. Bioorg Med Chem Lett. doi: 10.1016/j.bmcl.2006.03.043. in press. [DOI] [PubMed] [Google Scholar]
- 17.Fan X, Zhang X, Zhou L, Keith KA, Kern ER, Torrence PF. 5-(Dimethoxymethyl)-2′deoxyuridine: A novel gem diether nucleoside with anti-orthopoxvirus activity. J Med Chem. doi: 10.1021/jm0601710. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeSimone RW, Currie KS, Mitchell SA, Darrow JW, Pippin DA. Privileged structures: applications in drug discovery. Comb Chem High Throughput Screening. 2004;7(5):473. doi: 10.2174/1386207043328544. [DOI] [PubMed] [Google Scholar]
- 19.Horton DA, Bourne GT, Smythe ML. Exploring privileged structures: the combinatorial synthesis of cyclic peptides. Mol Diversity. 2002;5(4):289. doi: 10.1023/a:1021365402751. [DOI] [PubMed] [Google Scholar]
- 20.De Clercq E. Antivirals and antiviral strategies. Nat Rev Microbiol. 2004;2(9):704. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller G. Medicinal chemistry of target family-directed masterkeys. Drug Discovery Today. 2003;8(15):681. doi: 10.1016/s1359-6446(03)02781-8. [DOI] [PubMed] [Google Scholar]
- 22.Pulici M, Cervi G, Martina K, Quartieri F. Use of multicomponent, domino, and other one-pot syntheses on solid phase: powerful tools for the generation of libraries of diverse and complex compounds. Comb Chem High Throughput Screening. 2003;6(7):693. doi: 10.2174/138620703771981241. [DOI] [PubMed] [Google Scholar]
- 23.Dondoni A, Massi A. Decoration of dihydropyrimidine and dihydropyridine scaffolds with sugars via Biginelli and Hantzsch multicomponent reactions: an efficient entry to a collection of artificial nucleosides. Mol Diversity. 2003;6(3–4):261. doi: 10.1023/b:modi.0000006806.91483.a3. [DOI] [PubMed] [Google Scholar]
- 24.Shaabani A, Bazgir A, Bijanzadeh HR. A reexamination of Biginelli-like multicomponent condensation reaction: one-pot regioselective synthesis of spiro heterobicyclic rings. Mol Diversity. 2004;8(2):141. doi: 10.1023/b:modi.0000025613.35304.25. [DOI] [PubMed] [Google Scholar]
- 25.Bharadwaj AR, Scheidt KA. Catalytic multicomponent synthesis of highly substituted pyrroles utilizing a one-pot Sila-Stetter/Paal-Knorr strategy. Org Lett. 2004;6(14):2465. doi: 10.1021/ol049044t. [DOI] [PubMed] [Google Scholar]
- 26.Ono A, Okamoto T, Inada M, Nara H, Matsuda A. Nucleosides and nucleotides. 131. Synthesis and properties of oligonucleotides containing 5-formyl-2′-deoxyuridine. Chem Pharm Bull (Tokyo) 1994;42(11):2231. doi: 10.1248/cpb.42.2231. [DOI] [PubMed] [Google Scholar]
- 27.Park JS, Chang CT, Schmidt CL, Golander Y, De Clercq E, Descamps J, Mertes MP. Oxime and dithiolane derivatives of 5-formyl-2′-deoxyuridine and their 5′-phosphates: antiviral effects and thymidylate synthetase inhibition. J Med Chem. 1980;23(6):661. doi: 10.1021/jm00180a016. [DOI] [PubMed] [Google Scholar]
- 28.Kampf A, Pillar CJ, Woodford WJ, Mertes MP. Synthesis of 5-substituted 2′-deoxyuridines. J Med Chem. 1976;19(7):909. doi: 10.1021/jm00229a010. [DOI] [PubMed] [Google Scholar]
- 29.Selway JW. Antiviral activity of flavones and flavans. Prog Clin Biol Res. 1986;213:521. [PubMed] [Google Scholar]
- 30.Cheng HY, Lin CC, Lin TC. Antiviral properties of prodelphinidin B-2 3′-O-gallate from green tea leaf. Antiviral Chem Chemother. 2002;13(4):223. doi: 10.1177/095632020201300403. [DOI] [PubMed] [Google Scholar]
- 31.Bauer DJ, Selway JW, Batchelor JF, Tisdale M, Caldwell IC, Young DA. 4′,6-Dichloroflavan (BW683C), a new antirhinovirus compound. Nature. 1981;292(5821):369. doi: 10.1038/292369a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwashima M, Mori J, Ting X, Matsunaga T, Hayashi K, Shinoda D, Saito H, Sankawa U, Hayashi T. Antioxidant and antiviral activities of plastoquinones from the brown alga Sargassum micracanthum, and a new chromene derivative converted from the plastoquinones. Biol Pharm Bull. 2005;28(2):374. doi: 10.1248/bpb.28.374. [DOI] [PubMed] [Google Scholar]
- 33.Singh K, Singh J, Singh H. A synthetic entry into fused pyran derivatives through carbon transfer reactions of 1,3-oxazinanes and oxazolidines with carbon nucleophiles. Tetrahedron. 1996;52(45):14273. [Google Scholar]
- 34.Kern ER. In vitro activity of potential anti-poxvirus agents. Antiviral Res. 2003;57(1–2):35. doi: 10.1016/S0166-3542(02)00198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roy A, Schneller SW, Keith KA, Hartline CB, Kern ER. The 4′,4′-difluoro analog of 5′-noraristeromycin: A new structural prototype for possible antiviral drug development toward orthopoxvirus and cytomegalovirus. Bioorg Med Chem. 2005;13:4443. doi: 10.1016/j.bmc.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 36.Keith KA, Hitchcock MJ, Lee WA, Holy A, Kern ER. Evaluation of nucleoside phosphonates and their analogs and prodrugs for inhibition of orthopoxvirus replication. Antimicrob Agents Chemother. 2003;47(7):2193. doi: 10.1128/AAC.47.7.2193-2198.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pirrung MC, Pansare SV, Sarma KD, Keith KA, Kern ER. Combinatorial optimization of isatin-β-thiosemicarbazones as antipoxvirus agents. J Med Chem. 2005;48(8):3045. doi: 10.1021/jm049147h. [DOI] [PubMed] [Google Scholar]
- 38.Ali AN, Turner PC, Brooks MA, Moyer RW. The SPI-1 gene of rabbitpox virus determines host range and is required for hemorrhagic pock formation. Virology. 1994;202(1):305. doi: 10.1006/viro.1994.1347. [DOI] [PubMed] [Google Scholar]
- 39.De Clercq E. Cidofovir in the therapy and short-term prophylaxis of poxvirus infections. Trends Pharmacol Sci. 2002;23(10):456. doi: 10.1016/S0165-6147(02)02091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Clercq E. Cidofovir in the treatment of poxvirus infections. Antiviral Res. 2002;55(1):1. doi: 10.1016/S0166-3542(02)00008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Clercq E. Vaccinia virus inhibitors as a paradigm for the chemotherapy of poxvirus infections. Clin Microbiol Rev. 2001;14(2):382. doi: 10.1128/CMR.14.2.382-397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hruby DE, Ball LA. Cell-free synthesis of enzymatically active vaccinia virus thymidine kinase. Virology. 1981;113(2):594. doi: 10.1016/0042-6822(81)90187-2. [DOI] [PubMed] [Google Scholar]
- 43.Black ME, Hruby DE. Identification of the ATP-binding domain of vaccinia virus thymidine kinase. J Biol Chem. 1990;265(29):17584. [PubMed] [Google Scholar]
- 44.Black ME, Hruby DE. Quaternary structure of vaccinia virus thymidine kinase. Biochem Biophys Res Commun. 1990;169(3):1080. doi: 10.1016/0006-291X(90)92005-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Black ME, Hruby DE. Site-directed mutagenesis of a conserved domain in vaccinia virus thymidine kinase. Evidence for a potential role in magnesium binding. J Biol Chem. 1992;267(10):6801. [PubMed] [Google Scholar]
- 46.Black ME, Hruby DE. A single amino acid substitution abolishes feedback inhibition of vaccinia virus thymidine kinase. J Biol Chem. 1992;267(14):9743. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.