

Significance
Protein biosynthesis is most tightly controlled during translation initiation that involves numerous initiation factors and regulatory proteins. This complexity confounds conventional biochemical methods. Single-molecule approaches are ideally suited to address such questions. However, their application is hindered by the lack of fluorescently labeled components of the eukaryotic translation machinery. Here, we demonstrate an approach to label human 40S ribosomal subunits. As an extension of this approach, we used single-molecule fluorescence to demonstrate that 40S ribosomal subunits are recruited to the hepatitis C virus mRNA in a single-step process, and that components of a translational extract regulate the conformation of this complex.
Keywords: HCV IRES, translation initiation, human ribosomes, single-molecule FRET
Abstract
Translation initiation can occur by multiple pathways. To delineate these pathways by single-molecule methods, fluorescently labeled ribosomal subunits are required. Here, we labeled human 40S ribosomal subunits with a fluorescent SNAP-tag at ribosomal protein eS25 (RPS25). The resulting ribosomal subunits could be specifically labeled in living cells and in vitro. Using single-molecule Förster resonance energy transfer (FRET) between RPS25 and domain II of the hepatitis C virus (HCV) internal ribosome entry site (IRES), we measured the rates of 40S subunit arrival to the HCV IRES. Our data support a single-step model of HCV IRES recruitment to 40S subunits, irreversible on the initiation time scale. We furthermore demonstrated that after binding, the 40S:HCV IRES complex is conformationally dynamic, undergoing slow large-scale rearrangements. Addition of translation extracts suppresses these fluctuations, funneling the complex into a single conformation on the 80S assembly pathway. These findings show that 40S:HCV IRES complex formation is accompanied by dynamic conformational rearrangements that may be modulated by initiation factors.
Protein synthesis is a central process in health and disease (1, 2). The basic steps in translation have been mapped by genetic, biochemical, structural, and mechanistic studies. However, how translation is regulated and subverted, for example, during viral infection, remains poorly understood, especially in eukaryotes. All viruses compete for the cellular translation machinery to synthesize viral proteins required for virus proliferation. To that end, many viruses contain a structured internal ribosome entry site (IRES) in the 5′ untranslated region of their genome, which allows them to bypass the requirement for certain translation initiation factors. How IRESs achieve this goal remains unclear.
Recently, it has been shown that structurally and evolutionarily very diverse IRESs, such as hepatitis C virus (HCV) IRES, cricket paralysis virus (CrPV) IRES (3), and others (4), require ribosomal protein eS25 (RPS25) for efficient translation initiation, indicating that RPS25/IRES interactions could be a universal feature of IRES-mediated translation. RPS25 is located on the back of the head of the 40S ribosomal subunit, distal to the mRNA entry channel but proximal to different IRES RNAs as shown by cryo-EM structures of 80S:CrPV IRES and 80S:HCV IRES complexes. RPS25 is not essential for cap-dependent translation, suggesting that IRES/RPS25 interactions are required to bypass the requirement for the full set of initiation factors (3).
Translation initiation is a multistep process, with kinetics and dynamic interrogation refractory to conventional biochemical and biophysical methods. Single-molecule approaches provide insight into compositional and conformational dynamics of these asynchronous processes by following individual molecular events in real time. In bacteria, single-molecule methods allow direct observation of the multiple initiation pathways and conformational rearrangements that guide translation initiation (5, 6). The lack of fluorescently labeled components of translation initiation has prevented application of the single-molecule methods to study the mechanism of translation initiation in humans.
Here, we demonstrate an approach to create fluorescently labeled 40S ribosomal subunits from human cells. Using these subunits, we measure the kinetics and conformational pathway of 40S subunit recruitment to the HCV IRES. First, we created RPS25 KO cell lines using the clustered regularly interspaced short palindromic repeat (CRISPR)-Cas system. Next, we expressed RPS25 as a C-terminal fusion with mutant O6-alkylguanine DNA alkyltransferase (SNAP) as the sole source of the protein. We established biochemically that RPS25-SNAP is efficiently incorporated into functional 40S subunits, and rescues the defect in HCV IRES-mediated translation caused by RPS25 deletion. Using single-molecule fluorescence, we showed that Förster resonance energy transfer (FRET) between RPS25 and the base of domain II of the HCV IRES is dynamic. Conformational dynamics were altered by the presence of translational extract, thus indicating that conformational flexibility of RPS25 and domain II positions could be responsible for guiding downstream steps of translation initiation on HCV IRES. Finally, by measuring the kinetics of the 40S:HCV IRES interactions, we demonstrate that the rate of 40S subunit recruitment to the HCV IRES is not limited by the conformational flexibility of the free HCV IRES, and that upon 40S binding, the 40S:HCV IRES complexes are stable on the time scale of initiation. This study demonstrates the power of real-time observations of conformational and compositional dynamics during human translation initiation.
Results
To apply single-molecule approaches, fluorescently labeled ribosomal particles, translation factors, and tRNAs are required. Bacterial ribosomes have been fluorescently labeled by either protein- or nucleic acid-based attachment of dyes at specific locations. Mutants of ribosomal proteins containing single-Cys residues can be reacted with maleimide-conjugated dyes. Labeled and purified proteins are then incorporated into preassembled ribosomes lacking these specific proteins (7, 8). A second strategy exploits the introduction of metastable hairpins into surface-exposed loops of rRNA (9). Fluorescently labeled DNA oligonucleotides are then annealed to these hairpins, resulting in site-specific labeled ribosomal particles. This approach has been used successfully to generate fluorescently labeled 40S ribosomal subunits from Saccharomyces cerevisiae (10). However, these approaches are difficult to implement in mammalian ribosomes. Here, we pursued an alternative strategy by introducing a fluorescently labeled protein tag (SNAP-tag) into ribosomal proteins.
Generation of SNAP-Tagged RPS25.
Although most ribosomal proteins are essential, RPS25 is an exception. RPS25 is not essential in yeast, and siRNA or shRNA knockdown in mammalian cells does not disturb ribosome biogenesis and decreases cellular translation by only about 10% (3). Thus, we eliminated endogenous RPS25 using the CRISPR-Cas system (11) to create an RPS25 gene KO in the haploid cell line Hap1 (12). The guide RNA was designed to target the genomic sequence of exon 1, immediately upstream of the translation start site. By genotyping, two cell lines, termed CRISPR RPS25 KO #1 and #2, were isolated that had dissimilar deletions in the region around the AUG start codon (Fig. 1A). Immunoblotting confirmed that these two cell lines did not express endogenous RPS25 (Fig. 1B). To create RPS25-SNAP fusion protein-expressing cell lines, we used lentiviral transduction to introduce the RPS25-SNAP gene into the KO cells, creating KO + RPS25-SNAP cells. Expression of the 43-kDa RPS25-SNAP fusion protein was confirmed by immunoblotting with anti-RPS25 antibody. RPS25 abundances in the add-back cell lines corresponded to about 110% and 75% expression in Hap1 cells (Fig. 1B).
Fig. 1.

Expression of RPS25-SNAP in cell lines depleted of RPS25 using CRISPR-Cas9–mediated deletions in the RPS25 genomic sequence. (A) Genome sequences of the exonic and intronic sequences surrounding the ATG translation start codon (highlighted in red) from WT and CRISPR RPS25 KO cells. (B) Levels of RPS25 protein were undetectable by immunoblotting in two isolated RPS25 KO cell lines. Following lentivirus transduction with RPS25-SNAP, RPS25 was detectable by immunoblotting as a 43-kDa fusion protein. RPS6 and RPL13a were used as loading controls. MW, molecular weight. (C) RPS25 KO and add-back cell lines expressing RPS25-SNAP were lysed in the absence (lanes 1 and 4) or presence (lanes 2 and 5) of fluorescent SNAP649 dye. Cells were incubated in media containing 5 μM SNAP-fluorescein dye before lysis (lanes 3 and 6). Expression of RPS25-SNAP, RPS6, and RPL13a was detected by immunoblotting, and labeled RPS25 was detected by fluorescent scanning of the SDS/PAGE gel.
To detect RPS25-SNAP directly in living cells and in vitro, RPS25 KO and KO + RPS25-SNAP cells or cell lysates were treated with SNAP-fluorescein and SNAP-649 dyes. Total cell lysates were separated by SDS/PAGE, and gels were scanned for fluorescence. No fluorescence was detected in the RPS25 KO or in the KO RPS25-SNAP add-back cell lines in the absence of dye. However, RPS25-SNAP was detected in the presence of either dye (Fig. 1C) in KO + RPS25-SNAP lysates, indicating that RPS25-SNAP labeling is specific.
Intracellular Localization of RPS25-SNAP.
Expression of a tagged protein can cause it to misfold and lead to artifacts in its subcellular localization. Using confocal microscopy, we examined the cellular localization of RPS25-SNAP. RPS25-SNAP was visualized by adding a SNAP-fluorescein dye to live cells or by using an anti-SNAP antibody on fixed, permeabilized cells. Both RPS25-SNAP detection methods revealed a similar diffuse cytoplasmic staining pattern in KO + RPS25-SNAP cell lines, whereas no signal was detected in RPS25 KO cells (Figs. S1 and S2). Although this diffuse cytoplasmic pattern was also observed when stained for another ribosomal protein, RPS6, there is limited colocalization between RPS25-SNAP and RPS6. Lack of colocalization could be due to additional detection of free SNAP and minute amounts of RPS25-SNAP that are not incorporated into 40S ribosomal subunits (Fig. 2 and Fig. S3A).
Fig. 2.

SNAP-tag on RPS25 does not interfere with ribosome function. Cell lysates from KO #1 + RPS25-SNAP were treated with cycloheximide (Left) or puromycin (Right) and separated by sucrose gradient ultracentrifugation. Sucrose gradients were fractionated with the gradient fractions indicated on the x axis, and absorbance at 254 nm (black line) was continuously measured. Absorbance values are expressed as arbitrary units (AU). Proteins were precipitated from each gradient fraction and separated by 12% (wt/vol) SDS/PAGE. Gels were scanned for fluorescence, and fluorescence intensities were quantified and expressed as a percentage of the total fluorescence from all fractions (red line). RPS25-SNAP, RPS6, and RPL13a were detected by immunoblotting.
Incorporation of RPS25-SNAP into Actively Translating Ribosomes.
Cell lysates from KO + RPS25-SNAP cells were treated with an elongation inhibitor (cycloheximide or puromycin) and incubated with SNAP-649 dye. While cycloheximide prevents ribosome translocation (13, 14), the tRNA analog puromycin is incorporated into translating ribosomes and is used to separate the ribosomal subunits. Following separation of the cell lysates by sucrose gradient ultracentrifugation, proteins were separated by 12% (wt/vol) SDS/PAGE. The fluorescence scan of the gels revealed that RPS25-SNAP in cycloheximide-treated cell lysate mostly cosedimented with 40S ribosomal subunits and polysomes (Fig. 2 and Fig. S3A). In puromycin-treated cell lysates, RPS25-SNAP cosedimented with 40S ribosomal subunits. A similar sedimentation profile was observed when samples were immunoblotted for RPS6, another 40S subunit protein. RPL13a showed a slightly altered distribution, sedimenting in fractions 5–6 (corresponding to 60S and 80S) and the polysomes, which is consistent with its incorporation into the large subunit. We therefore concluded that the SNAP-tag does not interfere with incorporation of RPS25 into functional 40S and 80S ribosomal particles.
Requirement of RPS25-SNAP for Translation of the HCV IRES.
To examine if RPS25-SNAP recapitulates RPS25 function in IRES-mediated translation (3), we compared HCV IRES-mediated translation in RPS25-SNAP, RPS25-KO, and WT Hap1 cell lines using a dual-luciferase assay (15). To that end, a dual-luciferase plasmid containing the HCV IRES was electroporated into Hap1, RPS25 KO, and KO + RPS25-SNAP cell lines. RPS25 KO cells show an 80% decrease in IRES-mediated firefly expression compared with Hap1 cells (Fig. 3 and Fig. S3B). In the KO #1 + RPS25-SNAP cell line, where RPS25 is expressed above WT abundance (110%), HCV IRES-mediated translation was fully restored, whereas in KO #2 + RPS25-SNAP, where RPS25-SNAP abundance corresponds to 75% of WT, HCV-mediated translation was only partially rescued (Fig. 3 and Fig. S3B). Thus, IRES-mediated translation correlated with RPS25-SNAP abundance.
Fig. 3.

RPS25-SNAP restores HCV IRES-mediated translation in RPS25 KO cells. Hap1, RPS25 KO #1, and KO #1 + RPS25-SNAP cells were electroporated with a dual-luciferase construct containing the HCV IRES. Renilla and firefly luciferase were measured and plotted as the Renilla/firefly ratio on the y axis. The values from three independent experiments were averaged, and the error bars represent the SEM.
Dynamics of HCV IRES:40S Complex Formation.
We next used fluorescently labeled human 40S subunits to study the dynamics of translation initiation on the HCV IRES. The structure of the yeast ribosome showed that RPS25 sits at the head of the 40S ribosomal subunit, near RPS5 (16). A low-resolution cryo-EM structure, together with biochemical and cross-linking studies, locates RPS5 in close proximity to domain II of the HCV IRES (17, 18). Based on these results, we concluded that RPS25 and the base of domain II of HCV IRES could be within FRET distance (<70 Å). To test this hypothesis, we purified SNAP-547–labeled 40S ribosomal subunits from KO #1 + RPS25-SNAP cells. Ribosomes from this cell line were used in all subsequent experiments. HCV IRES RNA was transcribed by T7 RNA polymerase and dual-labeled with Cy5 and biotin by annealing cDNA oligonucleotides to unstructured regions upstream and downstream of the IRES, respectively (Fig. S4). As a result, Cy5 was placed at the base of the domain II and biotin was located 56 nt downstream of the start codon of HCV IRES. To observe single-molecule fluorescence of 40S:HCV IRES complexes, we formed complexes in bulk and immobilized them on a biotinylated glass surface via a neutravidin-biotin linker. The surface immobilization is dependent on biotin-neutravidin interactions, because preincubation with biotin or omission of the IRES or IRES biotinylation abrogated immobilization (Fig. S5). The brightness, lifetimes, and dwell behavior of SNAP-labeled ribosomal subunits were similar to or better than widely used cyanine dyes (Figs. S6 and S7).
The majority of immobilized 40S:HCV complexes showed FRET between the 40S ribosomal subunit and the HCV IRES. The percentage of molecules (>50%) showing FRET is consistent with the labeling efficiency of the HCV IRES, thus suggesting that FRET between 40S ribosomal subunits and HCV IRES exists in all cases when the 43S preinitiation complex was formed. The FRET intensity distribution is bimodal, with a major peak at 0.3 and a minor peak at 1.0 FRET efficiency (Fig. 4C). Interconversions between the two states were observed, thus demonstrating that the 40S:HCV complexes are dynamic and may adopt at least two conformations. The measured dwell times of the low and high FRET states were 75.7 ± 5.8 s (apparent rate klow→high = 0.013 s−1), and 36.5 ± 8.0 s (khigh→low = 0.027 s−1), respectively. The measured low FRET dwell time is the same as the photobleaching lifetime of SNAP dye (76.0 ± 8 s). Thus, the measurement reflects dye stability and puts an upper boundary on the low-to-high transition rate, where the conversion rate is much slower than the observed rate. In agreement, based on measured rates, the ratio of the low FRET to high FRET in the FRET intensity histogram is theoretically expected to be 1:2, rather than the observed 1:10. The slower low-to-high transition rate explains this discrepancy.
Fig. 4.
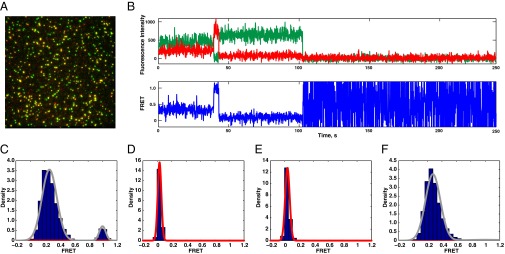
Conformational dynamics of 40S:HCV IRES complexes. The 40S:IRES complexes were formed in bulk, immobilized on the surface of the microscope slide, and illuminated with a 532-nm laser. (A) Composite image of 40S subunit (green) and HCV IRES (red) fluorescence channels collected at green illumination conditions. An area of approximately 1,000 μm2 is shown. (B) Example of a fluorescence trace. The anticorrelated behavior of the green and red signals is a signature of the FRET. The transition from low to high FRET occurs at a mark of ∼40 s. (C) 40S:HCV IRES FRET intensity histogram. Gaussian fits are shown in red, and the sum of individual fits is shown in gray (n = 146 molecules used to build distribution). (D) Control histogram of 40S subunit fluorescence bleed-through (n = 328). (E) Control histogram of 40S:HCV IRES complexes, where HCV IRES was labeled with scrambled Cy5 oligonucleotide (n = 128). (F) 40S:HCV IRES FRET intensity histogram in the presence of diluted translational extract (n = 111).
To examine the role of the 40S:HCV IRES complex conformational dynamics in IRES-mediated translation initiation, we repeated FRET measurements in the presence of translationally active cell lysate that contains all initiation factors. In the presence of lysate, the FRET distribution became unimodal and the high FRET state disappeared, indicating that low-to-high FRET transitions were suppressed by lysate components (Fig. 4F). A similar effect was observed when Mg2+ concentration was decreased twofold to 2.5 mM (Fig. S8).
Real-Time Formation of 40S:IRES Complexes.
The mechanism of 40S subunit recruitment to HCV IRES remains uncharacterized. HCV IRES recruitment may be a single binding step or a multistep process that involves conformational repositioning of IRES domains II and III concurrent with pseudoknot unwinding in domain IV. To determine pathways of 40S:HCV IRES complex formation, we observed 40S subunit binding to the HCV IRES in real time. Cy3-labeled and biotinylated HCV IRES was surface-immobilized, and 40S subunits labeled with SNAP-649 (Cy5-like) dye were delivered by rapid mixing. The 40S subunit arrival times were double exponential, with a main fast phase (80% of all arrival events) and a secondary slow phase that accounts for 20% of binding events (Fig. 5B); the slow phase was characterized by an association rate 10- to 20-fold slower than the fast phase. To improve fit robustness, the slow phase was approximated by linear regression, and only the fast phase will be referred to in the subsequent discussion. The rate of the fast phase was linearly dependent on 40S subunit concentration (20–80 nM), with a bimolecular rate constant of 1.1 μM−1⋅s−1. The linear dependence of association rates implies the lack of additional rate-limiting steps in the fast phase of HCV IRES recruitment. In agreement with static FRET measurements (Fig. S6), the lifetimes for 40S-IRES FRET were single exponentials and limited by photobleaching (Fig. 5C). Consistently, 40S-IRES FRET lifetimes were concentration-independent (Fig. 5E). Our results support a simple single-step mechanism for initial IRES binding to 40S subunits, without metastable intermediates.
Fig. 5.
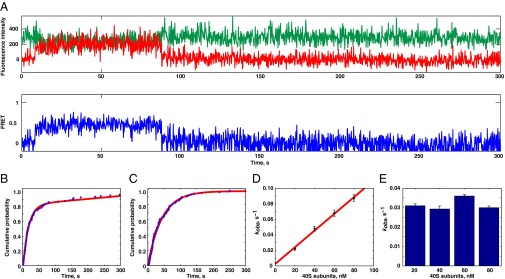
Kinetics of the 40S subunit recruitment to HCV IRES. (A) Example of a trace. Binding of the 40S subunit was detected by the appearance of FRET. (B) Representative 40S subunit arrival times distribution at 40 nM. Non-single exponential behavior is apparent. (C) Representative lifetime distribution at 40 nM 40S subunit. (D) Arrival rates were linearly dependent on the small subunit concentrations (E), whereas residency times were concentration-independent. kobs, measured reaction rate. Error bars represent error of the fit. The numbers of molecules used to obtain these fits are 75, 148, 162, and 157, correspondingly.
Discussion
Whereas single-molecule approaches have been used to examine pathways of translation initiation and elongation in bacteria (6, 19), translation dynamics in eukaryotes are still mostly unknown. Because translation in eukaryotes is more complex and involves numerous additional protein factors, it is essential to use single-molecule approaches to discriminate multiple parallel and asynchronous pathways. The main obstacle in the application of single-molecule approaches is the lack of fluorescently labeled reagents. Here, we demonstrated a method for site-specific fluorescent labeling of the 40S ribosomal subunits from human cells. To create an efficient labeling system, the endogenous RPS25 gene locus was disrupted using the CRISPR-Cas system and RPS25 was expressed as a SNAP fusion protein. The SNAP-tag allowed us to label RPS25 fluorescently in living cells and in vitro. We showed that the SNAP-tag does not interfere with incorporation of RPS25-SNAP into translating ribosomes, RPS25 localization, or RPS25 function in HCV IRES-mediated translation. SNAP-labeled 40S ribosomal subunits were suitable for single-molecule fluorescence measurements and have dye behavior similar to the cyanine dyes. This approach could be adopted to label other multisubunit components of translation, such as 60S, eukaryotic initiation factor 2 (eIF2), and eIF3.
Translation initiation is a kinetically controlled process. Many initiation factors with nanomolar dissociation constants are dynamic, rapidly cycling through bound and free states (20, 21), where progression into the downstream steps is determined by the forward and reverse rates of initial and subsequent reactions. Formation of the 40S:HCV IRES complex has previously been characterized only in terms of equilibrium constants. Here, we measured the rates of HCV IRES binding to 40S ribosomal subunits with single-molecule precision. Association rates were biphasic, with a minor slow phase. The presence of the second phase suggests that there was functional heterogeneity in HCV IRES populations. However, because the second phase is minor and significantly slower, it is unlikely to represent the biologically relevant pathway of 40S subunit recruitment. The apparent association rates of the fast phase were linearly dependent on 40S subunit concentration, indicating the absence of additional rate-limiting steps in the main pathway of 40S:IRES complex formation.
The previously measured equilibrium dissociation constant (KD = 2 nM) allowed us to estimate a 40S:HCV IRES complex dissociation rate, assuming a simple single-step mechanism of ∼0.002 s−1 (lifetime of 500 s). Hence, on the time scale of translation initiation (∼60 s) (22, 23), 40S subunit dissociation is insignificant and 40S subunit recruitment to HCV IRES interactions can be considered irreversible; thus, the efficiency of HCV IRES recruitment is solely determined by 40S subunit arrival rates.
Upon formation, the 40S:HCV IRES complex remains flexible. The observed changes in 40S:HCV IRES FRET efficiency correspond to large-scale rearrangements (∼30 Å) that occur on a slow time scale. There are several possibilities that might explain the observed changes in FRET levels. The HCV IRES is conformationally flexible (24), with individual domains moving relative to each other. The proposed model of HCV binding is based upon IRES flexibility and involves repositioning of domain II between solution and ribosome-bound molecules. Once bound to the 40S subunit and initiation factors, domain II plays a role in tRNA recruitment and the transition to elongation (25, 26). Here, we showed that the relative RPS25 and HCV IRES positions are dynamic, possibly reflecting the innate flexibility of the domain II position. Suppression of these conformational fluctuations in the presence of translation extracts shows that lysate components direct 40S:HCV IRES complexes into a single conformation that might be geared toward the forward steps of initiation. It is tempting to hypothesize that initiation factors are responsible for the observed effect. However, we cannot fully exclude the possibility that the observed FRET fluctuations are at least partially attributable to the SNAP-tag repositioning on the surface of the ribosome, due to the large size of the SNAP-tag used to label the 40S subunit fluorescently. In this case, binding of lysate components (e.g., initiation factors close to the RPS25) might restrict conformational flexibility of RPS25-SNAP fusion. Lysate components might thus suppress high FRET states either by directly interacting with RPS25, restricting mobility of the SNAP tag, or by remodeling the relative position of RPS25:HCV IRES complex. Studies with 40S complexes that harbor fluorescent tags at different ribosomal proteins will distinguish between these possibilities.
By single-molecule FRET between RPS25 and HCV IRES, we directly observed dynamics of 40S:HCV IRES complexes and described the kinetic pathway of 40S subunit recruitment to HCV IRES. This study creates a framework for future experiments that will reveal the dynamic pathways of human canonical and IRES-mediated translation initiation.
Materials and Methods
Cell Culture.
HeLa cells were grown in Dulbecco’s modified Eagle’s medium (Gibco, Life Technologies) supplemented with 10% (vol/vol) FBS, 2 mM l-glutamine (Gibco, Life Technologies) and 1× penicillin/streptomycin (Gibco, Life Technologies). RPS25 cDNA encoding the ORF was reverse-transcribed from total HeLa RNA using GCGCCGGATATCTCATGCATCTTCACCAGCAGC, amplified using this primer and GGCGCGAAGCTTATGCCGCCTAAGGACGACAAGAAG, and inserted into the HindIII and EcoRV sites of the p3xFLAG-CMV-7.1 Expression Vector (Sigma). Subsequently, the RPS25 sequence was amplified out of this vector using GGCGCGGCTAGCACCATGGACTACAAAGACCATGACGG and CGCGATATCTGCATCTTCACCAGCAGCTGG primers and inserted into the NheI and EcoRV sites of the pSNAPf vector to create the RPS25-SNAP fusion protein (New England Biolabs).
An RPS25 CRISPR sequence targeting the first exon of the RPS25 mRNA just upstream of the ATG start codon was designed as previously described (11). An RPS25 CRISPR Geneblock, TGTACAAAAAAGCAGGCTTTAAAGGAACCAATTCAGTCGACTGGATCCGGTACCAAGGTCGGGCAGGAAGAGGGCCTATTTCCCATGATTCCTTCATATTTGCATATACGATACAAGGCTGTTAGAGAGATAATTAGAATTAATTTGACTGTAAACACAAAGATATTAGTACAAAATACGTGACGTAGAAAGTAATAATTTCTTGGGTAGTTTGCAGTTTTAAAATTATGTTTTAAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGTATTCTCCGAGCTTCGCAAGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTTCTAGACCCAGCTTTCTTGTACAAAGTTGGCATTA, in which the RPS25 CRISPR target sequence (bold and underlined) was preceded by the U6 promoter sequence and followed by the guide RNA sequence (underlined) and the U6 termination signal (bold), was ordered from Integrated DNA Technologies, and cloned into the pCR-Blunt II-TOPO vector (Life Technologies). The RPS25 CRISPR vector was cotransfected with hCas9-expressing vector (Addgene 41815 hCas9 Church pcDNA3.3-Topo) into Hap1 cells grown in Iscove’s modified Dulbecco’s medium (IMDM; HyClone) supplemented with 10% (vol/vol) FBS, 2 mM l-glutamine, and 1× penicillin/streptomycin. Forty-eight hours after transfection, cells were subcloned and clonal cell lines were screened using the PCR primers AAGTTAGCTGCCGAGACCTG and ATAACACAGCAGGCACAGCGGC. Potential CRISPR KO cell lines were then genotyped for editing at the first exon of RPS25 using nested primers with the sequences GACACTTCGCACAACAGACC and ACAGCGCTATAGTATGCGTC.
Using the primers CACCATGCCGCCTAAGGACGA and TCATTAATTAACCTCGAGTTTAAACG, the RPS25-SNAP gene was amplified out of the pSNAPf vector and inserted into pENTR/d-TOPO (Life Technologies). Using Gateway cloning and LR Clonase II (Life Technologies), the RPS25-SNAP sequence was inserted into the pLenti CMV Puro DEST vector (w118-1) (27). The CRISPR RPS25-KO cell lines were lentivirally transduced with the RPS25-SNAP lentiviral vector. Transduced cells were selected with 1 μg/mL puromycin (InvivoGen).
Immunoblotting and Sucrose Gradient Analysis.
Following washing and harvesting of cells in cold PBS, total cell lysates were prepared by lysing cells in lysis buffer [150 mM NaCl, 1 mM EDTA, 100 mM Tris⋅HCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS] containing EDTA-free complete protease inhibitor mixture (Roche). After incubating cells on ice, debris was pelleted by centrifugation and protein concentration of the supernatant was determined by Bradford assay. Total cell lysate was separated by 12% (wt/vol) SDS/PAGE. Following the transfer onto an Invitrolon PVDF membrane (Invitrogen), standard immunoblotting techniques were used. Membranes were blocked in 5% (wt/vol) milk-PBS containing 0.1% Tween 20 (PBS/T), and PBS/T was used for washes. Incubation with primary antibodies in 5% (wt/vol) BSA (BSA-PBS/T) was carried out overnight at 4 °C using RPS25 (1:1,000 dilution, ab102940; Abcam) or for 1 h at room temperature (RT) using anti-RPS6 (1:2,000, no. 2217; Cell Signaling Technology) and anti-RPL13a (1:2,000, no. 2765; Cell Signaling Technology). Incubation with HRP-conjugated donkey anti-rabbit (sc-2313; Santa Cruz Biotechnology) secondary antibody was performed for 1 h at RT. Proteins were detected by chemiluminescence using Pierce ECL Western Blotting Substrate (Thermo Scientific). Protein expression levels in WT and add-back cell lines were quantified by measuring the integrated density on the exposed film. The RPS25 densities were normalized to the RPS6 loading control.
For live cell fluorescent labeling, cells were incubated for 30 min at 37 °C in IMDM containing 5 μM SNAP-fluorescein (S9107S; New England Biolabs). Excess dye was washed out by incubating cells for 30 min at 37 °C in IMDM without dye before cell harvest. For in vitro protein labeling, cells were harvested and lysed in lysis buffer. Following removal of debris by centrifugation, lysates were incubated with 5 μM SNAP-649 dye (S9159S; New England Biolabs) at 37 °C for 30 min.
Sucrose gradient analysis and sample processing followed the method described by Fuchs et al. (28). The only modification to this protocol was that 5 μM SNAP-649 was added to the lysis buffer. Cycloheximide prevents eukaryotic elongation factor 2-mediated ribosome translocation (13, 14) and brings translation to a halt. When puromycin is incorporated into translating ribosomes as a tRNA analog, it prematurely releases the newly synthesized peptide. Treatment of cell extract with puromycin under conditions of high ionic strength at 37 °C results in efficient separation of the ribosomal subunits. Following cell lysis on ice for 10 min, debris was pelleted at 8,400 × g at 4 °C for 5 min. To ensure efficient labeling reaction and to separate ribosomal subunits following puromycin treatment, lysates were incubated at 37 °C for 30 min before separation of cell lysates in 10–50% (wt/vol) sucrose gradients. Proteins from each fraction were precipitated with methanol and separated by 12% (wt/vol) SDS/PAGE. Gels were scanned for fluorescence using a Storm 860 fluorescent scanner (Molecular Dynamics), and intensities were quantified using ImageQuant (Molecular Dynamics).
Dual-Luciferase Assays.
A total of 2.5 μg of a dual-luciferase plasmid DNA, in which expression of the Renilla protein is cap-mediated and expression of the firefly cistron depends on the HCV IRES (15), was mixed with electroporation buffer (Teknova). A total of 7.5 × 105 cells were electroporated in 0.1 mm of electroporation cuvettes (BIORAD) using a BIORAD gene pulser (120 V, 1.5-ms pulse length, 10 pulses, 1.5 ms between pulses). Twenty-four hours after electroporation, luciferase activities were measured using the dual-luciferase kit (Promega).
Confocal Microscopy.
Cells were seeded at a density of 50,000 cells per well into a Permanox plastic eight-well NuncLab-Tek Chamber Slide System 48 h before fixation. Live cell protein labeling using SNAP-fluorescein was performed as described above. Cells were fixed for 10 min at RT using 4% (wt/vol) paraformaldehyde in 100 mM phosphate buffer (pH 7.4). Cells were permeabilized and blocked in PBS containing 3% (wt/vol) BSA, 1% saponin, and 1% Triton X-100 (Sigma). Immunostaining with anti-SNAP antibody (1:100, no. P9310S; New England Biolabs), anti-RPS6 (1:50, no. 2317; Cell Signaling), and Y10B (1:100, a gift from Joan Steitz, Yale University, New Haven, CT) was performed for 1 h at RT. Anti-IgG Alexa Fluor-conjugated antibodies of appropriate fluorescence and species reactivity were used for secondary detection, and samples were stained for 30 min at RT (1:300; Molecular Probes). Nuclei were stained with DAPI (1:500; Invitrogen), and Alexa Fluor 660-conjugated phalloidin was used for visualization of the actin cytoskeleton (1:50; Molecular Probes). Samples were mounted with Vectashield mounting medium (Vector Laboratories) and imaged with a Zeiss LSM 700 confocal microscope. Images were reconstructed using Volocity software (Improvision), and figures were assembled with Photoshop software (Adobe).
HCV RNA Purification and Labeling.
The HCV IRES, followed by the Firefly luciferase ORF, was amplified from a dual-luciferase plasmid (15) using the primers GGGAAGCTTTAATACGACTCACTATAGGGAGAGTCGACGGCGACACTCCACCATGAATC and CCCTCTAGAATTACACGGCGATCTTTCCGCCC. The amplified sequence was inserted into the HindIII and XbaI sites of pUC19. The sequence (CTTCTCGGCCTC) flanked by two SalI sites was inserted upstream of the HCV IRES. The resulting plasmid was linearized with NarI and in vitro-transcribed with T7 polymerase as described by McKenna (29). RNA was extracted with phenol-chloroform-isoamylalcohol (25:24:1 ratio) and chloroform, and was precipitated with ethanol and sodium acetate. The in vitro-transcribed RNA was purified by gel filtration on a Superdex 200 10/300 GL (GE Healthcare Life Sciences) column equilibrated with 10 mM Na-Pi (pH 6.5) and 100 mM NaCl. RNA was concentrated and buffer-exchanged in 10 mM Bis-Tris⋅HCl (pH 7.0) using an Amicon spin concentrator.
Cy5-GCTGTGAGGTGGTACTTAGTGAGG or Cy3-GCTGTGAGGTGGTACTTAGTGAGG and Ext-6-biotin (biotin-CTCTCTCGCCGGGCCTTTCTTTATG) oligonucleotides were coannealed to the 5′ and 3′ ends of HCV IRES mRNA, respectively, resulting in HCV IRES dual-labeled with biotin and cyanine dye. We found that the annealing efficiency of the fluorescent oligo was ∼50%. Decreased annealing efficiency could be due to limited accessibility of the annealing target or possibly due to formation of the alternative secondary structures in this region, as predicted by the Vienna RNA fold package (30). In both cases, the unlabeled RNA is dark and does not affect presented measurements.
Preparation of Fluorescently Labeled Human 40S Ribosomal Subunits.
A total of 12 × 150-mm dishes were grown to ∼80% confluency. Cells were washed with cold PBS, scraped in residual PBS, and pelleted. The cell pellet was resuspended in 500 μL of PBS and transferred into a 15-mL Dounce homogenizer. Following addition of 5 mL of lysis buffer [150 mM NaCl, 15 mM Tris⋅HCl (pH 7.5), 10 mM MgCl2, 1% Triton X-100, 1 mg/mL heparin, 2 mM DTT], cells were lysed by 10 strokes with the Dounce homogenizer. Lysates were transferred into a 15-mL conical tube and incubated on ice for 10 min. Lysates were cleared by centrifugation at 11,953 × g at 4 °C for 10 min in a Sorvall 5C centrifuge (DuPont). The cleared lysate was layered onto 4 mL of a 30% (wt/vol) sucrose cushion [500 mM KCl, 20 mM Tris⋅HCl (pH 7.5), 10 mM Mg(OAc)2, 30% (wt/vol) sucrose, 2 mM DTT, 0.1 mM EDTA] and spun at 30,000 rpm at 4 °C and for 14 h in an ultracentrifuge 70.1Ti rotor (Beckman Coulter).
The supernatant was removed, and the pellet was resuspended in 500 μL of buffer [500 mM KCl, 20 mM Tris⋅HCl (pH 7.5), 2 mM Mg(OAc)2, 75 mM NH4Cl, 250 mM sucrose, 2 mM DTT, 2 mM puromycin]. After transferring the resuspended ribosome pellet into a 1.7-mL tube, the sample was incubated on a nutator at 4 °C for 2 h. To label, the sample was incubated at 37 °C for 30 min with 2.5 μM SNAP dye. Insoluble material was pelleted for 20 min at 20,000 rpm at 4 °C before loading the sample onto a 10–30% (wt/vol) sucrose gradient [500 mM KCl, 20 mM Tris⋅HCl (pH 7.5), 6 mM Mg(OAc)2, 2 mM DTT] and spun at 20,000 rpm at 4 °C and for 16 h in an ultracentrifuge SW32Ti rotor (Beckman Coulter).
Fractions containing the 40S ribosomal subunits were pooled and concentrated into storage buffer [20 mM Tris⋅HCl (pH 7.5), 100 mM KCl, 2.5 mM MgCl2, 2 mM DTT, 6.8% (wt/vol) sucrose] using an Amicon-15 100,000-molecular weight cutoff concentrator. Sample concentration was determined by measuring the OD260, and labeled 40S ribosomal subunits were frozen in liquid nitrogen and stored at −80 °C.
Static Fluorescence Experiments.
HCV IRES was labeled by annealing fluorescent oligonucleotides to the unstructured regions upstream and downstream of the IRES. A twofold molar excess of the oligo was mixed with 0.5 μM RNA in 10 mM cacodylate-NaOH (pH 7.0), 100 mM KCl, and 1 mM EDTA. The reaction was incubated at 85 °C for 1 min and then slowly cooled to RT. MgCl2 was added to 4 mM to saturate the chelating capacity of EDTA. The 40S:HCV IRES complexes were assembled in 30 mM Hepes⋅KOH (pH 7.4), 100 mM KCl, and 5 mM MgCl2 buffer. One hundred nanomolar 40S subunits and HCV IRES containing mRNA were incubated for 15 min at 30 °C. The complexes were immobilized on the surface of the neutravidin-covered quartz slides at 100 pM for 5 min. Unbound complexes were washed with the reaction buffer supplemented with 2.5 mM trolox, 2.5 mM protocatechuic acid, and 0.06 U/μL protocatechuate-3,4-dioxygenase. Complexes were imaged at 532-nm excitation for 5 min at a 200-ms exposure using a home-built total internal reflection fluorescence system as described before (31). The translational HeLa extract (2× concentrate) was purchased from Pierce as a part of an in vitro translation kit (catalog no. 88881). The accessory proteins (energy regeneration system + polymerase) and buffer + NTPs supplied with the kit were not used. Rather, extract was diluted to a 1:10 working concentration with buffer containing 30 mM Hepes⋅KOH (pH 7.4), 100 mM KCl, 5.5 mM MgCl2, 1 mM GDPNP, 2.5 mM trolox, 2.5 mM protocatechuic acid, and 0.06 U/μL protocatechuate-3,4-dioxygenase, and added to immobilized complexes prior to measurements.
Measurement of 40S Subunit Kinetics.
HCV IRES labeled with Cy3 and biotin as described above was immobilized on the microscope slide surface at 33 pM for 5 min in 30 mM Hepes⋅KOH (pH 7.4), 100 mM KCl, and 5 mM MgCl2 buffer. Unbound IRES was washed with the reaction buffer supplemented with triplet-state quencher [2.5 mM trolox] and oxygen scavenging [2.5 mM protocatechuic acid and 0.06 U/μL protocatechuate-3,4-dioxygenase]. The 40S-SNAP-649 subunits in the same buffer as above were flown into the chamber by the pump concurrently with start of the movie acquisition. The movies were acquired for 12 min at a 200-ms exposure.
Supplementary Material
Acknowledgments
This study was supported by NIH Grants AI047365, AI099506, AI104557, and GM099687.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1421328111/-/DCSupplemental.
References
- 1.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell. 2009;136(4):731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spilka R, Ernst C, Mehta AK, Haybaeck J. Eukaryotic translation initiation factors in cancer development and progression. Cancer Lett. 2013;340(1):9–21. doi: 10.1016/j.canlet.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 3.Landry DM, Hertz MI, Thompson SR. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev. 2009;23(23):2753–2764. doi: 10.1101/gad.1832209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hertz MI, Landry DM, Willis AE, Luo G, Thompson SR. Ribosomal protein S25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol Cell Biol. 2013;33(5):1016–1026. doi: 10.1128/MCB.00879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marshall RA, Aitken CE, Puglisi JD. GTP hydrolysis by IF2 guides progression of the ribosome into elongation. Mol Cell. 2009;35(1):37–47. doi: 10.1016/j.molcel.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai A, et al. Heterogeneous pathways and timing of factor departure during translation initiation. Nature. 2012;487(7407):390–393. doi: 10.1038/nature11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zantema A, Maassen JA, Kriek J, Möller W. Preparation and characterization of fluorescent 50S ribosomes. Specific labeling of ribosomal proteins L7/L12 and L10 of Escherichia coli. Biochemistry. 1982;21(13):3069–3076. doi: 10.1021/bi00256a006. [DOI] [PubMed] [Google Scholar]
- 8.Hickerson R, Majumdar ZK, Baucom A, Clegg RM, Noller HF. Measurement of internal movements within the 30 S ribosomal subunit using Förster resonance energy transfer. J Mol Biol. 2005;354(2):459–472. doi: 10.1016/j.jmb.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Dorywalska M, et al. Site-specific labeling of the ribosome for single-molecule spectroscopy. Nucleic Acids Res. 2005;33(1):182–189. doi: 10.1093/nar/gki151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrov A, Puglisi JD. Site-specific labeling of Saccharomyces cerevisiae ribosomes for single-molecule manipulations. Nucleic Acids Res. 2010;38(13):e143. doi: 10.1093/nar/gkq390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carette JE, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477(7364):340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Obrig TG, Culp WJ, McKeehan WL, Hardesty B. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem. 1971;246(1):174–181. [PubMed] [Google Scholar]
- 14.Schneider-Poetsch T, et al. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010;6(3):209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lukavsky PJ, Otto GA, Lancaster AM, Sarnow P, Puglisi JD. Structures of two RNA domains essential for hepatitis C virus internal ribosome entry site function. Nat Struct Biol. 2000;7(12):1105–1110. doi: 10.1038/81951. [DOI] [PubMed] [Google Scholar]
- 16.Ben-Shem A, et al. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011;334(6062):1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 17.Boehringer D, Thermann R, Ostareck-Lederer A, Lewis JD, Stark H. Structure of the hepatitis C virus IRES bound to the human 80S ribosome: Remodeling of the HCV IRES. Structure. 2005;13(11):1695–1706. doi: 10.1016/j.str.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 18.Fukushi S, et al. Ribosomal protein S5 interacts with the internal ribosomal entry site of hepatitis C virus. J Biol Chem. 2001;276(24):20824–20826. doi: 10.1074/jbc.C100206200. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Petrov A, Tsai A, O’Leary SE, Puglisi JD. Coordinated conformational and compositional dynamics drive ribosome translocation. Nat Struct Mol Biol. 2013;20(6):718–727. doi: 10.1038/nsmb.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Leary SE, Petrov A, Chen J, Puglisi JD. Dynamic recognition of the mRNA cap by Saccharomyces cerevisiae eIF4E. Structure. 2013;21(12):2197–2207. doi: 10.1016/j.str.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciandrini L, Stansfield I, Romano MC. Ribosome traffic on mRNAs maps to gene ontology: Genome-wide quantification of translation initiation rates and polysome size regulation. PLOS Comput Biol. 2013;9(1):e1002866. doi: 10.1371/journal.pcbi.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vassilenko KS, Alekhina OM, Dmitriev SE, Shatsky IN, Spirin AS. Unidirectional constant rate motion of the ribosomal scanning particle during eukaryotic translation initiation. Nucleic Acids Res. 2011;39(13):5555–5567. doi: 10.1093/nar/gkr147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB. Rate-limiting steps in yeast protein translation. Cell. 2013;153(7):1589–1601. doi: 10.1016/j.cell.2013.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pérard J, Leyrat C, Baudin F, Drouet E, Jamin M. Structure of the full-length HCV IRES in solution. Nat Commun. 2013;4:1612. doi: 10.1038/ncomms2611. [DOI] [PubMed] [Google Scholar]
- 25.Kieft JS, Zhou K, Jubin R, Doudna JA. Mechanism of ribosome recruitment by hepatitis C IRES RNA. RNA. 2001;7(2):194–206. doi: 10.1017/s1355838201001790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukavsky PJ. Structure and function of HCV IRES domains. Virus Res. 2009;139(2):166–171. doi: 10.1016/j.virusres.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Campeau E, et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE. 2009;4(8):e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuchs G, Diges C, Kohlstaedt LA, Wehner KA, Sarnow P. Proteomic analysis of ribosomes: Translational control of mRNA populations by glycogen synthase GYS1. J Mol Biol. 2011;410(1):118–130. doi: 10.1016/j.jmb.2011.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna SA, et al. Purification and characterization of transcribed RNAs using gel filtration chromatography. Nat Protoc. 2007;2(12):3270–3277. doi: 10.1038/nprot.2007.480. [DOI] [PubMed] [Google Scholar]
- 30.Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–W74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marshall RA, Dorywalska M, Puglisi JD. Irreversible chemical steps control intersubunit dynamics during translation. Proc Natl Acad Sci USA. 2008;105(40):15364–15369. doi: 10.1073/pnas.0805299105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.