

Significance
For millions of years retroviruses, such as HIV in humans, have attacked vertebrates. Occasionally retroviruses infiltrate germ cells, incorporate themselves into the host’s genome, and transmit vertically to the host’s offspring as endogenous retroviruses (ERVs). Consequently, ERVs make up large portions of vertebrate genomes and represent a record of past host–retrovirus interactions. We developed pan-vertebrate ERV analyses to provide an overview of host–retrovirus interactions, generating insights into retroviral evolution, diversity, host-switching, and the factors influencing retroviral transmission. Astoundingly, we found over 36,000 ERV lineages across our sample of vertebrate diversity. The results provide knowledge about host–retrovirus coevolution, suggesting an unprecedented ability of retroviruses to switch between distantly related vertebrates and implying existence of additional, yet unidentified retroviruses.
Keywords: retrovirus, endogenous retrovirus, evolution, transmission, phylogenetics
Abstract
Although extensive research has demonstrated host-retrovirus microevolutionary dynamics, it has been difficult to gain a deeper understanding of the macroevolutionary patterns of host–retrovirus interactions. Here we use recent technological advances to infer broad patterns in retroviral diversity, evolution, and host–virus relationships by using a large-scale phylogenomic approach using endogenous retroviruses (ERVs). Retroviruses insert a proviral DNA copy into the host cell genome to produce new viruses. ERVs are provirus insertions in germline cells that are inherited down the host lineage and consequently present a record of past host–viral associations. By mining ERVs from 65 host genomes sampled across vertebrate diversity, we uncover a great diversity of ERVs, indicating that retroviral sequences are much more prevalent and widespread across vertebrates than previously appreciated. The majority of ERV clades that we recover do not contain known retroviruses, implying either that retroviral lineages are highly transient over evolutionary time or that a considerable number of retroviruses remain to be identified. By characterizing the distribution of ERVs, we show that no major vertebrate lineage has escaped retroviral activity and that retroviruses are extreme host generalists, having an unprecedented ability for rampant host switching among distantly related vertebrates. In addition, we examine whether the distribution of ERVs can be explained by host factors predicted to influence viral transmission and find that internal fertilization has a pronounced effect on retroviral colonization of host genomes. By capturing the mode and pattern of retroviral evolution and contrasting ERV diversity with known retroviral diversity, our study provides a cohesive framework to understand host–virus coevolution better.
Retroviruses [family Retroviridae (1)] are enveloped RNA viruses that infect vertebrate hosts. After cell entry and insertion of a DNA copy into the host cell genome, new viruses are synthesized using host cellular resources. The unique biology of retroviruses has facilitated major advances in molecular biology, notably the discovery of reverse transcriptase, insights into oncology, and applications as vectors (2), whereas ongoing epidemics arising from cross-species transfer of retroviruses illustrate their disease potential (3, 4). Screening for novel retroviruses is complicated by long periods of relative viral dormancy and limited pathogenicity in native hosts (5). Additionally, high rates of retrovirus evolution combined with deep evolutionary timescales separating major retroviral groups present considerable analytical challenges for the reconstruction of large-scale evolutionary relationships (6). Consequently, several major aspects of retrovirus biology await clarification: (i) retroviral origin and diversity, (ii) evolutionary patterns of host use, and (iii) mechanisms underlying retroviral transmission.
Here we address these three issues by using a strategy that alleviates the problems complicating evolutionary analyses of retroviral sequences and provide a framework for future exploration of the macroevolution of Retroviridae. We use endogenous retroviruses (ERVs), which have the same evolutionary origin as contemporary retroviruses (7, 8) and can be considered snapshots of retroviral evolution at the time of integration. Thus, screening vertebrate genomes for ERVs offers a valuable means to increase taxon sampling, permitting a deeper perspective into host–retroviral coevolution (9).
Using ERVs mined from a set of 65 vertebrate genomes, we first investigated retroviral origin and diversity by estimating retroviral phylogeny (Fig. 1) and quantifying ERV abundance across the different vertebrate hosts (Fig. 2). Of ∼94,000 ERVs detected from the 65 genomes spanning vertebrate diversity, we use ∼36,000 high-quality ERVs, which are relatively complete sequences with little to moderate mutational degradation and that receive a score of at least 300 from the RetroTector software (Tables S1 and S2) (10). To combat high sequence divergence and permit homology estimation among sequences across retroviral groups, we use a strategy to analyze conserved regions sampled from multiple locations across the retroviral genome (10, 11). In total, 28 conserved amino acid sequences were sampled, including six from gag [2 in matrix (MA); 2 in capsid (CA); 2 in nucleocapsid (NC), 2 from pro (protease, PR), and 20 from pol (11 in reverse transcriptase, RT, and 9 in integrase, IN)]. Retroviral locations of these sequences can be determined by cross-referencing the motif names provided in Hayward et al. (11) with the RetroTector documentation (10). Retroviral regions subject to rapid rates of evolution and increased instances of recombination, specifically those of the env gene, were excluded to maintain a high phylogenetic signal-to-noise ratio.
Fig. 1.
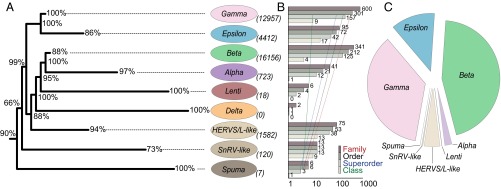
Retroviral evolution, host switching, and composition. (A) Retroviral tree schematic derived from reference retroviruses and ERVs sampled from 65 vertebrate host genomes (Tables S1 and S2). Reference retroviruses for the seven genera of Retroviridae (1), to which we apply the retrovirus “-like” ERV nomenclature (ref. 12 and Fig. S1), include, from the top, Gamma: murine leukemia virus (MLV), Epsilon: walleye dermal sarcoma virus (WDSV), Beta: mouse mammary tumor virus (MMTV), Alpha: avian leukosis virus (ALV), Lenti: HIV 1 (HIV1), Delta: human T-lymphotrophic virus 1 (HTLV1), and Spuma: simian foamy virus (SFV). The tree was rooted using C. elegans retrotransposon Cer1 (GenBank accession no. U15406) and additional gypsy/Ty3 sequences identified from the 65 analyzed vertebrate genomes. (B) Host switching estimated from the full retroviral phylogeny (Fig. S1). Bar graphs, numbers, and dashed lines indicate the frequency of switches and trends along the retrovirus phylogeny, with reference to switches between retroviral lineages and host classes, superorders, orders, and families, respectively, as shown in the key. (C) Abundance of classified ERVs in major retroviral lineages determined by phylogenetic analysis.
Fig. 2.
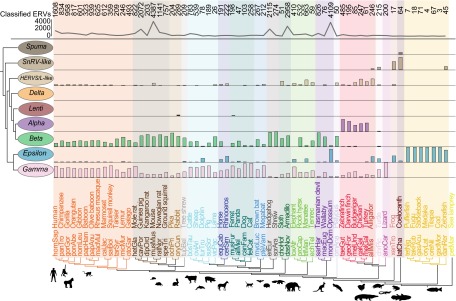
Distribution of major retroviral lineages present as ERVs within individual host genomes. Bar graphs indicate proportions of ERV groups (0–100%) within host genomes (Tables S1 and S2). The trend line in the upper panel indicates the number of classified ERVs for each genome along the host phylogeny.
Results and Discussion
Phylogenetics and Origins of Retroviridae.
The underlying tree topology recovered for Retroviridae (Fig. 1) is largely congruent with previous estimates based on considerably lower taxon sampling (12). However, we identify much greater sequence diversity toward the root of the tree (see below). Because ERVs do not follow standard retrovirus nomenclature (1), we adhere to previous practice in which ERVs are referred to as retrovirus “-like” (e.g., SnRV-like) with reference to phylogenetic analyses (12).
Despite the addition of a considerable amount of data, our results remain consistent with the hypothesis that the Spuma clade represents the most primitive retroviral group. However, recently sequenced primitive vertebrate lineages provide support for a new ERV group branching immediately after the Spuma clade, the SnRV-like clade. In addition to the snakehead fish retrovirus (SnRV) sequence, this clade is composed of turtle, coelacanth, alligator, and frog ERVs (Fig. 2). The precise branching order between the Spuma and SnRV-like clades must be interpreted with caution, because the node support separating them is relatively low. Furthermore, we identify several ERVs (from zebrafish, coelacanth, sea lamprey, and clawed frog) that lie more basal with respect to the Spuma clade (Fig. S1) but are derived with respect to sequences within the gypsy retrotransposons serving as outgroups in our analyses, which are found mainly in fish. This finding may offer key insights into early retrovirus evolution and suggests a possible marine origin, with subsequent diversification in early tetrapods. Of particular interest are the only ERVs identified in the most basal vertebrate lineage in our data, a jawless fish lineage represented by the sea lamprey (Petromyzon marinus), none of which could be classified according to the major retroviral groups (Fig. S1). Sea lampreys evolved ∼500 Mya (13), and if retroviruses emerged around that time, early sea lamprey ERVs now are likely eroded because of mutations. As a result, ERVs such as these either represent modern descendants of early retroviruses or are the product of more recent host switches.
A caveat for a possible marine origin of retroviruses is that sampled ray-finned fish mostly contain Epsilon-like ERVs (Fig. 2), which are relatively derived phylogenetically. Our data indicate that, to confirm the early evolutionary history of Retroviridae, additional primitive vertebrate lineages must be considered, particularly reptiles, amphibians, and fishes. Although the sea lamprey has not escaped retroviral activity, ERV abundance is lower relative to other vertebrates examined here. If this finding is a general feature of jawless fish confirmed by analysis of hagfish and additional lamprey genomes, it may reflect an unusually effective mechanism of retroviral restriction that could offer insights for the development of novel antiretrovirals.
Retrovirus Diversity and Host Distribution.
One of the most striking features emerging from our analyses is the vast diversity of ERVs (Fig. 1), especially compared with the 53 described species of retrovirus (1). ERVs lie distant from reference retroviruses over large swathes of the inferred tree (Fig. S1), suggesting that retrovirus lineages may be ephemeral and prone to rapid extinction over evolutionary timescales or, alternatively, that a considerable number of retroviruses remain to be identified. ERV abundance is dominated by two groups: Gamma-like ERVs and the most abundant Beta-like ERVs (Fig. 1). No jawed-vertebrate species examined has evolved to escape retroviral attack completely, but distinct broad-scale associations between different ERV groups and host clades are evident (Fig. 2 and Fig. S2). Epsilon-like ERVs make up almost the entire quotient in fish genomes (Fig. 2), whereas the coelacanth, amphibians, and reptiles have a diverse ERV makeup. At higher levels of vertebrate phylogeny, a strong trend toward Beta-like and Gamma-like ERVs presents a dichotomy in ERV distribution between mammals and more basal vertebrate orders. Despite our broad host taxon sampling, evidence of endogenous Delta-like retroviruses remains lacking. Furthermore, it is clear that Lenti and Spuma retroviruses either invade the germ line much more rarely than other retroviruses or do not persist long as ERVs. A possible alternative hypothesis could be that retroviral genera representing more recent phylogenetic groups have not had sufficient time to spread and colonize a wide diversity of hosts. However, this alternative hypothesis seems less plausible, because ancient ERVs from Lenti and Spuma lineages have been identified (14, 15). Nevertheless, it may be that historically Lenti and Spuma were more active in colonizing the germ line, with resultant ERVs now too fragmented for our analyses. For example, recent analyses detected fragmented Lenti-like sequences in several lemuriform primate genomes (16, 17), including that of the gray mouse lemur. Although we analyzed the gray mouse lemur genome (micMur, Tables S1 and S2), we did not identify any Lenti-like ERVs. Computational constraints currently prevent us from identifying all ERVs, especially those that are too fragmented. Instead, we focus on a broad overview of retroviral diversity across a wide sample of vertebrate diversity, using more complete high-quality ERV sequences. However, highly mutated and fragmented ERVs typically reflect more ancient, deeper host–retroviral associations, and their inclusion in future analyses may add further insight.
The number of high-quality ERVs in each host genome shows clear peaks for mouse, hedgehog, armadillo, guinea pig, and opossum (Fig. 2). Mouse ERVs are distributed throughout the phylogenetic tree (Fig. S1) and frequently are associated with rat and other rodent ERVs, as previously discussed for Gamma retroviruses (11). However, peaks in other ERV clades result largely from a small number of host-specific radiations of closely related ERVs, and the peaks largely disappear if these radiations are discounted from totals for each host taxon. The mechanisms behind these intense ERV bursts remain unclear and offer scope for future research.
Patterns in Retroviral Host Switching.
To examine evolutionary patterns of ERV host use, we quantified host switching across our phylogeny. We developed an algorithm to estimate the frequency of host switches for each major retroviral clade at the level of host family, order, superorder, and class (Fig. 1). Lower taxonomic levels are deemed redundant, because only two of the included host vertebrates are congeneric. We found that ERVs from divergent host groups typically are closely associated (Fig. S1). More specifically, host switching is incredibly common from the level of host superorder and below but is relatively infrequent between different vertebrate classes. These results confirm that retroviral interclass host switches are rare (11, 18), implying that key constraints act to restrict retroviruses to focal host classes. Although host taxon sampling remains limited by currently sequenced vertebrate genomes, sufficient host taxonomic overlap demonstrates that observed patterns are distinct from sampling features. Our results show an increasing tendency for host switching toward the tip of the tree, with Gamma, Beta, and Epsilon showing the highest number of switches (Fig. 1). Phylogenetically more basal ERV groups contain considerably fewer ERVs, suggesting constraints on the ability of these groups to diversify and exploit varied vertebrate hosts. However, it remains possible that the infectious retrovirus diversity for these groups is greater than currently appreciated, with low ERV abundance and host diversity reflecting an inferior capacity to colonize the germ line.
Host Factors Affecting Retrovirus Transmission.
We examined the effect of ecological variables and host life-history traits on retroviral transmission. Retroviruses have a low environmental persistence, and infection often is associated with bodily fluid transfer, so certain traits are predicted to predispose host species to a higher risk of retroviral spread (5). We tested whether higher abundances of retroviruses and ERVs were found in host species that (i) are predators, because of the increased likelihood of blood–blood transmission via oral or other injuries incurred during hunting and feeding on infected prey; (ii) exchange bodily fluids during sex; (iii) feed young using body fluids (e.g., lactation in mammals); (iv) potentially exchange body fluids during physical contests (e.g., injuries incurred during fighting); and (v) have higher fecundity (number of offspring), which may facilitate the fixation of newly established ERVs. In addition, we investigated if there are geographic hotspots of retroviral activity by incorporating global host range into our analyses (host explanatory variables are listed in Table S3). We analyzed whether these host traits explained the variation in total ERV abundance and the abundance of each major ERV clade using Bayesian phylogenetic mixed models (Figs. S3–S6 and Tables S4–S6), which provide a powerful means of investigating the relationships between multiple traits across species while accounting for nonindependence arising as the result of species coancestry (19). All the ecological and life-history traits we measured were highly evolutionarily conserved, apart from number of lifetime breeding attempts (Table S7).
We found that only one single life-history character correlates strongly with total ERV abundance: internal fertilization [parameter estimate of difference between internal and external fertilizers (β) = 3.06, confidence interval (CI) = 1.35–4.18, particle Markov chain Monte Carlo (pMCMC) = 0.001] (Fig. S3 and Table S5). The difference in overall abundance of ERVs between internal and external fertilizers was largely the result of differences in Gamma-like ERVs (β = 104.35, CI = 5.10–174.52, pMCMC < 0.0001) (Table S5) and Beta-like ERVs (β = 46.97, CI = 6.16–509.89, pMCMC < 0.0001) (Table S5), because the mode of fertilization had no effect on the abundance of Epsilon-like ERVs (β = −0.96, CI = −7.96–8.28, pMCMC = 0.92) (Table S5). However, although the mode of fertilization is directly related to transmission risk and occurs in the expected direction, these results must be interpreted with caution because there are only a few independent evolutionary origins of external fertilization: All included taxa except Xenopus and the ray-finned fishes (Actinoterygii) are internal fertilizers. It also is possible that additional, currently unmeasured ecological traits could underlie the observed pattern. Thus, the inclusion of additional hosts, particularly amphibians and additional internally fertilizing fishes, together with the analysis of further host traits will be useful in future studies.
We found that two other factors displayed group-specific effects. The number of lifetime breeding attempts had a significant positive relationship with the abundance of Gamma-like ERVs (β = 0.5, CI = 0.12–0.8, pMCMC = 0.008) (Fig. S4 and Table S5), indicating that the transmission and/or germline integration of at least some retroviruses may be higher in species that produce more offspring. In addition, as predicted because of the increased exposure to bodily fluids, the duration of weaning in mammals had a significant positive relationship with the number of Epsilon-like ERVs (β = 2.01, CI = 0.77–3.18, pMCMC = 0.001) (Fig. S5 and Table S6). However, contrary to expectations, the number of Beta-like ERVs was negatively related to the duration of weaning (β = −0.81, CI = −1.66 to −0.11, pMCMC = 0.02) (Fig. S5 and Table S6). Currently it is unclear why some ERV clades and not others are influenced by certain host characteristics, highlighting the need for further research in this area. Data for interspecies abundance did not support links between feeding and retrovirus transmission or between aggression and retrovirus transmission. Nevertheless, because ERVs from divergent hosts frequently occur as sister taxa in phylogenetic analyses, it remains possible that such life-history traits may be important for occasional cross-species transmission. It also remains an open question whether parasitic vectors mediate cross-species transmission of retroviruses, as they do for many other types of pathogens (20) and transposable elements (21).
Conclusions
As taxon sampling improves, comparative analysis of host traits is expected to offer further insights into the mechanisms of retroviral spread. Observed patterns in ERV distribution and abundance may change, particularly with greater sampling of host genomes from underrepresented vertebrate lineages. Current genome sampling is strongly biased toward mammalian taxa, which represent the smallest portion of vertebrate diversity [∼5,500 mammals, 6,600 amphibians, 9,100 reptiles, 10,000 birds, 31,000 fishes (22)] and do not appear to be common targets of all retroviruses. For example Alpha-like ERVs are recovered almost entirely from birds, whereas SnRV-like ERVs are recovered only from reptile, amphibian, and fish lineages (Fig. S2). Indeed, our ability to discern the SnRV-like and human endogenous retroviruses S and L (HERVS/L)-like groups in this study benefited greatly from including ERVs from several newly sequenced genomes belonging to more basal vertebrate lineages (e.g., coelacanth, turtle, alligator, frog). Because currently only a minute proportion of vertebrate diversity has been assayed for retroviral activity, major new retroviral clades may await discovery. That said, the profuse host switching detected could mean that the major outline of retroviral diversity is well sampled in this study. In either case, given the large evolutionary distances separating contemporary infectious retroviruses in our ERV phylogeny even with highly limited host sampling, it is possible that additional infectious retroviruses await discovery.
It is hoped that the techniques implemented in this study will facilitate additional evaluation of broad-scale patterns from ERV phylogenomic data because they allow the evolutionary signal to be separated from noise by stringent data filtering and enable more accurate establishment of sequence homology. Consequently, this methodology also is relevant to other big-data issues increasingly posed by large-scale phylogenetic datasets, particularly those focusing on highly diverse microbial systems.
ERVs offer a powerful resource for exploring host–retrovirus coevolution. Our analyses are based on individual representatives of vertebrate host species and provide a primary perspective into deep retroviral evolutionary phylogenomics. We predict that future studies using the approach described here and also within the framework of population genomics will generate further insights. Although most virology research has focused on humans and domestic and laboratory animals, our results show that a great number of retroviruses have invaded the germ line across vertebrate diversity over a long evolutionary timescale. Retroviral zoonoses present a pertinent issue as demonstrated by koala retrovirus (3) and HIV (4). Thus, analyses of ERV population genomics across diverse vertebrate species within a phylogenetic context may offer an important means of estimating extant retroviral diversity and potentially avoiding future retroviral transmission to humans (23).
One of our most remarkable findings is the dramatic occurrence of host generalism among retroviral groups, to an extent that is notable among parasitic associations. A better understanding of the mechanisms behind this widespread retroviral host switching is of major importance both for biomedicine and more generally for research into the evolution of host strategies to limit pathogens. In summary, these findings provide a framework for future studies of retroviral biology by illustrating the major patterns of retrovirus evolutionary ecology. These analyses will help place other research on retroviral biology in context and highlight key priorities for more targeted studies of host–retrovirus interactions and host–pathogen biology more widely.
Materials and Methods
ERV Detection and Phylogenetic Reconstruction.
We identified ∼94,000 ERVs by screening 65 vertebrate genomes (Tables S1 and S2), using the RetroTector software (10) as previously implemented (11). These genomes consist of assemblies and scaffolds available at the start of our analysis (hgdownload.soe.ucsc.edu/downloads.html) and represent major lineages from across vertebrate diversity. Variation in assembly quality is a recurrent issue for comparative studies of repetitive elements. As a measure of analysis quality, we include RetroTector ERV score distributions ≥300, representing high-quality ERVs, for each genome (Fig. S7). RetroTector determines ERV scores based on matches to conserved motifs and amino acid sequences across the retroviral genome corresponding to regions of key biological function from a diverse set of reference retroviral sequences. ERV scores below 300 generally imply insertions that either are heavily degraded by mutations or are fragmented and lack large segments of the proviral sequence (10). Consequently, focusing on ERVs with a RetroTector score of 300 and above maintains a baseline quality in our analyses, with a caveat that more fragmented ERVs, typically represented by older insertions, will not be included in downstream analyses. Approximately 36,000 high-quality ERV sequences were isolated during our screening of 65 vertebrate host genomes (summarized in Tables S1 and S2). Many of these ERVs consisted of sets sharing very high sequence similarity. Thus, to select single representatives among sets of very similar ERVs and thereby reduce total ERV numbers to a level that facilitated subsequent evolutionary analyses, a phylogenetic taxon reduction protocol was implemented. For each host genome, maximum-likelihood phylogenies were estimated for all high-quality ERVs using the program ExAML (version 1.0.0) (24), specifying the general time-reversible (GTR) model of sequence evolution, a gamma model of rate heterogeneity with four discrete rates, and a randomized starting tree estimated using parsimony in RAxML (version 7.3.6) (25). Following this step, nodes with a branch length below a previously determined threshold level of 0.07 mean substitutions per site were collapsed (11) recursively from tip to root, using a Perl script that invoked commands from the IO module of the Bio::Phylo package (version 0.56) (26). The collapsed tree then was parsed clade by clade, retaining the taxon containing the highest proportion of data for each clade and any taxa with a branch length above the threshold value. Then a new alignment was constructed including the retained ERVs from all analyzed host genomes together with a set of reference retroviral sequences. Additional information on quality control of detected ERVs, data selection, sequence alignment, and phylogenetic estimation are reported in Hayward et al. (11). The final alignment length was 1,658 nt sampled from across the retroviral genomes of 3,100 representative ERV sequences (Dataset S1) from the 65 vertebrate hosts and was used to construct our full retroviral phylogeny (Fig. S1 and Dataset S2). The full retroviral phylogeny was inferred using maximum likelihood in FastTree2 (version 2.1.7) (27), specifying the GTR model of nucleotide sequence evolution and the CAT (category) approximation to account for variation in rates across sites. A small number of extremely long-branched taxa apparent in initial output trees were removed from subsequent analyses. Such long-branch taxa may represent ERVs that have undergone a recombination breakpoint within the gag, pro, and pol region. The full retroviral phylogeny was rooted using Caenorhabditis elegans retrotransposon Cer1 (GenBank accession no. U15406) and additional gypsy/Ty3 sequences identified from the 65 analyzed vertebrate genomes. ERV taxa were color labeled according to host order in FigTree v1.4.0 (tree.bio.ed.ac.uk/software/figtree/). A host phylogeny was constructed based on current understanding of vertebrate evolution with reference to key publications in the field (13, 28–32).
Estimation of Host Switching.
Subtrees corresponding to each major retroviral group (Fig. 1) were pruned from the full ERV phylogeny (Fig. S1). From a table of host taxonomic identities for all tips in the tree, the number of host switches at family, order, superorder, and class levels was calculated for each subtree using a custom Perl script that called tree passing methods from the Bio::Phylo package (26). Specifically, the algorithm used a postorder tree traversal to assign tip host taxonomic identity to internal nodes in a depth-first manner, so that associations were carried sequentially from tips to the root. Host identity was determined for each node according to observed frequency at immediate daughter nodes. If several hosts shared equal frequency at a node, both were set as potential ancestral hosts. Subsequently, a preorder tree traversal was performed to resolve host identities and count host-switching events, starting at the root and progressing in a depth-first manner to the tips of the tree. Each change in taxonomic state between nodes was counted as a potential host switch, with any multiple host assignments at a daughter node clarified according to ancestral host identity. Reference sequences in the phylogeny, together with ERVs mined from the 65 host genomes, were all included in the analysis. Given that many of the ERVs in our phylogeny represent several closely related sequences, it is possible that host switches are underestimated, particularly for the large Gamma and Beta clades. However, as a consequence of the conservative inclusion of high-quality ERVs over sequences that have accumulated considerable mutations during a much longer evolutionary period, additional host-switching events at this level most likely would occur at the family or order level and thus support the observed patterns (Fig. 1).
Comparative Analyses of ERVs and Host Ecological Traits.
We investigated whether key host ecological traits, hypothesized to play important roles in retroviral transmission (Tables S1–S3), are associated with ERV distribution across host genomes. Ecological traits for each host taxon were collected from the Animal Diversity website (animaldiversity.ummz.umich.edu/) and from additional references in the literature (Table S3).
It is possible that our ability to detect associations between ecological and life-history variables and ERV abundances could be undermined by data from extant species being a poor indication of the evolutionary past when host species were colonized by ERVs. To examine whether life-history and ecological characteristics of host species explained the variation in ERV abundance, we estimated the degree to which extant measurements of host life-history and ecology could be used to infer values in the evolutionary past by estimating the phylogenetic signal in each of these variables. When traits are highly conserved over evolutionary time, enabling more accurate inferences about the evolutionary associations between traits, phylogenetic relationships between species explain a high degree of phenotypic variation. We measured the amount of phenotypic variation explained by phylogenetic relationships (phylogenetic heritability) as phylogenetic variance/total phenotypic variance (phylogenetic variance + residual variance) using Bayesian phylogenetic mixed models (BPMMs) with an intercept fitted as a fixed effect and the phylogeny fitted as a random effect. For binary (internal fertilization and mating system) and ordinal (aggression levels, meat included in diet) traits, residual variance cannot be estimated, and therefore it is not possible to estimate phylogenetic heritability. However, the intraclass correlation coefficient (ICC: for binary traits = /( + + π2/3); for ordinal traits = /( + +1) provides an estimate of the correlation in binary states between species in relation to their degree of ancestry, providing an analogous measure of phylogenetic heritability. We found that all the ecological and life-history traits we measured were evolutionarily conserved (Table S7), apart from the number of lifetime breeding attempts.
We analyzed variation in the number of ERVs using BPMMs with Markov chain Monte Carlo (MCMC) estimation in MCMCglmm (R version 3.0.1) (33, 34) and a Poisson error distribution and log link function. Before all analyses we Z-transformed (mean = 0, SD = 1) continuous fixed effects (35). The analyses included data from a taxonomically diverse range of host species. We took into account the nonindependence of data arising from the phylogenetic relationships between host species by fitting a phylogenetic covariance matrix as a random effect in all models (33, 34).
We conducted two sets of phylogenetic analyses. First, we examined whether the number of ERVs varied across biogeographical regions (seven-level fixed factor: Afrotropical, Australasia, Indomalaya, Nearctic, Neotropical, Oceanic, and Palearctic) to determine if there are hotspots of ERV evolution and accumulation. Four species in our dataset occurred in more than one geographical region, so we removed these species from the analysis. Second, we tested whether variation in the number of ERVs was explained by the life-history characteristics of species predicted to increase the likelihood of retroviral transmission. We were able to collect and analyze data on the mode of fertilization (two-level fixed factor: internal versus external), mating system (two-level fixed factor: monogamous versus promiscuous), frequency of aggressive interactions causing wounding (three-level fixed factor: low, medium, and high), number of lifetime breeding attempts (continuous fixed effect), number of offspring produced per breeding attempt (continuous fixed effect), and the amount of meat consumed within the diet (three-level fixed factor: none, occasional, and frequent). We also collected data on the length of the weaning period (continuous fixed effect), but we analyzed these data separately because they were restricted to mammals. When analyzing data on weaning period, we included all life-history characteristics found to be significantly associated with the number of ERVs to estimate the effects of weaning independently of other life-history correlates. We conducted all analyses on the sum total of the number of ERVs across all taxonomic classifications and on each of the major ERV groups (Gamma-like, Beta-like, and Epsilon-like) separately.
We ran each analysis for 1,000,000 iterations with a burn-in of 100,000 and a thinning interval of 100. This approach generated 10,000 posterior samples that we used to calculate the posterior mode, 95% CIs (lower CI–upper CI), and pMCMC (number of simulated cases that are >0 or <0 corrected for a finite number of MCMC samples). Terms were considered statistically significant when 95% CIs did not span 0 and pMCMC values were less than 0.05 (see ref. 34). Nonsignificant terms were removed from models until only significant terms remained, giving a minimal adequate model (MAM) (36).
We used inverse gamma priors for all R and G-side random effects (V = 1, ν = 0.002), which produced well-mixed chains, passed all convergence tests (see below) (37), and gave results that were almost identical to equivalent Frequentist models run with ASReml-R (version 3) (38). We checked the convergence of each analysis using two diagnostic tests in the R package “coda” (37). First, we ran each analysis three times and used the Gelman–Rubin statistic (potential scale reduction factor, PSR) to compare within- and between-chain variance (39). When convergence is met, PSR <1.1, and in all our analyses PSR was less than 1.01. Second, we used Geweke’s convergence diagnostic, which calculates Z-scores from mean parameter estimates ± SEs generated from the first 10% and the last 50% of the chain (40). If Z-scores follow an asymptotically standard normal distribution, the samples are considered to be drawn from a stationary distribution.
It was not possible to estimate branch lengths for the host phylogenetic tree, and therefore we arbitrarily set all branches to an equal length of 1, after which the tree was made ultrametric using FigTree1.4.0 (tree.bio.ed.ac.uk/software/figtree/). We tested the robustness of our results to this assumption by calculating branch lengths using two other methods: Grafen’s 1989 computation (41) with ρ set to 1 and Sanderson’s semiparametric method based on penalized likelihood (42). Both sets of branch length transformations were performed in the R package “ape” (version 3.1.1) (43). We reran all MAMs using the phylogenetic trees with the different branch lengths and found that the direction and significance of all effects were consistent across all models.
Supplementary Material
Acknowledgments
Analyses were performed using the Uppsala Multidisciplinary Center for Advanced Computational Science computer cluster (www.uppmax.uu.se). This work was funded by the Swedish Research Council Formas (P.J.) and the Medical Faculty at Uppsala University (P.J.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1414980112/-/DCSupplemental.
References
- 1.Stoye JP, et al. In: Retroviridae. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ, editors. Elsevier Academic; San Diego: 2012. pp. 477–495. [Google Scholar]
- 2.Kurth R, Bannert N. Retroviruses: Molecular Biology, Genomics and Pathogenesis. Caister Academic; Poole, UK: 2010. [Google Scholar]
- 3.Tarlinton RE, Meers J, Young PR. Retroviral invasion of the koala genome. Nature. 2006;442(7098):79–81. doi: 10.1038/nature04841. [DOI] [PubMed] [Google Scholar]
- 4.Sharp PM, Hahn BH. The evolution of HIV-1 and the origin of AIDS. Philos Trans R Soc Lond B Biol Sci. 2010;365(1552):2487–2494. doi: 10.1098/rstb.2010.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boeke JD, Stoye JP. Retrotransposons, Endogenous Retroviruses, and the Evolution of Retroelements. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Lab Press; Cold Spring Harbor, NY: 1997. pp. 343–436. [PubMed] [Google Scholar]
- 6.Lee A, Nolan A, Watson J, Tristem M. Identification of an ancient endogenous retrovirus, predating the divergence of the placental mammals. Philos Trans R Soc Lond B Biol Sci. 2013;368(1626):20120503. doi: 10.1098/rstb.2012.0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jern P, Coffin JM. Effects of retroviruses on host genome function. Annu Rev Genet. 2008;42:709–732. doi: 10.1146/annurev.genet.42.110807.091501. [DOI] [PubMed] [Google Scholar]
- 8.Stoye JP. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol. 2012;10(6):395–406. doi: 10.1038/nrmicro2783. [DOI] [PubMed] [Google Scholar]
- 9.Gifford R, Tristem M. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes. 2003;26(3):291–315. doi: 10.1023/a:1024455415443. [DOI] [PubMed] [Google Scholar]
- 10.Sperber GO, Airola T, Jern P, Blomberg J. Automated recognition of retroviral sequences in genomic data—RetroTector. Nucleic Acids Res. 2007;35(15):4964–4976. doi: 10.1093/nar/gkm515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayward A, Grabherr M, Jern P. Broad-scale phylogenomics provides insights into retrovirus-host evolution. Proc Natl Acad Sci USA. 2013;110(50):20146–20151. doi: 10.1073/pnas.1315419110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jern P, Sperber GO, Blomberg J. Use of endogenous retroviral sequences (ERVs) and structural markers for retroviral phylogenetic inference and taxonomy. Retrovirology. 2005;2:50. doi: 10.1186/1742-4690-2-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith JJ, et al. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat Genet. 2013;45(4):415–421. doi: 10.1038/ng.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katzourakis A, Gifford RJ, Tristem M, Gilbert MT, Pybus OG. Macroevolution of complex retroviruses. Science. 2009;325(5947):1512. doi: 10.1126/science.1174149. [DOI] [PubMed] [Google Scholar]
- 15.Katzourakis A, Tristem M, Pybus OG, Gifford RJ. Discovery and analysis of the first endogenous lentivirus. Proc Natl Acad Sci USA. 2007;104(15):6261–6265. doi: 10.1073/pnas.0700471104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gifford RJ, et al. A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proc Natl Acad Sci USA. 2008;105(51):20362–20367. doi: 10.1073/pnas.0807873105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilbert C, Maxfield DG, Goodman SM, Feschotte C. Parallel germline infiltration of a lentivirus in two Malagasy lemurs. PLoS Genet. 2009;5(3):e1000425. doi: 10.1371/journal.pgen.1000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin J, Herniou E, Cook J, O’Neill RW, Tristem M. Interclass transmission and phyletic host tracking in murine leukemia virus-related retroviruses. J Virol. 1999;73(3):2442–2449. doi: 10.1128/jvi.73.3.2442-2449.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harvey PH, Pagel MD. The Comparative Method in Evolutionary Biology. Oxford Univ Press; Oxford, UK: 1991. pp viii, 239 pp. [Google Scholar]
- 20.McGraw EA, O’Neill SL. Beyond insecticides: New thinking on an ancient problem. Nat Rev Microbiol. 2013;11(3):181–193. doi: 10.1038/nrmicro2968. [DOI] [PubMed] [Google Scholar]
- 21.Gilbert C, Schaack S, Pace JK, 2nd, Brindley PJ, Feschotte C. A role for host-parasite interactions in the horizontal transfer of transposons across phyla. Nature. 2010;464(7293):1347–1350. doi: 10.1038/nature08939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffmann M, et al. The impact of conservation on the status of the world’s vertebrates. Science. 2010;330(6010):1503–1509. doi: 10.1126/science.1194442. [DOI] [PubMed] [Google Scholar]
- 23.Groenen MA, et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491(7424):393–398. doi: 10.1038/nature11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stamatakis A, Aberer J. 2013. Novel parallelization schemes for large-scale likelihood-based phylogenetic inference. 2013 IEEE International Parallel & Distributed Processing Symposium (IPDPS) (IEEE Computer Society, New York), pp 1195–1204.
- 25.Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 26.Vos RA, Caravas J, Hartmann K, Jensen MA, Miller C. BIO:Phylo-phyloinformatic analysis using perl. BMC Bioinformatics. 2011;12:63. doi: 10.1186/1471-2105-12-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meredith RW, et al. Impacts of the Cretaceous terrestrial revolution and KPg extinction on mammal diversification. Science. 2011;334(6055):521–524. doi: 10.1126/science.1211028. [DOI] [PubMed] [Google Scholar]
- 29.Bininda-Emonds OR, et al. The delayed rise of present-day mammals. Nature. 2007;446(7135):507–512. doi: 10.1038/nature05634. [DOI] [PubMed] [Google Scholar]
- 30.Jetz W, Thomas GH, Joy JB, Hartmann K, Mooers AO. The global diversity of birds in space and time. Nature. 2012;491(7424):444–448. doi: 10.1038/nature11631. [DOI] [PubMed] [Google Scholar]
- 31.Amemiya CT, et al. The African coelacanth genome provides insights into tetrapod evolution. Nature. 2013;496(7445):311–316. doi: 10.1038/nature12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaffer HB, et al. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 2013;14(3):R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hadfield JD, Nakagawa S. General quantitative genetic methods for comparative biology: Phylogenies, taxonomies and multi-trait models for continuous and categorical characters. J Evol Biol. 2010;23(3):494–508. doi: 10.1111/j.1420-9101.2009.01915.x. [DOI] [PubMed] [Google Scholar]
- 34.Hadfield J. MCMC methods for multi-response generalised linear mixed models: The MCMCglmm R package. J Stat Softw. 2010;33(2):1–22. [Google Scholar]
- 35.Scheielzeth H. Simple means to improve the interpretability of regression coefficients. Methods in Ecology and Evolution. 2010;1(2):103–113. [Google Scholar]
- 36.Crawley MJ. 2002. Statistical Computing: An Introduction to Data Analysis Using S-Plus (Wiley, Hoboken, NJ), pp 772.
- 37.Plummer M, Best N, Cowles K, Vines K. Convergence diagnosis and output analysis for MCMC. R News. 2006;6(1):7–11. [Google Scholar]
- 38.Gilmour A, Gogel B, Cullis B, Thompson R. ASReml User Guide. VSN International; Hemel Hempstead: 2009. [Google Scholar]
- 39.Gelman A, Rubin D. Inference from iterative simulation using multiple sequences. Stat Sci. 1992;7(4):457–511. [Google Scholar]
- 40.Geweke J. Evaluating the accuracy of sampling-based approaches to calculating posterior moments. In: Bernardo J, Berger J, Dawid A, Smith A, editors. Bayesian Statistics. Clarendon; New York: 1992. [Google Scholar]
- 41.Grafen A. The phylogenetic regression. Philos Trans R Soc Lond B Biol Sci. 1989;326(1233):119–157. doi: 10.1098/rstb.1989.0106. [DOI] [PubMed] [Google Scholar]
- 42.Sanderson MJ. Estimating absolute rates of molecular evolution and divergence times: A penalized likelihood approach. Mol Biol Evol. 2002;19(1):101–109. doi: 10.1093/oxfordjournals.molbev.a003974. [DOI] [PubMed] [Google Scholar]
- 43.Paradis E, Claude J, Strimmer K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20(2):289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.