

Significance
Mesenchymal stem or stromal cells (MSCs) offer great promise as potential therapies for cancers and other diseases. However, applications of MSCs for cancer therapy are hindered by their protumor potential under certain conditions, considerable donor variations, and limited expandability. Here we report generation of induced pluripotent stem cells (iPSC)-derived MSCs with same tumor homing capacity but much less protumor potential using a modified protocol with high derivation efficiency. Starting from iPSCs with almost unlimited expandability, the protocol can be readily scaled up to provide huge amounts of MSCs with uniform biological properties for multicenter evaluation, large animal experiments, and potential clinical trial. Moreover, therapeutic transgenes can be inserted into safe-harbor loci of iPSCs before derivation of MSCs to eliminate insertional mutation and guarantee stable expression of transgenes during prolonged expansion.
Keywords: mesenchymal stem cells, cancer, iPS cells, tumor tropism, protumor effects
Abstract
Mesenchymal stem or stromal cells (MSCs) have many potential therapeutic applications including therapies for cancers and tissue damages caused by cancers or radical cancer treatments. However, tissue-derived MSCs such as bone marrow MSCs (BM-MSCs) may promote cancer progression and have considerable donor variations and limited expandability. These issues hinder the potential applications of MSCs, especially those in cancer patients. To circumvent these issues, we derived MSCs from transgene-free human induced pluripotent stem cells (iPSCs) efficiently with a modified protocol that eliminated the need of flow cytometric sorting. Our iPSC-derived MSCs were readily expandable, but still underwent senescence after prolonged culture and did not form teratomas. These iPSC-derived MSCs homed to cancers with efficiencies similar to BM-MSCs but were much less prone than BM-MSCs to promote the epithelial–mesenchymal transition, invasion, stemness, and growth of cancer cells. The observations were probably explained by the much lower expression of receptors for interleukin-1 and TGFβ, downstream protumor factors, and hyaluronan and its cofactor TSG6, which all contribute to the protumor effects of BM-MSCs. The data suggest that iPSC-derived MSCs prepared with the modified protocol are a safer and better alternative to BM-MSCs for therapeutic applications in cancer patients. The protocol is scalable and can be used to prepare the large number of cells required for “off-the-shelf” therapies and bioengineering applications.
The use of mesenchymal stromal or stem cells (MSCs) in cancer patients or cancer survivors is a promising strategy to improve treatment of advanced cancer (1) and to repair tissues damaged by cancers or by radical cancer therapies (2). Based on the unique homing capability of tissue-derived MSCs to stroma of various primary and metastatic cancers (3–6), MSCs have the potential to treat or even eliminate various cancers by delivering various anticancer agents (7–9). Because of their potential for differentiation (10, 11) and production of immunomodulatory, angiogenic, anti-apoptotic, anti-scarring, and prosurvival factors (12), MSCs have shown promising regeneration potential after radical cancer treatment in animal models, such as soft tissue reconstruction after disfiguring surgeries for head, neck, or breast cancers (13) and salivary gland regeneration for head and neck cancer patients treated with radiotherapy (14, 15). As one example, the combination of osteogenic potential and targeted delivery of anticancer agents make MSCs a promising option to treat tumor-induced osteolysis (16, 17). However, exogenous tissue-derived MSCs, including those from bone marrow, adipose tissues, and umbilical cord, have all shown a tendency to promote rather than inhibit cancers in many circumstances (18–23). Also, endogenous MSCs are a major source of reactive stromal cells that promote growth and metastasis of cancers (4, 24).
Moreover, MSCs have a limited proliferation potential and lose some of their important biological functions as they are expanded (25). Therefore, it is difficult to prepare large banks of the cells with uniform biological activities and/or transgene expression required for experiments in large animals and for potential clinical therapies. Another problem is that MSCs are being prepared with a variety of protocols in different laboratories from different donors. As a result, standardization of the cells has been extremely difficult and the data presented in different publications are difficult to compare. Hence large banks of reference cells are needed to advance the MSC research (26).
To address the limitations of expandability and standardization, we derived MSCs from induced pluripotent stem cells (iPSCs) with a modified protocol that can be expanded to provide large cell banks from a single cell clone. The protocol produces highly enriched MSC-like cells from iPSCs with high efficiency. The iPSC-derived MSCs (iPSC-MSCs) express the classical surface markers of MSCs, are capable of multilineage mesodermal differentiation and cancer homing, and can be expanded extensively, but do not preserve the pluoripotency of iPSCs. Surprisingly, iPSC-MSCs do not promote epithelial–mesenchymal transition (EMT), invasion, and stemness of cancer cells as is seen with bone marrow-derived MSCs (BM-MSCs). Consistent with these observations, the iPSC-MSCs express much lower levels than BM-MSCs of protumor factors including interleukine-6, prostaglandin E2, SDF1, and hyaluronan before and after exposure to tumor microenvironment. Our data indicated that iPSC-MSCs are a safe alternative to BM-MSCs for cancer therapy and other applications with better expandability and potential for genetic engineering.
Results
Derivation of MSC-Like Cells from Human iPS Cells.
Sánchez et al. (27) reported that inhibition of SMAD-2/3 signaling promoted derivation of MSCs from human embryonic stem cells (ESCs) but not from human iPSCs. To derive MSCs efficiently from human iPSCs, we modified their method by using chemically defined mTeSR1 medium (28) supplemented with the SMAD-2/3 inhibitor (SB-431542) and an atmosphere of 7.5% CO2 (29) to culture colonies of cells on Matrigel-coated plates. The cells were passaged at 80–90% confluency by lifting with Dispase. After about 25 d, most cells became larger with increased cytoplasm, and cells at the edge of the cell cluster became spindle-shaped, suggesting spontaneous differentiation (Fig. 1A). We then digested the cultures with trypsin to generate suspensions of single cells and transferred them to standard tissue culture plates. The cells were incubated in ESC–MSC medium (30) containing SB-431542, lifted at 80–90% confluency by trypsin about every 3 d, diluted 1:3, and passaged repeatedly under the same conditions (4431). During repeated passaging by trypsinization, more and more adherent cells gradually showed spindle-like morphology and appeared in whorls similar to MSCs and fibroblasts (Fig. 1A). After 45 d, quantitative RT-PCR (qRT-PCR) analysis indicated there was a marked decrease in the expression of the pluripotent genes Nanog and Oct4, the neuroectoderm marker Ecad, and the endoderm marker Foxa2 in the adherent cells. In contrast, there was a marked increase in the expression of the mesodermal marker CD140A/Pdgfra (Fig. 1B, P < 0.001). Flow cytometric analysis indicated that, similar to BM-MSCs, positive MSC markers were expressed by the vast majority of adherent cells (>99.6% for CD73, CD105, and CD166, and >88.4% for CD44 and CD90), whereas negative MSC markers including HLA-DR, CD11b, CD24, CD34, and CD45 were expressed by a very small fraction of these cells (<4.1%, Fig. 1C). When incubated in standard osteogenic media, the adherent cells were remarkably osteogenic, generating a fully differentiated monolayer of mineralizing MSCs within 10 d, about half of the time required for BM-MSCs (Fig. 1D). The adherent cells also generated cartilage in micromass cultures in the presence of both BMP2 and TGFβ (Fig. 1D). In contrast, when the cells were exposed to routine adipogenic conditions for a standard duration of 20 d, they were modestly responsive compared with BM-MSCs (Fig. 1D). qRT-PCR analysis indicated that in iPSC-MSCs the up-regulation of most markers for osteoblasts or chondrocytes after corresponding differentiation was significantly higher, whereas the up-regulation of most adipogenic markers was significantly lower compared with BM-MSCs (Fig. 1E). Because the adherent cells met the standard criteria of MSCs (32), they were subsequently referred to as iPSC-MSCs and designated as passage 0. As expected, telomerase activity in iPSC-MSCs was much higher than that in BM-MSCs (passage 4) but much lower than that in parent iPSCs (passage 39) (Fig. 1F). Consequently, the colony-forming unit–fibroblast (CFU–F)-forming efficiency of iPSC-MSCs was also much higher than that of BM-MSCs at passages 4 and 12 (Fig. 1G), indicating better expandability of iPSC-MSCs compared with BM-MSCs. The average population doubling time from passages 3–15 for iPSC-MSCs was significantly shorter than that of BM-MSCs (25.28 ± 2.92 vs. 33.91 ± 5.03 h, mean ± SEM, n = 3, P < 0.05), indicating that iPSC-MSCs propagate more rapidly than BM-MSCs. However, iPSC-MSCs were not immortal in culture. They underwent senescence and could not be expanded beyond 17 passages (64 population doublings), similar to BM-MSCs cultured under the same conditions that underwent senescence after 16 passages (48 population doublings) (Fig. 1H). Cytogenetic analysis indicated that iPSC-MSCs at passage 7 had a normal karyotype (Fig. S1), and no teratoma formation was observed in non-obese diabetic (NOD)/SCID mice inoculated with iPSC-MSCs after 4 mo. The data indicated therefore that we have developed an efficient and safe protocol to derive MSCs from human iPSCs.
Fig. 1.

Characterization of iPSC-MSCs. (A) Derivation and morphology of MSC-like cells from human iPSCs. (B) qRT-PCR analysis of relative expression of marker genes for pluripotency and each germ layer in iPSCs, BM-MSCs, and iPSC-MSCs (**P < 0.01 vs. iPSCs; ND, not detected). (C) Flow cytometry analysis of surface markers in iPSC-MSCs. (D and E) Multilineage differentiation of iPSC-MSCs. After 2 wk of corresponding induction, differentiated iPSC-MSCs were stained for mineralization with Alizarin Red, for chondrocytes with Toluidine Blue, or for lipid drops with Oil Red, respectively (D), and analyzed for expression of marker genes of osteoblasts, chondrocytes, or adipocytes, respectively, by qRT-PCR in comparison with identically differentiated BM-MSCs (E). (F) Telomerase activities in iPSCs, BM-MSCs, and iPSC-MSCs. (G) CFU–F-forming assay. *P < 0.05. (H) Growth curves of BM-MSCs and iPSC-MSCs (n = 3). iPSC-MSCs ceased expanding after 17 passages (64 population doublings) and BM-MSCs after 16 passages (48 population doublings).
Human iPSC-MSCs Were Capable of Homing to Tumors.
MSCs from various tissues have a unique tumor-homing capacity that enables them to serve as vehicles for gene therapy of advanced cancers. The tumor tropism of these MSCs is mediated by multiple chemokine receptors such as CXCR4 and CXCR6 (4–6), CD44 (33), and VEGFR1 (3) and integrins such as ITGA6 and ITGB1 (34, 35). The expression of VEGFR1 was dramatically higher in iPSC-MSCs than in BM-MSCs, whereas the expression of other homing-related genes was comparable between iPSC-MSCs and BM-MSCs (Fig. 2A). In vitro transwell migration experiments showed that there was significantly increased migration of BM-MSCs to MDA-MB231 cells, a line of triple-negative human breast cancer cells, compared with control human embryonic kidney 293T cells. Similar results were reported previously (8). The migration of iPSC-MSCs to MDA-MB231 cells was similarly significantly increased compared with that of 293T cells or medium alone (Fig. 2B, P < 0.01), and was comparable to that of BM-MSCs to the MDA-MB231 cells (P > 0.1). To confirm the in vivo tumor tropism of iPSC-MSCs, we generated human cancer xenograft models of LoVo colorectal cancer cells and MDA-MB231 breast cancer cells. After tumor establishment, BM-MSCs or iPSC-MSCs transduced with CMV-copGFP lentivirus were injected into tumor-bearing mice intravenously. To quantify the homing of MSCs to cancer, we developed individual standard curves of CMV qPCR for BM-MSCs or iPSC-MSCs carrying CMV-copGFP by adding varying amounts of genomic DNA (gDNA) from corresponding cells to gDNAs of LoVo or MDA-MB-231 tumor tissues from mice without infusion of MSCs (Fig. 2C, R2 > 0.97). Sixteen hours after MSC infusion, qPCR of CMV promoter sequence indicated that infused BM-MSCs and iPSC-MSCs homed to LoVo or MDA-MB-231 tumors with comparable efficiencies (Fig. 2 D and E, P > 0.05). Consistent with these observations, GFP+ cells were found in sections of LoVo or MDA-MB-231 tumor samples from mice infused with BM-MSCs or iPSC-MSCs carrying CMV-copGFP (Fig. 2F). Taken together, these data indicated that iPSC-MSCs are capable of homing to cancer similarly to BM-MSCs.
Fig. 2.

The tumor tropism of iPSC-MSCs. (A) qRT-PCR analysis of genes related to tumor homing in MSCs. (B) In vitro migration of MSCs toward 293T or MDA-MB-231 cells in transwells. (C) Standard curve for qPCR assays of MSCs carrying CMV-copGFP added into LoVo or MDA-MB-231 cancer xenografts. Values indicate ∆Ct for primers for CMV promoter and mouse/human GAPDH genes on same samples; n = 3. (D and E) Estimated percentage of homed MSCs carrying CMV-copGFP in all tumor cells in the LoVo or MDA-MB-231 cancer xenograft model based on qPCR of CMV promoter; n = 5. (F) Homing of i.v. infused GFP-MSCs to established s.c. LoVo or MDA-MB-231 cancer xenografts was confirmed by immunofluorescence staining of copGFP in frozen sections.
iPSC-MSCs Had Less Potential than BM-MSCs to Promote Epithelial–Mesenchymal Transition, Invasion, and Cancer Stem Cell Expansion.
Interactions between carcinoma cells and MSCs promote metastasis and/or expansion of the cancer stem cells by enhancing EMT (19). We first compared the potential of iPSC-MSCs and BM-MSCs to enhance EMT of cocultured cancer cells. Cancer cells were transduced with CMV-copGFP lentiviruses, cocultured with MSCs, and then sorted by FACS. In LoVo cancer cells, 12 h of cocultures with BM-MSCs significantly decreased the expression of the epithelial maker E-cadherin (ECAD) and significantly increased expression of mesenchymal makers fibronectin1 (FN1), N-cadherin (NCAD), vimentin (VIM), and metalloproteinase 2 (MMP2) as well as the pro-EMT factors ZEB1, ZEB2, and TWIST1 (Fig. 3A). In contrast, coculture with iPSC-MSCs did not significantly decrease expression of ECAD and had either no significant effects on or produced much smaller increases of the expression of these mesenchymal markers or pro-EMT genes in LoVo cells (Fig. 3A). The results demonstrated therefore that the iPSC-MSCs had less potential to promote EMT than BM-MSCs. To determine whether the iPSC-MSCs promoted invasion of cancer cells, an invasion assay using collagen IV-coated Boyden chambers was used. After coculture with BM-MSCs for 3 d, invasion of LoVo, HCC1806, and MCF7 human cancer cells was significantly increased (Fig. 3B, P < 0.05). In contrast, there was no significant increase after coculture with iPSC-MSCs (Fig. 3B, P > 0.05). To test whether the iPSC-MSCs promoted expansion of cancer stem cells (CSCs), we examined the aldehyde dehydrogenase (ALDH)+ population in LoVo colorectal cancer cells that are enriched for CSCs (19). After coculture with BM-MSCs for 5 d, there was significant expansion of the ALDH+ cells (Fig. 3C). Coculture with iPSC-MSCs had no significant effect. Similar effects of iPSC-MSCs on expression of EMT-related genes and ALDH+ population were observed in HCC1806, another line of human triple-negative breast cancer cells (Fig. S2). Consistent results were obtained with another assay for cancer stem cells: the mammosphere-forming capacity that is characteristic of breast cancer stem cells in cultures of MCF7 and HCC1806 cells (36). After coculture for 3 d, mammosphere formation of FACS-isolated breast cancer cells was significantly increased by BM-MSCs but not by iPSC-MSCs (Fig. 3D). Coinoculation with BM-MSCs remarkably increased the tumor-initiating ability and the tumor growth of multiple types of cancer cells including HCC1806 (19); however, we found that the tumor-initiating ability and the weights of tumors of HCC1806 cells coinoculated with iPSC-MSCs were significantly lower than those coinoculated with BM-MSCs when either 5 × 104 or 5 × 103 HCC1806 cancer cells were injected (P < 0.05 for tumor weight, Fig. 3E). Collectively, these data indicated that our iPSC-MSCs are less prone to support the growth and invasion of cancers than BM-MSCs.
Fig. 3.

The effects of iPSC-MSC on EMT, invasion, and cancer stem cells of cocultured cancer cells. (A–D) After coculture with GFP-labeled BM-MSCs or iPS-MSCs, cancer cells were isolated by FACS and subjected to (A) qRT-PCR analysis of genes related to EMT and invasion. (B) Invasion assay with collagen IV-coated Boyden chambers. (C) Representative flow cytometry analysis of ALDH+ population and the percentage of ALDH+ cells (mean ± SEM of three independent tests). (D) Mammosphere-forming assay of breast cancer cells. (E) Weights of tumors derived from HCC1806 cells injected into SCID mice with BM- or iPSC-MSCs at 6 wk.
Activity of IL1R-PGE2-IL6 Pathway Was Marginal in iPSC-MSCs.
For cancer cells such as LoVo and HCC1806 that express a high level of interleukin-1 (IL1), the protumor effect of MSCs is mediated mainly by the IL1 receptor (IL1R)/prostaglandin E2 (PGE2) pathway (19). Our iPSC-MSCs compared with BM-MSCs expressed much lower levels of mRNA for IL1R type 1 (IL1R1), the signal transducer of IL1 pathway (37), and prostaglandin E synthase (PTGES/mPGES1). The much lower levels of expression of these two genes were not significantly affected by treatment with 0.25 ng/mL IL1β, tumor-conditioned medium (TCM) from LoVo cells or coculture with LoVo cells (Fig. 4A). The basal level of expression of cyclooxygenase-2 (Cox2/PTGS2), another key PGE2 synthase, was about the same in BM-MSCs and iPSC-MSCs cultured in αMEM, but there was less up-regulation of Cox2 in iPSC-MSCs than in BM-MSCs after treatment with IL1 or LoVo TCM or coculture with LoVo cells (Fig. 4A, P < 0.05). As expected from these observations, the level of PGE2 in the medium of iPSC-MSCs was lower than that in BM-MSCs after treatment with IL1, LoVo TCM, or LoVo cells (Fig. 4B, P < 0.01). As a consequence, the expression of Interleukin-6 (IL6), a major protumor factor regulated by both IL1 and PGE2, was much lower in iPSC-MSCs than in BM-MSCs under all culture conditions (Fig. 4A, P < 0.05). These data indicate that the iPSC-MSCs are insensitive to IL1 and hence have much less potential than BM-MSCs to promote the growth and invasion of IL1-expressing cancer cells.
Fig. 4.
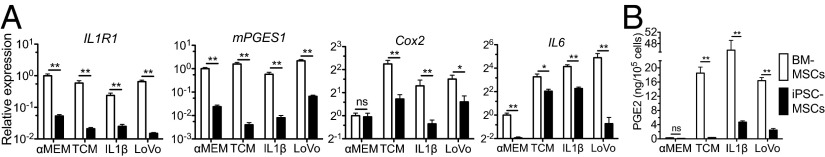
ILR-PGE2-IL6 pathway in iPSC-MSCs. (A) qRT-PCR analysis of genes of the ILR-PGE2-IL6 pathway in MSCs cultured with αMEM, IL1, LoVo conditioned medium (TCM), or cells for 3 d. (B) ELISA of PGE2 in 3-d medium of MSCs cultured under above conditions.
TGFβ Signaling and Production of Related Protumor Factors Was Less in iPSC-MSCs than in BM-MSCs.
TGFβ signaling is also essential for the protumor effects of BM-MSCs. It increases expression of multiple protumor factors such as stromal cell-derived factor 1 (SDF1/CXCL12), plasminogen activator inhibitor type 1 (PAI1/SERPINE1), and IL6 (4, 38, 39). When cultured in αMEM or TCM from MDA-MB-231 cancer cells or treated with 1 ng/mL TGFβ1, iPSC-MSCs expressed lower levels than BM-MSCs of the TGFβ receptor type 2 (TGFBR2) and of the TGFβ target genes inhibitor of differentiation 3 (ID3) (40), IL6, and SDF1 (P < 0.05, Fig. 5A). The expression of TGFβ receptor type 1 (TGFBR1) was not significantly different between these two types of MSCs cultured in αMEM or TCM, but was significantly lower in iPSC-MSCs than in BM-MSCs when both cells were treated with TGFβ1 (P < 0.05, Fig. 5A). The expression of PAI1, another TGFβ target gene (41) with protumor activities, was not significantly different between these two types of MSCs cultured in αMEM, but was significantly lower in iPSC-MSCs than in BM-MSCs when treated with TCM or TGFβ1 (P < 0.05, Fig. 5A). Western blot analysis confirmed that the level of phospho-Smad3 was significantly lower in iPSC-MSCs than in BM-MSCs when cultured in αMEM or TCM or treated with TGFβ1, whereas the level of phospho-Smad2 was undetectable in both MSCs cultured in αMEM or TCM but was significantly lower in iPSC-MSCs than in BM-MSCs when treated with TGFβ1 (P < 0.05, Fig. 5B). These data indicate that decreased TGFβ signaling also contributes to the lack of significant protumor effects in iPSC-MSCs.
Fig. 5.

TGFβ-SDF1 pathway in iPSC-MSCs. (A) qRT-PCR analysis of TGFβ receptors (TGFBR1 and TGFBR2) and TGFβ target genes (ID3, IL6, PAI1, and SDF1) in MSCs cultured with αMEM, TGFβ, or MDA-MB-231–conditioned medium (TCM) for 3 d. (B) Western blot analysis of levels of phospho-Smad2 and phospho-Smad3 in MSCs cultured under above conditions, normalized to GAPDH (n = 3).
iPSC-MSCs Compared with BM-MSCs Produced Less Hyaluronan and TSG6 and Did Not Up-Regulate Lysyl Oxidase in Cocultures with Cancer Cells.
One essential mechanism of the protumor effects of BM-MSCs is the up-regulation of lysyl oxidase (LOX) in adjacent cancer cells by triggering the CD44-signaling pathway with hyaluronan (HA) to promote EMT and metastasis (42). Tumor necrosis factor α-induced protein 6 (TSG6), a secreted protein highly expressed by BM-MSCs (43), enhances or induces the binding of HA to cell-surface CD44 (44). In iPSC-MSCs, the expression of TSG6 and dominant HA synthases (HAS1 and HAS2) was dramatically lower than that in BM-MSCs with or without coculture with LoVo cancer cells (Fig. 6A, P < 0.01). Consistent with this observation, the amount of HA secreted into medium by iPSC-MSCs was significantly lower than by BM-MSCs at passages 5 and 15 (Fig. 6B, P < 0.01). As expected, in cocultures with HCC1806 or LoVo cancer cells, iPSC-MSCs were less effective than BM-MSCs in up-regulating the LOX mRNA in cancer cells (Fig. 6C, P < 0.01). Therefore, the results indicated that decreased up-regulation of LOX contributes to the lack of significant pro-EMT and proinvasion effects of iPSC-MSCs.
Fig. 6.

The expression of HASs and TSG6 and the HA production in MSCs and the induction of LOX in cancer cells. (A) qRT-PCR analysis of HAS1-3 and TSG6 in MSCs cultured alone or with LoVo cells for 3 d. (B) Enzyme immunoassay (EIA) analysis of HA in 6-d medium of BM-MSCs or iPSC-MSCs at passages 5 and 15. (C) qRT-PCR analysis of LOX in cancer cells cultured alone or with MSCs for 3 d.
Discussion
The differentiation of human iPSCs to MSCs has been reported to be much less efficient than the differentiation of ESCs to MSCs (20 vs. 40% CD73+) (27). Also, flow cytometric sorting is generally necessary to isolate iPSC-derived MSCs, a procedure that is expensive, technically challenging, and may cause damage to cells. We modified the differentiation protocol by initially inhibiting Smad2/3 signaling in iPSCs cultured with chemically defined mTeSR1 medium (28) and then passaging cells by trypsinization (31) repeatedly (30) in 7.5% CO2 (29), conditions that were previously reported to improve the differentiation toward MSCs. During the repeated passaging by trypsinization, we used standard tissue culture plastic dishes instead of gelatin-coated plates. The early introduction of the tissue culture plastic dishes probably accelerated selection for MSC-like cells because the dishes are pretreated under proprietary conditions to increase oxygenated derivatives on the surface of the plastic and thereby make them more hydrophilic and increase the adherence of vertebrate fibroblasts and similar cells (45). The selection by adherence on the treated plastic also met one of the minimal defining criteria for human MSCs (32). This modified protocol achieved highly efficient enrichment of iPSC-MSCs (>99.6% were positive for CD73, CD105, and CD166) and eliminated the need of flow cytometric sorting. The iPSC-MSCs expanded more rapidly and to a greater extent than BM-MSCs, but still eventually underwent senescence similar to BM-MSCs. Therefore, they were less likely to cause tumors or malignancies in patients than cells that are immortal in culture (46). Also, the iPSC-MSCs did not form teratomas in mice.
The immunosuppressive, anti-inflammatory, and differentiation properties of MSCs derived from ESCs or iPSCs have been examined by several laboratories (27, 47). However, no analysis of the tumor-homing and anti- or protumor properties of ESC- or iPSC-derived MSCs has been reported. We found that our iPSC-MSCs can home to tumors with the same efficiency as BM-MSCs, but do not promote EMT, invasion, or the stemness of cancer cells as BM-MSCs do. BM-MSCs and cancer cells interact through multiple mechanisms, and the molecular profiles but not the tissue of origin of cancer cells appear to determine their interaction with MSCs. For cancer cells producing a high level of IL1 such as in LoVo colon cancer cells and HCC1806 breast cancer cells, BM-MSCs are activated by tumor-derived IL1 to produce protumor factors such as PGE2 and IL6 to promote cancer progression (19). For cancer cells producing a low level of IL1, such as MDA-MB-231 (widely metastatic) and MCF7 (noninvasive) breast cancer cells, (i) hyaluronan produced by MSCs activates the CD44 pathway in cancers to induce LOX expression and promote the EMT and invasion of cancer cells (42); and (ii) TGFβ produced by cancer cells or tumor stromal cells promotes expression of protumor factors such as IL6 and SDF1 by BM-MSCs (4, 38). Intriguingly, the expression of multiple genes related to these three pathways, including receptors for IL1 and TGFβ, IL6, SDF1, and the synthases of PGE2 and hyaluronan, as well as the production of PGE2 and hyaluronan, was dramatically lower in iPSC-MSCs with or without exposure to tumor microenvironment. Together, all of the factors may contribute to the significant decrease of protumor potential of iPSC-MSCs. The protumor effects of MSCs happen rapidly as indicated by significant up-regulation of pro-EMT genes in cancer cells cocultured with BM-MSCs for 12 h (19), suggesting that the protumor risk may compromise the efficacy of anticancer agents delivered by MSCs and is difficult to circumvent by transducing MSCs with suicide genes.
The other advantage of iPSC-MSCs is that transgenes can be inserted into safe-harbor loci of iPS cells to eliminate the risk of insertional mutation and to guarantee the stable expression of transgenes over prolonged expansion and differentiation (48). Subsequently, MSCs can be derived from the safely engineered iPS cells. This approach is not feasible for MSCs from bone marrow or other tissues because of their limited expandability and because correctly targeted clones from a single cell need to be established and then extensively expanded for therapeutic applications.
In summary, compared with BM-MSCs, iPSC-MSCs developed with our modified protocol have the same tumor tropism but much less protumor potential. They can also be readily genetically engineered and the protocol can be scaled up to produce large numbers of the cells. Hence, iPSC-MSCs prepared with the modified protocol provide a promising alternative to BM-MSCs for therapy of cancer patients or survivors and for other applications including bioengineering.
Materials and Methods
Detailed information is provided in SI Materials and Methods.
CY2 iPSCs were maintained and expanded in Matrigel-coated plates in the feeder-free mTeSR1 medium (Stemcell Technologies). For MSC derivation, iPSCs were first cultured in the mTESR1 medium supplemented with 10 μM TGFβ inhibitor SB-431542 (44) (Sigma-Aldrich) in Matrigel-coated plates at 37 °C and 7.5% CO2 (29) and passaged at 80–90% confluence by 2 mg/mL of Dispase. When most cells at the edge of a cell cluster became spindle-shaped in about 25 d, they were trypsinized into single cells and cultured in standard tissue-culture plastic dishes with modified human ESC–MSC medium (30) in the presence of SB-431542. The medium was changed daily, and the cells were passaged at 80∼90% confluence at the ratio of 1:3 about every 3 d and analyzed for expression of MSC surface markers by flow cytometry weekly. When the majority of cells were positive for MSC surface markers, they were designated passage 0 iPSC-MSCs and seeded at 500 cells/cm2 in 20% (vol/vol) FBS αMEM medium and harvested at 70–80% confluence for further experiments.
The bone marrow MSCs (donor #7075L) were from our National Institutes of Health-funded MSC distribution center (medicine.tamhsc.edu/irm/msc-distribution.html) and cultured under the same conditions as iPSC-MSCs.
The multilineage differentiation of iPS-MSCs was performed using standard published conditions for BM-MSCs (48). The qRT-PCR was done as reported (49). Telomerase activity was measured with a Quantitative Kit (Allied Biotech, MT3010). A CFU–F culture assay and migration assays of MSCs were performed as reported (37). Invasion assays were performed with 8-μm porous membranes coated with collagen IV. ALDH activity was examined with the ALDEFLUOR kit (Stemcell Technologies). For mammosphere assays, breast cancer cells were transduced with CMV-copGFP and cultured alone or with BM-MSCs or iPSC-MSCs for 3 d, isolated by FACS, and then incubated for 7 d at 500 cells/well in ultra low-attachment 12-well plates.
All animal work was approved by the joint Scott & White and Texas A&M Health Science Center Institutional Animal Care and Use Committee. When s.c. xenograft models of LoVo or MDA-MB-231 cancers were established, BM-MSCs or iPSC-MSCs transduced with CMV-copGFP lentiviruses were injected via tail vein, and tumors were harvested 16 h later for section and qPCR assay of CMV promoter (50). For tumor initiating and growth assay, HCC1806 cancer cells were coinjected with BM-MSCs or iPSC-MSCs into the fourth mammary fat pad of NOD/SCID mice, and tumors were harvested 6 wk after inoculation.
Supplementary Material
Acknowledgments
We thank Drs. Mahendra Rao and Jizhong Zou (National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases) for providing iPS cell lines and the iPSC culture protocol.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1423008112/-/DCSupplemental.
References
- 1.Droujinine IA, Eckert MA, Zhao W. To grab the stroma by the horns: From biology to cancer therapy with mesenchymal stem cells. Oncotarget. 2013;4(5):651–664. doi: 10.18632/oncotarget.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zimmerlin L, Park TS, Zambidis ET, Donnenberg VS, Donnenberg AD. Mesenchymal stem cell secretome and regenerative therapy after cancer. Biochimie. 2013;95(12):2235–2245. doi: 10.1016/j.biochi.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaturvedi P, et al. Kshitiz Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013;123(1):189–205. doi: 10.1172/JCI64993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quante M, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–272. doi: 10.1016/j.ccr.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song C, Li G. CXCR4 and matrix metalloproteinase-2 are involved in mesenchymal stromal cell homing and engraftment to tumors. Cytotherapy. 2011;13(5):549–561. doi: 10.3109/14653249.2010.542457. [DOI] [PubMed] [Google Scholar]
- 6.Jung Y, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795. doi: 10.1038/ncomms2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JH, et al. Dental follicle cells and cementoblasts induce apoptosis of ameloblast-lineage and Hertwig’s epithelial root sheath/epithelial rests of Malassez cells through the Fas-Fas ligand pathway. Eur J Oral Sci. 2012;120(1):29–37. doi: 10.1111/j.1600-0722.2011.00895.x. [DOI] [PubMed] [Google Scholar]
- 8.Loebinger MR, Eddaoudi A, Davies D, Janes SM. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. 2009;69(10):4134–4142. doi: 10.1158/0008-5472.CAN-08-4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grisendi G, et al. Adipose-derived mesenchymal stem cells as stable source of tumor necrosis factor-related apoptosis-inducing ligand delivery for cancer therapy. Cancer Res. 2010;70(9):3718–3729. doi: 10.1158/0008-5472.CAN-09-1865. [DOI] [PubMed] [Google Scholar]
- 10.Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276(5309):71–74. doi: 10.1126/science.276.5309.71. [DOI] [PubMed] [Google Scholar]
- 11.Pittenger MF, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 12.Meirelles LdaS, Fontes AM, Covas DT, Caplan AI. Mechanisms involved in the therapeutic properties of mesenchymal stem cells. Cytokine Growth Factor Rev. 2009;20(5-6):419–427. doi: 10.1016/j.cytogfr.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Donnenberg VS, Zimmerlin L, Rubin JP, Donnenberg AD. Regenerative therapy after cancer: What are the risks? Tissue Eng Part B Rev. 2010;16(6):567–575. doi: 10.1089/ten.teb.2010.0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin CY, et al. Cell therapy for salivary gland regeneration. J Dent Res. 2011;90(3):341–346. doi: 10.1177/0022034510386374. [DOI] [PubMed] [Google Scholar]
- 15.Sumita Y, et al. Bone marrow-derived cells rescue salivary gland function in mice with head and neck irradiation. Int J Biochem Cell Biol. 2011;43(1):80–87. doi: 10.1016/j.biocel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fritz V, et al. Antitumoral activity and osteogenic potential of mesenchymal stem cells expressing the urokinase-type plasminogen antagonist amino-terminal fragment in a murine model of osteolytic tumor. Stem Cells. 2008;26(11):2981–2990. doi: 10.1634/stemcells.2008-0139. [DOI] [PubMed] [Google Scholar]
- 17.Li X, et al. Human placenta-derived adherent cells prevent bone loss, stimulate bone formation, and suppress growth of multiple myeloma in bone. Stem Cells. 2011;29(2):263–273. doi: 10.1002/stem.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karnoub AE, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 19.Li HJ, Reinhardt F, Herschman HR, Weinberg RA. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012;2(9):840–855. doi: 10.1158/2159-8290.CD-12-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S, et al. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011;71(2):614–624. doi: 10.1158/0008-5472.CAN-10-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barkholt L, et al. Risk of tumorigenicity in mesenchymal stromal cell-based therapies: Bridging scientific observations and regulatory viewpoints. Cytotherapy. 2013;15(7):753–759. doi: 10.1016/j.jcyt.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Gu J, et al. Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-β/Smad pathway. PLoS ONE. 2012;7(12):e52465. doi: 10.1371/journal.pone.0052465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klopp AH, et al. Omental adipose tissue-derived stromal cells promote vascularization and growth of endometrial tumors. Clin Cancer Res. 2012;18(3):771–782. doi: 10.1158/1078-0432.CCR-11-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kidd S, et al. Origins of the tumor microenvironment: Quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS ONE. 2012;7(2):e30563. doi: 10.1371/journal.pone.0030563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larson BL, Ylostalo J, Lee RH, Gregory C, Prockop DJ. Sox11 is expressed in early progenitor human multipotent stromal cells and decreases with extensive expansion of the cells. Tissue Eng Part A. 2010;16(11):3385–3394. doi: 10.1089/ten.tea.2010.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Viswanathan S, et al. Soliciting strategies for developing cell-based reference materials to advance MSC research and clinical translation. Stem Cells Dev. 2014;23(11):1157–1167. doi: 10.1089/scd.2013.0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sánchez L, et al. Enrichment of human ESC-derived multipotent mesenchymal stem cells with immunosuppressive and anti-inflammatory properties capable to protect against experimental inflammatory bowel disease. Stem Cells. 2011;29(2):251–262. doi: 10.1002/stem.569. [DOI] [PubMed] [Google Scholar]
- 28.Ludwig TE, et al. Feeder-independent culture of human embryonic stem cells. Nat Methods. 2006;3(8):637–646. doi: 10.1038/nmeth902. [DOI] [PubMed] [Google Scholar]
- 29.Olivier EN, Bouhassira EE. Differentiation of human embryonic stem cells into mesenchymal stem cells by the “raclure” method. Methods Mol Biol. 2011;690:183–193. doi: 10.1007/978-1-60761-962-8_13. [DOI] [PubMed] [Google Scholar]
- 30.Lai RC, Choo A, Lim SK. Derivation and characterization of human ESC-derived mesenchymal stem cells. Methods Mol Biol. 2011;698:141–150. doi: 10.1007/978-1-60761-999-4_11. [DOI] [PubMed] [Google Scholar]
- 31.Lian Q, et al. Derivation of clinically compliant MSCs from CD105+, CD24-differentiated human ESCs. Stem Cells. 2007;25(2):425–436. doi: 10.1634/stemcells.2006-0420. [DOI] [PubMed] [Google Scholar]
- 32.Dominici M, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 33.Spaeth EL, et al. Mesenchymal CD44 expression contributes to the acquisition of an activated fibroblast phenotype via TWIST activation in the tumor microenvironment. Cancer Res. 2013;73(17):5347–5359. doi: 10.1158/0008-5472.CAN-13-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee RH, et al. The CD34-like protein PODXL and alpha6-integrin (CD49f) identify early progenitor MSCs with increased clonogenicity and migration to infarcted heart in mice. Blood. 2009;113(4):816–826. doi: 10.1182/blood-2007-12-128702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ip JE, et al. Mesenchymal stem cells use integrin beta1 not CXC chemokine receptor 4 for myocardial migration and engraftment. Mol Biol Cell. 2007;18(8):2873–2882. doi: 10.1091/mbc.E07-02-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cioce M, et al. Mammosphere-forming cells from breast cancer cell lines as a tool for the identification of CSC-like- and early progenitor-targeting drugs. Cell Cycle. 2010;9(14):2878–2887. [PubMed] [Google Scholar]
- 37.Sims JE, et al. Interleukin 1 signaling occurs exclusively via the type I receptor. Proc Natl Acad Sci USA. 1993;90(13):6155–6159. doi: 10.1073/pnas.90.13.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shangguan L, et al. Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells. 2012;30(12):2810–2819. doi: 10.1002/stem.1251. [DOI] [PubMed] [Google Scholar]
- 39.Hogan NM, et al. Impact of mesenchymal stem cell secreted PAI-1 on colon cancer cell migration and proliferation. Biochem Biophys Res Commun. 2013;435(4):574–579. doi: 10.1016/j.bbrc.2013.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Chambers RC, Leoni P, Kaminski N, Laurent GJ, Heller RA. Global expression profiling of fibroblast responses to transforming growth factor-beta1 reveals the induction of inhibitor of differentiation-1 and provides evidence of smooth muscle cell phenotypic switching. Am J Pathol. 2003;162(2):533–546. doi: 10.1016/s0002-9440(10)63847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boehm JR, Kutz SM, Sage EH, Staiano-Coico L, Higgins PJ. Growth state-dependent regulation of plasminogen activator inhibitor type-1 gene expression during epithelial cell stimulation by serum and transforming growth factor-beta1. J Cell Physiol. 1999;181(1):96–106. doi: 10.1002/(SICI)1097-4652(199910)181:1<96::AID-JCP10>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 42.El-Haibi CP, et al. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. Proc Natl Acad Sci USA. 2012;109(43):17460–17465. doi: 10.1073/pnas.1206653109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee RH, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5(1):54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lesley J, et al. TSG-6 modulates the interaction between hyaluronan and cell surface CD44. J Biol Chem. 2004;279(24):25745–25754. doi: 10.1074/jbc.M313319200. [DOI] [PubMed] [Google Scholar]
- 45.Ramsey WS, Hertl W, Nowlan ED, Binkowski NJ. Surface treatments and cell attachment. In Vitro. 1984;20(10):802–808. doi: 10.1007/BF02618296. [DOI] [PubMed] [Google Scholar]
- 46.Prockop DJ, Keating A. Relearning the lessons of genomic stability of human cells during expansion in culture: Implications for clinical research. Stem Cells. 2012;30(6):1051–1052. doi: 10.1002/stem.1103. [DOI] [PubMed] [Google Scholar]
- 47.de Peppo GM, et al. Engineering bone tissue substitutes from human induced pluripotent stem cells. Proc Natl Acad Sci USA. 2013;110(21):8680–8685. doi: 10.1073/pnas.1301190110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zou J, et al. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: Functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117(21):5561–5572. doi: 10.1182/blood-2010-12-328161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hai B, et al. Wnt/β-catenin signaling regulates postnatal development and regeneration of the salivary gland. Stem Cells Dev. 2010;19(11):1793–1801. doi: 10.1089/scd.2009.0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moulay G, Boutin S, Masurier C, Scherman D, Kichler A. Polymers for improving the in vivo transduction efficiency of AAV2 vectors. PLoS One. 2010;5(12):e15576. doi: 10.1371/journal.pone.0015576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.