

Abstract
Oncogenic mutations in Ras deregulate cell death and proliferation to cause cancer in a significant number of patients. Although normal Ras signaling during development has been well elucidated in multiple organisms, it is less clear how oncogenic Ras exerts its effects. Furthermore, cancers with oncogenic Ras mutations are aggressive and generally resistant to targeted therapies or chemotherapy. We identified the exocytosis component Sec15 as a synthetic suppressor of oncogenic Ras in an in vivo Drosophila mosaic screen. We found that oncogenic Ras elevates exocytosis and promotes the export of the pro-apoptotic ligand Eiger (Drosophila TNF). This blocks tumor cell death and stimulates overgrowth by activating the JNK-JAK-STAT non-autonomous proliferation signal from the neighboring wild-type cells. Inhibition of Eiger/TNF exocytosis or interfering with the JNK-JAK-STAT non-autonomous proliferation signaling at various steps suppresses oncogenic Ras-mediated overgrowth. Our findings highlight important cell-intrinsic and cell-extrinsic roles of exocytosis during oncogenic growth and provide a new class of synthetic suppressors for targeted therapy approaches.
Keywords: Eiger, TNF, Exocytosis, Oncogenic Ras, Tumors
INTRODUCTION
Activating mutations of the oncogenic Ras gene are highly prevalent in human cancers (Bos, 1989). However, targeted therapy strategies have not yielded the desired effects and the available chemotherapy regimens are not effective at treating the most aggressive Ras cancers (Asghar et al., 2010; Lièvre et al., 2010). Genetic studies in fruit flies and worms have led to the identification of Ras effectors and the characterization of the Ras/Raf/mitogen activated protein kinases (Ras/MAPK) signaling cascade during normal development (Nishida and Gotoh, 1993; Simon et al., 1991; Sternberg and Horvitz, 1991). This pathway was found to be conserved in mammals (Alessi et al., 1994; Cowley et al., 1994; Kolch et al., 1991; Mansour et al., 1994; Qiu et al., 1995). However, targeting components of the Ras/MAPK signaling cascade only partly inhibits overgrowth (Blum and Kloog, 2005), suggesting that oncogenic Ras drives tumorigenesis via additional signaling events. Moreover, blocking the Ras/MAPK pathway causes death of normal cells, thus making this approach not suitable for targeted therapy and placing an impetus on the need to identify the additional signaling events that oncogenic Ras specifically triggers to promote tumor development. Identification of mutations that synthetically suppress Ras tumor growth could not only broaden our understanding of cancer biology, but could also lead to the discovery of novel therapeutic targets. Indeed, several laboratories have conducted RNA interference (RNAi)-based synthetic suppressor screens in tissue culture settings and have identified important vulnerabilities of oncogenic Ras cells (Barbie et al., 2009; Luo et al., 2009; Sarthy et al., 2007; Scholl et al., 2009; Steckel et al., 2012; Strebhardt and Ullrich, 2006). Additional whole-animal synthetic suppressor screens could be particularly informative for revealing the role of oncogenic Ras in processes that are important for regulating growth in vivo.
Expression of oncogenic Ras (RasV12) in Drosophila imaginal discs gives rise to overgrowth (Karim and Rubin, 1998). Generating patches of labeled RasV12-expressing cells surrounded by wild-type cells allows putative host-tumor cell interactions to occur and permits genetic screens to identify mutations that can either enhance or suppress the growth of RasV12-expressing tumors (Chi et al., 2010; Pagliarini and Xu, 2003). Here, we report the characterization of one of the synthetic suppressor mutations, sec15, a mutation in a gene that encodes a component of the exocytosis machinery. Sec15 is a subunit of the evolutionarily conserved multiprotein complex termed the Sec6/Sec8 complex or the exocyst complex, which was originally identified in yeast (Finger et al., 1998; Finger and Novick, 1998; Guo et al., 1999; Heider and Munson, 2012; Novick et al., 1980; TerBush and Novick, 1995). The exocyst complex regulates secretion in eukaryotes by controlling the delivery of vesicles to the cell plasma membrane (Grindstaff et al., 1998; Guo et al., 1999; Heider and Munson, 2012; Jafar-Nejad et al., 2005; Mehta et al., 2005; TerBush and Novick, 1995). In contrast to core exocyst components, which are broadly required for normal exocyst function and cell viability, Sec15 regulates the delivery of specific cargo proteins and is dispensable for cell viability in Drosophila (Mehta et al., 2005).
In addition to being essential for cellular organization in all eukaryotes, vesicle transport has recently been found to play important roles in regulating signal transduction. For example, transport of endocytosed cell surface molecules to signaling targets on endosomes allows signal transduction to occur, whereas targeting these molecules to the lysosome for degradation attenuates or suppresses signaling (Seto et al., 2002). Transcytosis of vesicles facilitates the establishment of morphogen gradients, which are crucial for conveying proliferation and cell fate specification cues during development (Seto et al., 2002). Exocytosis has been previously found to mediate signal transduction by sending signaling molecules including neurotransmitters and ligands to neighboring cells (Li and Chin, 2003). By studying how sec15 suppresses RasV12, we show that exocytosis also regulates signal transduction within a cell by clearing signaling ligands. We found that RasV12 cells clear Eiger (also known as TNF) by exocytosis to downregulate pro-apoptotic Janus NH2-terminal kinase (JNK, also known as Bsk – FlyBase) signaling (Igaki et al., 2009; Moreno et al., 2002) and thus evade cell death. We have previously shown that JNK activation triggered by cell polarity defects could stimulate non-autonomous JAK-STAT signaling for proliferation (Wu et al., 2010). Here, we show that oncogenic Ras elevates exocytosis to hijack this process in order to promote overgrowth. Exocytosis-dependent accumulation of Eiger/TNF results in JNK activation in surrounding wild-type cells, which in turn, non-autonomously stimulates JAK-STAT signaling to promote the proliferation of RasV12 cells. These findings provide new mechanistic insights into the long known ability of oncogenic Ras cells to avoid cell death and promote growth, and also highlight the importance of exocytosis in signal transduction and cancer biology.
RESULTS
sec15 synthetically interacts with oncogenic Ras
In Drosophila, GFP-marked mosaic clones of cells expressing RasV12 overgrow to develop into tumors (Pagliarini and Xu, 2003). The overgrowth phenotype can be readily ascertained by visualizing fluorescent signal intensity in third instar whole larvae (Fig. 1A,C) or by examining clone size in dissected eye-antenna imaginal discs (Fig. 1E,G). Furthermore, RasV12 tumors caused pupal lethality (98.4%, N=62; Fig. 1K). We induced sec15 or RasV12 single mutant clones or RasV12, sec15 double mutant clones and examined the growth of these mutant clones in similarly aged third instar eye-antenna discs. We found that the sec15 mutation did not disrupt cell proliferation (supplementary material Fig. S1A,B), and the size of sec15 mutant clones was comparable to that of wild-type clones (Fig. 1A,B,E,F), consistent with the reported cell viability of the sec15 null mutation (Mehta et al., 2005). In addition, sec15 null mutant cells persisted into the adult eye (Fig. 1I,J). The viability of sec15 mutant cells is not due to maternal protein deposition, as Sec15 protein level was dramatically reduced in mutant clone cells (supplementary material Fig. S2A). However, the sec15 mutation dramatically suppressed the overgrowth phenotype of RasV12 clones (Fig. 1C,D,G,H; 77.4% of the double mutants showed strong suppression similar to that shown in Fig. 1D,H; N=53). Furthermore, the sec15 mutation rescued the lethality of the animals bearing RasV12 tumors (76% viable, N=125, compared with 1.6% viable, N=62 for RasV12 animals; Fig. 1L). Moreover, RNA interference (RNAi)-mediated knockdown of Sec15 in RasV12, scrib− cells suppressed tumor growth and invasion (supplementary material Fig. S8A-D). Finally RNAi knockdown of two core exocyst components, Sec6 and Sec8 showed a similar effect on RasV12-mediated overgrowth (supplementary material Fig. S1C-N). The sec15 mutant or Sec15 or Sec8 RNAi alone had no detectable effect on growth, whereas Sec6 RNAi alone showed a reduction in clone sizes (supplementary material Fig. S1C-N). Taken together, we conclude that the sec15 mutation synthetically suppresses RasV12 tumor growth.
Fig. 1.
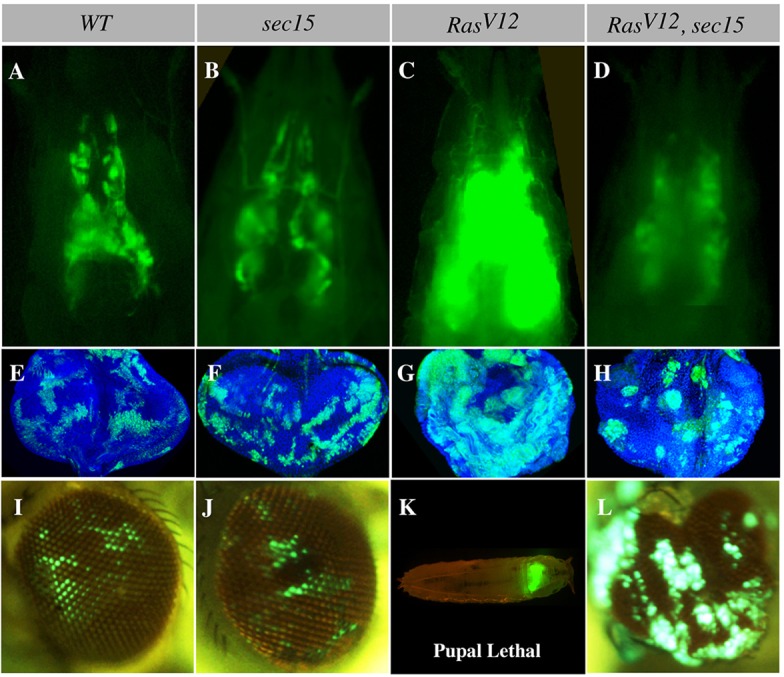
RasV12 and sec15 synthetic lethal interaction. (A-D) Intact third instar larval cephalic regions showing wild-type (WT), sec15, RasV12 and RasV12, sec15 double mutant eye-antenna disc clones. Wild-type (A) and sec15 (B) clones are comparable in size, whereas RasV12 clones (C) overgrow to develop tumors. The sec15 mutation suppresses RasV12 tumor growth (D). (E-H) Similarly aged eye discs bearing wild-type, sec15, RasV12 and RasV12, sec15 double mutant clones. Wild-type (E) and sec15 (F) clones show a comparable sparse pattern, whereas RasV12 clones (G) overgrow to form large contiguous tumors. However, RasV12, sec15 double mutant clones are sparse and smaller (H). (I-L) Adult eyes showing lethality and contribution of larval clones to the eye field. Wild-type (I) and sec15 (J) clones similarly persist into the adult eye and do not cause animal lethality. RasV12 clones (K) are pupal lethal but animal lethality is suppressed in animals bearing RasV12, sec15 double mutant clones, and cells of this genotype now contribute to the adult eye (L).
Oncogenic Ras stimulates the exocyst
Interestingly, it was previously observed that RNAi depletion of exocytosis proteins also suppresses HRasV12-mediated tumor growth in human cells (Issaq et al., 2010). We thus investigated the mechanism underlying this phenomenon. The observation that sec15 can selectively suppress RasV12 tumor growth suggested that oncogenic Ras could regulate the exocyst to promote growth. We first analyzed the abundance of exocyst proteins in RasV12 tumor clones relative to that of wild-type cells. We examined Sec15 protein levels in RasV12 tumors and extended this analysis to include Sec6, Sec8 and Rab11. Rab11 interacts with Sec15 to initiate assembly of the exocyst and the exocytosis of endocytosed molecules (Wu et al., 2005; Zhang et al., 2004). The previously published Sec15, Sec6 and Sec8 antibodies showed negligible staining in corresponding mutant and RNAi knockdown clones (supplementary material Fig. S2A-C). Sec15, Sec6 and Sec8 proteins were specifically upregulated in RasV12clones (Fig. 2A-C,E,G). Similarly, Rab11 was upregulated in RasV12 cells (Fig. 2D,F). The increased levels of exocyst proteins in RasV12 cells were subsequently confirmed by western blotting (Fig. 2H). Exocyst proteins displayed no obvious subcellular localization defects in RasV12 cells (Fig. 2E-G), indicating that oncogenic Ras stimulates exocyst proteins but does not affect their respective subcellular localization.
Fig. 2.

RasV12 cells upregulate exocyst proteins. (A-G) RasV12 clones stained with antibodies against exocyst proteins (Sec15, Sec6, Sec8 and Rab11) and co-stained with DAPI (blue) to mark nuclei. Lower panels show Sec15, Sec6, Sec8 and Rab11 channels alone. Sec15 is upregulated in RasV12 cells (A). Similarly, Sec6 (B), Sec8 (C) and Rab11 (D) are upregulated in RasV12 clones but not in neighboring wild-type cells. Green dotted lines denote clone boundaries. (E-G) x/z confocal projection images of RasV12 clones (green) stained with anti-Sec8, anti-Rab11 and anti-Sec15, and with DAPI. Sec8 (E), Rab11 (F) and Sec15 (G) proteins display no obvious localization defects but show increased abundance in RasV12 cells. (H) Lysate from discs bearing wild-type (WT) or RasV12 clones immunoblotted with anti-Sec6, anti-Sec8, anti-Sec15 or anti-Tubulin antibodies (loading control). Sec6, Sec8 and Sec15 protein abundance is increased in RasV12 lysates.
The observation that Rab11 is upregulated along with core exocyst proteins suggested that oncogenic Ras elevates the secretion of endocytosed molecules. To begin to address this possibility, we sought to examine the transport of endocytosed molecules in the mutant cells by performing Dextran dye pulse-chase experiments. After the dye pulse phase we found that RasV12 cells had no problem taking up the dye as they contained more dye (dye-positive punctae) than control cells [4.05±1.47 for RasV12 versus 2.29±0.65 for wild-type cells (means±s.d.); Fig. 3A,B,G], indicating that endocytosis is generally not inhibited in RasV12 cells. Examining dye abundance in mutant versus control cells after the dye chase period provided a crude measure of secretion within these cells. Interestingly, RasV12 cells were significantly more efficient at dye clearance after the dye chase phase. RasV12 cells showed fewer dye-positive punctae compared with wild-type cells (the pulse:chase ratios of dye-positive punctae for RasV12 and wild-type cells were 4.63±0.27 and 1.53±1.12, respectively; Fig. 3D,E,G). The sec15 mutation abrogated the increased dye clearance effect of oncogenic Ras [2.74±1.98 (after the pulse), 1.26±0.29 (after chase) and 2.18±0.34 (pulse:chase ratio); Fig. 3C,F,G]. Taken together, the above data indicate that oncogenic Ras stimulates the exocyst.
Fig. 3.

RasV12 cells stimulate secretion through Sec15. (A-F) Projections of confocal images showing wild-type (WT), RasV12 or RasV12, sec15 mutant clones (green, middle panels) after pulse (A-C) and followed by a chase period (D-F) of Dextran dye (red, lower panels). Samples are co-stained with DAPI (blue) to mark nuclei. Middle panels show the clone channel alone. The dotted green lines in the lower panels denote clone boundaries. RasV12 cells (B) show no problem in dye uptake. Cellular clearance of the dye is accelerated in RasV12 cells (E, lower panel) compared with that of similarly treated wild-type control cells (D, lower panel). The sec15 mutation suppresses the ability of RasV12 cells to clear out the dye (C,F). (G) Quantification of A-F. The number of dye-positive vesicles after each experimental phase (pulse and chase) was scored in multiple clones for each genotype. Pulse:chase ratios were derived from averages of dye-positive vesicle numbers after each experimental phase. Error bars represent standard deviation from the mean for each genotype analyzed. In this and all subsequent figures, numbers within bars indicate the number of cells analyzed per corresponding genotype.
Oncogenic Ras promotes the interaction of Eiger/TNF with the exocyst
We next sought to investigate the significance of exocyst stimulation vis-à-vis tumor growth. One way in which Ras activation contributes to tumorigenesis is by allowing the cell to evade cell death (Cox and Der, 2003; Downward, 1998; Wolfman et al., 2002; Wu et al., 2009). In Drosophila, the tumor necrosis factor (TNFα) homolog Eiger triggers cell death (Igaki et al., 2009; Moreno et al., 2002). Similar to vertebrate TNFs, Eiger is produced as a transmembrane protein and is subsequently cleaved to yield a soluble ligand (Black et al., 1997; Brandt et al., 2004; Jue et al., 1990; Kauppila et al., 2003; Moss et al., 1997). Eiger/TNF is endocytosed to activate JNK signaling on endosomes, which triggers cell death (Igaki et al., 2009; Moreno et al., 2002). Preventing endocytosis impinges on Eiger/TNF signaling (Igaki et al., 2009). Because endocytosis is not generally inhibited in RasV12 cells and sec15 suppresses oncogenic Ras-mediated overgrowth, we hypothesized that oncogenic Ras could promote an interaction between the exocyst and Eiger/TNF in order to accelerate cellular clearance of Eiger/TNF, hence preventing Eiger/TNF signaling from directly killing tumor cells. We first examined whether Eiger/TNF and Sec15 proteins interact in RasV12 cells using two independent and complementary approaches, immunostaining and biochemistry. We found that Eiger/TNF colocalized with Sec15 in RasV12 cells (Fig. 4A-C). Sectioning of isosurface-rendered images from confocal projections revealed that indeed Sec15 coats Eiger/TNF-positive vesicles (Fig. 4D), suggesting that Eiger/TNF is a Sec15 cargo protein. Moreover, we performed anti-Eiger or anti-GFP control pulldown experiments on lysate prepared from eye-antenna discs bearing RasV12 clones and found that Sec15 and other exocyst proteins, Sec5 and Sec8, specifically co-precipitated with Eiger/TNF (Fig. 4M). We conclude that oncogenic Ras promotes the interaction of the exocyst with Eiger/TNF.
Fig. 4.

Sec15 interacts with Eiger/TNF. (A-D) RasV12 cells counterstained with anti-Sec15 (red) and anti-Eiger (green). Sec15 and Eiger colocalize (A; side panels represent magnified view from white box). B and C are single channels for Sec15 and Eiger from A. (D) Cross-section images of isosurface-rendered confocal projection images showing Eiger (green) inside Sec15-coated (red) vesicles. (E-H) RasV12 cells counterstained with anti-Rab11 (red) and anti-Eiger (green). Rab11 and Eiger colocalize (E; side panels represent magnified view from white box). F and G are single channels for Rab11 and Eiger from E. (H) Cross-section of isosurface-rendered confocal projection showing Eiger (green) inside Rab11-coated (red) vesicles. (I-L) RasV12 cells co-expressing Rab5-GFP (red) counterstained with anti-Eiger (green). Rab5-GFP and Eiger colocalize (I; side panels represent magnified view from white box). J and K are single channels for Rab5-GFP and Eiger, respectively. (L) Cross-section of isosurface-rendered confocal projections showing Eiger (green) inside Rab5-GFP-coated (red) vesicles. (M) Anti-Eiger or anti-GFP (control) immunoprecipitation experiments with lysate derived from tissues bearing RasV12 clones. Immunoprecipitates (IP) were blotted with antibodies against Sec15, Sec8, Sec5 and α-Tubulin (loading control).
Oncogenic Ras promotes Eiger/TNF exocytosis to prevent tumor cell death
Endocytosed molecules destined for secretion are shuttled to the cell cortex in Rab11-positive endosomes (Dollar et al., 2002; Sonnichsen et al., 2000; Ullrich et al., 1996). We thus tested whether Eiger/TNF localizes to early (Rab5-positive) endosomes and to recycling (Rab11-positive) endosomes. We found that Eiger/TNF colocalized with both Rab5 and Rab11 endosomes (Fig. 4E-L). Together with the Eiger/TNF-exocyst interaction data above, these observations supported the notion that Eiger/TNF is being cleared out of RasV12 tumors by exocytosis. To directly test this, we examined Eiger/TNF protein distribution in similarly aged RasV12 or RasV12, sec15 mutant clones. We found that Eiger/TNF accumulated at the margins of RasV12 clones (Fig. 5A-C), consistent with the Eiger/TNF protein being exocytosed.
Fig. 5.

Sec15-dependent Eiger exocytosis suppresses JNK-induced cell death in RasV12 cells but promotes JNK activation in the surrounding wild-type cells. (A-Y) Comparable z-sections showing RasV12 or RasV12, sec15 double mutant clones or RasV12, eiger-RNAi clones (green) stained with anti-Eiger (red) or anti-phospho-JNK (pJNK, red) to detect JNK signaling activity or anti-active caspase 3 to detect apoptotic cells, and with DAPI (blue) to mark DNA. Gray panels show Eiger, phospho-JNK or caspase 3 channels alone. The dotted green lines denote clone boundaries. C,J and Q are z-section views. Eiger is concentrated at the boundary of RasV12 clones (A-C) but not at the boundary of RasV12, sec15 (D,E) or RasV12, eiger-RNAi (F,G) clones. Eiger is moderately retained in sec15 clones (V,W) but is present at higher levels in RasV12, sec15 double mutant clones (D,E). Wild-type cells surrounding RasV12 clones (H-J), but not wild-type cells surrounding RasV12, sec15 (K,L) or RasV12, eiger-RNAi (M,N) clones show elevated phospho-JNK levels. RasV12, sec15 double mutant clones show elevated phospho-JNK levels (K,L). Correspondingly, cell death can be detected in wild-type cells surrounding RasV12 clones (O-Q) but not in wild-type cells surrounding RasV12, sec15 double mutant clones. Instead, cell death is concentrated inside the double mutant clones (R,S). (T,U) Confocal projection showing discs containing RasV12, sec15, bsk-DN triple mutant clones stained with DAPI to mark nuclei and stained for active caspase-3 to detect apoptotic cells. The caspase channel alone is shown in U. (X,Y) sec15 mutant cells stained with DAPI and anti-phospho-JNK. (Z) Quantification of caspase-positive cells in RasV12 single or RasV12, sec15 double or RasV12, sec15, bsk-DN triple mutant clones. Error bars denote standard deviations from the mean for each genotype analyzed. P values are derived from a two-tailed distribution with two-sample equal variance t-test. Scale bars: 5 μm.
Importantly, examination of RasV12, sec15 double mutant clones revealed that Eiger/TNF no longer accumulated at the boundary, but rather accumulated within the clones (Fig. 5D,E). The sec15 mutant clones showed a moderate retention of Eiger/TNF (Fig. 5V,W). We then depleted Eiger/TNF specifically in the RasV12 cells and found that this prevented the accumulation of Eiger/TNF that is otherwise seen as a ring around the clones (Fig. 5F,G).
Furthermore, the cellular release of soluble TNF from its precursor transmembrane form and its resulting activity are mediated by the TNF-converting enzyme, Tace (Black et al., 1997; Blobel, 1997; Moss et al., 1997). Eiger/TNF contains a Tace cleavage site equivalent to TNFα cleavage site in vertebrates (Kauppila et al., 2003; Narasimamurthy et al., 2009). We first verified that Drosophila Tace plays a similar role in Eiger/TNF signaling. First, we found that RNAi knockdown of the sole Drosophila Tace in the developing eye suppressed the Eiger/TNF-mediated small-eye phenotype (supplementary material Fig. S3A-D). Similar results were obtained in wing imaginal discs (data not shown). We subsequently tested for a role for Tace in Eiger/TNF non-autonomous signaling using the hanging-eye phenotype model (Narasimamurthy et al., 2009). We found that coexpression of eiger and Tace in cells rescued by expression of the dominant-negative JNK allele bsk-DN in the developing eye (GMR>egr,Tace,bsk-DN) produced a hanging-eye phenotype (supplementary material Fig. S3F,F′). The majority of GMR>egr,Tace,bsk-DN animals die in late pupal stages (73%, N=157) and show a ring of necrotic tissue around the eyes (supplementary material Fig. S3F″). Interestingly, the GMR>egr,bsk-DN animals did not have a dramatic hanging-eye phenotype (supplementary material Fig. S3E). These data indicate that Tace plays an important role in the secretion and signaling of Eiger. Finally, we knocked down Tace by RNAi in the RasV12 cells and found that this abolished Eiger/TNF accumulation around the RasV12 clones (supplementary material Fig. S4). Collectively, these data indicate that the pool of Eiger/TNF seen around RasV12 clones originates from RasV12 cells, and Sec15 plays an important role in Eiger/TNF protein export.
Eiger/TNF normally activates JNK signaling to trigger cell death (Igaki et al., 2009; Moreno et al., 2002). Correspondingly, we found that JNK activity was elevated inside the RasV12, sec15 double mutant clones compared with that of RasV12 or sec15 mutant clones (Fig. 5H,L,X,Y). Increased JNK signaling correlated with increased ectopic cell death inside the RasV12, sec15 double mutant clones compared with RasV12 single mutant clones, as measured by activated caspase-3 immunoreactivity, a cell death indicator (58.74±19.48%; N=257 versus 6.99±4.32%; N=699 caspase-positive cells, respectively; mean±s.d.; Fig. 5O-Q versus Fig. 5R,S; Fig. 5Z). Furthermore, concomitant suppression of JNK signaling, by expressing the dominant-negative JNK allele bsk-DN, restored viability to RasV12, sec15 clones (3.85±5.8%; N=408 caspase-positive cells; Fig. 5T,U,Z). We conclude that Sec15-dependent exocytosis of Eiger/TNF downregulates JNK to protect RasV12 cells from apoptosis.
Eiger/TNF activates JAK-STAT signaling non-autonomously
Although blocking JNK signaling in RasV12, sec15 double mutant clones suppresses cell death, it fails to fully restore RasV12 tumor growth (supplementary material Fig. S5A,B). This suggests that Sec15 contributes to RasV12 tumor growth by an additional mechanism. Similar to vertebrate TNF, Eiger can act non-autonomously (Cordero et al., 2010; Pérez-Garijo et al., 2013). In addition, it has been shown that JNK signaling can non-autonomously stimulate the proliferation of adjacent cells (Ryoo et al., 2004; Wu et al., 2010), thus we wondered whether the Sec15-dependent accumulation of Eiger/TNF in the surrounding wild-type cells activates JNK to promote RasV12 tumor growth. We tested whether JNK is activated in the wild-type cells surrounding RasV12 clones. JNK activity was elevated in wild-type cells surrounding RasV12tumors (Fig. 5H-J), but not in wild-type cells surrounding the Eiger/TNF-secretion-defective RasV12, sec15 double mutant clones or the Eiger/TNF RNAi-depleted RasV12 clones (Fig. 5K-N). In agreement with this and previous findings (Karim and Rubin, 1998), we could detect activated caspase-positive cells around RasV12 tumors (Fig. 5O-Q). Therefore, increased Eiger/TNF secretion by RasV12 cells leads to Eiger/TNF accumulation and activation of JNK signaling in surrounding wild-type cells. This is consistent with earlier reports of Eiger/TNF activating JNK non-autonomously (Cordero et al., 2010; Pérez-Garijo et al., 2013).
We have previously shown that JNK activation triggered by cell polarity defects can induce the expression of JAK-STAT ligands encoded by the unpaired (upd) genes. Upd ligands cooperate with RasV12 through non-autonomous JAK-STAT activation (Wu et al., 2010). Blocking JNK signaling in RasV12 cells has no obvious effect on their proliferation (Igaki et al., 2006), supporting the possibility that the observed JNK activation in surrounding wild-type cells could promote RasV12 tumor growth by a non-cell autonomous mechanism. We thus wondered whether Eiger/TNF-exocytosis-mediated JNK activation in the surrounding cells promotes the growth of RasV12 cells. We first examined Upd expression levels using the Upd-lacZ reporter line (Sun et al., 1995) and found that Upd was particularly up-regulated in wild-type cells surrounding RasV12 clones (supplementary material Fig. S6D). Next, we used a JAK-STAT activity GFP reporter (Bach et al., 2007), which includes the promoter fragment of Socs36E, a transcriptional target of JAK-STAT signaling (Karsten et al., 2002), to examine JAK-STAT activity level in RasV12 cells. JAK-STAT was upregulated in RasV12 cells and in the surrounding Upd-producing wild-type cells (Fig. 6A). This increase was more robust in larger RasV12 clones than in smaller ones (supplementary material Fig. S6B, arrow versus boxed area). Similarly, Stat92E immunoreactivity, which is an indicator of JAK-STAT pathway activation (Chen et al., 2002; Johansen et al., 2003), was elevated in RasV12 discs (supplementary material Fig. S6F). In a complementary experiment, we found that the transcription level of Socs36E was elevated in RasV12 discs compared with that of wild-type control discs (Fig. 6F), consistent with the STAT-GFP and Stat92E results. This indicates that JAK-STAT signaling is activated in RasV12 cells.
Fig. 6.

Eiger/TNF exocytosis activates JAK-STAT signaling through JNK. (A) The JAK-STAT activity GFP-reporter line shows pathway activation in RasV12 clones. The lower panel shows STAT-GFP alone. (B) dTAK1-rescued RasV12 clones (red) induced in dtak1 hemizygous mutant discs do not show JAK-STAT GFP (green) induction. Lower panel shows STAT-GFP alone. The red outlines show clone boundaries in A and B. (C,D) RasV12 or RasV12, dom-RNAi double mutant clones stained with DAPI to mark nuclei and with antibody against the mitotic marker, phospho-histone-3 (PH3). The majority of RasV12 clones are phospho-histone-3 positive (C). The lower panel of C represents a magnified view of DAPI and phospho-histone-3 from the boxed area in the upper panel. RasV12, dom-RNAi clones (D) show reduced phospho-histone-3 staining. The lower panel shows the phospho-histone-3 channel alone. The green outlines show clone boundaries in D. Scale bars: 5 μm. (E) Quantification of C and D. (F) RT-PCR experiment measuring Socs36E levels relative to expression levels of the housekeeping gene Rp49. Normalized fold differences in Socs36E expression levels between RasV12 and wild-type (WT) or between RasV12, eiger-RNAi (eiger RNAi-knockdown specifically in RasV12 clones) and RasV12 or between RasV12, sec15 double mutant and RasV12 clones are shown. Experiments were performed in triplicate and standard deviations were derived from the coefficient variations of experimental and control samples.
To test whether the observed activation of JAK-STAT in RasV12 cells is due to increased Eiger/TNF exocytosis to neighboring wild-type cells and the resulting JNK activation there, we first blocked Eiger/TNF exocytosis to the surrounding wild-type cells and examined JAK-STAT activation. We found that depleting Eiger/TNF protein specifically in RasV12 cells (RasV12, egr-RNAi clones) or preventing the secretion of Eiger/TNF (RasV12, sec15 double mutant cells) or reducing the protein dosage of the Eiger/TNF receptor (Wengen) all suppressed JAK-STAT activity compared to that of RasV12 cells (Fig. 6F; supplementary material Fig. S6E-G). Consistent with these results, wild-type cells surrounding RasV12, sec15 double mutant clones failed to up-regulate Upd (supplementary material Fig. S6H). Next, we tested whether directly blocking JNK activation specifically in the surrounding wild-type cells would suppress JAK-STAT activation in RasV12 clones. To prevent JNK activation specifically in wild-type cells surrounding RasV12 clones, we induced RasV12 clones in JNKKK dtak1 (Tak1 – FlyBase) mutant discs, but rescued JNKKK/dTAK1 function with a UAS-dTAK1 transgene only in the RasV12 clones (i.e. clones of cells co-expressing RasV12 and dTAK1 in dtak1-null mutant discs). These clones, hereafter referred to as dtak1//RasV12 clones, did not show STAT-GFP expression (Fig. 6B), mimicking RasV12, egr-RNAi clones. Finally, we removed the surrounding wild-type cells by expressing RasV12 throughout the developing eye discs [Eyeless-Gal4>RasV12, (ey>RasV12)] and examined JAK-STAT activation. These discs did not show broad STAT-GFP induction. Instead, we detected isolated patches of STAT-GFP-positive cells near caspase-positive cells (supplementary material Fig. S6C). Taken together, these findings indicate that it is the activation of JNK signaling specifically in the surrounding wild-type cells that non-cell autonomously stimulates JAK-STAT signaling in RasV12 cells.
JAK-STAT signaling promotes oncogenic Ras-mediated overgrowth
Finally, we sought to investigate whether this Eiger/TNF exocytosis-triggered JNK-JAK-STAT signaling relay contributes to RasV12 tumor growth. If this is the case, then it should be expected that (1) depleting Eiger/TNF in RasV12 cells (RasV12, egr-RNAi), (2) inducing RasV12 clones in eiger mutant animals, (3) directly blocking JNK in wild-type cells surrounding RasV12 cells (RasV12//dtak1, juxtaposed dtak1 and RasV12 mutant cells) or (4) preventing JAK-STAT activation in RasV12 cells would result in suppression of overgrowth. Indeed, RasV12, egr-RNAi or RasV12 clones induced in eiger mutant animals showed reduced growth compared with that of mosaic RasV12 clones (Fig. 7A-C,G). Moreover, RasV12, Tace-RNAi clones, which display no Eiger/TNF accumulation around the clones, showed reduced growth compared with that of RasV12 clones. Tace-RNAi alone showed no effect on growth (supplementary material Fig. S3C,G-J). Similarly, RasV12//dtak1 mutant cells showed reduced growth compared to that of controls (Fig. 7E-G).
Fig. 7.
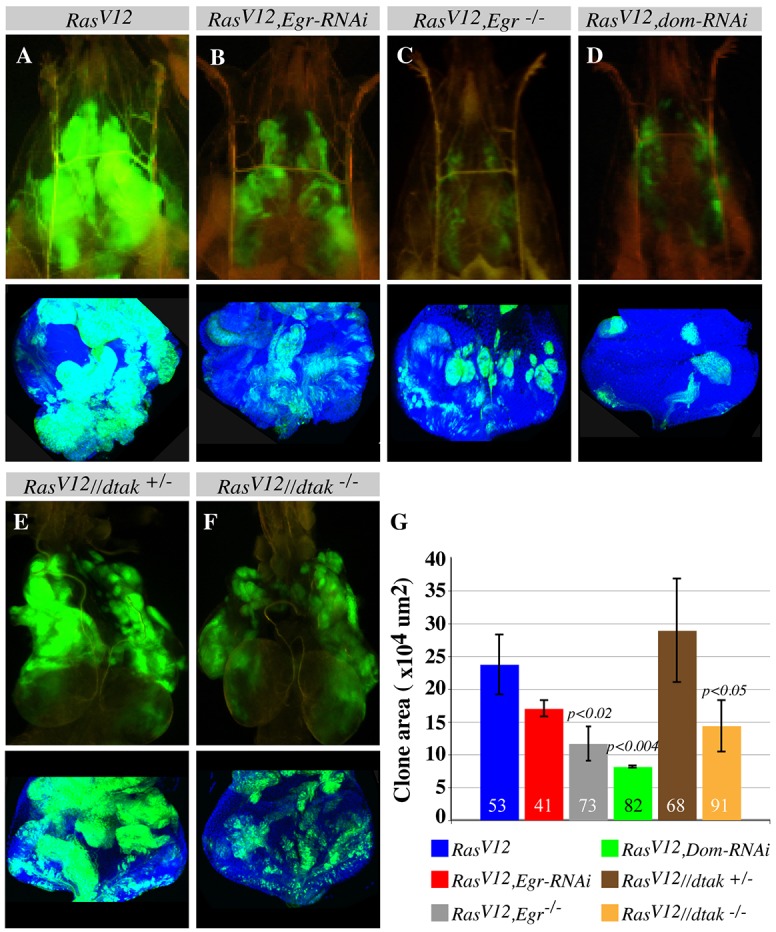
Eiger/TNF exocytosis activation of JAK-STAT signaling promotes RasV12 tumor overgrowth. (A-F) Clones (green) from similarly aged intact third instar larvae (A-D), dissected cephalic regions (E,F) or dissected eye discs (lower panels). RasV12 clones overgrow (A). eiger/Tnf RNAi knockdown specifically in the RasV12 clones (B) or generating RasV12 clones in eiger mutant animals (C) or blocking JAK-STAT signaling by using Domeless RNAi (dom-RNAi) (D) suppresses the overgrowth of RasV12 clones. Dissected cephalic regions (E,F) or dissected eye discs (lower panels) show that dTAK1-rescued RasV12 clones induced in dtak1 hemizygous males (RasV12//dtak−/−) (F), exhibit reduced growth compared with dTAK1-rescued RasV12 clones induced in the siblings (dtak1 heterozygous females; RasV12//dtak+/−) (E). (G) Quantification of A-F. Confocal stacks from similarly aged animals of the indicated genotypes and the image analysis software Imaris were used to measure clone (green) sizes. Error bars show s.d. The numbers in the bars indicate the sample size for the corresponding genotype.
Furthermore, removing the surrounding wild-type cells by expressing RasV12 throughout the eye discs using eyFLP, Act>y+>Gal4, UAS-RasV12 or ey>RasV12 results only in moderate growth compared to the more pronounced overgrowth of mosaic RasV12 tissues (supplementary material Fig. S6I,J and see figure S1 in Wu et al., 2010). Consistent with reduced growth, ey>RasV12 discs do not cause animal lethality, whereas the overgrowth of mosaic RasV12 clones kills the animal during pupal stages (Fig. 1K; supplementary material Fig. S6K). We then tested whether preventing JAK-STAT activation in RasV12 cells, by co-expressing an RNAi transgene against the JAK-STAT receptor, Domeless, (RasV12, dom-RNAi clones) suppresses RasV12 tumor growth. RasV12, dom-RNAi clones showed a reduced mitotic potential as determined by the percentage of phosphorylated-histone-3-positive cells (6.3±5%, N=214 for RasV12, dom-RNAi clones and 49.5±29.5%, N=214 for RasV12; mean±s.d.; Fig. 6C,D,E) and correspondingly showed reduced tumor growth (Fig. 7A,D,G). Similarly, blocking JAK-STAT activity in ey>RasV12 (ey>RasV12, dom-RNAi) suppresses growth (supplementary material Fig. S5K,L). This suggests that the patches of dying cells contribute to the growth of RasV12 cells. Taken together, the above findings indicate that the accumulation of Eiger/TNF and the resulting activation of JNK signaling in the surrounding wild-type cells non-autonomously promote RasV12 tumor growth through JAK-STAT activation (Fig. 8).
Fig. 8.
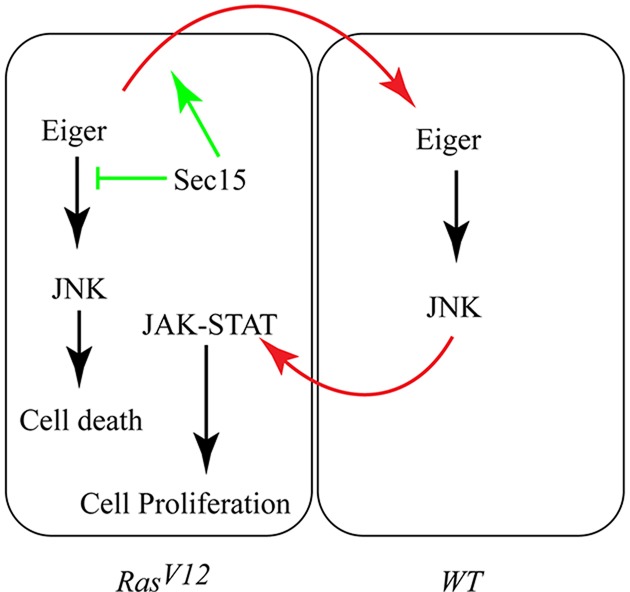
Oncogenic Ras promotes overgrowth by stimulating Eiger/TNF exocytosis and activating JNK-JAK-STAT non-autonomous proliferation signaling. Eiger normally activates JNK to trigger cell death. JAK-STAT signaling on the other end promotes cell proliferation (black arrows). Sec15 blocks Eiger-induced cell death (green inhibition arrow) by promoting (top green arrow) the exocytosis of Eiger to the surrounding wild-type cells (top red arrow). This results in JNK activation in the surrounding wild-type (WT) cells, which non-autonomously activates the JAK-STAT pathway (bottom red arrow) to promote oncogenic Ras tumor growth.
DISCUSSION
Earlier efforts to understand Ras signaling during normal development in model organisms led to the elucidation of the Ras/MAPK signaling cascade (Nishida and Gotoh, 1993; Simon et al., 1991; Sternberg and Horvitz, 1991). However, the signaling events that are elicited by oncogenic Ras to drive overgrowth are complex and not fully understood (reviewed by Mitin et al., 2005). Here, we used a Drosophila model and searched for mutations that can block RasV12 overgrowth in an in vivo synthetic screen. We identified sec15, a mutation in a gene encoding a component of the exocytosis machinery, as a synthetic suppressor of RasV12 tumor growth. Studying the underlying cause of this phenomenon led to the unexpected discovery of both cell-intrinsic and cell-extrinsic mechanisms that promote RasV12 overgrowth.
Oncogenic Ras stimulates Eiger/TNF exocytosis
We found that RasV12 tumors elevate exocytosis to promote rapid clearance of Eiger/TNF and hence avert JNK-mediated cell death. Oncogenic Ras could stimulate the exocyst through the Ral-specific guanine nucleotide exchange factors (RalGEFs). It has been shown that oncogenic Ras activates RalGEFs, which in turn activate the Ras-like small GTPases RalA and RalB (Lim et al., 2005; Urano et al., 1996; White et al., 1996; Wolthuis et al., 1998). Moreover, RalGEF-Ral signaling stimulates gene expression (Neel et al., 2011). It is therefore likely that Ral mediates the observed increased levels of exocyst proteins in RasV12 cells. Consistent with this, Sec15 transcription is increased in RasV12 tissues (supplementary material Fig. S7A). Furthermore, activated Rals interact with and stimulate the exocyst through Exo84 and Sec5 (Hamad et al., 2002; Moskalenko et al., 2002), suggesting that RalGEF-Ral signaling could be the underlying means by which RasV12 cells generally stimulate exocytosis. We cannot rule out the possibility that the exocytosis of other molecules is elevated.
Eiger/TNF exocytosis is mediated by the Sec15-Eiger/TNF interaction. Consistent with this, sec15 mutant cells retain Eiger/TNF. In contrast to RasV12, sec15 double mutant cells, sec15 mutant cells do not undergo apoptosis. This is likely owing to the fact that oncogenic Ras stimulates Eiger/TNF expression (supplementary material Fig. S7B) in addition to promoting Eiger/TNF exocytosis. The Sec15-Eiger/TNF interaction is interesting as it might represent a novel role of the exocyst in cargo selection. It is tempting to speculate that the cytosolic domain of Eiger/TNF could mediate its interaction with the exocyst. In the future it will be interesting to investigate the nature of this interaction, map out the relevant domains and elucidate a sorting mechanism. The finding that oncogenic Ras hijacks the exocytosis machinery to clear up pro-apoptotic ligands provides a new way by which tumor cells can evade cell death and an additional explanation of how exocytosis can modulate signal transduction.
Oncogenic Ras usurps the JNK-JAK-STAT non-autonomous signal to promote overgrowth
We have previously shown that JNK activation triggered by cell polarity defects can stimulate non-autonomous JAK-STAT signaling for proliferation and that JAK-STAT signaling can cooperate with RasV12 (Wu et al., 2010). Here, we discovered that one of the effects of oncogenic Ras itself is to hijack this non-autonomous JNK-JAK-STAT proliferation signaling cascade. We propose that Eiger/TNF, including Eiger/TNF from the nearby fast-secreting RasV12 cells, is endocytosed but rapidly cleared out of RasV12 cells by the exocyst. Elevated exocytosis in RasV12 cells creates an imbalance such that the amount of secreted Eiger/TNF is higher than that being internalized. This not only causes a RasV12 cell to retain less Eiger/TNF, but the endocytosis and exocytosis cycle also permits neighboring RasV12 cells to relay Eiger/TNF, resulting in the eventual accumulation of Eiger/TNF in the surrounding wild-type cells. Consistent with this, depleting Eiger/TNF or interfering with the exocyst specifically in RasV12 cells abolished Eiger/TNF accumulation around RasV12 clones. Eiger/TNF from RasV12 cells activates JNK signaling in the surrounding wild-type cells. JNK activation stimulates JAK-STAT signaling in RasV12 cells to promote tumor overgrowth. Congruent with this model, JNK activity and the JAK-STAT ligand Upd are upregulated in wild-type cells surrounding RasV12 clones. Preventing Eiger/TNF secretion (RasV12, sec15 double mutant clones or RasV12, egr-RNAi clones) suppresses JNK activation, Upd stimulation and JAK-STAT activation in the surrounding wild-type cells. Moreover, blocking JNK signaling specifically in wild-type cells surrounding RasV12 clones prevents JAK-STAT activation in RasV12 cells and suppresses tumor growth.
Interestingly, oncogenic Ras or wound-induced JNK activation does not cause invasion, in contrast to JNK induced by the cell polarity mutation scrib. This could be due to context-dependent JNK activation. However, we found that Sec15-RNAi suppresses the growth and invasion phenotypes of RasV12, scrib double mutant clones (supplementary material Fig. S8A-D) and rescues animal lethality (supplementary material Fig. S8C-E), highlighting the importance of exocytosis in promoting tumor growth and invasion.
Another striking result of our study is the realization of the importance of the interaction between RasV12 and the surrounding non-RasV12 cells in oncogenic Ras-mediated growth. This is best illustrated by the fact that a tissue in which oncogenic Ras is uniformly expressed grows less than a tissue containing both wild-type and oncogenic Ras-expressing cells. This is consistent with accumulating data from rodents and patients indicating that signaling events emanating from host/stromal cells promote tumor development (Bremnes et al., 2011; Lu et al., 2013; Mueller and Fusenig, 2004; Sounni and Noel, 2012). Additional significance is derived from recent studies of various solid tumors showing heterogeneity within each tumor (Diaz et al., 2012; Gerlinger et al., 2012; Gerstung et al., 2011; Marusyk and Polyak, 2010).
Our study reveals that an important effect of oncogenic Ras is the increased exocytosis of Eiger/TNF. Indeed, the fact that blocking exocytosis of Eiger/TNF can suppress oncogenic Ras-mediated growth highlights the importance of Eiger/TNF exocytosis in cancer biology. Exocytosis proteins are conserved and have been implicated in diverse human cancers types, including Ras cancers (Cheng et al. 2004, 2005; Issaq et al., 2010; Neel et al., 2011; Palmer et al., 2002), suggesting that a similar tumor-promoting mechanism could be at play in vertebrates. Because sec15 null mutation does not cause cell lethality and the gene is evolutionarily conserved, it suggests a new type of potential therapeutic targets.
MATERIALS AND METHODS
Fly lines
Animals were aged at 25°C on standard medium. The following fly lines were used in this study: (1) y w; FRT82B; (2) y w; FRT82B, UAS-RasV12/TM6B; (3) y w; FRT40A, UAS-RasV12; (4) y w; FRT82B, sec151 (H. Bellen, Baylor College of Medicine, Houston, TX, USA); (5) y w; UAS-RasV12; FRT82B, sec151/TM6B; (6) UAS-Sec15-RNAi (VDRC); (7) FRT40,UAS-RasV12;UAS-eiger-RNAi/TM6B; (8) w;UAS-RasV12; FRT82B,Stat92E06346/TM6B; (9) w;UAS-RasV12; FRT82B,UAS-dom (IR)/TM6B; (10) y w,ey-Flp1;act>y+>GAL4,UAS-GFP.S65T;FRT82B,tub-GAL80; (11) y w,ey-Flp1;act>y+>GAL4,UAS-myrRFP;FRT82B,tub-GAL80; (12) y w,ey-Flp1;tub-GAL80,FRT40A;act>y+>GAL4,UAS-GFP.S65T; (13) w;10XSTAT-GFP.1; (14) UAS-UAS-BskDN;eyFLP5,Act>y+>Gal4, UAS-GFP/Cyo;FRT82B, Tub-Gal80/TM6B; (15) Eyeless-GAL4 (Bloomington); (16) w,dTak1; ey-Flp5,act>y+>GAL4,UAS-GFP;FRT82B,tub-GAL80; (17) w,dTak1; ey-Flp5, act>y+>GAL4,UAS-myrRFP;FRT82B,tub-GAL80; (18) UAS-dTAK1; FRT82B, UAS-RasV12; (19) upd-lacZ; UAS-RasV12; sec151, FRT82B/Sb; (20) UAS-RasV12, UAS-Sec15-RNAi; 10XSTAT-GFP.1/TM6B; (21) ey-FLP5, act>y+>GAL4,UAS-GFP; (22) Sec6-RNAi (VDRC); (23) Sec8-RNAi (VDRC); (24) Wgn22 (P. Barker, McGill University, Montreal, Canada); (25) UAS-TACE-RNAi; (26) UAS-TACE; (27) UAS-Rab5:GFP (Bloomington); (28) GMR-Gal4; (29) UAS-Eigerw; and (30) UAS-Bsk-DN.
Staining and imaging
Eye-antenna discs were dissected, fixed and stained as described previously (Igaki et al., 2006; Pagliarini and Xu, 2003; Wu et al., 2010). Tumor and adult eye size analyses were carried out on a Leica MZ FLIII fluorescence stereomicroscope equipped with a camera. Samples were examined by confocal microscopy with a Zeiss LSM510 Meta system. Images were analyzed and processed with Imaris (Bitplane) and Illustrator (Adobe) software, respectively. The following primary antibodies were used: guinea pig anti-Sec6 at 1:1000 [U. Tepass (University of Toronto, Canada) and H. Bellen]; guinea pig anti-Sec8 at 1:1000 (U. Tepass and H. Bellen); guinea pig anti-Sec15 at 1:1000 (H. Bellen); rat anti-Rab11 at 1:1000 (S. Cohen, University of Kansas, USA); rabbit anti-Rab11 at 1:1000 (D. Ready, Purdue University, IN, USA); rabbit anti-Stat92E at 1:1000 (S. Hou); rabbit anti-phospho-histone-3 at 1:1000; rabbit anti-cleaved caspase-3 at 1:500; rabbit anti-Eiger polyclonal antibody R1 at 1:50 (T. Igaki, Kobe University, Japan); mouse anti-phospho-JNK monoclonal antibody G9 at 1:250. Secondary antibodies were from Invitrogen. TUNEL staining was performed using the Apoptag Red kit from Chemicon.
Isosurface rendering
Projections of serial confocal sections were used to generate three-dimensional graphical images using the image analysis software Imaris. The clipping plane function was used to section across the resulting graphical images.
Vesicle trafficking studies
The whole central nervous system, including the attached eye discs, was dissected in Schneider medium and incubated in Schneider medium supplemented with 0.5 mg/ml Dextran-Alexa-546 for 2 h. Half of the samples were immediately washed and fixed. Eye discs were separated and mounted in DAPI-Vectashield. The remaining half of the samples were washed and incubated in Schneider medium for an additional 2 h (chase), rinsed in PBS, fixed and mounted as above.
Western blots and immunoprecipitation
Third instar imaginal discs bearing wild-type or RasV12-expressing cells were homogenized in lysis buffer (50 mM HEPES pH 7.5, 150 mM KCl, 5 mM MgCl2, supplemented with protease inhibitor tablets; Roche). Samples were separated by SDS-PAGE, transferred onto nitrocellulose membrane and blotted with anti-Sec15, anti-Sec6, anti-Sec8. Lysates were diluted 1:100 and blotted with anti-α-Tubulin antibodies as a loading control. For immunoprecipitation experiments, 500 µl of lysate obtained from discs containing RasV12 clones was pre-cleared with Protein A-agarose beads for 1 h at 4°C, split equally into two tubes and incubated either with 2 μl of anti-Eiger or 2 μl of anti-GFP antibodies for 4 h at 4°C. Lysates were then incubated with Protein A-agarose beads for 1 h at room temperature. For pull downs, beads were precipitated and washed three times in modified lysis buffer containing 0.5% Triton X-100. Samples were separated by SDS-PAGE, transferred onto nitrocellulose membrane and blotted with antibodies against Sec15, Sec8, Sec5 and α-Tubulin.
Real-time polymerase chain reaction
Total RNA from eye-antenna imaginal discs containing wild-type or mutant clones was isolated using a Trizol RNA extraction method. The SuperScript III First-Strand Synthesis System kit was used to synthesize cDNA. Real-time PCR was carried out on an Applied Biosystems machine using the SYBR Green fast kit following the manufacturer's instructions. Relative gene expression was obtained from triplicate runs normalized to Rp49 (RpL32 – FlyBase) as the endogenous control. The following primers were used: Rp49, 5′-GGCCCAAGATCGTGAAGAAG-3′ and 5′-ATTTG-TGCGACAGCTTAGCATATC-3′; Socs36E, 5′-GTCGCTGCCAGTCAG-CAA-3′ and 5′-TGCTCCCATTGAAAGTGCTTT-3′; Sec15, 5′-CATTA-TGCTGTCCATCAACACCTT-3′ and 5′-CAGCGTTGCAGTAAAACCT-CATT-3′; eiger, 5′-GATGGTCTGGATTCCATTGC-3′ and 5′-TAGTCT-GCGCCAACATCATC-3′.
Clones of mutant cells in the eye-antennal discs were generated using standard MARCM system (Lee and Luo, 2001) at 25°C. Hanging-eye phenotype experiments were carried out at 29°C. Immunostaining and immunoprecipitation experiments were performed as described previously (Chabu and Doe, 2008; Pagliarini and Xu, 2003).
Supplementary Material
Acknowledgements
We thank U. Tepass, H. Bellen, D. Ready, S. Cohen and the Vienna Drosophila RNAi Center for kindly providing reagents, and S. Landrette and T. Klein (Yale University, CT, USA) for comments.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
C.C. and T.X. designed the research, C.C. performed experiments and analyzed the data. C.C. and T.X. wrote the manuscript.
Funding
C.C. is funded by a National Institutes of Health/National Cancer Institute (NIH/NCI) post-doctoral grant [grant number 1F32CA142118-01A1]. This work was also supported by a grant from NIH/NCI to T.X. [R01 CA69408-17] T.X. is a Howard Hughes Medical Institute Investigator. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.108092/-/DC1
References
- Alessi D. R., Saito Y., Campbell D. G., Cohen P., Sithanandam G., Rapp U., Ashworth A., Marshall C. J. and Cowley S. (1994). Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 13, 1610-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghar U., Hawkes E. and Cunningham D. (2010). Predictive and prognostic biomarkers for targeted therapy in metastatic colorectal cancer. Clin. Colorectal Cancer 9, 274-281 10.3816/CCC.2010.n.040 [DOI] [PubMed] [Google Scholar]
- Bach E. A., Ekas L. A., Ayala-Camargo A., Flaherty M. S., Lee H., Perrimon N. and Baeg G.-H. (2007). GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr. Patterns 7, 323-331 10.1016/j.modgep.2006.08.003 [DOI] [PubMed] [Google Scholar]
- Barbie D. A., Tamayo P., Boehm J. S., Kim S. Y., Moody S. E., Dunn I. F., Schinzel A. C., Sandy P., Meylan E., Scholl C. et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108-112 10.1038/nature08460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black R. A., Rauch C. T., Kozlosky C. J., Peschon J. J., Slack J. L., Wolfson M. F., Castner B. J., Stocking K. L., Reddy P., Srinivasan S. et al. (1997). A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385, 729-733 10.1038/385729a0 [DOI] [PubMed] [Google Scholar]
- Blobel C. P. (1997). Metalloprotease-disintegrins: links to cell adhesion and cleavage of TNF alpha and Notch. Cell 90, 589-592 10.1016/S0092-8674(00)80519-X [DOI] [PubMed] [Google Scholar]
- Blum R. and Kloog Y. (2005). Tailoring Ras-pathway--inhibitor combinations for cancer therapy. Drug Resist. Updat. 8, 369-380 10.1016/j.drup.2005.11.002 [DOI] [PubMed] [Google Scholar]
- Bos J. L. (1989). ras oncogenes in human cancer: a review. Cancer Res. 49, 4682-4689. [PubMed] [Google Scholar]
- Brandt S. M., Dionne M. S., Khush R. S., Pham L. N., Vigdal T. J. and Schneider D. S. (2004). Secreted bacterial effectors and host-produced Eiger/TNF drive death in a Salmonella-infected fruit fly. PLoS Biol. 2, e418 10.1371/journal.pbio.0020418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremnes R. M., Al-Shibli K., Donnem T., Sirera R., Al-Saad S., Andersen S., Stenvold H., Camps C. and Busund L.-T. (2011). The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: emphasis on non-small cell lung cancer. J. Thorac. Oncol. 6, 824-833 10.1097/JTO.0b013e3182037b76 [DOI] [PubMed] [Google Scholar]
- Chabu C. and Doe C. Q. (2008). Dap160/intersectin binds and activates aPKC to regulate cell polarity and cell cycle progression. Development 135, 2739-2746 10.1242/dev.024059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.-W., Chen X., Oh S.-W., Marinissen M. J., Gutkind J. S. and Hou S. X. (2002). mom identifies a receptor for the Drosophila JAK/STAT signal transduction pathway and encodes a protein distantly related to the mammalian cytokine receptor family. Genes Dev. 16, 388-398 10.1101/gad.955202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K. W., Lahad J. P., Kuo W.-L., Lapuk A., Yamada K., Auersperg N., Liu J., Smith-McCune K., Lu K. H., Fishman D. et al. (2004). The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat. Med. 10, 1251-1256 10.1038/nm1125 [DOI] [PubMed] [Google Scholar]
- Cheng K. W., Lahad J. P., Gray J. W. and Mills G. B. (2005). Emerging role of RAB GTPases in cancer and human disease. Cancer Res. 65, 2516-2519 10.1158/0008-5472.CAN-05-0573 [DOI] [PubMed] [Google Scholar]
- Chi C., Zhu H., Han M., Zhuang Y., Wu X. and Xu T. (2010). Disruption of lysosome function promotes tumor growth and metastasis in Drosophila. J. Biol. Chem. 285, 21817-21823 10.1074/jbc.M110.131714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero J. B., Macagno J. P., Stefanatos R. K., Strathdee K. E., Cagan R. L. and Vidal M. (2010). Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev. Cell 18, 999-1011 10.1016/j.devcel.2010.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley S., Paterson H., Kemp P. and Marshall C. J. (1994). Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77, 841-852 10.1016/0092-8674(94)90133-3 [DOI] [PubMed] [Google Scholar]
- Cox A. D. and Der C. J. (2003). The dark side of Ras: regulation of apoptosis. Oncogene 22, 8999-9006 10.1038/sj.onc.1207111 [DOI] [PubMed] [Google Scholar]
- Diaz L. A. Jr, Williams R. T., Wu J., Kinde I., Hecht J. R., Berlin J., Allen B., Bozic I., Reiter J. G., Nowak M. A. (2012). The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486, 537-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollar G., Struckhoff E., Michaud J. and Cohen R. S. (2002). Rab11 polarization of the Drosophila oocyte: a novel link between membrane trafficking, microtubule organization, and oskar mRNA localization and translation. Development 129, 517-526. [DOI] [PubMed] [Google Scholar]
- Downward J. (1998). Ras signalling and apoptosis. Curr. Opin. Genet. Dev. 8, 49-54 10.1016/S0959-437X(98)80061-0 [DOI] [PubMed] [Google Scholar]
- Finger F. P. and Novick P. (1998). Spatial regulation of exocytosis: lessons from yeast. J. Cell Biol. 142, 609-612 10.1083/jcb.142.3.609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger F. P., Hughes T. E. and Novick P. (1998). Sec3p is a spatial landmark for polarized secretion in budding yeast. Cell 92, 559-571 10.1016/S0092-8674(00)80948-4 [DOI] [PubMed] [Google Scholar]
- Gerlinger M., Rowan A. J., Horswell S., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., Tarpey P. et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883-892 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstung M., Eriksson N., Lin J., Vogelstein B. and Beerenwinkel N. (2011). The temporal order of genetic and pathway alterations in tumorigenesis. PLoS ONE 6, e27136 10.1371/journal.pone.0027136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grindstaff K. K., Yeaman C., Anandasabapathy N., Hsu S.-C., Rodriguez-Boulan E., Scheller R. H. and Nelson W. J. (1998). Sec6/8 complex is recruited to cell-cell contacts and specifies transport vesicle delivery to the basal-lateral membrane in epithelial cells. Cell 93, 731-740 10.1016/S0092-8674(00)81435-X [DOI] [PubMed] [Google Scholar]
- Guo W., Grant A. and Novick P. (1999). Exo84p is an exocyst protein essential for secretion. J. Biol. Chem. 274, 23558-23564 10.1074/jbc.274.33.23558 [DOI] [PubMed] [Google Scholar]
- Hamad N. M., Elconin J. H., Karnoub A. E., Bai W., Rich J. N., Abraham R. T., Der C. J. and Counter C. M. (2002). Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 16, 2045-2057 10.1101/gad.993902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heider M. R. and Munson M. (2012). Exorcising the exocyst complex. Traffic 13, 898-907 10.1111/j.1600-0854.2012.01353.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T., Pagliarini R. A. and Xu T. (2006). Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 16, 1139-1146 10.1016/j.cub.2006.04.042 [DOI] [PubMed] [Google Scholar]
- Igaki T., Pastor-Pareja J. C., Aonuma H., Miura M. and Xu T. (2009). Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell 16, 458-465 10.1016/j.devcel.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issaq S. H., Lim K.-H. and Counter C. M. (2010). Sec5 and Exo84 foster oncogenic ras-mediated tumorigenesis. Mol. Cancer Res. 8, 223-231 10.1158/1541-7786.MCR-09-0189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafar-Nejad H., Andrews H. K., Acar M., Bayat V., Wirtz-Peitz F., Mehta S. Q., Knoblich J. A. and Bellen H. J. (2005). Sec15, a component of the exocyst, promotes notch signaling during the asymmetric division of Drosophila sensory organ precursors. Dev. Cell 9, 351-363 10.1016/j.devcel.2005.06.010 [DOI] [PubMed] [Google Scholar]
- Johansen K. A., Iwaki D. D. and Lengyel J. A. (2003). Localized JAK/STAT signaling is required for oriented cell rearrangement in a tubular epithelium. Development 130, 135-145 10.1242/dev.00202 [DOI] [PubMed] [Google Scholar]
- Jue D. M., Sherry B., Luedke C., Manogue K. R. and Cerami A. (1990). Processing of newly synthesized cachectin/tumor necrosis factor in endotoxin-stimulated macrophages. Biochemistry 29, 8371-8377 10.1021/bi00488a025 [DOI] [PubMed] [Google Scholar]
- Karim F. D. and Rubin G. M. (1998). Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 125, 1-9. [DOI] [PubMed] [Google Scholar]
- Karsten P., Häder S. and Zeidler M. P. (2002). Cloning and expression of Drosophila SOCS36E and its potential regulation by the JAK/STAT pathway. Mech. Dev. 117, 343-346 10.1016/S0925-4773(02)00216-2 [DOI] [PubMed] [Google Scholar]
- Kauppila S., Maaty W. S. A., Chen P., Tomar R. S., Eby M. T., Chapo J., Chew S., Rathore N., Zachariah S., Sinha S. K. et al. (2003). Eiger and its receptor, Wengen, comprise a TNF-like system in Drosophila. Oncogene 22, 4860-4867 10.1038/sj.onc.1206715 [DOI] [PubMed] [Google Scholar]
- Kolch W., Heidecker G., Lloyd P. and Rapp U. R. (1991). Raf-1 protein kinase is required for growth of induced NIH/3T3 cells. Nature 349, 426-428 10.1038/349426a0 [DOI] [PubMed] [Google Scholar]
- Lee T. and Luo L. (2001). Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 24, 251-254 10.1016/S0166-2236(00)01791-4 [DOI] [PubMed] [Google Scholar]
- Li L. and Chin L. S. (2003). The molecular machinery of synaptic vesicle exocytosis. Cell. Mol. Life Sci. 60, 942-960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lièvre A., Blons H. and Laurent-Puig P. (2010). Oncogenic mutations as predictive factors in colorectal cancer. Oncogene 29, 3033-3043 10.1038/onc.2010.89 [DOI] [PubMed] [Google Scholar]
- Lim K.-H., Baines A. T., Fiordalisi J. J., Shipitsin M., Feig L. A., Cox A. D., Der C. J. and Counter C. M. (2005). Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 7, 533-545 10.1016/j.ccr.2005.04.030 [DOI] [PubMed] [Google Scholar]
- Lu J., Ye X., Fan F., Xia L., Bhattacharya R., Bellister S., Tozzi F., Sceusi E., Zhou Y., Tachibana I. et al. (2013). Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of jagged-1. Cancer Cell 23, 171–185 10.1016/j.ccr.2012.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Emanuele M. J., Li D., Creighton C. J., Schlabach M. R., Westbrook T. F., Wong K.-K. and Elledge S. J. (2009). A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137, 835-848 10.1016/j.cell.2009.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour S. J., Matten W. T., Hermann A. S., Candia J. M., Rong S., Fukasawa K., Vande Woude G. F. and Ahn N. G. (1994). Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265, 966-970 10.1126/science.8052857 [DOI] [PubMed] [Google Scholar]
- Marusyk A. and Polyak K. (2010). Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta 1805, 105-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta S. Q., Hiesinger P. R., Beronja S., Zhai R. G., Schulze K. L., Verstreken P., Cao Y., Zhou Y., Tepass U., Crair M. C. et al. (2005). Mutations in Drosophila sec15 reveal a function in neuronal targeting for a subset of exocyst components. Neuron 46, 219-232 10.1016/j.neuron.2005.02.029 [DOI] [PubMed] [Google Scholar]
- Mitin N., Rossman K. L. and Der C. J. (2005). Signaling interplay in Ras superfamily function. Curr. Biol. 15, R563 10.1016/j.cub.2005.07.010 [DOI] [PubMed] [Google Scholar]
- Moreno E., Yan M. and Basler K. (2002). Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 12, 1263-1268 10.1016/S0960-9822(02)00954-5 [DOI] [PubMed] [Google Scholar]
- Moskalenko S., Henry D. O., Rosse C., Mirey G., Camonis J. H. and White M. A. (2002). The exocyst is a Ral effector complex. Nat. Cell Biol. 4, 66-72 10.1038/ncb728 [DOI] [PubMed] [Google Scholar]
- Moss M. L., Jin S.-L., Milla M. E., Burkhart W., Carter H. L., Chen W.-J., Clay W. C., Didsbury J. R., Hassler D., Hoffman C. R. et al. (1997). Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 385, 733-736 10.1038/385733a0 [DOI] [PubMed] [Google Scholar]
- Mueller M. M. and Fusenig N. E. (2004). Friends or foes - bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 4, 839-849 10.1038/nrc1477 [DOI] [PubMed] [Google Scholar]
- Narasimamurthy R., Geuking P., Ingold K., Willen L., Schneider P. and Basler K. (2009). Structure-function analysis of Eiger, the Drosophila TNF homolog. Cell Res. 19, 392-394 10.1038/cr.2009.16 [DOI] [PubMed] [Google Scholar]
- Neel N. F., Martin T. D., Stratford J. K., Zand T. P., Reiner D. J., Der C. J. (2011). The RalGEF-Ral effector signaling network: the road less traveled for anti-ras drug discovery. Genes Cancer 2, 275-287 10.1177/1947601911407329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida E. and Gotoh Y. (1993). The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem. Sci. 18, 128-131 10.1016/0968-0004(93)90019-J [DOI] [PubMed] [Google Scholar]
- Novick P., Field C. and Schekman R. (1980). Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 21, 205-215 10.1016/0092-8674(80)90128-2 [DOI] [PubMed] [Google Scholar]
- Pagliarini R. A. and Xu T. (2003). A genetic screen in Drosophila for metastatic behavior. Science 302, 1227-1231 10.1126/science.1088474 [DOI] [PubMed] [Google Scholar]
- Palmer R. E., Lee S. B., Wong J. C., Reynolds P. A., Zhang H., Truong V., Oliner J. D., Gerald W. L. and Haber D. A. (2002). Induction of BAIAP3 by the EWS-WT1 chimeric fusion implicates regulated exocytosis in tumorigenesis. Cancer Cell 2, 497-505 10.1016/S1535-6108(02)00205-2 [DOI] [PubMed] [Google Scholar]
- Pérez-Garijo A., Fuchs Y. and Steller H. (2013). Apoptotic cells can induce non-autonomous apoptosis through the TNF pathway. Elife 2, e01004 10.7554/eLife.01004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu R.-G., Chen J., Kirn D., McCormick F. and Symons M. (1995). An essential role for Rac in Ras transformation. Nature 374, 457-459 10.1038/374457a0 [DOI] [PubMed] [Google Scholar]
- Ryoo H. D., Gorenc T. and Steller H. (2004). Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev. Cell 7, 491-501 10.1016/j.devcel.2004.08.019 [DOI] [PubMed] [Google Scholar]
- Sarthy A. V., Morgan-Lappe S. E., Zakula D., Vernetti L., Schurdak M., Packer J. C. L., Anderson M. G., Shirasawa S., Sasazuki T. and Fesik S. W. (2007). Survivin depletion preferentially reduces the survival of activated K-Ras-transformed cells. Mol. Cancer Ther. 6, 269-276 10.1158/1535-7163.MCT-06-0560 [DOI] [PubMed] [Google Scholar]
- Scholl C., Fröhling S., Dunn I. F., Schinzel A. C., Barbie D. A., Kim S. Y., Silver S. J., Tamayo P., Wadlow R. C., Ramaswamy S. et al. (2009). Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 137, 821-834 10.1016/j.cell.2009.03.017 [DOI] [PubMed] [Google Scholar]
- Seto E. S., Bellen H. J. and Lloyd T. E. (2002). When cell biology meets development: endocytic regulation of signaling pathways. Genes Dev. 16, 1314-1336 10.1101/gad.989602 [DOI] [PubMed] [Google Scholar]
- Simon M. A., Bowtell D. D. L., Dodson G. S., Laverty T. R. and Rubin G. M. (1991). Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 67, 701-716 10.1016/0092-8674(91)90065-7 [DOI] [PubMed] [Google Scholar]
- Sonnichsen B., De Renzis S., Nielsen E., Rietdorf J. and Zerial M. (2000). Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 149, 901-914 10.1083/jcb.149.4.901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounni N. E. and Noel A. (2012). Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 59, 85-93 10.1373/clinchem.2012.185363 [DOI] [PubMed] [Google Scholar]
- Steckel M., Molina-Arcas M., Weigelt B., Marani M., Warne P. H., Kuznetsov H., Kelly G., Saunders B., Howell M., Downward J. et al. (2012). Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 22, 1227-1245 10.1038/cr.2012.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg P. W. and Horvitz H. R. (1991). Signal transduction during C. elegans vulval induction. Trends Genet. 7, 366-371 10.1016/0168-9525(91)90257-Q [DOI] [PubMed] [Google Scholar]
- Strebhardt K. and Ullrich A. (2006). Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 6, 321-330 10.1038/nrc1841 [DOI] [PubMed] [Google Scholar]
- Sun Y. H., Tsai C. J., Green M. M., Chao J. L., Yu C. T., Jaw T. J., Yeh J. Y. and Bolshakov V. N. (1995). White as a reporter gene to detect transcriptional silencers specifying position-specific gene expression during Drosophila melanogaster eye development. Genetics 141, 1075-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TerBush D. R. and Novick P. (1995). Sec6, Sec8, and Sec15 are components of a multisubunit complex which localizes to small bud tips in Saccharomyces cerevisiae. J. Cell Biol. 130, 299-312 10.1083/jcb.130.2.299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O., Reinsch S., Urbe S., Zerial M. and Parton R. G. (1996). Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 135, 913-924 10.1083/jcb.135.4.913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano T., Emkey R. and Feig L. A. (1996). Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J. 15, 810-816. [PMC free article] [PubMed] [Google Scholar]
- White M. A., Vale T., Camonis J. H., Schaefer E. and Wigler M. H. (1996). A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J. Biol. Chem. 271, 16439-16442 10.1074/jbc.271.28.16439 [DOI] [PubMed] [Google Scholar]
- Wolfman J. C., Palmby T., Der C. J. and Wolfman A. (2002). Cellular N-Ras promotes cell survival by downregulation of Jun N-terminal protein kinase and p38. Mol. Cell. Biol. 22, 1589-1606 10.1128/MCB.22.5.1589-1606.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolthuis R. M. F., Zwartkruis F., Moen T. C. and Bos J. L. (1998). Ras-dependent activation of the small GTPase Ral. Curr. Biol. 8, 471-474 10.1016/S0960-9822(98)70183-6 [DOI] [PubMed] [Google Scholar]
- Wu S., Mehta S. Q., Pichaud F., Bellen H. J. and Quiocho F. A. (2005). Sec15 interacts with Rab11 via a novel domain and affects Rab11 localization in vivo. Nat. Struct. Mol. Biol. 12, 879-885 10.1038/nsmb987 [DOI] [PubMed] [Google Scholar]
- Wu Y., Zhuang Y., Han M., Xu T. and Deng K. (2009). Ras promotes cell survival by antagonizing both JNK and Hid signals in the Drosophila eye. BMC Dev. Biol. 9, 53 10.1186/1471-213X-9-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M., Pastor-Pareja J. C. and Xu T. (2010). Interaction between RasV12 and scribbled clones induces tumour growth and invasion. Nature 463, 545-548 10.1038/nature08702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-M., Ellis S., Sriratana A., Mitchell C. A. and Rowe T. (2004). Sec15 is an effector for the Rab11 GTPase in mammalian cells. J. Biol. Chem. 279, 43027-43034 10.1074/jbc.M402264200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.