

Abstract
Lysosomal acid lipase (LAL) deficiency is an under-recognized lysosomal disease caused by deficient enzymatic activity of LAL. In this report we describe two affected female Mexican siblings with early hepatic complications. At two months of age, the first sibling presented with alternating episodes of diarrhea and constipation, and later with hepatomegaly, elevated transaminases, high levels of total and low-density lipoprotein cholesterol, and low levels of high-density lipoprotein. Portal hypertension and grade 2 esophageal varices were detected at four years of age. The second sibling presented with hepatomegaly, elevated transaminases and mildly elevated low-density lipoprotein and low high-density lipoprotein at six months of age. LAL activity was deficient in both patients. Sequencing of LIPA revealed two previously unreported heterozygous mutations in exon 4: c.253C>A and c.294C>G. These cases highlight the clinical continuum between the so-called Wolman disease and cholesteryl ester storage disease, and underscore that LAL deficiency represents a single disease with a degree of clinical heterogeneity.
Keywords: Cholesteryl ester storage disease, Liver fibrosis, Dyslipidemia, Liver steatosis, Wolman disease
Core tip: Lysosomal acid lipase deficiency is a rare genetic disorder related to the metabolism of cholesterol and triglycerides inside the lysosome. In this report, we present the findings from two siblings with no lysosomal acid lipase activity caused by two previously unidentified mutations in exon 4 of LIPA. The patients had early hepatic presentation, including severe cirrhosis and esophageal varices in the elder sibling, underscoring the significant morbidity that can occur at all ages of lysosomal acid lipase deficiency and highlighting possible compensatory mechanisms in liver function in children.
INTRODUCTION
Lysosomal acid lipase deficiency (LALD; OMIM #278000) is an autosomal recessive disease caused by mutations in LIPA at chromosomal locus 10q23.31, which encodes a hydrolase involved in the degradation of lysosomal cholesterol esters and triglycerides. Clinically, LALD has been historically reported as one of two principal phenotypic presentations: early onset, often called Wolman disease (WD), and late onset, termed cholesteryl ester storage disease (CESD)[1].
The early presentation of LALD has an estimated prevalence of 1/350000 in infants, with symptoms such as diarrhea, massive hepatosplenomegaly, malabsorption, cachexia and adrenal calcifications, which typically develop within the first three months of life. Liver cirrhosis results in fatal liver failure before one year of age[1]. The exact prevalence of LALD in children and adults is not yet established, and ranges from 1 in 150000 to 300000 in Caucasians[1] and 1 in 40000 in German newborns[2]. The clinical signs and symptoms are heterogeneous, including hepatic steatosis leading to hepatomegaly and hepatic fibrosis and cirrhosis, splenomegaly, type II hyperlipoproteinemia, and accelerated atherosclerosis. Although some patients remain asymptomatic until adulthood, infants and children can have significant morbidity and early mortality as a result of liver cirrhosis and liver failure[3].
A review analyzing 135 CESD patients published in the literature found that the mean age at presentation was 5 years (range: 1-44 years), with hepatomegaly as the most common manifestation[3]. In that review, Bernstein et al[3] noted that the age at presentation was between 0 and 2 years in the most severely affected cases, corresponding to 27% of patients and highlighting the significance of liver morbidity (cirrhosis, fibrosis, and failure). Another recent review showed that all 71 pediatric LALD cases had hepatomegaly, with 63% presenting symptoms before five years of age[4]. The early signs of portal hypertension and its correlation to hepatic fibrosis and the progression of disease are unknown. Although liver manifestations of the disease usually predominate, dyslipidemia and associated cardiac complications have also been reported[1,3]. The principal causes of death reported in CESD patients were liver failure and bleeding of esophageal varices[3]. However, bleeding of esophageal varices was reported in only 8% of patients[3], and this complication is primarily described in Mexican patients[5].
In this report, we describe the cases of two Mexican siblings with LALD that was attributed to previously undescribed mutations in exon 4 of LIPA. These siblings presented symptoms within the first year of life and developed portal hypertension before four years of age. Both cases demonstrated early clinical signs and symptoms and relatively rapid clinical progression of the disease compared to previously reported cases.
CASE REPORT
Sibling 1
This report describes the case of a 9-year-old girl with healthy, non-consanguineous parents from Mexico, who was apparently normal at birth and met normal development milestones. She became symptomatic at two months of age, presenting with alternating episodes of diarrhea and constipation, and reduced weight and height. Hepatomegaly was detected by abdominal radiography at six months of age during the evaluation for diarrhea. An initial laboratory workup indicated leukocytosis (16000/mL) and eosinophilia (2700/mL), and a bone marrow analysis showed an increase of myeloid series, with augmented cellularity and the presence of megakaryocytes, without immature cells. The patient then underwent additional testing to rule out a hematologic malignancy. Further workup included karyotype in peripheral blood, reported as 46, XX[20].
Laboratory assessments at two years of age showed: hemoglobin, 14.1 g/dL; leukocytes, 13200/mL; eosinophilia, 3200/mL (24.6% of white blood cell differential count); platelets, 330000; abnormal liver enzymes and dyslipidemia (Table 1) with evidence of chronic Epstein-Barr virus infection. Serology for hepatitis A, B and C was negative. Hepatosplenomegaly was confirmed by abdominal ultrasound.
Table 1.
Hepatic function test, lipid profile and coagulation values in patients
| Parameter (unit) |
Sibling 1 |
Sibling 2 |
Reference value | ||
| 2 yr | 9 yr | 6 mo | 4 yr | ||
| AST (U/L) | 229 | 192 | 61 | 221 | 8-50 |
| ALT (U/L) | 344 | 193 | 56 | 181 | 7-45 |
| Alkaline phosphatase (U/L) | 365 | 545 | 328 | 402 | 4 yr: 169-372 |
| 9 yr: 212-468 | |||||
| Lactate dehydrogenase (U/L) | 258 | 281 | 288 | 236 | 4-6 yr: 145-345 |
| 7-9 yr: 143-290 | |||||
| Total bilirubin (mg/dL) | 0.8 | 0.42 | 0.2 | 0.42 | 0.1-0.9 |
| Indirect bilirubin (mg/dL) | 0.7 | 0.2 | 0.1 | 0.28 | |
| Total cholesterol (mg/dL) | 343.0 | 211.0 | 165.0 | 221.0 | < 170 |
| Triglycerides (mg/dL) | 316.0 | 118.0 | 154.0 | 181.0 | < 90 |
| LDL-cholesterol (mg/dL) | 277.1 | 157.4 | 123.0 | 154.8 | < 100 (optimal) |
| HDL-cholesterol (mg/dL) | 22.3 | 30.0 | 30.0 | 30.0 | ≥ 60 |
| Trombin time (min) | 21.87 | 23.1 | 21.1 | 20.8 | 15-22 |
| Prothrombin time (min) | 13.9 | 14.8 | 12.0 | 12.4 | 11-15 |
| pTT (min) | 30.2 | 35.1 | 27.8 | 32.0 | 25-33 |
| INR (%) | 1.15 (92.8%) | 1.3 (58.4%) | 1 (100.8%) | 1.1 (72.8%) | 0.8-1.1 (89%-129%) |
A computed tomography scan at four years of age revealed an enlarged liver, with a longitudinal diameter of 18 cm and spleen length of 13.4 cm, without adrenal calcifications. The patient was transferred to the pediatric gastroenterology department in our hospital for further investigation for metabolic disease. She presented gastrointestinal bleeding and endoscopy revealed grade 2 (Sohendra) esophageal varices.
When the patient was five years of age, a hepatic biopsy showed multinodular microvesicular steatosis in 10% of hepatocytes, with irregular small nodules separated by gross bands of porto-portal fibrosis with no zonal preference, ballooning degeneration and pseudoacinar regeneration, and no stasis of bilirubin or deposition of copper or iron (Figure 1A, B). The portal triads were expanded by fibrous tissue, with sparse duct proliferation. Cells with foamy cytoplasm were observed in the portal space and in the fibrous tissue, which demonstrated diastase resistance and Periodic acid-Schiff positivity. Kupffer sinusoidal cells were immunopositive for CD68 (Figure 1C) and immunonegative for Hepar-1 and pan-cytokeratin, with fine granules containing autofluorescent ceroid-like material (Figure 1D).
Figure 1.
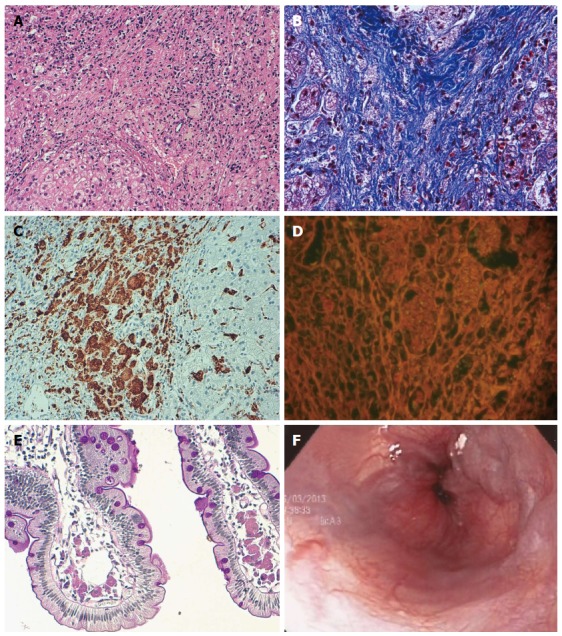
Liver histopathology and endoscopy images from sibling 1. A: Presence of ballooning and pseudoacinar regeneration, with foamy cells (HE, magnification × 400); B: Presence of portal triads expanded by fibrosis tissue and foamy cells (Masson’s tricromic, magnification × 400); C: Positive CD68 immunoreactivity in cells (CD68 immunostaining); D: Autofluorescence of foamy cells in portal spaces (Masson’s tricromic, magnification × 400); E: Duodenal biopsy shows histiocytes with ceroid material in the lamina propria of the villi; F: Endoscopy showing esophageal varices.
Doppler ultrasound at six years of age showed a homogenous parenchyma, with normal echogenicity and without intrahepatic lesions or increased flux in portal circulation.
A duodenal biopsy performed when the patient was seven years of age showed preserved architecture of duodenal mucosa, but with groups of histiocytes with fluorescent ceroid material occupying about 40% of the lamina propria of villi (Figure 1E) that were associated with lymphocytes. A liver biopsy also demonstrated the same changes, with minimal steatosis that was decreased from the biopsy performed at five years. The histology indicated micronodular cirrhosis, widened portal spaces and fibrotic tissue with histiocytes, with an absence of a central vein and nodules of hepatic regeneration.
The pathologic findings of the biopsies were interpreted as a possible diagnosis of Niemann-Pick disease. Thus, sphingomyelinase activity was measured by mass spectrometry, and quantified as 2.3 μmol/L per hour (reference value ≥ 2 μmol/L per hour). A skin biopsy was taken to determine cholesterol esterification and filipin staining in cultured fibroblasts to eliminate Niemann-Pick type C disease (performed at Mayo Clinic, United States), revealing low-density lipoprotein esterification that was 2% of normal control cells, similar to patients with Niemann-Pick type C disease, but with normal filipin staining.
The patient was not given any lipid-lowering drugs; recent laboratory test results are shown in Table 1. Associated with the progression of her liver disease and the associated portal hypertension and splenomegaly, the patient’s platelet count decreased to 96000 (reference range: 150000-500000). Endoscopy performed at nine years of age revealed continued presence of grade 2 esophageal varices (Figure 1F) and one medium gastric varix; the esophageal varices were ligated. The last Doppler ultrasound showed a longitudinal liver diameter of 14.7 cm, increased liver echogenicity, micronodular pattern, spleen longitudinal length of 14.6 cm, and increase in portal flux and collateral circulation with porto-systemic shunts (splenorenal shunt and umbilical vein recanalization) (Figure 2). The patient is currently being treated with propranolol (2 mg/kg per day) for gastrointestinal bleeding prophylaxis and vitamin K (10 mg/d) for prolonged bleeding times.
Figure 2.
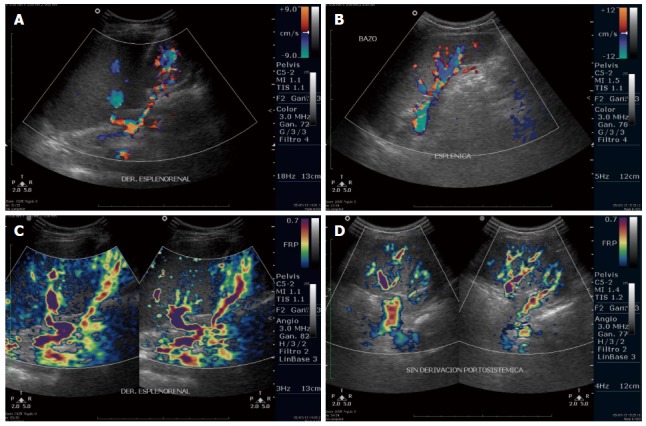
Comparison of Doppler ultrasound images from sibling 1 (A,C) and sibling 2 (B,D). A: Splenic vein with a spontaneous splenorenal shunt; B: Splenic vein with collateral vessels at the hilum, without porto-systemic shunts; C: Dilated and tortuous vessels, collateral circulation and a spontaneous splenorenal shunt; D: Absence of a splenorenal shunt.
Sibling 2
Given the suspicion of a probable lysosomal storage disease in sibling 1, we began the study of her now 4-year-old sister at six months of age. The initial physical examination revealed reduced height and hepatomegaly, which was confirmed by an abdominal ultrasound showing the liver with a longitudinal diameter of 8.4 cm and increased echogenicity, and a spleen length of 7.1 cm. Laboratory tests revealed alterations in liver function and lipid profile (Table 1); however, a bone marrow aspirate indicated no evidence of storage disease. Sphingomyelinase activity was 2.8 μmol/L per hour. A recent endoscopy showed no varices, but Doppler ultrasound demonstrated increased hepatic flux, without porto-systemic shunts (Figure 2).
LAL enzymatic and molecular analyses
Following exclusion of Niemann-Pick diseases, a diagnosis of LALD was considered for both siblings. LAL activity was determined using a fluorescence-based whole blood assay with 4-methylumbelliferone[6], which revealed < 0.02 pmol/punch*h (normal range: 24.00-134.00 pmol/punch*h) in dried blood and 0.000 nmol/punch*h in whole blood (normal range: 0.027-0.152 nmol/punch*h). LAL activity from dried blood samples was 40.06 pmol/punch*h in the father, and 19.53 pmol/punch*h (low normal) in the mother.
LIPA sequencing
Genomic DNA was extracted from dried blood spots, and exons 1-10 of LIPA were amplified by polymerase chain reaction and labeled with Big Dye Terminator (Applied Biosystems of Thermo Fisher Scientific Inc., Waltham, MA, United States) and resolved by capillary electrophoresis on a 3730XL Genetic Analyzer (Applied Biosystems). Sequencing revealed a previously reported single nucleotide polymorphism within intron 5 (rs2297472; c.539-5C>T), along with two previously unreported missense mutations in exon 4: c.253C>A (p.Gln85Lys) and c.294C>G (p.Asn98Lys) (Figure 3). Subsequent sequencing of exon 4 in the parents showed that the father was a carrier of the c.294C>G mutation, and the mother carried the c.253C>A mutation. This confirmed the trans-state of mutations in the siblings.
Figure 3.
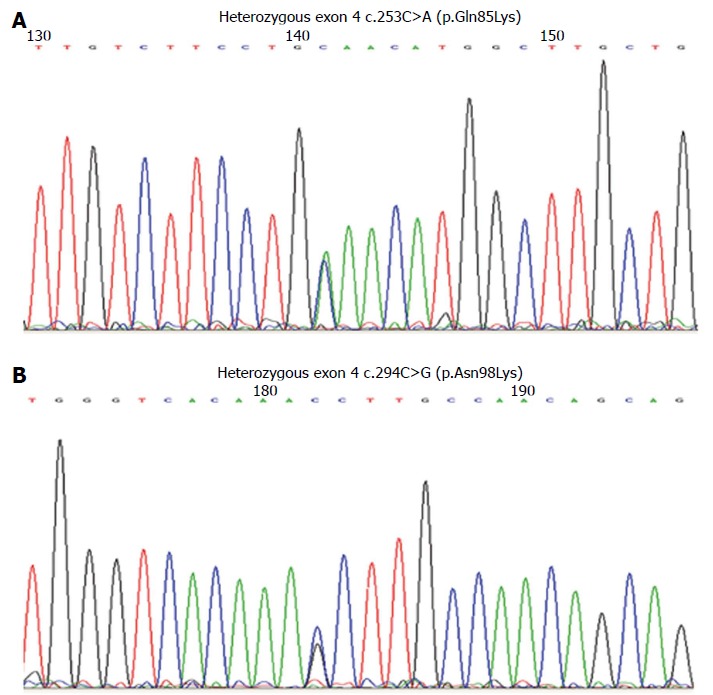
Electropherograms showing the mutations in exon 4 of the LIPA gene. A: Mutation in exon 4: c.253C>A (p.Gln85Lys); B: Mutation in exon 4: c.294C>G (p.Asn98Lys).
Bioinformatics analyses using the Polyphen-2 and Sorting Intolerant From Tolerant (SIFT) predictive tools indicated that the exon 4 mutations are likely to be pathogenic, which was also indirectly demonstrated by the undetectable level of LAL activity in whole blood and the very low incorporation of low-density lipoprotein cholesterol in fibroblasts. These mutations were not found in other patients with LALD studied at the Massachusetts General Hospital, in the 1092 control individuals of diverse ethnic background studied in the 1000 Genomes Project (http://www.1000genomes.org), or in the 6500 samples from NHLBI GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS), suggesting that they are very rare.
DISCUSSION
There are several previous reports of severe LALD in patients of Mexican origin. A Mexican family was studied at Baylor College of Medicine in Texas with three affected members who developed esophageal varices, hepatic failure and pulmonary complications early in life[7-10]. Another Mexican female studied at the same institute represented the first case of hepatic transplantation in CESD after developing several complications[11]. In Mexico, two infants were diagnosed with Wolman disease by the characteristic adrenal calcification at autopsy[12,13], and another case was diagnosed by the presence of crystal structures upon electron microscopy[14]. Considering the severity of hepatic failure, it was thought that Mexican patients had more severe alleles or genes that modulated CESD[5]. Only two reports included a molecular diagnosis: a male infant affected by LALD (Mexican father and American mother) had Trp95X/fs219 mutations[15]; and a female infant with a sibling that died at three months had compound heterozygosity for one new mutation: p.Gln98His/p.Gly342Arg[16].
The mutations found at amino acids 85 and 98 in exon 4 reported here are novel and were associated with undetectable whole blood enzyme activity, though a different mutation has been reported at amino acid 85 (p.Gln85Arg)[3,17]. Although structural features of the LAL protein are not well defined, exon 4 may be functionally important as the majority of mutations are within this exon[3,17] and its deletion causes LALD[3]. The absence of detectable activity is an interesting finding because these siblings had early non-fatal complications, suggesting that LALD is a continuous phenotype with some cases presenting overlapping features of Wolman disease and CESD, and indicating that enzymatic activity cannot predict the phenotype[3]. Studies of the natural history of LALD and correlating genotype and phenotype are urgently needed.
An exon 8 splice junction mutation (E8SJM) is present in the majority of LALD cases[1]. A review of the 55 published cases of LALD with molecular analysis of LIPA demonstrated that 89% of patients had the E8SJM mutation in at least one allele[3]. Ethnicity influences the prevalence of LALD, as a recent screen showed an increased frequency of E8SJM carriers in Caucasian and Hispanic populations (1:300) compared to African American, Asian and Ashkenazi groups[18]. The cases reported here demonstrate that despite a high prevalence of the E8SJM allele, LALD can originate from alternate mutations. In the Human Mutation Database, there are only 48 LIPA mutations reported to date in infant (19 mutations) and child/adult (27 mutations) cases[17]. Thus, further studies of carrier frequency of mutations in the full LIPA gene are required.
Clinicians, particularly pediatric gastroenterologists/hepatologists, should be aware of LALD in view of the frequent reported pediatric presentations[3] and the potential for misdiagnosis. Children can have all the complications related to the progression of cirrhosis, such as portal hypertension, ascites, esophageal/gastric varices, coagulopathy and bleeding; but these complications may not follow the adult pattern. Therefore, the detection of fibrosis, which progresses to cirrhosis, is a priority for the assessment of hepatic dysfunction, the need for hepatic transplantation and prognosis. Hepatic biopsy is still the most reliable assessment of progressive liver disease, so it is necessary to develop and validate noninvasive techniques that correlate with histopathology in order to identify fibrosis and to evaluate response to treatment, such as the recently developed dry blood spot assay for diagnostic confirmation of LALD[6]. It is also necessary to explore the use of Doppler ultrasound for identifying subtle changes in portal flow, and the presence of compensatory changes like recanalization of umbilical vein and splenic renal shunts. Magnetic resonance spectroscopy can be used to quantify intrahepatic fat in LALD[19], but cannot evaluate the presence of fibrosis.
The progression to fibrosis in sibling 1 corresponded with an increase in alanine aminotransferase levels, altering the ratio with aspartate aminotransferase. The patient also presented with a porto-systemic shunt (Figure 2) that likely maintained stable portal hypertension and decreased the risk of variceal bleeding, despite the extensive fibrosis. It is not clear if these changes are the natural course of hepatic compromise in pediatric LALD patients. Other cases of LALD in Mexican patients demonstrate a rapid progression to hepatic failure[7-10].
Treatment for LALD has been limited to the use of lipid-lowering drugs or hepatic transplantation, which was performed in nine patients with mixed-results but with no long-term follow-up[3]. Sebelipase alfa (Synageva BioPharma Corp., Lexington, MA, United States), a recombinant human LAL, is under development for use as an enzymatic replacement therapy. The initial trial in adults showed a decrease in serum lipids and liver volume and normalization of liver transaminases[20,21], and a phase 3 global trial is currently underway[22]. Timely diagnosis, as well as incorporation of LAL activity in newborn screening, will be needed to optimize the time to begin enzymatic replacement therapy. Further research is needed for evaluating additional therapies for CESD patients, such as bone marrow and hepatic transplantation.
CONCLUSION
LALD is an underdiagnosed pediatric disease requiring an increased awareness and early diagnosis, particularly in Caucasian and Hispanic individuals. This report presents the cases of two siblings carrying two previously unreported mutations in exon 4 of LIPA that likely resulted in a deficiency of LAL activity with early symptomatology and complications.
ACKNOWLEDGMENTS
We would like to thank Radhika Tripuraneni, MD, MPH, for critical reading of the manuscript, Angelica Toriz-Ortiz, MD, for ultrasound imaging, and the medical staff of the Endoscopy and Pathology Department of CMN “20 de Noviembre”, along with all the personnel involved in the care of the patients.
This work was presented in abstract form at 2013’s National Week of Gastroenterology (Semana Nacional de Gastroenterologia de la AMG) in Veracruz, Mexico and at IX Congress of SLEIMPN in Medellín, Colombia.
COMMENTS
Case characteristics
Two siblings were affected by lysosomal acid lipase deficiency with early manifestations and hepatic complications, with the elder presenting with bleeding episodes, esophageal varices and portal hypertension.
Clinical diagnosis
The two cases presented similar characteristics: sibling 1 presented with gastrointestinal manifestations (alternating diarrhea and constipation episodes), hepatomegaly, reduced height and weight; sibling 2 presented with reduced height and hepatomegaly on physical examination.
Differential diagnosis
Other lysosomal storage diseases with hepatomegaly and Niemann-Pick cells, such as acid sphingomyelinase-deficiency (Niemann-Pick B), Niemann-Pick C, and Tangier disease.
Laboratory diagnosis
High levels of total cholesterol, low-density lipoprotein, aspartate and alanine aminotransferases, and low levels of high-density lipoprotein, normal bilirubin and undetectable lysosomal acid lipase activity in blood.
Imaging diagnosis
For both cases, abdominal ultrasound showed hepatomegaly and Doppler ultrasound showed abnormal portal flux velocities. Endoscopy in sibling 1 revealed esophageal varices.
Pathological diagnosis
In sibling 1, liver biopsy at five years of age showed microvesicular steatosis, foamy cells, fibrosis and cirrhosis.
Treatment
Both siblings were treated with a beta-blocker. Endoscopic variceal ligation was performed in sibling 1.
Related reports
Only one case of a Mexican infant with lysosomal acid lipase deficiency included LIPA mutation analysis. Other reports demonstrate that patients with the late presentation of this condition show severe hepatic disease with rapid progression.
Term explanation
Trans-state of mutations refers to mutations present in different alleles inherited from each parent.
Experiences and lessons
Limitations with enzymatic and molecular diagnosis along with a poor awareness of rare diseases can lead to a wrong diagnosis, as demonstrated in the patients reported here. New biochemical genetics techniques are available to achieve a correct diagnosis.
Peer review
The authors highlight the presentation of lysosomal acid lipase deficiency with hepatic complications and the importance of newly identified gene mutations. They also noted that enzyme levels were very low in the mother, a carrier of the mutation, suggesting that function is also impacted in a heterozygous state.
Footnotes
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 18, 2014
First decision: July 21, 2014
Article in press: September 30, 2014
P- Reviewer: Hoare M, Kovacs SJ, Liaskou E, Mohn A S- Editor: Qi Y L- Editor: Wang TQ E- Editor: Ma S
References
- 1.Grabowski GA, Du H. Lysosomal Acid Lipase Deficiencies: The Wolman Disease/Cholesteryl Ester Storage Disease Spectrum. The Online Metabolic and Molecular Bases of Inherited Diseases. New York: McGraw-Hill; 2012. [Google Scholar]
- 2.Muntoni S, Wiebusch H, Jansen-Rust M, Rust S, Seedorf U, Schulte H, Berger K, Funke H, Assmann G. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol. 2007;27:1866–1868. doi: 10.1161/ATVBAHA.107.146639. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58:1230–1243. doi: 10.1016/j.jhep.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 4.Zhang B, Porto AF. Cholesteryl ester storage disease: protean presentations of lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr. 2013;56:682–685. doi: 10.1097/MPG.0b013e31828b36ac. [DOI] [PubMed] [Google Scholar]
- 5.Assmnann G, Seedorf U. Acid Lipase Deficiency: Wolman Disease and Cholesteryl Ester Storage Disease. The Online Metabolic and Molecular Bases of Inherited Diseases. New York: McGraw-Hill; 2009. [Google Scholar]
- 6.Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413:1207–1210. doi: 10.1016/j.cca.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 7.Beaudet AL, Lipson MH, Ferry GD, Nichols BL. Acid lipase in cultured fibroblasts: cholesterol ester storage disease. J Lab Clin Med. 1974;84:54–61. [PubMed] [Google Scholar]
- 8.Beaudet AL, Ferry GD, Nichols BL, Rosenberg HS. Cholesterol ester storage disease: clinical, biochemical, and pathological studies. J Pediatr. 1977;90:910–914. doi: 10.1016/s0022-3476(77)80557-x. [DOI] [PubMed] [Google Scholar]
- 9.Michels VV, Driscoll DJ, Ferry GD, Duff DF, Beaudet AL. Pulmonary vascular obstruction associated with cholesteryl ester storage disease. J Pediatr. 1979;94:621–623. doi: 10.1016/s0022-3476(79)80033-5. [DOI] [PubMed] [Google Scholar]
- 10.Cagle PT, Ferry GD, Beaudet AL, Hawkins EP. Pulmonary hypertension in an 18-year-old girl with cholesteryl ester storage disease (CESD) Am J Med Genet. 1986;24:711–722. doi: 10.1002/ajmg.1320240416. [DOI] [PubMed] [Google Scholar]
- 11.Ferry GD, Whisennand HH, Finegold MJ, Alpert E, Glombicki A. Liver transplantation for cholesteryl ester storage disease. J Pediatr Gastroenterol Nutr. 1991;12:376–378. doi: 10.1097/00005176-199104000-00016. [DOI] [PubMed] [Google Scholar]
- 12.Peña-Alonso YR, Ramón-García G. Enfermedad de Wolman en una niña mexicana. Bol Med Hos Infant Mex. 1994;51:660–664. [Google Scholar]
- 13.Ramón-García G, Díaz-Ponce H, Díaz-Pérez C, Delgado-González E. [A 3-month-old girl with fever, abdominal distension, vomiting, and jaundice] Gac Med Mex. 2000;136:361–367. [PubMed] [Google Scholar]
- 14.Fernández-Aragón M, Cervantes-Bustamante R, De León-Bojorge B, Zárate-Mondragón F, Mata-Rivera N, Barrios EM, Campos MG, Ramírez-Mayans JA. [Cholesterol ester storage disease] Rev Gastroenterol Mex. 2004;69:171–175. [PubMed] [Google Scholar]
- 15.Anderson RA, Bryson GM, Parks JS. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesterol ester storage disease. Mol Genet Metab. 1999;68:333–345. doi: 10.1006/mgme.1999.2904. [DOI] [PubMed] [Google Scholar]
- 16.Gómez-Nájera M, Barajas-Medina H, Gallegos-Rivas MC, Mendez-Sashida P, Goss K, Sims KB, Radhikatripuraneni , Valles-Ayoub Y. New Diagnostic Method for Lysosomal Acid Lipase Deficiency and the Need to Recognize its Manifestation in Infants (Wolman Disease) J Pediatr Gastroenterol Nutr. 2013:Epub ahead of print. doi: 10.1097/MPG.0000000000000175. [DOI] [PubMed] [Google Scholar]
- 17.Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott SA, Liu B, Nazarenko I, Martis S, Kozlitina J, Yang Y, Ramirez C, Kasai Y, Hyatt T, Peter I, et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology. 2013;58:958–965. doi: 10.1002/hep.26327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thelwall PE, Smith FE, Leavitt MC, Canty D, Hu W, Hollingsworth KG, Thoma C, Trenell MI, Taylor R, Rutkowski JV, et al. Hepatic cholesteryl ester accumulation in lysosomal acid lipase deficiency: non-invasive identification and treatment monitoring by magnetic resonance. J Hepatol. 2013;59:543–549. doi: 10.1016/j.jhep.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balwani M, Breen C, Enns GM, Deegan PB, Honzík T, Jones S, Kane JP, Malinova V, Sharma R, Stock EO, et al. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology. 2013;58:950–957. doi: 10.1002/hep.26289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valayannopoulos V, Malinova V, Honzík T, Balwani M, Breen C, Deegan PB, Enns GM, Jones SA, Kane JP, Stock EO, et al. Sebelipase alfa over 52 weeks reduces serum transaminases, liver volume and improves serum lipids in patients with lysosomal acid lipase deficiency. J Hepatol. 2014;61:1135–1142. doi: 10.1016/j.jhep.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Synageva BioPharma Corp. A Multicenter Study of SBC-102 (Sebelipase Alfa) in Patients With Lysosomal Acid Lipase Deficiency/ARISE (Acid Lipase Replacement Investigating Safety and Efficacy. Active clinical trial, information. 14 April. 2014. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01757184. [Google Scholar]