

Abstract
Patatin-like phospholipase domain-containing 3 (PNPLA3 or adiponutrin) displays anabolic and catabolic activities in lipid metabolism, and has been reported to be significantly associated with liver fat content. Various studies have established a strong link between the 148 isoleucine to methionine protein variant (I148M) of PNPLA3 and liver diseases, including nonalcoholic fatty liver disease (NAFLD). However, detailed demographic and ethnic characteristics of the I148M variant and its role in the development of nonalcoholic fatty liver fibrosis have not been fully elucidated. The present review summarizes the current knowledge on the association between the PNPLA3 I148M variant and NAFLD, and especially its role in the development of nonalcoholic fatty liver fibrosis. First, we analyze the impact of demographic and ethnic characteristics of the PNPLA3 I148M variant and the presence of metabolic syndrome on the association between PNPLA3 I148M and NAFLD. Then, we explore the role of the PNPLA3 I148M in the development of nonalcoholic fatty liver fibrosis, and hypothesize the underlying mechanisms by speculating a pro-fibrogenic network. Finally, we briefly highlight future research that may elucidate the specific mechanisms of the PNPLA3 I148M variant in fibrogenesis, which, in turn, provides a theoretical foundation and valuable experimental data for the clinical management of nonalcoholic fatty liver fibrosis.
Keywords: PNPLA3 I148M variant, Polymorphism, Nonalcoholic fatty liver disease, Nonalcoholic fatty liver fibrosis
Core tip: In this review, we summarize the association between the PNPLA3 I148M variant and nonalcoholic fatty liver disease (NAFLD), and especially its role in nonalcoholic fatty liver fibrosis. The variant is associated with NAFLD, but is predominant in women, not in men. The association may vary among different ethnic populations, but is not affected by the presence of metabolic syndrome. We speculate there is a pro-fibrogenic network that the PNPLA3 I148M variant may promote the development of fibrogenesis by activating the hedgehog signaling pathway, which, in turn, leads to the activation and proliferation of hepatic stellate cells, and excessive generation and deposition of extracellular matrix.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is defined histologically or by proton magnetic resonance spectroscopy as hepatic fat accumulation (steatosis) exceeding 5% in the absence of excessive ethanol consumption, drugs, toxins, infectious diseases or any other specific etiologic factors of liver disease[1]. NAFLD embraces a morphological spectrum of hepatic diseases, ranging from nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH). NASH is the late stage of NAFLD, in which hepatic inflammation and fibrosis co-exist. In a proportion of patients, NASH can progress towards cirrhosis and even hepatocellular carcinoma (HCC)[2]. With a general prevalence of 25%-30%, NAFLD currently represents the most common cause of liver dysfunction, and is now the most prevalent liver disorder in Western countries[3].
It is well known that metabolic risk factors such as obesity, insulin resistance, type 2 diabetes mellitus and dyslipidemia are deeply associated with the pathophysiology of NAFLD[2,4]. Moreover, genetic mutations also play a significant role in predisposition to the development and progression of NAFLD[5].
In 2008, a single nucleotide polymorphism (SNP) in the patatin-like phospholipase domain-containing 3 (PNPLA3, also known as adiponutrin) gene, or rs738409 polymorphism, which represents a substitution from cytosine to guanine that results in a switch from isoleucine to methionine at residue 148 (I148M), was reported to be significantly associated with liver fat content[6].
Since then, extensive investigations of the association between the PNPLA3 I148M variant (or rs738409 polymorphism) and NAFLD have been carried out, and various studies have established a strong link between the PNPLA3 I148M variant and the development and progression of NAFLD, including nonalcoholic fatty liver fibrosis[7-12]. These results indicate that this variant may be a potential modifier of NAFLD, especially nonalcoholic fatty liver fibrosis. However, detailed demographic and ethnic characteristics of the I148M variant and its role in the development of nonalcoholic fatty liver fibrosis, along with the specific molecular mechanisms, have not been fully elucidated. Therefore, the present review summarizes the current knowledge on the association between the PNPLA3 I148M variant and NAFLD, and the variant’s role in the development of nonalcoholic fatty liver fibrosis.
EXPRESSION AND FUNCTION OF THE PNPLA3 GENE AND PNPLA3 I148M VARIANT
The PNPLA3 gene is located in the long branch of human chromosome 22, and encodes a transmembrane polypeptides chain containing 481 amino acids[13]. PNPLA3 protein is highly expressed on the endoplasmic reticulum and lipid membranes of hepatocytes as well as adipose tissue[14], and changes in the expression are closely associated with the nutrient status[15]. Rae-Whitcombe and colleagues reported that the promoter region of the PNPLA3 gene is regulated by glucose and insulin[16]. Consistently, PNPLA3 mRNA levels have been demonstrated to decrease after fasting and increased by refeeding in mice[17]. The nutritional regulation of PNPLA3 has further been confirmed, as the gene is shown to be regulated by sterol regulatory element binding protein 1c (SREBP-1c) in mouse liver and human hepatocytes[17-19], which responds in turn to insulin and glucose.
With the conserved patatin domain at its N-terminal, the PNPLA3 protein demonstrates a predominant triglyceride hydrolase activity with mild lysophosphatidic acid acyltransferase activity[13]. It has been shown that substitution of methionine for isoleucine at residue 148 does not alter the orientation of the catalytic dyad, but the longer side chain of methionine restricts access of substrate to the catalytic serine at residue 47[20]. The size of the substrate-access entry site is significantly reduced in mutants, which limits the access of palmitic acid to the catalytic dyad[21]. Recently, Kumari and colleagues determined that the PNPLA3 I148M variant induced an increase in lipogenic activity, leading to increased hepatic triglyceride synthesis[22]. Similarly, Li et al[23] generated transgenic mice over-expressing PNPLA3 in the liver and observed that the PNPLA3 I148M variant exerted three effects on hepatic triglyceride metabolism: increased synthesis of fatty acids and triglyceride; impaired hydrolysis of triglyceride; and depletion of triglyceride long-chain polyunsaturated fatty acids. These findings suggest that the increase in hepatic triglyceride levels associated with the PNPLA3 I148M variant is induced by multiple changes in triglyceride metabolism. These previous studies showed that acid modification within the catalytic patatin domain of the PNPLA3 protein acts as a kind of “gain of function” mutation enhancing the accumulation of lipids in the hepatocytes.
PNPLA3 I148M VARIANT AND NAFLD
In 2008, Romeo and colleagues first reported a genome-wide association study to explore the genes associated with susceptibility to NAFLD[6]. They demonstrated that the PNPLA3 148M allele was robustly associated with increased liver fat content, and the association remained highly significant after adjusting for body mass index (BMI), diabetes status, ethanol use, as well as global and local ancestry. In addition, the PNPLA3 I148M variant was also found to be associated with elevated serum aminotransferase levels[6,24] and increased computed tomography-measured hepatic steatosis and histological NAFLD[25]. A series of subsequent candidate gene studies[26,29,36,37] have verified the association between the PNPLA3 I148M variant and NAFLD.
PNPLA3I148M variant is associated with NAFLD in adults
The PNPLA3 I148M variant is reported to be dose-dependently associated with increased levels of serum triglyceride, alanine aminotransferase (ALT) and aspartate aminotransferase (AST)[26]. In addition, numerous studies have demonstrated that the PNPLA3 I148M variant is associated with liver fat content[27-29]. These findings confirm that the PNPLA3 I148M variant is associated not only with fat accumulation in the liver, but also with liver injury since aminotransferases are the most sensitive liver function parameters. Liver injury is believed to be triggered by lipotoxicity, which results from hepatic fat accumulation. It has been previously reported that liver necrosis induced by intracellular lipotoxicity parallels liver fat accumulation[30], and the degree of steatosis correlates with the severity of liver injury in NAFLD[31].
Currently, liver biopsy is used as the gold standard for diagnosis of NAFLD. Although it is expensive and not ethically feasible, especially in uninvestigated patients, there are still some studies on NAFLD based on histological diagnosis. The PNPLA3 I148M variant has been confirmed to be strongly associated with an increased risk of histological NAFLD[32]. In a case-control study, patients who were homozygotes of the 148M allele had higher steatosis scores (33.3% ± 4.0%) compared with heterozygotes of the 148IM allele (26.3% ± 3.5%) and 148I allele (14.9% ± 3.9%), indicating that the variant was significantly associated with the degree of liver steatosis[27].
PNPLA3 I148M variant is associated with pediatric NAFLD
NAFLD is not only a disease affecting the adult population, but also a leading liver disease in children worldwide[8,33]. A study of Hispanic children and adolescents in the Unites States showed that the 148M allele was associated with higher liver fat content and lower HDL cholesterol levels[34]. This is consistent with the observation that Hispanic children who were homozygotes of the 148MM allele were susceptible to increased hepatic fat when dietary carbohydrate intake was high[35]. In addition, a study of obese Taiwanese children also showed that the PNPLA3 I148M variant was associated with an increase in ALT levels and an increased risk of NAFLD[36,37].
In a more extensive study of pediatric patients with biopsy-proven NAFLD, the PNPLA3 I148M variant was associated with the severity of steatosis, hepatocellular ballooning and lobular inflammation, and the presence of NASH and fibrosis, but not with BMI, adiposity, lipid levels, insulin resistance and ALT levels[8]. In another large study[7] to determine the association between SNPs and the histological severity of NAFLD, 223 children with histologically confirmed NAFLD were investigated. It was observed that the 148M allele was associated with an earlier presentation of the disease, but not with histological severity. However, the association was marginal in the multivariate analysis (P = 0.045).
Therefore, although the currently available findings suggest that the PNPLA3 I148M variant confers genetic susceptibility to liver injury in children at a young age, most subjects studied were obese children or pediatric NAFLD patients, and the samples were relatively small in most of the studies. Thus, well-designed large studies that include pediatric NAFLD patients and matched healthy children are required to confirm the association. At the very least, a meta-analysis would offer valuable information.
ASSOCIATION BETWEEN PNPLA3 I148M VARIANT AND NAFLD IS AFFECTED BY GENDER AND PROBABLY BY ETHNICITY, BUT NOT BY METABOLIC SYNDROME
The PNPLA3 I148M variant shows a potential sexual dimorphism on NAFLD susceptibility[9,32]. In a gender-specific analysis of a NASH cohort, Speliotes et al[32] observed that the effect of the PNPLA3I148M variant on histological NAFLD was higher in women than in men. Indeed, a meta-regression analysis showed a negative correlation between male gender and the effect of the PNPLA3 I148M variant on liver fat content[9].
The above findings suggest a predominant association between the PNPLA3 I148M variant with NAFLD in women, but not in men. It is known that estrogen levels are different in men and women, and estrogen is a critical hormone involved in lipid metabolism. Therefore, the gender differences may mainly result from the variations in the hormone, the variation in the gene, or the interaction of the two. However, it is necessary to test whether there is a true and reproducible interaction between estrogen and PNPLA3 I148M in well-defined population-based cohorts.
The prevalence of NAFLD differs among different populations. Hispanics have been demonstrated to have a higher prevalence of hepatic steatosis compared with European-Americans, whereas African-Americans have a lower prevalence[38]. In addition, Asian-Indian men have more liver fat and are more insulin-resistant than BMI- and age-matched white individuals[39].
Romeo and colleagues found that the frequencies of the 148M allele matched the prevalence of NAFLD in the Dallas Heart Study, and Hispanics had a higher frequency of the 148M allele (49%) compared with European Americans (23%) and African Americans (17%)[6]. Another study by Wagenknecht et al[40] suggested that the PNPLA3 I148M variant contributed to the variation in NAFLD across multiple ethnicities. These findings indicate that the PNPLA3 I148M variant may explain ethnic differences in the prevalence of NAFLD, and that some of the ethnic variations in NAFLD are genetic.
On the other hand, in a study of 144 biopsy-proven NAFLD patients and 198 controls in Malaysia, the PNPLA3 I148M variant was associated with susceptibility to NAFLD (OR = 2.34; 95%CI: 1.69-3.24)[12]. However, the association remained similar in three ethnic groups, namely Chinese (OR = 1.94; 95%CI: 1.12-3.37), Indian (OR = 3.51; 95%CI: 1.69-7.26) and Malay (OR = 2.05; 95%CI: 1.25-3.35), which indicates no effect of ethnicity on the association between the PNPLA3 I148M variant and NAFLD. Nevertheless, these three ethnic groups all belong to Asian populations, which may be different from Hispanics, Europeans and Africans in terms of association between the PNPLA3 I148M variant and NAFLD.
NAFLD is now considered the hepatic manifestation of metabolic syndrome[41]. Insulin resistance in adipose tissue induces an excess of free fatty acid supply to the liver, which may lead to lipotoxicity, oxidative stress, and apoptosis[42]. Whether the association between the PNPLA3 I148M variant and NAFLD is confounded by the presence of metabolic syndrome has been investigated. Although a few studies have suggested an association between the PNPLA3 I148M variant and metabolic syndrome, such as insulin resistance[26,43], other studies have failed to reveal the association[9,32,44,45]. For example, in a study of 592 cases with European ancestry, there were no associations of the PNPLA3 I148M variant with BMI, triglyceride levels, high- and low-density lipoprotein levels, or diabetes[32]. Moreover, a study of 330 German subjects showed that the PNPLA3 I148M variant was strongly associated with fatty liver, but not with insulin resistance or estimates of liver injury[44]. In addition, in a study of 218 French type 2 diabetic patients, the PNPLA3 I148M variant was not correlated with visceral obesity and was inversely associated with carotid intima media thickness, suggesting that fatty liver associated with the PNPLA3 I148M variant may not be linked to metabolic disorders[45]. Indeed, in a recent meta-analysis, all included studies showed a lack of a significant difference among genotypes for BMI, glucose and insulin levels, and homeostasis model assessment of insulin resistance[9].
Furthermore, there appears to be no association between the PNPLA3 I148M variant and metabolic syndrome in children. In a study of obese children and adolescents, the PNPLA3 I148M variant was associated with increased levels of ALT and AST, but not with glucose tolerance and insulin sensitivity[46]. The PNPLA3I148M variant also conferred susceptibility to hepatic steatosis in obese youths, but without increasing insulin resistance[47].
In summary, the PNPLA3 I148M variant is associated with NAFLD both in adults and children, and the association is affected by gender and ethnicity, but not by the presence of metabolic syndrome (Table 1).
Table 1.
Studies evaluating the association between the PNPLA3 I148M variant and nonalcoholic fatty liver disease
| Ref. | Population/Ethnicity country | n | Age | Diagnosis criteria | Key findings |
| Wang et al[26] | Asian | 879 | Adult | US | Increase in TG, ALT and AST |
| Tai Wan | |||||
| Sookoian et al[27] | Caucasian | 266 | Adult | US | Increased liver fat and liver Injury |
| Argentina | Liver biopsy | ||||
| Kollerits et al[28] | Italy/Austria/United States | 4290 | Adult | NA | Increase in ALT and AST |
| Xu et al[29] | Chinese | 651 | Adult | US | Increased ALT, GGT and related to development and progression of NAFLD |
| China | |||||
| Speliotes et al[32] | Caucasian | 1597 | Adult | Liver biopsy | Increased risk of histological NAFLD, but not associated with metabolic syndrome |
| United States | |||||
| Rotman et al[7] | Caucasian | 1117 | Adult | Liver biopsy | Earlier presentation of NAFLD in pediatric patients |
| United States | Pediatric | ||||
| Valenti et al[8] | Caucasian | 149 | Pediatric | Liver biopsy | Associated with steatosis, NASH and fibrosis |
| Italian | |||||
| Goran et al[34] | Hispanic | 327 | Pediatric | MRS | Higher liver fat and lower HDL-C |
| United States | |||||
| Davis et al[35] | Hispanic | 153 | Pediatric | MRI | Increased liver fat when dietary carbohydrate intake |
| United States | |||||
| Lin et al[36] | Asian | 520 | Pediatric | US | Increased ALT and risk of NAFLD |
| Tai Wan | |||||
| Viitasalo et al[37] | Caucasian | 481 | Pediatric | NA | Increase in ALT |
| Finland | |||||
| Sookoian et al[9] | Meta-analysis | A negative correlation between male sex and the variant on liver fat, and a lack of significant difference among genotypes for metabolic syndrome | |||
| Romeo et al[6] | Hispanic/ | 9229 | Adult | H-MRS | Hispanics have a higher frequency of the 148M allele than European Americans and African Americans |
| European American/ | |||||
| African American | |||||
| United States | |||||
| Zain et al[12] | Chinese, Indian and Malay | 342 | Adult | Liver biopsy | No effect of ethnicity on the association between the variant and NAFLD |
| Malaysia | |||||
| Browning et al[38] | White/Black/Hispanic | 2287 | Adult | H-MRS | Frequency of hepatic steatosis varied with ethnicity and gender |
| United States | |||||
| Petersen et al[39] | Caucasian/ | 482 | Pediatric | Proton MRS | Asian-Indians have increased liver fat and prevalence of insulin resistance compared with all other ethnic groups |
| Eastern Asian/ | Adult | ||||
| Asian-Indian/Black/ | |||||
| Hispanic | |||||
| United States | |||||
| Wagenknecht et al[40] | Hispanic American/ | 1214 | Adult | Abdominal | Hispanic Americans have a higher frequency of the 148M allele than African Americans |
| African American | CT scanning | ||||
| United States | |||||
| Kantartzis et al[44] | Caucasian | 330 | Adult | H-MRS | Higher liver fat but not insulin sensitivity, lipids, or liver enzymes |
| Germany | MRT | ||||
| Petit et al[45] | Caucasian | 218 | Adult | H-MRS | Not associated with BMI or visceral fat area |
| France | |||||
| Romeo et al[46] | Caucasian | 475 | Pediatric | US | Increased ALT and AST, but not glucose tolerance and insulin sensitivity |
| Italy | |||||
| Santoro et al[47] | Caucasian/ | 85 | Pediatric | MRI | Increase susceptibility to hepatic steatosis, but without increasing insulin resistance |
| Hispanic/ | |||||
| African American | |||||
| United States |
PNPLA3: Patatin-like phospholipase domain-containing 3; NAFLD: Nonalcoholic fatty liver disease; NASH: Nonalcoholic steatohepatitis; US: Ultrasonography; H-MRS: Hydrogen magnetic resonance spectroscopy; MRI: Magnetic resonance imaging; MRT: Magnetic resonance tomography; CT: Computed tomography; TG: Triglyceride; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; GGT: Gamma-glutamyl transferase; HDL-C: High-density lipoprotein cholesterol; LDL-C: Low-density lipoprotein cholesterol; BMI: Body mass index; NA: Not available.
ROLE OF THE PNPLA3 I148M VARIANT IN NONALCOHOLIC FATTY LIVER FIBROSIS
Nonalcoholic fatty liver fibrosis represents a necessary pathological pathway that patients with NAFLD undergo and then progress to cirrhosis, HCC and end-stage liver disease, and poses a noteworthy economic burden worldwide. Liver fibrosis is a reversible wound-healing response to continuous chronic liver injuries[48], and the most characteristic hallmark is the excessive production and accumulation of intrahepatic extracellular matrix (ECM), including fibronectin, type I collagen, proteoglycan, etc., which eventually lead to hepatic structural change and dysfunction. Therefore, whether or not to control or reverse liver fibrosis affects the prognosis of patients to a great extent. However, challenges remain, as the underlying specific pathogenesis of liver fibrosis is still unclear.
Various studies have established that the PNPLA3 I148M variant is significantly associated with the development of fibrogenesis and the severity of nonalcoholic fatty liver fibrosis[7,8,10,11,32,49]. In 2010, Valenti and colleagues demonstrated that the PNPLA3 I148M variant influenced both the presence of NASH (OR = 1.5; 95%CI: 1.12-2.04) and the severity of liver fibrosis (OR = 1.5; 95%CI: 1.09-2.12) in a large series of 591 biopsied patients with NAFLD independently of the degree of obesity, diabetes and steatosis[11]. In addition, Rotman et al[7] carried out a study in a large cohort of 894 adults and 223 children with histopathological markers of NAFLD, and confirmed that the PNPLA3 I148M variant was associated with portal (P < 0.001) and lobular inflammation (P = 0.005), Mallory-Denk bodies (P = 0.020), and fibrosis (P < 0.001). Furthermore, in an observational cross-sectional study of 899 European patients with chronic liver diseases, there was a prominent association between the PNPLA3 I148M variant and enhanced liver stiffness by using a non-invasive transient elastography[10]. This association between the PNPLA3 I148M variant and the severity of fibrosis in patients with histologically confirmed NAFLD was replicated in a case-control analysis (OR = 3.37; 95%CI: 2.85-3.97; P < 0.001)[32]. More importantly, consistent with previous findings in adults, a prospective study of 149 consecutive Caucasian children and adolescents with biopsy-proven NAFLD showed stronger evidence that the PNPLA3 I148M variant significantly influenced the occurrence of fibrosis (P = 0.01) irrespective of confounding factors[8].
Recently, a meta-analysis established a significant association between the PNPLA3 I148M variant and advanced nonalcoholic fatty liver fibrosis[49]. In a dominant model, patients with PNPLA3 148MM or 148IM exhibited a significantly increased risk of developing advanced fibrosis compared with 148II carriers (OR = 1.29; 95%CI: 1.21-1.38). In line with the dominant model, a recessive model yielded a similar strength of the association (OR = 1.32; 95%CI: 1.20-1.45)[49]. Therefore, there is little doubt that there exists an association between the PNPLA3 I148M variant and nonalcoholic fatty liver fibrosis. Continuous research on the strategies for potential prevention or even curative intervention of nonalcoholic fatty liver fibrosis is warranted.
However, the specific mechanism of the PNPLA3 I148M variant in the development and progression of nonalcoholic fatty liver fibrosis is still not clear. Up to now, abnormal activation of hepatic stellate cells (HSCs) characterized by retinoid loss was considered as the key contributor to fibrogenesis irrespective of the underlying disease[50]. The hedgehog (Hh) signaling pathway, consisting of Hh ligands, transmembrane protein receptors patched and smoothened, and Gli family transcription factors, is one of the most classic signaling pathways participating in the process of cell differentiation and proliferation during embryonic development[51,52].
Recent studies have shown a strong association between the Hh signaling pathway and the development and progression of nonalcoholic fatty liver fibrosis[53-55]. Guy et al[53] demonstrated that the activation of the Hh pathway paralleled histological severity of injury and liver fibrosis in a cross-sectional immunohistochemical study of a large cohort of biopsy-proven adult NAFLD patients. Moreover, a study of 56 children with NAFLD at the University of California, San Diego, United States, also showed significant associations between sonic Hh grade, the numbers of Hh-ligand-producing cells, Hh-responsive cells, and fibrosis stage[55]. In addition, it has been reported that the Hh signaling pathway regulates the HSC-to-myofibroblast transition[56,57], the expansion of hepatic progenitor cells[53,54], and the expression of cholangiocyte chemokines[58,59]. Meanwhile, cholangiocytes and hepatic progenitor cells can activate the Hh signaling pathway by generating Hh ligands[59] and increasing the expression of Gli2[54] (a Hh-regulated target gene), which, in turn, activates HSCs. Based on the available evidence, it is speculated that cross-talk between the Hh signaling pathway and activated HSCs, as well as progenitor cells and cholangiocytes, forms a pro-fibrogenic network together and leads to excessive generation and deposition of ECM and eventually fibrogenesis. Accordingly, we hypothesize that the PNPLA3 I148M variant promotes the development of fibrogenesis by activating the Hh signaling pathway, which, in turn, leads to the activation and proliferation of HSCs, and excessive generation and deposition of ECM (Figure 1). To test this hypothesis, future studies are needed to established PNPLA3 I148M transgenic mouse models, which can be used to establish transgenic mouse models of nonalcoholic fatty liver fibrosis. With such models, the role of the PNPLA3 I148M variant in nonalcoholic fatty liver fibrosis and the underlying mechanisms can be further explored. Consequently, the association between the PNPLA3 I148M variant and the Hh signaling pathway, and the precise mechanisms at molecular, cellular and genetic levels by which the PNPLA3 I148M variant participates in the development of fibrogenesis are expected to be elucidated, which will lay a theoretical foundation and provide valuable experimental data for the clinical management of nonalcoholic fatty liver fibrosis.
Figure 1.
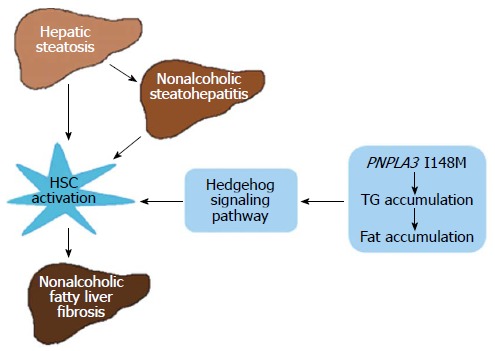
Simplified schematic model showing the hypothetical molecular mechanism by which the PNPLA3 I148M variant participates in the development and progression of nonalcoholic fatty liver fibrosis. The hedgehog signaling pathway links PNPLA3 with the activation of hepatic stellate cells, which is considered as the central part of fibrogenesis. PNPLA3 protein exhibits activities of lysophosphatidic acid acyltransferase and acylglycerol hydrolase to maintain the TG balance in the liver. The “gain function” of the PNPLA3 I148M variant causes Triglyceride (TG) accumulation in the liver, which accelerates the progression of nonalcoholic fatty liver disease. HSC: Hepatic stellate cell.
CONCLUSION
The PNPLA3 I148M variant is associated with NAFLD, but is predominant in women, not in men. The association may vary among different ethnic populations, but is not affected by the presence of metabolic syndrome. The PNPLA3 I148M variant may promote the development of fibrogenesis by activating the Hh signaling pathway, which, in turn, leads to the activation and proliferation of HSCs, and excessive generation and deposition of ECM. Further studies are needed to understand the underlying mechanisms.
Footnotes
Supported by National Natural Science Foundation of China No. 81170337/H0304.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 13, 2014
First decision: September 15, 2014
Article in press: October 21, 2014
P- Reviewer: Fan JG, Fouad YM, Morales-Gonzalez JA, Mikolasevic I S- Editor: Qi Y L- Editor: Cant MR E- Editor: Liu XM
References
- 1.Gao X, Fan JG. Diagnosis and management of non-alcoholic fatty liver disease and related metabolic disorders: consensus statement from the Study Group of Liver and Metabolism, Chinese Society of Endocrinology. J Diabetes. 2013;5:406–415. doi: 10.1111/1753-0407.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Bhala N, Jouness RI, Bugianesi E. Epidemiology and natural history of patients with NAFLD. Curr Pharm Des. 2013;19:5169–5176. doi: 10.2174/13816128113199990336. [DOI] [PubMed] [Google Scholar]
- 4.Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10:330–344. doi: 10.1038/nrgastro.2013.41. [DOI] [PubMed] [Google Scholar]
- 5.Erickson SK. Nonalcoholic fatty liver disease. J Lipid Res. 2009;50 Suppl:S412–S416. doi: 10.1194/jlr.R800089-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology. 2010;52:894–903. doi: 10.1002/hep.23759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valenti L, Alisi A, Galmozzi E, Bartuli A, Del Menico B, Alterio A, Dongiovanni P, Fargion S, Nobili V. I148M patatin-like phospholipase domain-containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology. 2010;52:1274–1280. doi: 10.1002/hep.23823. [DOI] [PubMed] [Google Scholar]
- 9.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53:1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 10.Krawczyk M, Grünhage F, Zimmer V, Lammert F. Variant adiponutrin (PNPLA3) represents a common fibrosis risk gene: non-invasive elastography-based study in chronic liver disease. J Hepatol. 2011;55:299–306. doi: 10.1016/j.jhep.2010.10.042. [DOI] [PubMed] [Google Scholar]
- 11.Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, Mozzi E, Roviaro G, Vanni E, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209–1217. doi: 10.1002/hep.23622. [DOI] [PubMed] [Google Scholar]
- 12.Zain SM, Mohamed R, Mahadeva S, Cheah PL, Rampal S, Basu RC, Mohamed Z. A multi-ethnic study of a PNPLA3 gene variant and its association with disease severity in non-alcoholic fatty liver disease. Hum Genet. 2012;131:1145–1152. doi: 10.1007/s00439-012-1141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pingitore P, Pirazzi C, Mancina RM, Motta BM, Indiveri C, Pujia A, Montalcini T, Hedfalk K, Romeo S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim Biophys Acta. 2014;1841:574–580. doi: 10.1016/j.bbalip.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 14.He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, Cohen JC, Hobbs HH. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285:6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baulande S, Lasnier F, Lucas M, Pairault J. Adiponutrin, a transmembrane protein corresponding to a novel dietary- and obesity-linked mRNA specifically expressed in the adipose lineage. J Biol Chem. 2001;276:33336–33344. doi: 10.1074/jbc.M105193200. [DOI] [PubMed] [Google Scholar]
- 16.Rae-Whitcombe SM, Kennedy D, Voyles M, Thompson MP. Regulation of the promoter region of the human adiponutrin/PNPLA3 gene by glucose and insulin. Biochem Biophys Res Commun. 2010;402:767–772. doi: 10.1016/j.bbrc.2010.10.106. [DOI] [PubMed] [Google Scholar]
- 17.Liu YM, Moldes M, Bastard JP, Bruckert E, Viguerie N, Hainque B, Basdevant A, Langin D, Pairault J, Clément K. Adiponutrin: A new gene regulated by energy balance in human adipose tissue. J Clin Endocrinol Metab. 2004;89:2684–2689. doi: 10.1210/jc.2003-031978. [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, Hobbs HH. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci USA. 2010;107:7892–7897. doi: 10.1073/pnas.1003585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubuquoy C, Robichon C, Lasnier F, Langlois C, Dugail I, Foufelle F, Girard J, Burnol AF, Postic C, Moldes M. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J Hepatol. 2011;55:145–153. doi: 10.1016/j.jhep.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 20.Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. Characterization of the human patatin-like phospholipase family. J Lipid Res. 2006;47:1940–1949. doi: 10.1194/jlr.M600185-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Xin YN, Zhao Y, Lin ZH, Jiang X, Xuan SY, Huang J. Molecular dynamics simulation of PNPLA3 I148M polymorphism reveals reduced substrate access to the catalytic cavity. Proteins. 2013;81:406–414. doi: 10.1002/prot.24199. [DOI] [PubMed] [Google Scholar]
- 22.Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, Wongsiriroj N, Nagy HM, Ivanova PT, Scott SA, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, Castro-Perez J, Cohen JC, Hobbs HH. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122:4130–4144. doi: 10.1172/JCI65179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan X, Waterworth D, Perry JR, Lim N, Song K, Chambers JC, Zhang W, Vollenweider P, Stirnadel H, Johnson T, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet. 2008;83:520–528. doi: 10.1016/j.ajhg.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang CW, Lin HY, Shin SJ, Yu ML, Lin ZY, Dai CY, Huang JF, Chen SC, Li SS, Chuang WL. The PNPLA3 I148M polymorphism is associated with insulin resistance and nonalcoholic fatty liver disease in a normoglycaemic population. Liver Int. 2011;31:1326–1331. doi: 10.1111/j.1478-3231.2011.02526.x. [DOI] [PubMed] [Google Scholar]
- 27.Sookoian S, Castaño GO, Burgueño AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res. 2009;50:2111–2116. doi: 10.1194/jlr.P900013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kollerits B, Coassin S, Kiechl S, Hunt SC, Paulweber B, Willeit J, Brandstätter A, Lamina C, Adams TD, Kronenberg F. A common variant in the adiponutrin gene influences liver enzyme values. J Med Genet. 2010;47:116–119. doi: 10.1136/jmg.2009.066597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J, Xin YN, Lü WH, Lin ZH, Zhang DD, Zhang M, Dong QJ, Jiang XJ, Xuan SY. [Polymorphism rs738409 in PNPLA3 is associated with inherited susceptibility to non-alcoholic fatty liver disease] Zhonghua Ganzangbing Zazhi. 2013;21:619–623. doi: 10.3760/cma.j.issn.1007-3418.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 30.Wanless IR, Shiota K. The pathogenesis of nonalcoholic steatohepatitis and other fatty liver diseases: a four-step model including the role of lipid release and hepatic venular obstruction in the progression to cirrhosis. Semin Liver Dis. 2004;24:99–106. doi: 10.1055/s-2004-823104. [DOI] [PubMed] [Google Scholar]
- 31.Chalasani N, Wilson L, Kleiner DE, Cummings OW, Brunt EM, Unalp A. Relationship of steatosis grade and zonal location to histological features of steatohepatitis in adult patients with non-alcoholic fatty liver disease. J Hepatol. 2008;48:829–834. doi: 10.1016/j.jhep.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Speliotes EK, Butler JL, Palmer CD, Voight BF, Hirschhorn JN. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology. 2010;52:904–912. doi: 10.1002/hep.23768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pacifico L, Nobili V, Anania C, Verdecchia P, Chiesa C. Pediatric nonalcoholic fatty liver disease, metabolic syndrome and cardiovascular risk. World J Gastroenterol. 2011;17:3082–3091. doi: 10.3748/wjg.v17.i26.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goran MI, Walker R, Le KA, Mahurkar S, Vikman S, Davis JN, Spruijt-Metz D, Weigensberg MJ, Allayee H. Effects of PNPLA3 on liver fat and metabolic profile in Hispanic children and adolescents. Diabetes. 2010;59:3127–3130. doi: 10.2337/db10-0554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis JN, Lê KA, Walker RW, Vikman S, Spruijt-Metz D, Weigensberg MJ, Allayee H, Goran MI. Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am J Clin Nutr. 2010;92:1522–1527. doi: 10.3945/ajcn.2010.30185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin YC, Chang PF, Hu FC, Yang WS, Chang MH, Ni YH. A common variant in the PNPLA3 gene is a risk factor for non-alcoholic fatty liver disease in obese Taiwanese children. J Pediatr. 2011;158:740–744. doi: 10.1016/j.jpeds.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 37.Viitasalo A, Pihlajamaki J, Lindi V, Atalay M, Kaminska D, Joro R, Lakka TA. Associations of I148M variant in PNPLA3 gene with plasma ALT levels during 2-year follow-up in normal weight and overweight children: the PANIC Study. Pediatr Obes. 2014:Epub ahead of print. doi: 10.1111/ijpo.234. [DOI] [PubMed] [Google Scholar]
- 38.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 39.Petersen KF, Dufour S, Feng J, Befroy D, Dziura J, Dalla Man C, Cobelli C, Shulman GI. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci USA. 2006;103:18273–18277. doi: 10.1073/pnas.0608537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wagenknecht LE, Palmer ND, Bowden DW, Rotter JI, Norris JM, Ziegler J, Chen YD, Haffner S, Scherzinger A, Langefeld CD. Association of PNPLA3 with non-alcoholic fatty liver disease in a minority cohort: the Insulin Resistance Atherosclerosis Family Study. Liver Int. 2011;31:412–416. doi: 10.1111/j.1478-3231.2010.02444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 42.Day CP. From fat to inflammation. Gastroenterology. 2006;130:207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 43.Sevastianova K, Kotronen A, Gastaldelli A, Perttilä J, Hakkarainen A, Lundbom J, Suojanen L, Orho-Melander M, Lundbom N, Ferrannini E, et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am J Clin Nutr. 2011;94:104–111. doi: 10.3945/ajcn.111.012369. [DOI] [PubMed] [Google Scholar]
- 44.Kantartzis K, Peter A, Machicao F, Machann J, Wagner S, Königsrainer I, Königsrainer A, Schick F, Fritsche A, Häring HU, et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes. 2009;58:2616–2623. doi: 10.2337/db09-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petit JM, Guiu B, Masson D, Duvillard L, Jooste V, Buffier P, Terriat B, Bouillet B, Brindisi MC, Loffroy R, et al. Specifically PNPLA3-mediated accumulation of liver fat in obese patients with type 2 diabetes. J Clin Endocrinol Metab. 2010;95:E430–E436. doi: 10.1210/jc.2010-0814. [DOI] [PubMed] [Google Scholar]
- 46.Romeo S, Sentinelli F, Cambuli VM, Incani M, Congiu T, Matta V, Pilia S, Huang-Doran I, Cossu E, Loche S, et al. The 148M allele of the PNPLA3 gene is associated with indices of liver damage early in life. J Hepatol. 2010;53:335–338. doi: 10.1016/j.jhep.2010.02.034. [DOI] [PubMed] [Google Scholar]
- 47.Santoro N, Kursawe R, D’Adamo E, Dykas DJ, Zhang CK, Bale AE, Calí AM, Narayan D, Shaw MM, Pierpont B, et al. A common variant in the patatin-like phospholipase 3 gene (PNPLA3) is associated with fatty liver disease in obese children and adolescents. Hepatology. 2010;52:1281–1290. doi: 10.1002/hep.23832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiang DJ, Roychowdhury S, Bush K, McMullen MR, Pisano S, Niese K, Olman MA, Pritchard MT, Nagy LE. Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS One. 2013;8:e69114. doi: 10.1371/journal.pone.0069114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singal AG, Manjunath H, Yopp AC, Beg MS, Marrero JA, Gopal P, Waljee AK. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. Am J Gastroenterol. 2014;109:325–334. doi: 10.1038/ajg.2013.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ray K. Liver: hepatic stellate cells hold the key to liver fibrosis. Nat Rev Gastroenterol Hepatol. 2014;11:74. doi: 10.1038/nrgastro.2013.244. [DOI] [PubMed] [Google Scholar]
- 51.Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30:303–312. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 52.Omenetti A, Choi S, Michelotti G, Diehl AM. Hedgehog signaling in the liver. J Hepatol. 2011;54:366–373. doi: 10.1016/j.jhep.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, Diehl AM. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. 2012;55:1711–1721. doi: 10.1002/hep.25559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friedman SL. Liver fibrosis in 2012: Convergent pathways that cause hepatic fibrosis in NASH. Nat Rev Gastroenterol Hepatol. 2013;10:71–72. doi: 10.1038/nrgastro.2012.256. [DOI] [PubMed] [Google Scholar]
- 55.Swiderska-Syn M, Suzuki A, Guy CD, Schwimmer JB, Abdelmalek MF, Lavine JE, Diehl AM. Hedgehog pathway and pediatric nonalcoholic fatty liver disease. Hepatology. 2013;57:1814–1825. doi: 10.1002/hep.26230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi SS, Omenetti A, Witek RP, Moylan CA, Syn WK, Jung Y, Yang L, Sudan DL, Sicklick JK, Michelotti GA, et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1093–G1106. doi: 10.1152/ajpgi.00292.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swiderska-Syn M, Syn WK, Xie G, Krüger L, Machado MV, Karaca G, Michelotti GA, Choi SS, Premont RT, Diehl AM. Myofibroblastic cells function as progenitors to regenerate murine livers after partial hepatectomy. Gut. 2014;63:1333–1344. doi: 10.1136/gutjnl-2013-305962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Omenetti A, Syn WK, Jung Y, Francis H, Porrello A, Witek RP, Choi SS, Yang L, Mayo MJ, Gershwin ME, et al. Repair-related activation of hedgehog signaling promotes cholangiocyte chemokine production. Hepatology. 2009;50:518–527. doi: 10.1002/hep.23019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Omenetti A, Diehl AM. Hedgehog signaling in cholangiocytes. Curr Opin Gastroenterol. 2011;27:268–275. doi: 10.1097/MOG.0b013e32834550b4. [DOI] [PMC free article] [PubMed] [Google Scholar]