

Abstract
AIM: To characterize an alcohol and high fat diet induced chronic pancreatitis rat model that mimics poor human dietary choices.
METHODS: Experimental rats were fed a modified Lieber-DeCarli alcohol (6%) and high-fat (65%) diet (AHF) for 10 wk while control animals received a regular rodent chow diet. Weekly behavioral tests determined mechanical and heat sensitivity. In week 10 a fasting glucose tolerance test was performed, measuring blood glucose levels before and after a 2 g/kg bodyweight intraperitoneal (i.p.) injection of glucose. Post mortem histological analysis was performed by staining pancreas and liver tissue sections with hematoxylin and eosin. Pancreas sections were also stained with Sirius red and fast green to quantify collagen content. Insulin-expressing cells were identified immunohistochemically in separate sections. Tissue staining density was quantified using Image J software. After mechanical and heat sensitivity became stable (weeks 6-10) in the AHF-fed animals, three different drugs were tested for their efficacy in attenuating pancreatitis associated hypersensitivity: a Group II metabotropic glutamate receptor specific agonist (2R,4R)-4-Aminopyrrolidine-2,4-dicarboxylate (APDC, 3 mg/kg, ip; Tocris, Bristol, United Kingdom), nociceptin (20, 60, 200 nmol/kg, ip; Tocris), and morphine sulfate (3 mg/kg, μ-opioid receptor agonist; Baxter Healthcare, Deerfield, IL, United States).
RESULTS: Histological analysis of pancreas and liver determined that unlike control rats, AHF fed animals had pancreatic fibrosis, acinar and beta cell atrophy, with steatosis in both organs. Fat vacuolization was significantly increased in AHF fed rats (6.4% ± 1.1% in controls vs 23.8% ± 4.2%, P < 0.05). Rats fed the AHF diet had reduced fasting glucose tolerance in week 10 when peak blood glucose levels reached significantly higher concentrations than controls (127.4 ± 9.2 mg/dL in controls vs 161.0 ± 8.6 mg/dL, P < 0.05). This concurred with a 3.5 fold higher incidence of single and small 2-10 cell insulin-positive cell clusters (P < 0.05). Insulin expressing islet of Langerhans cells appeared hypertrophied while islet number and area measurements were not different from controls. Weekly behavioral tests determined that mechanical and heat sensitivities were significantly increased by 4 wk on AHF diet compared to controls. Hypersensitivity was attenuated with efficacy similar to morphine with single dose treatment of either metabotropic glutamate receptor 2/3 agonist APDC, or nociceptin, the endogenous ligand for opioid-receptor-like 1 receptor.
CONCLUSION: The AHF diet induces a chronic alcoholic pancreatitis in rats with measurable features resembling clinical patients with chronic pancreatitis and type 3c diabetes mellitus.
Keywords: Lieber-DeCarli diet, Orphanin FQ receptor, Metabotropic glutamate receptor, Liver steatosis, Pain, Behavior, Glucose tolerance, Type 3c diabetes mellitus
Core tip: Chronic pancreatitis is a progressive and potentially fatal disease caused by persistent unresolved inflammation and pancreatic fibrosis. It can be accompanied by intractable abdominal pain and progress to type 3c diabetes mellitus (T3cDM) and pancreatic cancer. Animal models of acute pancreatitis are typically chemically induced, invasive, of short duration, and have a high mortality rate. This study characterizes a diet-induced chronic rat model closely mimicking poor human dietary choices to investigate therapies for alcoholic chronic pancreatitis with developing T3cDM. The efficacy of acute opioid and non-opioid pharmacological interventions are compared to morphine in pain-related behavior tests.
INTRODUCTION
Chronic pancreatitis is a progressive and potentially fatal disease caused by persistent unresolved inflammation and pancreatic fibrosis typically accompanied by intractable abdominal pain, particularly during the early phase[1,2]. Other symptoms include weight loss due to insufficiency of the exocrine pancreas, type 3c diabetes mellitus (T3cDM), and an increased risk of developing pancreatic cancer[3-13]. Factors that increase susceptibility to the diverse etiology of chronic pancreatitis include lifestyle choices such as regular alcohol consumption, particularly binge drinking, hyperlipidemia/hypertriglyceridemia caused by a diet high in saturated fat, and smoking, as well as heritable factors[1,12,14-21]. Although 50%-70% of patients with chronic pancreatitis regularly consume alcohol, only around 10% of patients with alcohol use disorders develop this disease, indicating other contributing factors[1,22-25]. Worldwide the incidence of chronic pancreatitis is higher in the male population, partially explained by the recently identified X chromosome linked genetic polymorphism in the claudin-2 gene[19]. Chief clinical complaint of afflicted patients is intractable pain and currently recommended pain management therapies range from non-narcotic analgesics increasing to strong opiates depending on the patient’s complaints[25-27]. Concurrent analysis of amino acids in plasma and serum of patients with chronic pancreatitis identified a doubling of the glutamate concentration, the main excitatory neurotransmitter of the nervous system and a known facilitator of pain, while other amino acid levels are unaffected or decreased[28,29]. Knowledge of the underlying pathophysiology of chronic pancreatitis resulting in this severe abdominal pain is still limited and is needed for the development of better pharmacological treatments. Presently employed animal models poorly reproduce the clinical etiology of chronic pancreatitis as they are highly invasive, requiring laparotomy surgery and/or utilize repetitive dosing with caustic chemicals. The abdominal hypersensitivity induced with these experimental procedures is better suited for modeling acute pancreatitis[30,31].
Chronic pancreatitis is a multifactorial disease. Clinical observations indicate that diets high in fat facilitate the progression of alcoholic chronic pancreatitis[12,18,21,25]. Animal studies modeling chronic pancreatitis have reported limited or no success when ethanol is used alone[32-36]. The combination of fatty acids and ethanol is required to induce noticeable cell damage to cultured pancreatic cells while the application of individual compounds alone is unable to injure these cells[37-39]. Recently, we established a rat model of chronic pancreatitis using a modified Lieber-DeCarli diet with increased dietary saturated fat content (28%)[40,41]. In the present study, the alcoholic chronic pancreatitis rat model was modified by further increasing the dietary saturated fat content to 65%. Unique to the current model is developing T3cDM, recognized by the American Diabetes Association and the National Institutes of Health, is an underdiagnosed secondary disease associated with exocrine pancreatic damage[6,8,12,13]. Clinically, between 5%-10% of patients with diabetes are diagnosed with T3cDM[6,13]. This is the first animal model of chronic pancreatitis that develops T3cDM. The model is utilized here to quantify chronic pancreatitis induced pain related behaviors that persist at least 10 wk for analgesic testing.
Using the alcohol and high fat diet (AHF) induced rat chronic pancreatitis model, the efficacy of acute pharmacological activation of three inhibitory metabotropic Gi protein-coupled receptors (GPCR) was tested. The agonists activate either Group II metabotropic glutamate receptor 2 and receptor 3 (mGluR2/3), μ-opioid receptor (MOR), or the opioid receptor like-1 receptor (ORL-1). Signaling through mGluR2/3 receptors was initiated by glutamate binding[42,43], while the opioid receptors ORL-1 and μ-opioid receptor MOR are activated by the endogenous neuropeptides nociceptin (also called orphanin FQ[44,45]), enkephalins and beta-endorphins, respectively[46,47]. Their activation results in downstream signaling events that activate voltage-gated potassium channels to inhibit voltage-gated calcium channels. Thus, activating these metabotropic receptors on peripheral nociceptors activated by noxious stimuli inhibits the release of neuropeptide modulators and glutamate, the main excitatory neurotransmitter in the nervous system[48-50].
Agonists for mGluR2/3 have been shown to be analgesic in animal models of somatic inflammatory and neuropathic pain[51-55]. Information about ORL-1 activation indicates that intrathecal and peripheral applications of nociceptin attenuate hypersensitivity in inflammatory and neuropathic rodent pain models[56-59], while intracerebroventricular injections produce hyperalgesia[60]. The efficacy of their activation in reducing hypersensitivity in the chronic alcoholic pancreatitis model in the present study is compared to morphine, the customary opiate utilized for experimental comparisons. Using the AHF rat chronic pancreatitis model with developing T3cDM, we provide the first evidence that peripheral activation of inhibitory GPCR-mediated signaling cascades through mGluR2/3 or ORL-1 are similarly efficacious to morphine in reversing hypersensitivity induced in this chronic visceral pain model.
MATERIALS AND METHODS
Ethics statement
All animal procedures were conducted according to the guidelines for the ethical treatment of experimental animals published by the Internal Association for the Study of Pain and approved by the University of Kentucky Institutional Animal Care and Use Committee (IACUC#2007-0113).
Induction of alcohol pancreatitis
Experiments were performed using a total of 16 male Fischer rats weighing 230-250 g (Harlan Laboratories, Indianapolis, IN). Animals were housed individually on a 12/12 h reverse light cycle so that behavioral assays were conducted during their active night phase. Food and water were given ad libitum and animals were divided into two groups: (1) controls (n = 7) received regular low soy rodent chow (Teklab #8626, Harlan, Indianapolis, IN) and (2) alcohol and high fat (AHF) diet fed animals (n = 9) received a modified Lieber-DeCarli diet. Along with the 6% ethanol liquid diet, a daily portion of 8 g lard was provided. The liquid diet (1000 g) contained 14% LD101A (TestDiet, Richmond, IN), 9% maltodextrin, 10% apple juice, 6% ethanol, and 3.3% corn oil. The AHF liquid diet was initially introduced without ethanol and corn oil. On a weekly basis the ethanol content of the food was increased from 4%-6% and finally corn oil was also added, resulting in a liquid maintenance diet containing 6% ethanol and 30% corn oil in AHF fed animals. Throughout this time AHF fed animals also received the 8 g lard supplement daily. Consumed liquid diet and lard were measured daily and averaged throughout the experimental time period to calculate the percentage of consumed dietary fat. Average daily liquid diet consumption was 20 g, of which 27.6% was nutritional value and 72.6% was water. Of this 5.48 g with nutritional value, 28% was fat (1.5 g). Animals consumed an average of 6 g of lard daily, resulting in 7.5 g total fat consumption per day out of the total 11.5 g of nutritional diet. Thus, total daily dietary fat consumption was about 65%.
Behavioral testing
Weekly behavioral assays to determine mechanical and heat sensitivity were performed on all animals. The abdomen was shaved at least 24 h prior to testing. Alcohol was removed 4 h prior to behavioral testing and drug application to minimize potential interference of alcohol on motor control and interactions with the compound of interest. Previous studies had shown that alcohol withdrawal in this experimental setting did not interfere with outcomes of these behavioral assays[41].
Animals were placed in individual Plexiglas boxes on an elevated metal screen mesh (3 mm2 holes). Mechanical sensitivity of the abdomen and hindpaws was tested by probing the animal through the mesh from below using two different experimental methods. Abdominal referred sensitivity was characterized as previously reported by Vera-Portocarrero et al[61] (2003). Briefly, the upper right abdominal quadrant overlaying the pancreas was probed 10 times each in ascending order with 2 different von Frey filaments eliciting 1.0 or 6.0 g bending force equivalent to 9.8 or 58.8 mN. This assessed behavior in response to both sub- and supra-threshold mechanical stimuli, i.e., touch and poke, to identify the time course for development of chronic mechanical hypersensitivity. Stimuli were applied at 10 s intervals and the number of abdominal withdrawals counted. A single trial to determine the number of responses to application of 10 stimuli with a single von Frey filament was performed per animal per time point. Mechanical hypersensitivity thresholds of the hindpaw footpads were subsequently determined while the animals were still on the elevated mesh screen table. For this measure, the up-down method was used incorporating a series of 8 von Frey filaments exerting calibrated bending forces (0.4, 0.6, 1.0, 2.0, 4.0, 6.0, 8.0, 15.0 g or 3.9, 5.9, 9.8, 19.6, 39.2, 58.8, 98.0 mN[62]).
Heat sensitivity was determined using a hot plate analgesiometer apparatus set at 50 °C (Columbus Instruments, Columbus, OH). Animals were placed individually on the hotplate (254 mm × 254 mm heated surface) surrounded by a 30 cm high Plexiglas barrier and the latency until a nocifensive response occurred, such as shaking or licking the paw, jumping, or running was recorded. Animals were immediately removed after the initial response and a cut-off time of 25 s prevented burn injury. The heat test was performed 3 times at 20 min intervals and the latencies averaged.
Drug treatments
After mechanical and heat sensitivity developed in the AHF-fed animals, the efficacy of three different drugs in attenuation of pancreatitis associated hypersensitivity was determined to identify contributing signaling pathways: a Group II metabotropic glutamate receptor specific agonist, (2R,4R)-4-Aminopyrrolidine-2,4-dicarboxylate (APDC, 3 mg/kg, ip; Tocris, Bristol, United Kingdom), nociceptin (20, 60, 200 nmol/kg, ip; Tocris), and morphine sulfate (3 mg/kg, subcutaneous (s.c.); Baxter Healthcare, Deerfield, IL, United States). All drugs were diluted in sterile saline and tested after hypersensitivity became stable (weeks 6-10). Mechanical and heat sensitivity were determined 24 h before treatment and 1, 4, and 24 h after intraperitoneal (ip) injection of a single dose of drug or vehicle (saline) was administered. Efficacy of drug treatments was tested once a week in the same animals since no long-lasting changes for acute use of these drugs is reported nor noted in the present study.
Glucose tolerance test
Prior to euthanasia, a glucose tolerance test was performed with all animals[63]. Rats were fasted for 6 h and then given glucose (2 g/kg body weight, ip) in distilled sterile water (25% w/v solution). Glucose levels in tail vein blood samples were measured prior to and at several timepoints after giving glucose (15, 30, 60, 120, 180 min) using a blood glucose meter and appropriate test strips (FreeStyle, Abbott Diabetes Care, Alameda, CA).
Histology
Liver and pancreas samples were collected prior to perfusion of the animal and immerse fixed overnight in 4% paraformaldehyde in phosphate buffered saline, transferred into 70% ethanol, and paraffin embedded. Sections were cut (5 μm), rehydrated, stained for collagen fibers with Sirius red (Electron Microscopy Sciences, #26357-02), and counterstained with fast green. Other sections were stained with Sirius red and hematoxylin and eosin (HE) using routine histological protocols. Liver and pancreas samples were analyzed after acquiring five random images per slide using ACT software and a Nikon microscope for analysis using Image J. A red color threshold macro was used to measure the image areas with fibrous collagen stained red by Sirius red in 5 random fast green stained sections per group. The area quantified is presented as a ratio of the total tissue area. Islets of Langerhans were identified optically in HE and Sirius red stained sections. Images were taken of all Islets, their sizes measured, and their ratio normalized to total tissue area. Quantification of white space among the lobules of condensed pancreas tissue was performed taking 5 random images in HE and Sirius red stained tissue sections, quantifying the ratio of unstained to stained tissue area. A threshold macro determining white areas was used to assess the fat content leached out in paraffin processing in HE and Sirius red stained tissue, and the ratio of unstained white area to total tissue area per image was determined.
Immunofluorescent tissue staining
Deparaffinized and rehydrated pancreas tissue on slides was incubated overnight in a primary rabbit antibody against insulin (H-86, sc-9168; Santa Cruz Biotechnology, Santa Cruz, CA) 1:400 at room temperature. Tissue was washed, incubated in a goat anti-rabbit secondary antibody conjugated to Alexa Fluor 488 (Life Technologies, Grand Island, NY) 1:1000 for 2 h at room temperature, washed and coverslipped in VectaShield hard set mounting medium with DAPI (Vectorlabs, Burlingame, CA). Fluorescent staining of all proliferating insulin-positive cells in images of whole pancreas sectioned head to tail was analyzed using a Nikon microscope with Metamorph software. Post-processing and quantitative analyses were conducted using Photoshop and Image J. Colocalization of DAPI and insulin was used to count the number of cells in small clusters of under 10 cells and the counts normalized to total tissue area in each pancreatic section. Graph Pad Prism and Excel Microsoft software were used for statistical analysis and graphing of the data.
Statistical analysis
Data are expressed as mean ± SE. Statistical significance of behavioral and immunohistochemical data was determined using analysis of variance tests and Student t test with the significance level set at P < 0.05. Post hoc comparison included Student t tests to test statistical differences between control and experimental groups before and after drug treatments.
RESULTS
Chronic consumption of AHF diet induces histopathological changes in pancreas and liver indicative of alcoholic pancreatitis
Adult male Fischer rats were fed the alcohol and high fat diet (AHF) ad libitum for 10 wk. The liquid diet contains 6% (wt/vol) ethanol and 3.3% (wt/vol) corn oil supplemented with a daily dose of 6 g saturated fat resulting in the daily consumed diet to contain 65% fat, modified from Lieber-DeCarli and others[35,41,64,65]. Control animals received standard rat chow pellets (Harlan Teklad 8626). Post mortem histopathological analysis of the pancreas using Sirius red, hematoxylin, and eosin staining revealed acinar atrophy, fibrosis, and necrosis in all of the tissue samples from AHF diet fed rats (Figure 1A and B). Pancreata of AHF fed rats developed enlarged interlobular spaces, and cellular atrophy. Pancreatic interlobular spaces, measured as the percent white area to stained tissue, were significantly increased in AHF fed rats (Figure 1C; control = 8.9% ± 1.0%, n = 4; AHF = 15.7% ± 2.0%, n = 6; P < 0.05) indicating cell shrinkage and/or necrosis. Parenchymal fibrosis was determined histologically as the ratio of the area of pancreatic tissue collagen proteins stained with Sirius red to staining of non-collagenous proteins with fast green[66-68]. Small amounts of interstitial collagen were detected in control tissue samples while the ratio of collagenous and non-collagenous protein staining was significantly increased in AHF fed animals (Figure 1D, F; control = 3.6% ± 0.6%; AHF = 8.8% ± 1.7%; P < 0.05). Collagen bundles were increased in particular and clearly visible surrounding pancreatic ducts, lobes, and blood vessels in tissue samples from the AHF experimental group, while only diffuse staining localized within and among the acinar cells was observed in control animals. In the liver, chronic AHF diet consumption produced clearly visible steatosis when tissue was stained with Sirius red, hematoxylin, and eosin. Alcoholic fatty liver was quantified by measuring the unstained intracellular area in liver tissue samples remaining of the severe fat vacuolization after graded alcohol removal of the paraffin. Fat vacuolization was significantly increased in AHF fed animals compared to controls (Figure 1G-I; control = 6.4% ± 1.1%; AHF = 23.8% ± 4.2%; P < 0.05).
Figure 1.
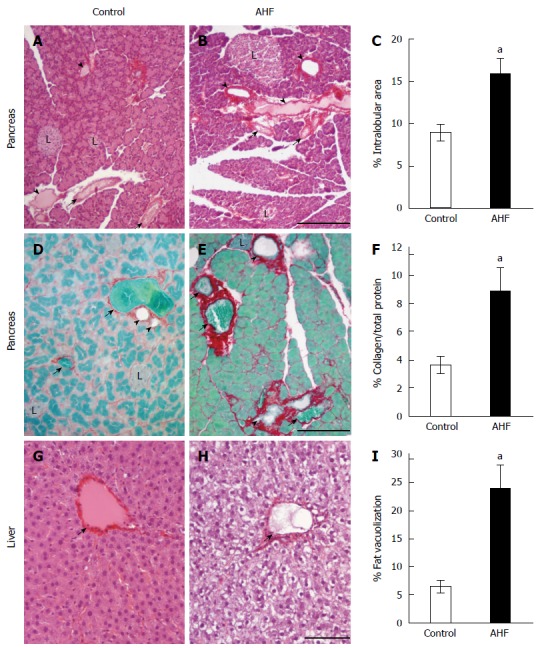
Histopathological analysis of the effects of alcohol and high fat diet fat diet on pancreas and liver. A: Micrograph of pancreas from a control regular chow fed rat stained with hematoxylin and eosin (HE); B: Micrograph of pancreas from an alcohol and high fat diet (AHF) fed rat stained with HE; C: Quantification of intralobular spaces (area percentage) determined there was a significant increase in pancreata from AHF fed rats; D: Micrograph of pancreas from a control rat fed regular chow with Sirius red stained collagen fibrosis and fast green counterstain; E: Micrograph of pancreas from an AHF fed rat with Sirius red stained collagen fibrosis and fast green counterstain; F: Quantification of extracellular collagen deposits (stained red) as a percent of total tissue area. A significant increase in collagen staining was detected in pancreata of AHF fed rats; G: Micrograph of liver from a control rat fed regular chow stained with HE; H: Micrograph of liver from an AHF fed rat stained with HE; I: Quantification of intracellular fat vacuolization (area percentage). Unstained areas are fat vacuoles that were significantly increased in liver samples from AHF fed rats. aP < 0.05, AHF vs control. L: Islet of Langerhans; Solid arrow head: Pancreatic duct; Small arrow: Blood vessel. A, B: Scalebar 50 μm; D, E: Scalebar 100 μm; G, H: Scalebar 100 μm.
Immunohistochemical analysis of insulin expression in pancreas tissue stained from animals fed (A) regular chow or (B) AHF diet determined there was prominent proliferation of insulin immunoreactive cells evident in AHF fed animals (Figure 2A-C). After chronic AHF diet for 10 wk the pancreas had atrophy of insulin producing cells in the islets and an increase of spurious proliferating insulin expressing cells. The number of single to small cell clusters of 10 or less was 3.5 fold higher in AHF fed animals (1.29 ± 0.19 per mm2) compared to controls (0.33 ± 0.09 per mm2, P < 0.05) (Figure 2C).
Figure 2.
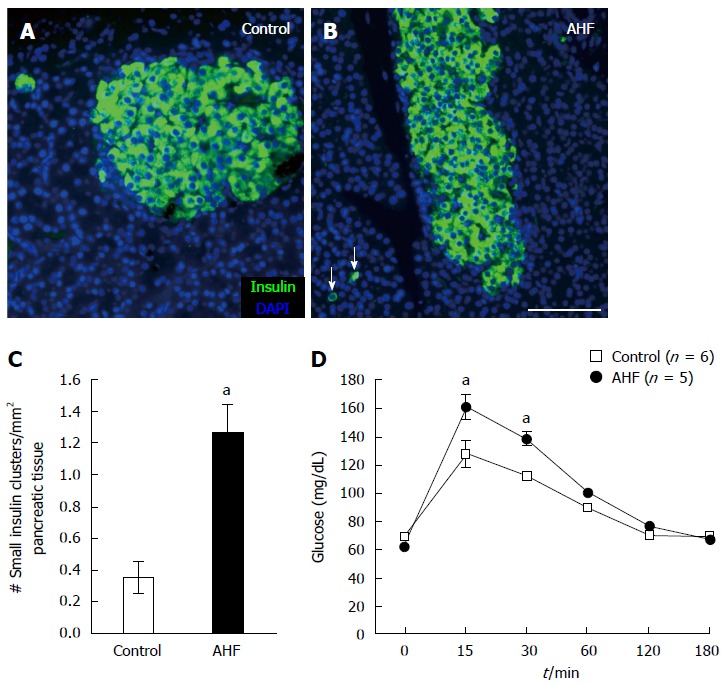
Chronic alcohol and high fat diet induced insulin cell proliferation and decreased glucose tolerance. A: Micrograph of insulin immunoreactivity in pancreas from a control rat fed regular chow; B: Micrograph of insulin immunoreactivity in pancreas from a rat fed alcohol and high fat (AHF). In tissue samples from AHF fed rats insulin producing cells in the islets are atrophied and an increase of spurious proliferating insulin expressing cells (arrows) is detected in the pancreas; C: Quantification of the number of single cells or small insulin cell clusters per mm2 pancreas tissue detected a significant increase in AHF fed rats; D: Intraperitoneal glucose tolerance test at 10 wk determined that glucose tolerance was reduced in fasted AHF fed animals. Blood glucose levels of AHF fed animals peaked to significantly higher levels 15 and 30 min post injection. aP < 0.05, AHF vs control. A-B: Scalebar 100 μm.
Chronic AHF diet consumption decreases glucose tolerance
Weekly monitoring of blood glucose levels in naïve control and AHF fed rats did not detect significant differences throughout the experiment. Unfasted blood glucose concentrations remained constant at 85.1 ± 3.1 mg/dL in controls and 83.4 ± 4.6 mg/dL in AHF fed rats. Glucose tolerance testing following ip injection of 2 g glucose per kg bodyweight was performed in week 10 after fasting animals for 6 h to measure metabolic efficiency of glucose clearance from the blood stream (Figure 2D). Animals fed AHF had significantly higher peak blood glucose levels 15 and 30 min after glucose injection (control 15 min: 127.4 ± 9.2 mg/dL, 30 min: 112.0 ± 3.9 mg/dL; AHF 15 min: 161.0 ± 8.6 mg/dL, 30 min: 138.4 ± 4.4 mg/dL, P < 0.05), indicating reduced glucose tolerance.
Chronic AHF diet consumption causes mechanical and heat hypersensitivity
A chief complaint of clinical patients with chronic pancreatitis is intractable pain. The pain is commonly focused in the upper abdomen but can radiate to the back with some patients reporting overall sensitivity. In the animal model, mechanical and heat sensitivity were monitored weekly (Figure 3). Abdominal sensitivity was assayed by testing the reflexive responses to low intensity (1 g = 9.8 mN force; Figure 3A) and higher intensity (6 g = 58.8 mN force; Figure 3B) mechanical stimuli applied with von Frey filaments[61]. The Y-axis indicates the average number of abdominal withdrawals in response to the bending force stimulations (10 ×). Low intensity (1 g) mechanical stimulation applied on the abdomen elicited an average of 3 of 10 responses from control rats fed a standard chow diet (Figure 3A, open squares). Baseline responses to low intensity and higher intensity stimulation on the abdomen skin were not different between groups [1 g: control = 2.3 ± 0.5; AHF = (1.8 ± 0.5 responses)/10; N.S.; 6 g: control = 5.8 ± 0.5; AHF = (5.5 ± 0.5 responses)/10; N.S.]. Responses increased significantly during the experiment’s duration in the AHF group (1 g: P < 0.01; 6 g: P < 0.0001). In week 5, AHF fed rats responded with almost twice as many abdominal withdrawals to low intensity mechanical stimulation than controls [control = 3.6 ± 0.2; AHF = (6.2 ± 0.7 responses)/10; P < 0.05; Figure 3A]. Similarly, the responses to 6 g mechanical stimulation of the abdomen was significantly increased in AHF animals in week 5 [control = 6.6 ± 0.4; AHF = (9.2 ± 0.3 responses)/10; P < 0.05]. Higher intensity (6 g) mechanical stimulation of the abdomen consistently elicited an average of 6 of 10 responses from control rats while AHF fed animals responded every time after week 5. Mechanical and heat sensitivity of control animals did not change during the study.
Figure 3.
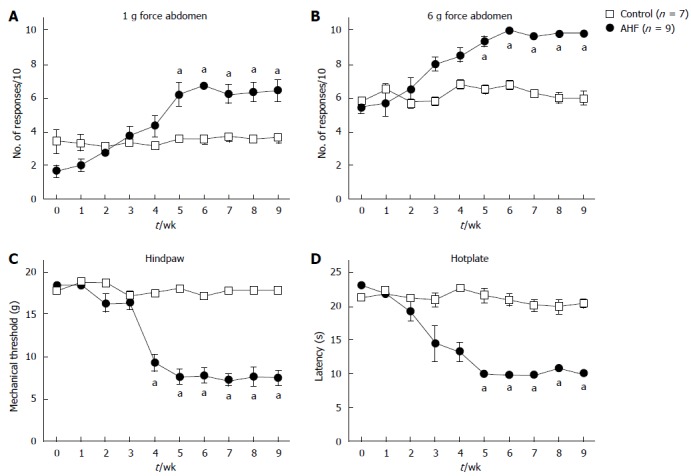
Time course of nociceptive behaviors in rats with chronic pancreatitis evoked by mechanical and heat stimuli. A, B: Abdominal sensitivity is determined by the number of abdominal withdrawals in response to von Frey filament bending force stimulations (× 10). Responses to (A) low intensity (1 g) and (B) high intensity (6 g) mechanical stimulation elicited significantly more responses in alcohol and high fat (AHF) fed rats starting in week 5; C: Mechanical thresholds of the hindpaws were determined with von Frey filaments using the up-down method. In week 4 mechanical sensitivity thresholds of AHF fed rats were significantly reduced and did not recover while on the AHF diet; D: Heat sensitivity was determined by measuring the response latency (s) in the hotplate test (50 °C). In week 4, AHF fed rats demonstrated reduced response latencies in the hotplate test which did not recover during the 10 wk experimental time course. Open squares: control animals. Solid circles: AHF fed animals. aP < 0.05, AHF vs control.
Similarly, mechanical thresholds for eliciting hindpaw withdrawal in AHF fed rats decreased significantly in week 4 from 18.72 g force to 9.3 ± 1.0 g force (P < 0.05; Figure 3C). This referred hypersensitivity of the hindpaw plantar surface was characterized by determining the mechanical withdrawal threshold using the up-down method[62]. The mechanical thresholds of the hindpaws of control animals did not change throughout the experiment (17.8 ± 0.6 g force). Hindpaw mechanical thresholds for AHF fed rats remained decreased at 7.6 ± 0.9 g force (P < 0.01) through the experiment end at week 10.
Heat sensitivity was determined indirectly by measuring the latency (s) to an escape response in the hotplate test (50 °C) (Figure 3D). Response latencies on the hotplate, i.e., the time until animals displayed signs of nociception such as shaking the paw, licking, agitation, were not different between groups at baseline (control = 21.2 ± 0.1 s; AHF = 22.9 ± 0.5 s; N.S.). In week 4 hotplate sensitivity was significantly decreased in the AHF fed group (control = 22.6 ± 0.2 s; AHF = 13.3 ± 1.4 s; P < 0.05; Figure 3D). The reduced latencies did not recover during the 10 wk experimental time course. At the end of the experiment (10 wk), control animals spend 18.3 ± 0.4 s on the hotplate before showing signs of distress while AHF animals reacted within 10.0 ± 0.3 s (P < 0.05).
Analgesic effects of APDC on AHF diet induced hypersensitivity
Glutamate is a widely expressed neuropeptide within the nervous system. When binding to group II metabotropic glutamate receptors, mGluR2 and mGluR3, Gi-protein coupled receptors, their activation inhibits adenylyl cyclase. This initiates inhibitory signaling cascades that result in reduced neuronal activity and decrease the release of further glutamate[49,50]. The mGluR2/3 agonist APDC has been shown to reduce inflammation induced mechanical and heat hypersensitivity in mouse models of somatic pain[51,52]. Here we used a single dose of 3 mg/kg APDC (ip) to decrease mechanical and heat hypersensitivity induced by chronic pancreatitis, a concentration previously shown to optimally reduce hypersensitivity in acute somatic inflammatory animal models. Within 1 h of APDC treatment, reversal was noted of the increased nociceptive response evoked by low intensity abdominal stimulation in AHF fed rats (Figure 4A). Responses to low intensity mechanical stimulation of the abdomen were 3.8 ± 1.0, instead of 6.8 ± 0.9 responses to 10 stimuli observed at baseline. Responses of the controls 1 h after APDC treatment (1 h: 3.5 ± 0.3 responses) remained the same (-24 h: 3.0 ± 0.4 responses; Figure 4A). Responses of AHF fed rats to higher intensity mechanical stimulation of the abdomen were similarly reduced at the 1 h timepoint [-24 h: (10 ± 0 responses)/10; 1 h: (7.0 ± 0.7 responses)/10; P < 0.05; Figure 4B].
Figure 4.
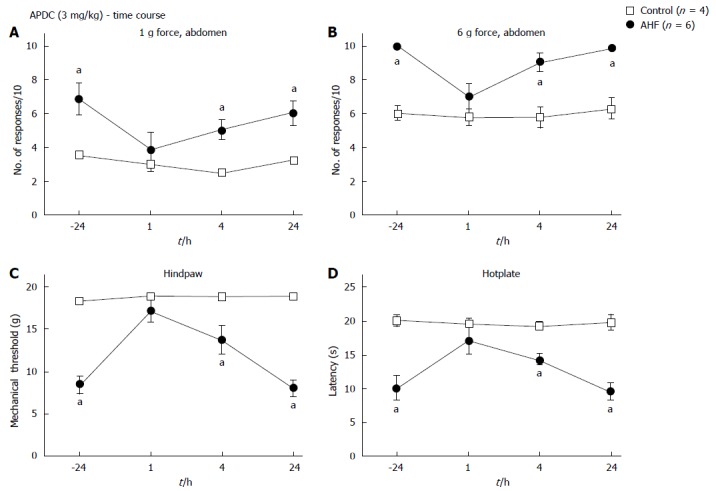
Time course of nociceptive responses to mechanical and heat stimuli after a single systemic treatment with (2R,4R)-4-Aminopyrrolidine-2,4-dicarboxylate. A, B: Animals were treated with a single ip dose of (2R,4R)-4-Aminopyrrolidine-2,4-dicarboxylate (APDC) (3 mg/kg). APDC reversed the increase in nociception responses 1 h post treatment evoked by (A) low intensity (1 g) abdominal stimulation and (B) high intensity (6 g) abdominal stimulation; C: Alcohol and high fat (AHF) fed rats had increased hindpaw mechanical withdrawal thresholds; D: Response latencies of AHF fed rats increased 1 h after APDC treatment in the hotplate test, but sensitivity of the controls was not altered. aP < 0.05, AHF vs control.
Mechanical thresholds of the hindpaws (-24 h: 8.4 ± 1.0 g force; 1h: 17.0 ± 1.3 g force; AHF n = 6, P < 0.05; Figure 4C) and latencies on the hotplate (-24 h: 10.2 ± 0.5 s; 1 h: 17.3 ± 1.0 s; AHF n = 6, P < 0.05) improved significantly to values indistinguishable from control animals (Figure 4D). The effect of APDC was abolished 24 h after treatment.
Nociceptin treatment reverses AHF diet induced hypersensitivity
Nociceptin, an endogenous opioid-related neuropeptide, binds to the opioid receptor like-1 receptor (ORL-1) whose peripheral activation and activation in the spinal cord has been shown to be anti-nociceptive[57,58,69]. Both control and AHF fed animals received single ip injections of vehicle only, 20, 60, or 200 nmol/kg nociceptin. Nociceptive responses to mechanical and heat stimulation 1 h after treatment with a single ip dose of nociceptin in animals with AHF induced pancreatitis were compared to controls.
Dose-dependent improvement was observed after the single dose of nociceptin for responses to low intensity mechanical stimulation of the abdomen (Figure 5A), responses to higher intensity mechanical stimulation of the abdomen (Figure 5B), and mechanical thresholds on the hindpaws (Figure 5C). In AHF fed animals 20 nmol/kg nociceptin decreased abdominal hypersensitivity to low intensity (1 g) mechanical stimulation [vehicle: control = 2.8 ± 0.5; AHF = (6.3 ± 0.7 responses)/10; P < 0.05; 20 nmol nociceptin: control = 3.5 ± 0.3; AHF = (4.7 ± 0.4 responses)/10; n.s.; Figure 5A]. Abdominal withdrawal of AHF animals to higher intensity (6 g) mechanical stimulation decreased from 9.8 ± 0.3 to 6.5 ± 0.4 responses after treatment with the highest dose of nociceptin. This was not different from vehicle treated control animals [(6.5 ± 0.5 responses)/10; Figure 5B]. Treatment of AHF fed animals with 20 nmol/kg nociceptin increased mechanical thresholds on the hindpaws so that they were no longer different from controls while not altering the sensitivity in control animals (vehicle: control = 18.2 ± 0.5; AHF = 6.3 ± 0.7 g force; P < 0.05; 20 nmol: control = 18.7 ± 0.0; AHF = 15.7 ± 1.6 g force; P < 0.05; Figure 5C).
Figure 5.
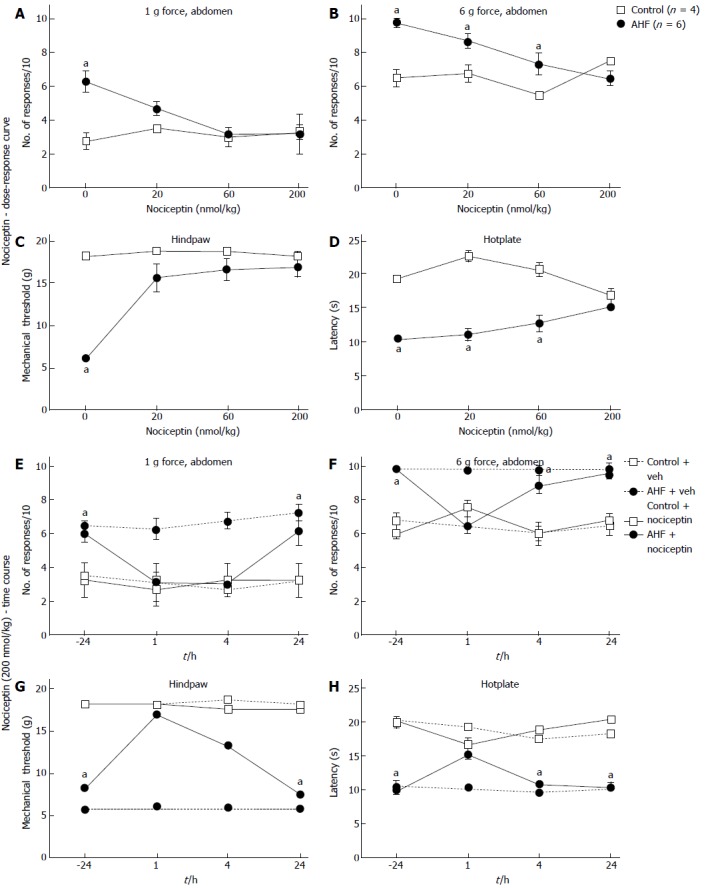
Dose-response curves and time course of nociceptive responses to mechanical and heat stimuli after a single systemic treatment with nociceptin. A, B: Comparisons of the number of responses to mechanical stimulation on the abdomen skin 1 h after treatment with nociceptin (ip) for control and alcohol and high fat (AHF) fed rats. Dose-dependent responses were reduced to (A) low intensity (1 g) and (B) high intensity (6 g) mechanical stimulation of the abdomen; C: Nociceptin increased hindpaw mechanical withdrawal threshold in AHF fed rats; D: Hotplate response latencies of AHF fed animals improved with the highest dose of nociception. In control animals nociceptin did not alter mechanical and heat sensitivity. Vehicle treatments had no effect on mechanical or heat sensitivity of animals from either group; E-H: Mechanical and heat responses in animals treated with either a single systemic injection of nociceptin (200 nmol/kg in saline) or with the saline vehicle; E: Abdominal withdrawals in response to low intensity (1 g) mechanical stimulation decreased to control levels at the 1 and 4 h post injection timepoints of AHF fed rats. Mechanical hypersensivity of vehicle treated AHF diet fed rats did not improve; F: Responses of AHF fed rats to high intensity (6 g) abdominal mechanical stimulation were decreased to control values only 1 h after injection of nociception; G: Hindpaws mechanical withdrawal thresholds of AHF fed rats recovered to control values for up to 4 h post injection; H: Hotplate response latencies of AHF fed rats improved only at the 1 h time point after nociceptin treatment. aP < 0.05, AHF vs control.
Thus, a dose of 200 nmol nociceptin per kg body weight was determined to be the most effective in reducing sensitized nociceptive responses in all four behavioral assays in AHF fed rats (Figure 5E-H). With this dose, abdominal withdrawals of AHF fed rats in response to low intensity mechanical stimulation decreased to control levels at the 1 and 4 h timepoints. Responses of AHF fed animals to mechanical stimulation of the abdomen at 1 h were significantly decreased [1 g: -24 h = 6.0 ± 0.5; 1 h = (3.2 ± 1.1 responses)/10; P < 0.05; 6 g: -24 h = 9.8 ± 0.2; 1 h = (6.5 ± 0.4 responses)/10; P < 0.05; Figure 5E, F] and returned to pre-treatment levels 24 h after the injection [1 g: 24 h = (6.2 ± 0.9 responses)/10; 6 g: 24 h = (9.5 ± 0.3 responses)/10]. Nociceptive responses of AHF fed rats to higher intensity abdominal mechanical stimulation were decreased to control values only at the 1 h time point after nociceptin treatment (Figure 5F). Mechanical withdrawal thresholds of the hindpaws of AHF fed animals also increased significantly from 8.4 ± 1.0 g force prior to treatment to 17.0 ± 1.3 g force 1 h after treatment (Figure 5G). Four hours after nociceptin treatment, the mechanical thresholds of AHF fed animals with chronic pancreatitis decreased again, returning to their previous hypersensitive level within 24 h (4 h = 13.3 ± 2.2; 24 h = 7.5 ± 1.0 g force; Figure 5G). Nociceptin treatments had no effect on mechanical sensitivity in control animals. In control animals the 200 nmol/kg dose increased hypersensitivity but was not significant [(7.5 ± 0.3 responses)/10]. Vehicle treatments did not alter nociceptive responses of naïve or AHF fed animals.
Hotplate response latencies of AHF animals were increased dose dependently from 10.4 ± 0.2 s (vehicle) to 15.1 ± 0.4 s (200 nmol/kg nociceptin), but the improvement was significant only at the highest dose of nociceptin (Figure 5D). After systemic injection of 200 nmol/kg nociceptin, for AFH animals, the hotplate response latency increased from 10.1 ± 0.7 s prior to treatment to 15.1 ± 0.4 s 1 h afterwards (P < 0.05; Figure 5H). This improvement remained significantly lower than in untreated control (20.0 ± 0.9 s; P < 0.05). Heat responses of control animals slightly decreased 1 h after injection of 200 nmol/kg nociceptin (vehicle: 19.2 ± 0.3 s; 200 nmol: 16.7 ± 1.0 s; Figure 5H). The ability of nociceptin to reverse hypersensitivity of AHF fed animals was optimal at 1 h after which it decreased and was completely absent 24 h after injection.
Comparison to effects of morphine on AHF diet induced hypersensitivity
Many studies have demonstrated the efficacy of morphine in reversal of acute and chronic pancreatitis associated hypersensitivity in animal models as well as its use for pain management of chronic pancreatitis patients with pain. Single dose s.c. treatment with 3 mg/kg morphine sulfate solution resulted in significant reduction of mechanical and heat hypersensitivity in AHF fed rats (Figure 6). Responses of AHF fed rats to low intensity mechanical stimulation of the abdomen [-24 h: (5.5 ± 0.3 responses)/10; 1 h: 1.8 ± 0.5 responses /10; 4 h: (2.0 ± 0.8 responses)/10; AHF n = 4; P < 0.05; Figure 6A] was significantly reduced for up to 4 h compared to responses before treatment. Similarly, responses of AHF fed rats to higher intensity mechanical stimulation of the abdomen [-24 h: (9.0 ± 0.6 responses)/10; 1 h: (2.3 ± 0.8 responses)/10; 4 h: (6.0 ± 0.4 responses)/10; AHF n = 4; P < 0.05; Figure 6B] was significantly reduced for up to 4 h compared to responses before treatment. Naïve animals responded to higher intensity mechanical stimulation of the abdomen with 6.5 ± 0.6 responses/10 which was significantly reduced 1 h after morphine treatment to 3.3 ± 0.5 responses/10 (P < 0.05; Figure 6B). Mechanical thresholds of the hindpaws in the AHF diet group improved after morphine treatment for up to 4 h (-24 h: 7.6 ± 1.0 g force; 1h: 18.7 ± 0 g force; 4h: 18.2 ± 0.4 g force; AHF n = 4, P < 0.05; Figure 6C). No changes in naïve animals were measured after morphine treatment for either on the hindpaw nociceptive withdrawal thresholds for low intensity mechanical stimulation or the responses to low threshold (1 g) abdominal stimulation. Morphine treatment reversed heat sensitivity of AHF fed animals back to response levels recorded in naïve animals for up to 4 h (-24 h: 10.7 ± 0.1 s; 1h: 20.4 ± 0.7 s; 4h: 18.2 ± 0.7 s; AHF n = 4, P < 0.05; Figure 6D). Responses of naïve rats on the hotplate were not affected by the 3 mg/kg morphine. Animals were tested during their active period and did not appear somnolent for any of the testing. After 24 h no effects of morphine were detected in any of the tests.
Figure 6.
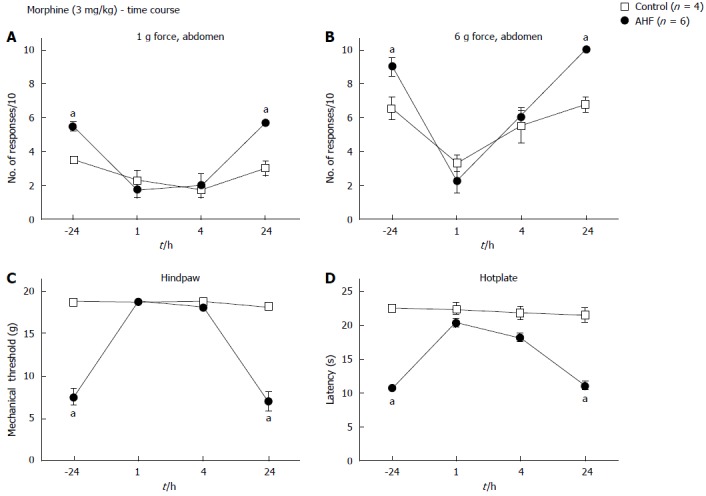
Time course of nociceptive responses to mechanical and heat stimuli after a single systemic treatment with morphine. Animals were injected with morphine (3 mg/kg, subcutaneous) and responses to mechanical and heat stimuli measured. Morphine treatment of alcohol and high fat (AHF) fed rats decreased the number of responses to (A) low (1 g) and (B) high intensity (6 g) mechanical stimulation of the abdominal skin for up to 4 h. Abdominal withdrawal responses of controls evoked by high intensity (6 g) mechanical stimulation were reduced 1 h after treatment compared to before morphine. Hindpaw (C) mechanical withdrawal thresholds and (D) response latencies on the hot plate test of AHF fed rats were improved at 1 and 4 h after morphine treatment. Controls were unaffected by the low dose morphine. aP < 0.05, AHF vs control.
DISCUSSION
Here we present a non-invasive dietary animal model of alcoholic chronic pancreatitis induced in 3 wk but persisting for at least 10 wk. The model replicates poor dietary choices of clinical patients diagnosed with alcoholic chronic pancreatitis. Recent estimates reported that 51%-67% of Americans consume 3-5 alcoholic beverages per day which concurs with a decrease in diet quality and an increase of dietary fat intake[70-72]. Concurrently, the Food and Agriculture Organization (FAO) of the United Nations reported a steady increase of the average daily dietary fat consumption per capita in the United States from 140 g in 1990/92 to 161 g in 2005/7 while it averaged 131 g fat in the developed world[73]. Comparison of nutritional profiles identified that patients with chronic alcoholic pancreatitis consumed significantly more saturated fat and animal protein compared to patients suffering from alcohol consumption induced cirrhosis of the liver[21,74,75]. Other studies have shown that drinkers consume significantly more dietary fat and calories on drinking days[71,72,76]. Exactly this combination is described as inducing chronic alcoholic pancreatitis, with alcohol being the leading and diet the secondary cause[77,78]. Animal models of alcoholic chronic pancreatitis and fatty liver disease have shown that increased dietary fat consumption results in higher hepatic triglycerides and associated fat vacuolization[64,65,79]. Hence, the AHF rat model of chronic pancreatitis described here closely mimics high-risk human dietary choices that are part of the etiology of chronic alcoholic pancreatitis with liver steatosis.
Presently favored rodent models of pancreatitis are highly invasive and poorly reproduce disease progression seen in clinical patients. They often require laparotomy surgery to obstruct the pancreatic duct, or direct instill chemical irritants such as the aromatic nitro compound trinitrobenzene sulfonic acid or ethanol into the pancreatic duct. Other models utilize the repeated supraphysiological ip injections of caerulein, an analog of the gastrointestinal peptide hormone cholecystokinin, in combination with an ip lipopolysaccharide injection or a single dose of the highly toxic organotin dibutyltin chloride into the tail vein[30,31,80]. All of these models induce a rapid immune response that is focused primarily on the pancreas. By chemically modifying cell surface proteins an inflammatory cell reaction is induced which ultimately results in acinar cell death with or without prior dysregulated digestive enzyme release. Thus, other current models of pancreatitis are better suited for study of acute pancreatitis[31,81,82]. Animals can recover in these acute models within 48-72 h as in most cases of acute pancreatitis. However, in the AHF model the pathological and functional symptomatology of chronic alcoholic pancreatitis persists. Regeneration of the damaged tissue is impaired by the continued alcohol consumption, facilitating the establishment of chronic pancreatitis[83].
Chronic pancreatitis is a multifactorial disease caused by a combination of regular alcohol consumption, a low quality diet high with high fat content producing hyperlipidemia, smoking, and hereditary factors[12,19,21,23,75,84]. The AHF model of chronic pancreatitis focuses on the combined effects of alcohol and a diet high in fat on the pancreatitis and associated comorbidities. Chronic consumption of alcohol results in its metabolism not only in the liver, but also in the pancreas. Alcohol is typically metabolized via acetaldehyde into acetyl which is used for energy production. In the results presented here, liver steatosis induced by alcohol consumption[40,85] was visible not only in the centrilobular region of the liver where lipids are predominantly stored in fatty vacuoles[86], but was detected throughout the large area of sampled tissue in the AHF fed animals indicating severity of disease progression. Metabolism of dietary alcohol and fat converges on the same enzymes, so that when high amounts of alcohol and fatty acids are consumed, cellular signaling molecules anandamide and arachidonic acid, and cytotoxic acetaldehydes accumulate within liver and pancreas that are able to directly activate ion channels such as Transient Receptor Potential Cation Channel, Subfamily A, Member 1 (TRPA1); Transient Receptor Potential Cation Channel, Subfamily V, Member 1 (TRPV1); and Transient Receptor Potential Cation Channel, Subfamily V, Member 4 (TRPV4) localized in the cell membranes of nociceptive sensory neurons innervating the pancreas as well as on local cells to induce cellular damage[84,87-90]. In the pancreas acetaldehydes and fatty acid ethyl esters can further induce fragility of pancreatic zymogen granules and cause cellular damage to pancreatic cells[7,84,90,91]. Resulting cellular atrophy and necrosis induce separation of pancreatic lobules as well as peri- and intralobular fibrosis, indicators of severe damage to parenchymal tissue, were visible throughout the examined pancreatic tissue of AHF fed animals. Cell damage and necrosis directly and indirectly activate the pancreatic local immune stellate cells, and activate nociceptors innervating the pancreas, amplifying pain sensation and pancreatic tissue damage through neurogenic inflammation[89,92,93].
Alcoholic chronic pancreatitis not only results in damage to the exocrine pancreas but can also impair and dysregulate insulin metabolism in the endocrine pancreas which in patients results in the development of diabetes mellitus type 3c[10,13]. In the present study, 10 wk of AHF diet resulted in decreased glucose tolerance in rats. While no changes in islet of Langerhans number and size distribution were detected (data not shown), fasting glucose tolerance was impaired, cellular atrophy of insulin cells was evident histologically, and a 3.5 fold increase of small clusters of insulin producing cells were detected in AHF fed rats. This suggested a compensatory reaction to beta cell atrophy and decreased effectiveness. In a Korean study increased alcohol consumption coincided with increased dietary fat which concurred with decreased pancreatic beta cell function[75]. Low-density lipoproteins, metabolic products of saturated fats, have been shown to reduce beta-cell function, decreasing glucose-stimulated insulin secretion and causing beta-cell proliferation in primary cell culture experiments[94].
Exposure of cultured beta cells to combined fatty acids and ethanol has been shown to decrease their mitochondrial activity and adenosine triphosphate (ATP) production and increase released reactive oxygen species, indicators of their decreased function[38]. Platelet-derived growth factor as well as interleukin-1β, two pro-inflammatory cytokines and signaling molecules that are increased in pancreatitis, have been reported to induce beta-cell proliferation[95]. The proliferation of small beta-cell groups observed in AHF fed pancreas tissue suggest a regenerative mechanism is active in the rats to compensate for ethanol and fatty acid induced cell damage.
As in our previous study using a similar protocol to induce chronic pancreatitis, animals fed AHF diet developed mechanical hypersensitivity of the abdomen and hindpaws as well as increased heat sensitivity. Alcohol metabolism in chronic alcoholism is not confined to visceral organs but occurs in every tissue. As a result of cellular damage produced by chronic alcoholism, resulting oxidative stress can also be measured in skeletal muscle[96]. Cellular damage releases molecules such as ATP and incompletely metabolized alcohol products activate and sensitize nociceptors throughout the body, resulting in hypersensitivity that is not confined to the abdominal region. Activity induced central sensitization further exacerbates hypersensitivity to noxious and normally innocuous stimuli. Clinically, 85% of chronic pancreatitis patients require treatment for pain at some point during disease progression and are treated depending on severity of the pain with non-narcotic analgesics and as pain increases, ever increasing doses of opiates as well as pancreatic enzyme therapy. In extreme cases, nerve blocks and eventually surgical intervention may be required[26,27]. While opioids are efficacious in patients as well as in animal models of chronic pancreatitis, even acute doses have severe side effects and may lead to overdose. Long term use of morphine can cause addiction and constipation, its efficacy to reduce pain decreases with duration of use, while ongoing pancreatic inflammation is not improved. Yet, despite the side effects, prescription rates for opioids have increased almost exponentially in the last decade and non-addictive alternatives are very much needed[97,98]. We therefore investigated the hypothesis that activation of other Gi-coupled GPCRs would decrease nociceptive signaling.
Initial focus was on the inhibitory mGluR2/3 signaling pathway. Glutamate is the main excitatory neurotransmitter that depolarizes neurons by binding to its ionotropic and metabotropic receptors. However, glutamate initiates inhibitory signaling cascades when bound to the group II metabotropic glutamate receptors, mGluR2 and mGluR3 to dampen cellular excitability[99,100]. Activation of these Gi-coupled GPCRs negatively modulate adenylate cyclase, alter synaptic connectivity, and decrease release of glutamate[101]. Group II mGluRs are expressed throughout the nervous system, in primary sensory neurons, predominantly presumed nociceptors, in the dorsal horn of the spinal cord, as well as in visceral organs such as beta-cells of the pancreas[102-105]. After acute single dose treatment with the mGluR2/3 agonist APDC in the present study, the hypersensitized responses to mechanical and heat stimuli of AHF animals were no longer significantly different from those of controls. This attenuation was of short duration (1 h) suggesting that systemic administration of APDC may have activated mGluR2/3 receptors on peripheral nociceptive sensory neurons, thus reducing nociception. Previous studies have reported efficacious use of group II agonists as analgesics without any apparent side effects in animal models of neuropathic and acute inflammatory pain without altering nociceptive responses of control animals[51,52,54,106-108]. Systemic use of mGluR2/3 agonists is able to activate receptors on peripheral sensory neurons to decrease their activity and central release of glutamate, thus interrupting neurogenic inflammation and facilitating tissue regeneration. Due to their expression within the limbic system, mGluR2/3 receptors have been studied in connection with schizophrenia, alcohol and drug addiction, and anxiety[109-111]. The potential ability of a single drug to be used not only for pain management in alcoholic chronic pancreatitis, but also to support patients’ alcohol cessation and dietary management would be of immense benefit, simplifying dosing and regular use of pharmacological therapy. Prolonged use of a different mGluR2/3 agonist, LY379268, for pain management in somatic pain models showed that repeated daily systemic dosing quickly resulted in desensitization and lost ability to reverse hypersensitivity[53,55]. Long-term experiments utilizing lower doses or novel mGluR2/3 agonists for repeated treatments are needed to investigate its efficacy in reversing alcoholic chronic pancreatitis associated hypersensitivity.
Nociceptin, also called orphanin FQ, is an endogenous opioid-like peptide that does not bind to the classical opioid receptors but to ORL-1. Like mGluR2/3, activation of this Gi-coupled GPCR also inhibits adenylate cyclase and voltage-gated Ca2+-channels while activating voltage-gated K+-channels which results in decreased neuronal activity and decreased glutamate release[44,45,112]. In the AHF diet induced chronic pancreatitis animal group, acute systemic treatments with nociceptin successfully reversed the induced mechanical and heat hypersensitivity in AHF fed animals. Unlike morphine, these treatments did not alter nociceptive responses of control animals. Morphine decreases mechanical and heat sensitivity concentration-dependently in control animals. In the present experiments morphine decreased mechanical sensitivity in control rats while response latencies in the hotplate test were not affected. The Fischer F344 rat strain is known for having higher anxiety compared to other rat strains[113]. Animals were acclimated to the experimental set-up for mechanical sensitivity assays whereas the hotplate test posed a novel environment which may have altered the effects of morphine in control Fischer F344 rats.
Nociceptin and downstream signaling pathways initiated by binding to the ORL-1 receptor have been enigmatic. Initial reports described that intracerebroventricular injections induced hyperalgesia and high doses inhibited locomotion[44,45]. Subsequent studies using intrathecal injections demonstrated analgesic properties of ORL-1 activation by reducing acute inflammatory and nerve injury induced neuropathic hypersensitivity[56,57,69,114]. Intrathecal nociceptin reduced peripheral release of vasodilatory neuropeptides co-expressed with ORL-1, thus decreasing edema after acute inflammatory insult by decreasing dorsal root potentials and glutamatergic transmission[115-117]. Due to the absence of reliable antibodies for ORL-1, its localization still relies on in situ hybridization studies. Its mRNA has been localized throughout nociceptive signaling pathways, including small diameter, presumably nociceptive sensory neurons, within the superficial dorsal horn as well as around the central canal, and in multiple regions throughout the brain[116,118]. Reduction of hypersensitivity in AHF animals after systemic treatment with nociceptin was most likely achieved by ORL-1 activation within the peripheral nervous system and resulting decrease in neuronal activity and glutamate release. While activation of ORL-1 within the spinal cord is predicted to also be able to attenuate chronic pancreatitis induced hypersensitivity, repeated use of systemic nociceptin may result in increased crossing of the blood brain barrier which can be compromised in cases of severe peripheral inflammation thus promoting pro-nociceptive effects similar to intracerebroventricular injections[119,120]. Combined treatment using morphine and nociceptin provided synergistic analgesia thus diminishing the amount of morphine needed to attenuate pain sensation and reducing the risk of sensitization to morphine in a previous study[2]. This opens the possible therapeutic option of employing the nociceptin-NOP system for more efficient pain management of chronic pancreatitis-associated pain while reducing side effects.
In summary, the Gi-coupled GPCR signaling pathways downstream from mGluR2/3 and ORL-1 receptors investigated here, presented good alternatives to presently employed opioid pain therapies for the treatment of chronic pancreatitis induced pain. Likewise, these studies suggest these agents have potential as adjuvant therapy to reduce the dosage of morphine derivatives and related side effects.
COMMENTS
Background
Chronic pancreatitis is a progressive and potentially fatal disease caused by persistent unresolved inflammation and pancreatic fibrosis. Pancreatitis can be accompanied by severe intractable abdominal pain. Currently recommended pain management therapies range from non-narcotic analgesics to strong opiates with their accompanying side-effects, but are not effective as long term treatments. Presently employed animal models poorly reproduce the clinical etiology of chronic pancreatitis as they are highly invasive, requiring laparotomy surgery and/or utilize repetitive dosing with caustic chemicals. The abdominal hypersensitivity induced with these experimental procedures is better suited for modeling acute pancreatitis.
Research frontiers
To date no satisfactory pain therapy is available for treatment of chronic pancreatitis nor is there a suitable experimental model for the study of chronic pancreatitis resembling the clinical syndrome with which to test potential therapeutics. Knowledge of the underlying pathophysiology of chronic pancreatitis resulting in this severe abdominal pain is needed for the development of better pharmacological interventions.
Innovations and breakthroughs
The animal model of chronic pancreatitis is induced with a 6% alcohol and high fat diet similar to poor human dietary choices. It produces hypersensitivity to touch with fine filaments on the abdomen and has characteristics of type 3c diabetes mellitus (T3cDM), i.e., abnormal insulin histology and intolerance to glucose. Reduction of hypersensitivity induced by the model was demonstrated with the pharmacological activation of three inhibitory metabotropic Gi protein-coupled receptors. Analgesia was demonstrated with single doses of drugs activating inhibitory glutamate and opiate-like receptors, mGluR2/3 and ORL-1, respectively, in comparisons to morphine.
Applications
These therapies for the treatment of chronic pancreatitis associated pain are suggested as good alternatives to presently employed opioid pain therapies. Used as adjuvant therapy with morphine derivatives for more effective relief of chronic pancreatitis pain, these agents would have potential to reduce the opiate dosage and related side effects.
Terminology
Intractable pancreatic pain cannot be alleviated with current therapies. Using the 6% alcohol and high fat diet (AHF) induced rat chronic pancreatitis model, two therapies were tested, including nociceptin, an endogenous ligand for the opioid-receptor-like 1 receptor, and (2R,4R)-4-Aminopyrrolidine-2,4-dicarboxylate, an activator of inhibitory mGluR2/3 glutamate receptors. These therapies reduced the hypersensitivity that develops on the abdominal skin of the rats with chronic pancreatitis as effectively as morphine.
Peer review
This study demonstrated that the AHF diet replicating high risk human dietary choices induced a chronic alcoholic pancreatitis in rats with histological features and developing glucose intolerance resembling clinical patients with chronic pancreatitis and T3cDM. So, this paper has novelty and innovation.
Footnotes
Supported by National Institutes of Health, No. NINDS R01 NS39041.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 20, 2014
First decision: August 6, 2014
Article in press: September 30, 2014
P- Reviewer: Zhang ZM S- Editor: Ma YJ L- Editor: A E- Editor: Wang CH
References
- 1.Coté GA, Yadav D, Slivka A, Hawes RH, Anderson MA, Burton FR, Brand RE, Banks PA, Lewis MD, Disario JA, et al. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin Gastroenterol Hepatol. 2011;9:266–273; quiz e27. doi: 10.1016/j.cgh.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nøjgaard C, Becker U, Matzen P, Andersen JR, Holst C, Bendtsen F. Progression from acute to chronic pancreatitis: prognostic factors, mortality, and natural course. Pancreas. 2011;40:1195–1200. doi: 10.1097/MPA.0b013e318221f569. [DOI] [PubMed] [Google Scholar]
- 3.Malka D, Hammel P, Maire F, Rufat P, Madeira I, Pessione F, Lévy P, Ruszniewski P. Risk of pancreatic adenocarcinoma in chronic pancreatitis. Gut. 2002;51:849–852. doi: 10.1136/gut.51.6.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malka D, Hammel P, Sauvanet A, Rufat P, O’Toole D, Bardet P, Belghiti J, Bernades P, Ruszniewski P, Lévy P. Risk factors for diabetes mellitus in chronic pancreatitis. Gastroenterology. 2000;119:1324–1332. doi: 10.1053/gast.2000.19286. [DOI] [PubMed] [Google Scholar]
- 5.Howes N, Neoptolemos JP. Risk of pancreatic ductal adenocarcinoma in chronic pancreatitis. Gut. 2002;51:765–766. doi: 10.1136/gut.51.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardt PD, Brendel MD, Kloer HU, Bretzel RG. Is pancreatic diabetes (type 3c diabetes) underdiagnosed and misdiagnosed? Diabetes Care. 2008;31 Suppl 2:S165–S169. doi: 10.2337/dc08-s244. [DOI] [PubMed] [Google Scholar]
- 7.Apte MV, Pirola RC, Wilson JS. Pancreas: alcoholic pancreatitis--it’s the alcohol, stupid. Nat Rev Gastroenterol Hepatol. 2009;6:321–322. doi: 10.1038/nrgastro.2009.84. [DOI] [PubMed] [Google Scholar]
- 8.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2010;33 Suppl 1:S62–S69. doi: 10.2337/dc10-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358. doi: 10.1016/j.bpg.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Cui Y, Andersen DK. Pancreatogenic diabetes: special considerations for management. Pancreatology. 2011;11:279–294. doi: 10.1159/000329188. [DOI] [PubMed] [Google Scholar]
- 11.Cui Y, Andersen DK. Diabetes and pancreatic cancer. Endocr Relat Cancer. 2012;19:F9–F26. doi: 10.1530/ERC-12-0105. [DOI] [PubMed] [Google Scholar]
- 12.Andersen DK, Andren-Sandberg Å, Duell EJ, Goggins M, Korc M, Petersen GM, Smith JP, Whitcomb DC. Pancreatitis-diabetes-pancreatic cancer: summary of an NIDDK-NCI workshop. Pancreas. 2013;42:1227–1237. doi: 10.1097/MPA.0b013e3182a9ad9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ewald N, Bretzel RG. Diabetes mellitus secondary to pancreatic diseases (Type 3c)--are we neglecting an important disease? Eur J Intern Med. 2013;24:203–206. doi: 10.1016/j.ejim.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 14.Dufour MC, Adamson MD. The epidemiology of alcohol-induced pancreatitis. Pancreas. 2003;27:286–290. doi: 10.1097/00006676-200311000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Rosendahl J, Bödeker H, Mössner J, Teich N. Hereditary chronic pancreatitis. Orphanet J Rare Dis. 2007;2:1. doi: 10.1186/1750-1172-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieb JG, Forsmark CE. Review article: pain and chronic pancreatitis. Aliment Pharmacol Ther. 2009;29:706–719. doi: 10.1111/j.1365-2036.2009.03931.x. [DOI] [PubMed] [Google Scholar]
- 17.Affronti J. Chronic pancreatitis and exocrine insufficiency. Prim Care. 2011;38:515–537; ix. doi: 10.1016/j.pop.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Nitsche C, Simon P, Weiss FU, Fluhr G, Weber E, Gärtner S, Behn CO, Kraft M, Ringel J, Aghdassi A, et al. Environmental risk factors for chronic pancreatitis and pancreatic cancer. Dig Dis. 2011;29:235–242. doi: 10.1159/000323933. [DOI] [PubMed] [Google Scholar]
- 19.Whitcomb DC. Genetic risk factors for pancreatic disorders. Gastroenterology. 2013;144:1292–1302. doi: 10.1053/j.gastro.2013.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.National Institute of Health. Pancreatitis. NIH Publication. 2008. pp. No. 08–1596. Available from: http://digestive.niddk.nih.gov/ddiseases/pubs/pancreatitis/index.aspx. [Google Scholar]
- 21.Rajesh G, Girish BN, Vaidyanathan K, Balakrishnan V. Diet, nutrient deficiency and chronic pancreatitis. Trop Gastroenterol. 2013;34:68–73. doi: 10.7869/tg.2012.100. [DOI] [PubMed] [Google Scholar]
- 22.Ammann RW, Buehler H, Bruehlmann W, Kehl O, Muench R, Stamm B. Acute (nonprogressive) alcoholic pancreatitis: prospective longitudinal study of 144 patients with recurrent alcoholic pancreatitis. Pancreas. 1986;1:195–203. [PubMed] [Google Scholar]
- 23.Apte MV, Wilson JS, Korsten MA. Alcohol-related pancreatic damage: mechanisms and treatment. Alcohol Health Res World. 1997;21:13–20. [PMC free article] [PubMed] [Google Scholar]
- 24.Thrower EC, Gorelick FS, Husain SZ. Molecular and cellular mechanisms of pancreatic injury. Curr Opin Gastroenterol. 2010;26:484–489. doi: 10.1097/MOG.0b013e32833d119e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goulden MR. The pain of chronic pancreatitis: a persistent clinical challenge. Br J Pain. 2013;7:8–22. doi: 10.1177/2049463713479230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gachago C, Draganov PV. Pain management in chronic pancreatitis. World J Gastroenterol. 2008;14:3137–3148. doi: 10.3748/wjg.14.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burton F, Alkaade S, Collins D, Muddana V, Slivka A, Brand RE, Gelrud A, Banks PA, Sherman S, Anderson MA, et al. Use and perceived effectiveness of non-analgesic medical therapies for chronic pancreatitis in the United States. Aliment Pharmacol Ther. 2011;33:149–159. doi: 10.1111/j.1365-2036.2010.04491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adrych K, Smoczynski M, Stojek M, Sledzinski T, Slominska E, Goyke E, Smolenski RT, Swierczynski J. Decreased serum essential and aromatic amino acids in patients with chronic pancreatitis. World J Gastroenterol. 2010;16:4422–4427. doi: 10.3748/wjg.v16.i35.4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Girish BN, Rajesh G, Vaidyanathan K, Balakrishnan V. Zinc status in chronic pancreatitis and its relationship with exocrine and endocrine insufficiency. JOP. 2009;10:651–656. [PubMed] [Google Scholar]
- 30.Aghdassi AA, Mayerle J, Christochowitz S, Weiss FU, Sendler M, Lerch MM. Animal models for investigating chronic pancreatitis. Fibrogenesis Tissue Repair. 2011;4:26. doi: 10.1186/1755-1536-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lerch MM, Gorelick FS. Models of acute and chronic pancreatitis. Gastroenterology. 2013;144:1180–1193. doi: 10.1053/j.gastro.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 32.Singh M, LaSure MM, Bockman DE. Pancreatic acinar cell function and morphology in rats chronically fed an ethanol diet. Gastroenterology. 1982;82:425–434. [PubMed] [Google Scholar]
- 33.Letko G, Nosofsky T, Lessel W, Siech M. Transition of rat pancreatic juice edema into acute pancreatitis by single ethanol administration. Pathol Res Pract. 1991;187:247–250. doi: 10.1016/S0344-0338(11)80779-X. [DOI] [PubMed] [Google Scholar]
- 34.Siech M, Heinrich P, Letko G. Development of acute pancreatitis in rats after single ethanol administration and induction of a pancreatic juice edema. Int J Pancreatol. 1991;8:169–175. doi: 10.1007/BF02924430. [DOI] [PubMed] [Google Scholar]
- 35.Kono H, Nakagami M, Rusyn I, Connor HD, Stefanovic B, Brenner DA, Mason RP, Arteel GE, Thurman RG. Development of an animal model of chronic alcohol-induced pancreatitis in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1178–G1186. doi: 10.1152/ajpgi.2001.280.6.G1178. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Guo M, Hu B, Liu R, Wang R, Tang C. Does chronic ethanol intake cause chronic pancreatitis?: evidence and mechanism. Pancreas. 2008;37:189–195. doi: 10.1097/MPA.0b013e31816459b7. [DOI] [PubMed] [Google Scholar]
- 37.Norton ID, Apte MV, Lux O, Haber PS, Pirola RC, Wilson JS. Chronic ethanol administration causes oxidative stress in the rat pancreas. J Lab Clin Med. 1998;131:442–446. doi: 10.1016/s0022-2143(98)90145-7. [DOI] [PubMed] [Google Scholar]
- 38.Dembele K, Nguyen KH, Hernandez TA, Nyomba BL. Effects of ethanol on pancreatic beta-cell death: interaction with glucose and fatty acids. Cell Biol Toxicol. 2009;25:141–152. doi: 10.1007/s10565-008-9067-9. [DOI] [PubMed] [Google Scholar]
- 39.Siech M, Zhou Z, Zhou S, Bair B, Alt A, Hamm S, Gross H, Mayer J, Beger HG, Tian X, et al. Stimulation of stellate cells by injured acinar cells: a model of acute pancreatitis induced by alcohol and fat (VLDL) Am J Physiol Gastrointest Liver Physiol. 2009;297:G1163–G1171. doi: 10.1152/ajpgi.90468.2008. [DOI] [PubMed] [Google Scholar]
- 40.DeCarli LM, Lieber CS. Fatty liver in the rat after prolonged intake of ethanol with a nutritionally adequate new liquid diet. J Nutr. 1967;91:331–336. doi: 10.1093/jn/91.3_Suppl.331. [DOI] [PubMed] [Google Scholar]
- 41.Yang H, McNearney TA, Chu R, Lu Y, Ren Y, Yeomans DC, Wilson SP, Westlund KN. Enkephalin-encoding herpes simplex virus-1 decreases inflammation and hotplate sensitivity in a chronic pancreatitis model. Mol Pain. 2008;4:8. doi: 10.1186/1744-8069-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bischofberger J, Schild D. Glutamate and N-acetylaspartylglutamate block HVA calcium currents in frog olfactory bulb interneurons via an mGluR2/3-like receptor. J Neurophysiol. 1996;76:2089–2092. doi: 10.1152/jn.1996.76.3.2089. [DOI] [PubMed] [Google Scholar]
- 43.Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- 44.Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour JL, Guillemot JC, Ferrara P, Monsarrat B. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- 45.Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Civelli O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- 46.Waterfield AA, Leslie FM, Lord JA, Ling N, Kosterlitz HW. Opioid activities of fragments of beta-endorphin and of its leucine65-analogue. Comparison of the binding properties of methionine- and leucine-enkephalin. Eur J Pharmacol. 1979;58:11–18. doi: 10.1016/0014-2999(79)90334-0. [DOI] [PubMed] [Google Scholar]
- 47.Kosterlitz HW. Opioid peptides and their receptors. Prog Biochem Pharmacol. 1980;16:3–10. doi: 10.1016/b978-0-08-028021-9.50005-8. [DOI] [PubMed] [Google Scholar]
- 48.Malmberg AB, Yaksh TL. The effect of morphine on formalin-evoked behaviour and spinal release of excitatory amino acids and prostaglandin E2 using microdialysis in conscious rats. Br J Pharmacol. 1995;114:1069–1075. doi: 10.1111/j.1476-5381.1995.tb13315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang FR, Bradford HF, Ling EA. Metabotropic glutamate receptors in the control of neuronal activity and as targets for development of anti-epileptogenic drugs. Curr Med Chem. 2009;16:2189–2204. doi: 10.2174/092986709788612710. [DOI] [PubMed] [Google Scholar]
- 50.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang D, Gereau RW. Peripheral group II metabotropic glutamate receptors (mGluR2/3) regulate prostaglandin E2-mediated sensitization of capsaicin responses and thermal nociception. J Neurosci. 2002;22:6388–6393. doi: 10.1523/JNEUROSCI.22-15-06388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang D, Gereau RW. Peripheral group II metabotropic glutamate receptors mediate endogenous anti-allodynia in inflammation. Pain. 2003;106:411–417. doi: 10.1016/j.pain.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 53.Jones CK, Eberle EL, Peters SC, Monn JA, Shannon HE. Analgesic effects of the selective group II (mGlu2/3) metabotropic glutamate receptor agonists LY379268 and LY389795 in persistent and inflammatory pain models after acute and repeated dosing. Neuropharmacology. 2005;49 Suppl 1:206–218. doi: 10.1016/j.neuropharm.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 54.Du J, Zhou S, Carlton SM. Group II metabotropic glutamate receptor activation attenuates peripheral sensitization in inflammatory states. Neuroscience. 2008;154:754–766. doi: 10.1016/j.neuroscience.2008.03.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zammataro M, Chiechio S, Montana MC, Traficante A, Copani A, Nicoletti F, Gereau RW. mGlu2 metabotropic glutamate receptors restrain inflammatory pain and mediate the analgesic activity of dual mGlu2/mGlu3 receptor agonists. Mol Pain. 2011;7:6. doi: 10.1186/1744-8069-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hao JX, Xu IS, Wiesenfeld-Hallin Z, Xu XJ. Anti-hyperalgesic and anti-allodynic effects of intrathecal nociceptin/orphanin FQ in rats after spinal cord injury, peripheral nerve injury and inflammation. Pain. 1998;76:385–393. doi: 10.1016/S0304-3959(98)00071-2. [DOI] [PubMed] [Google Scholar]
- 57.Kolesnikov YA, Pasternak GW. Peripheral orphanin FQ/nociceptin analgesia in the mouse. Life Sci. 1999;64:2021–2028. doi: 10.1016/s0024-3205(99)00149-6. [DOI] [PubMed] [Google Scholar]
- 58.Fu X, Zhu ZH, Wang YQ, Wu GC. Regulation of proinflammatory cytokines gene expression by nociceptin/orphanin FQ in the spinal cord and the cultured astrocytes. Neuroscience. 2007;144:275–285. doi: 10.1016/j.neuroscience.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 59.Agostini S, Eutamene H, Broccardo M, Improta G, Petrella C, Theodorou V, Bueno L. Peripheral anti-nociceptive effect of nociceptin/orphanin FQ in inflammation and stress-induced colonic hyperalgesia in rats. Pain. 2009;141:292–299. doi: 10.1016/j.pain.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 60.Suaudeau C, Florin S, Meunier JC, Costentin J. Nociceptin-induced apparent hyperalgesia in mice as a result of the prevention of opioid autoanalgesic mechanisms triggered by the stress of an intracerebroventricular injection. Fundam Clin Pharmacol. 1998;12:420–425. doi: 10.1111/j.1472-8206.1998.tb00966.x. [DOI] [PubMed] [Google Scholar]
- 61.Vera-Portocarrero LP, Lu Y, Westlund KN. Nociception in persistent pancreatitis in rats: effects of morphine and neuropeptide alterations. Anesthesiology. 2003;98:474–484. doi: 10.1097/00000542-200302000-00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 63.Williams DB, Wan Z, Frier BC, Bell RC, Field CJ, Wright DC. Dietary supplementation with vitamin E and C attenuates dexamethasone-induced glucose intolerance in rats. Am J Physiol Regul Integr Comp Physiol. 2012;302:R49–R58. doi: 10.1152/ajpregu.00304.2011. [DOI] [PubMed] [Google Scholar]
- 64.Lieber CS, DeCarli LM. Quantitative relationship between amount of dietary fat and severity of alcoholic fatty liver. Am J Clin Nutr. 1970;23:474–478. doi: 10.1093/ajcn/23.4.474. [DOI] [PubMed] [Google Scholar]
- 65.Tsukamoto H, Towner SJ, Yu GS, French SW. Potentiation of ethanol-induced pancreatic injury by dietary fat. Induction of chronic pancreatitis by alcohol in rats. Am J Pathol. 1988;131:246–257. [PMC free article] [PubMed] [Google Scholar]
- 66.Reber PU, Patel AG, Toyama MT, Ashley SW, Reber HA. Feline model of chronic obstructive pancreatitis: effects of acute pancreatic duct decompression on blood flow and interstitial pH. Scand J Gastroenterol. 1999;34:439–444. doi: 10.1080/003655299750026489. [DOI] [PubMed] [Google Scholar]
- 67.Demols A, Van Laethem JL, Quertinmont E, Degraef C, Delhaye M, Geerts A, Deviere J. Endogenous interleukin-10 modulates fibrosis and regeneration in experimental chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1105–G1112. doi: 10.1152/ajpgi.00431.2001. [DOI] [PubMed] [Google Scholar]
- 68.van Westerloo DJ, Florquin S, de Boer AM, Daalhuisen J, de Vos AF, Bruno MJ, van der Poll T. Therapeutic effects of troglitazone in experimental chronic pancreatitis in mice. Am J Pathol. 2005;166:721–728. doi: 10.1016/S0002-9440(10)62293-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kamei J, Ohsawa M, Kashiwazaki T, Nagase H. Antinociceptive effects of the ORL1 receptor agonist nociceptin/orphanin FQ in diabetic mice. Eur J Pharmacol. 1999;370:109–116. doi: 10.1016/s0014-2999(99)00112-0. [DOI] [PubMed] [Google Scholar]
- 70.Breslow RA, Smothers BA. Drinking patterns and body mass index in never smokers: National Health Interview Survey, 1997-2001. Am J Epidemiol. 2005;161:368–376. doi: 10.1093/aje/kwi061. [DOI] [PubMed] [Google Scholar]
- 71.Breslow RA, Guenther PM, Juan W, Graubard BI. Alcoholic beverage consumption, nutrient intakes, and diet quality in the US adult population, 1999-2006. J Am Diet Assoc. 2010;110:551–562. doi: 10.1016/j.jada.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Food and Agriculture Organization of the United Nations. FAO Statistics Division 2010. Available from: http://faostat.fao.org/
- 73.Noel-Jorand MC, Bras J. A comparison of nutritional profiles of patients with alcohol-related pancreatitis and cirrhosis. Alcohol Alcohol. 1994;29:65–74. [PubMed] [Google Scholar]
- 74.Chung HK, Cho Y, Shin MJ. Alcohol use behaviors, fat intake and the function of pancreatic β-cells in non-obese, healthy Korean males: findings from 2010 Korea National Health and Nutrition Examination Survey. Ann Nutr Metab. 2013;62:129–136. doi: 10.1159/000345587. [DOI] [PubMed] [Google Scholar]
- 75.Manari AP, Preedy VR, Peters TJ. Nutritional intake of hazardous drinkers and dependent alcoholics in the UK. Addict Biol. 2003;8:201–210. doi: 10.1080/1355621031000117437. [DOI] [PubMed] [Google Scholar]
- 76.Sarles H. An international survey on nutrition and pancreatitis. Digestion. 1973;9:389–403. doi: 10.1159/000197468. [DOI] [PubMed] [Google Scholar]
- 77.Durbec JP, Sarles H. Multicenter survey of the etiology of pancreatic diseases. Relationship between the relative risk of developing chronic pancreaitis and alcohol, protein and lipid consumption. Digestion. 1978;18:337–350. doi: 10.1159/000198221. [DOI] [PubMed] [Google Scholar]
- 78.Sarles H, Lebreuil G, Tasso F, Figarella C, Clemente F, Devaux MA, Fagonde B, Payan H. A comparison of alcoholic pancreatitis in rat and man. Gut. 1971;12:377–388. doi: 10.1136/gut.12.5.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schwartz ES, La JH, Scheff NN, Davis BM, Albers KM, Gebhart GF. TRPV1 and TRPA1 antagonists prevent the transition of acute to chronic inflammation and pain in chronic pancreatitis. J Neurosci. 2013;33:5603–5611. doi: 10.1523/JNEUROSCI.1806-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zaninovic V, Gukovskaya AS, Gukovsky I, Mouria M, Pandol SJ. Cerulein upregulates ICAM-1 in pancreatic acinar cells, which mediates neutrophil adhesion to these cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G666–G676. doi: 10.1152/ajpgi.2000.279.4.G666. [DOI] [PubMed] [Google Scholar]
- 81.Gumy C, Chandsawangbhuwana C, Dzyakanchuk AA, Kratschmar DV, Baker ME, Odermatt A. Dibutyltin disrupts glucocorticoid receptor function and impairs glucocorticoid-induced suppression of cytokine production. PLoS One. 2008;3:e3545. doi: 10.1371/journal.pone.0003545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schneider KJ, Scheer M, Suhr M, Clemens DL. Ethanol administration impairs pancreatic repair after injury. Pancreas. 2012;41:1272–1279. doi: 10.1097/MPA.0b013e31824bde37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Apte M, Pirola R, Wilson J. New insights into alcoholic pancreatitis and pancreatic cancer. J Gastroenterol Hepatol. 2009;24 Suppl 3:S51–S56. doi: 10.1111/j.1440-1746.2009.06071.x. [DOI] [PubMed] [Google Scholar]
- 84.Lieber CS, DeCarli LM, Feinman L, Hasumura Y, Korsten M, Matsuzaki S, Teschke R. Effect of chronic alcohol consumption on ethanol and acetaldehyde metabolism. Adv Exp Med Biol. 1975;59:185–227. doi: 10.1007/978-1-4757-0632-1_14. [DOI] [PubMed] [Google Scholar]
- 85.Petersen P. Ultrastructure of periportal and centrilobular hepatocytes in human fatty liver of various aetiology. Acta Pathol Microbiol Scand A. 1977;85:421–427. doi: 10.1111/j.1699-0463.1977.tb00444.x. [DOI] [PubMed] [Google Scholar]
- 86.Bang S, Kim KY, Yoo S, Kim YG, Hwang SW. Transient receptor potential A1 mediates acetaldehyde-evoked pain sensation. Eur J Neurosci. 2007;26:2516–2523. doi: 10.1111/j.1460-9568.2007.05882.x. [DOI] [PubMed] [Google Scholar]
- 87.Schwartz ES, Christianson JA, Chen X, La JH, Davis BM, Albers KM, Gebhart GF. Synergistic role of TRPV1 and TRPA1 in pancreatic pain and inflammation. Gastroenterology. 2011;140:1283–1291.e1-e2. doi: 10.1053/j.gastro.2010.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang LP, Ma F, Abshire SM, Westlund KN. Prolonged high fat/alcohol exposure increases TRPV4 and its functional responses in pancreatic stellate cells. Am J Physiol Regul Integr Comp Physiol. 2013;304:R702–R711. doi: 10.1152/ajpregu.00296.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Haber PS, Apte MV, Moran C, Applegate TL, Pirola RC, Korsten MA, McCaughan GW, Wilson JS. Non-oxidative metabolism of ethanol by rat pancreatic acini. Pancreatology. 2004;4:82–89. doi: 10.1159/000077608. [DOI] [PubMed] [Google Scholar]
- 90.Haber PS, Apte MV, Applegate TL, Norton ID, Korsten MA, Pirola RC, Wilson JS. Metabolism of ethanol by rat pancreatic acinar cells. J Lab Clin Med. 1998;132:294–302. doi: 10.1016/s0022-2143(98)90042-7. [DOI] [PubMed] [Google Scholar]
- 91.McCarroll JA, Phillips PA, Park S, Doherty E, Pirola RC, Wilson JS, Apte MV. Pancreatic stellate cell activation by ethanol and acetaldehyde: is it mediated by the mitogen-activated protein kinase signaling pathway? Pancreas. 2003;27:150–160. doi: 10.1097/00006676-200308000-00008. [DOI] [PubMed] [Google Scholar]
- 92.Vera-Portocarrero L, Westlund KN. Role of neurogenic inflammation in pancreatitis and pancreatic pain. Neurosignals. 2005;14:158–165. doi: 10.1159/000087654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rütti S, Ehses JA, Sibler RA, Prazak R, Rohrer L, Georgopoulos S, Meier DT, Niclauss N, Berney T, Donath MY, et al. Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology. 2009;150:4521–4530. doi: 10.1210/en.2009-0252. [DOI] [PubMed] [Google Scholar]
- 94.Sjöholm A. Intracellular signal transduction pathways that control pancreatic beta-cell proliferation. FEBS Lett. 1992;311:85–90. doi: 10.1016/0014-5793(92)81373-t. [DOI] [PubMed] [Google Scholar]
- 95.Fernández-Solà J, García G, Elena M, Tobías E, Sacanella E, Estruch R, Nicolás JM. Muscle antioxidant status in chronic alcoholism. Alcohol Clin Exp Res. 2002;26:1858–1862. [PubMed] [Google Scholar]
- 96.Pade PA, Cardon KE, Hoffman RM, Geppert CM. Prescription opioid abuse, chronic pain, and primary care: a Co-occurring Disorders Clinic in the chronic disease model. J Subst Abuse Treat. 2012;43:446–450. doi: 10.1016/j.jsat.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 97.Mehendale AW, Goldman MP, Mehendale RP. Opioid overuse pain syndrome (OOPS): the story of opioids, prometheus unbound. J Opioid Manag. 2013;9:421–438. doi: 10.5055/jom.2013.0185. [DOI] [PubMed] [Google Scholar]
- 98.Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 99.Blackshaw LA, Page AJ, Young RL. Metabotropic glutamate receptors as novel therapeutic targets on visceral sensory pathways. Front Neurosci. 2011;5:40. doi: 10.3389/fnins.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moldrich RX, Beart PM. Emerging signalling and protein interactions mediated via metabotropic glutamate receptors. Curr Drug Targets CNS Neurol Disord. 2003;2:109–122. doi: 10.2174/1568007033482922. [DOI] [PubMed] [Google Scholar]
- 101.Carlton SM, Hargett GL, Coggeshall RE. Localization of metabotropic glutamate receptors 2/3 on primary afferent axons in the rat. Neuroscience. 2001;105:957–969. doi: 10.1016/s0306-4522(01)00238-x. [DOI] [PubMed] [Google Scholar]
- 102.Carlton SM, Hargett GL. Colocalization of metabotropic glutamate receptors in rat dorsal root ganglion cells. J Comp Neurol. 2007;501:780–789. doi: 10.1002/cne.21285. [DOI] [PubMed] [Google Scholar]
- 103.Brice NL, Varadi A, Ashcroft SJ, Molnar E. Metabotropic glutamate and GABA(B) receptors contribute to the modulation of glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia. 2002;45:242–252. doi: 10.1007/s00125-001-0750-0. [DOI] [PubMed] [Google Scholar]
- 104.Volpi C, Fazio F, Fallarino F. Targeting metabotropic glutamate receptors in neuroimmune communication. Neuropharmacology. 2012;63:501–506. doi: 10.1016/j.neuropharm.2012.05.024. [DOI] [PubMed] [Google Scholar]
- 105.Valli MJ, Schoepp DD, Wright RA, Johnson BG, Kingston AE, Tomlinson R, Monn JA. Synthesis and metabotropic glutamate receptor antagonist activity of N1-substituted analogs of 2R,4R-4-aminopyrrolidine-2,4-dicarboxylic acid. Bioorg Med Chem Lett. 1998;8:1985–1990. doi: 10.1016/s0960-894x(98)00352-7. [DOI] [PubMed] [Google Scholar]
- 106.Chiechio S, Nicoletti F. Metabotropic glutamate receptors and the control of chronic pain. Curr Opin Pharmacol. 2012;12:28–34. doi: 10.1016/j.coph.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 107.Osikowicz M, Mika J, Przewlocka B. The glutamatergic system as a target for neuropathic pain relief. Exp Physiol. 2013;98:372–384. doi: 10.1113/expphysiol.2012.069922. [DOI] [PubMed] [Google Scholar]
- 108.Vinson PN, Conn PJ. Metabotropic glutamate receptors as therapeutic targets for schizophrenia. Neuropharmacology. 2012;62:1461–1472. doi: 10.1016/j.neuropharm.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Spooren W, Lesage A, Lavreysen H, Gasparini F, Steckler T. Metabotropic glutamate receptors: their therapeutic potential in anxiety. Curr Top Behav Neurosci. 2010;2:391–413. doi: 10.1007/7854_2010_36. [DOI] [PubMed] [Google Scholar]
- 110.Moussawi K, Kalivas PW. Group II metabotropic glutamate receptors (mGlu2/3) in drug addiction. Eur J Pharmacol. 2010;639:115–122. doi: 10.1016/j.ejphar.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl) 2010;210:137–147. doi: 10.1007/s00213-010-1806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Langen B, Fink H. Anxiety as a predictor of alcohol preference in rats? Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:961–968. doi: 10.1016/j.pnpbp.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 113.Chen Y, Sommer C. Activation of the nociceptin opioid system in rat sensory neurons produces antinociceptive effects in inflammatory pain: involvement of inflammatory mediators. J Neurosci Res. 2007;85:1478–1488. doi: 10.1002/jnr.21272. [DOI] [PubMed] [Google Scholar]
- 114.Dong XW, Williams PA, Jia YP, Priestley T. Activation of spinal ORL-1 receptors prevents acute cutaneous neurogenic inflammation: role of nociceptin-induced suppression of primary afferent depolarization. Pain. 2002;96:309–318. doi: 10.1016/S0304-3959(01)00460-2. [DOI] [PubMed] [Google Scholar]
- 115.Pettersson LM, Sundler F, Danielsen N. Expression of orphanin FQ/nociceptin and its receptor in rat peripheral ganglia and spinal cord. Brain Res. 2002;945:266–275. doi: 10.1016/s0006-8993(02)02817-2. [DOI] [PubMed] [Google Scholar]
- 116.Mika J, Li Y, Weihe E, Schafer MK. Relationship of pronociceptin/orphanin FQ and the nociceptin receptor ORL1 with substance P and calcitonin gene-related peptide expression in dorsal root ganglion of the rat. Neurosci Lett. 2003;348:190–194. doi: 10.1016/s0304-3940(03)00786-9. [DOI] [PubMed] [Google Scholar]
- 117.Houtani T, Nishi M, Takeshima H, Sato K, Sakuma S, Kakimoto S, Ueyama T, Noda T, Sugimoto T. Distribution of nociceptin/orphanin FQ precursor protein and receptor in brain and spinal cord: a study using in situ hybridization and X-gal histochemistry in receptor-deficient mice. J Comp Neurol. 2000;424:489–508. [PubMed] [Google Scholar]
- 118.Brooks TA, Hawkins BT, Huber JD, Egleton RD, Davis TP. Chronic inflammatory pain leads to increased blood-brain barrier permeability and tight junction protein alterations. Am J Physiol Heart Circ Physiol. 2005;289:H738–H743. doi: 10.1152/ajpheart.01288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ronaldson PT, Davis TP. Targeting blood-brain barrier changes during inflammatory pain: an opportunity for optimizing CNS drug delivery. Ther Deliv. 2011;2:1015–1041. doi: 10.4155/tde.11.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tian JH, Xu W, Fang Y, Mogil JS, Grisel JE, Grandy DK, Han JS. Bidirectional modulatory effect of orphanin FQ on morphine-induced analgesia: antagonism in brain and potentiation in spinal cord of the rat. Br J Pharmacol. 1997;120:676–680. doi: 10.1038/sj.bjp.0700942. [DOI] [PMC free article] [PubMed] [Google Scholar]