

Abstract
AIM: To determine the role of NOB1, a regulator of cell survival in yeast, in human colorectal cancer cells.
METHODS: Lentivirus-mediated small interfering RNA (siRNA) was used to inhibit NOB1 expression in RKO human colorectal cancer cells in vitro and in vivo in a mouse xenograft model. The in vitro and in vivo knockdown efficacy was determined using both Western blot and quantitative reverse transcription polymerase chain reaction (qRT-PCR). qRT-PCR was also used to analyze the downstream signals following NOB1 knockdown. Cell growth and colony formation assays were used to determine the effect of NOB1 inhibition on RKO proliferation and their ability to form colonies. Endonuclease activity, as evaluated by terminal deoxytransferase-mediated dUTP nick end labeling (TUNEL), and annexin V staining were used to determine the presence of apoptotic cell death prior to and following NOB1 inhibition. Cell cycle analysis was used to determine the effect of NOB1 inhibition on RKO cell cycle. A cDNA microarray was used to determine global differential gene expression following NOB1 knockdown.
RESULTS: Virus-mediated siRNA inhibition of NOB1 resulted in (1) the down-regulation of NOB1 expression in RKO cells for both the mRNA and protein; (2) inhibition of NOB1 expression both in vitro and in vivo experimental systems; (3) cell growth inhibition via significant induction of cell apoptosis, without alteration of the cell cycle distribution; and (4) a significant decrease in the average weight and volume of xenograft tumors in the NOB1-siRNA group compared to the control scr-siRNA group (P = 0.001, P < 0.05). Significantly more apoptosis was detected within tumors in the NOB1-siRNA group than in the control group. Microarray analysis detected 2336 genes potentially regulated by NOB1. Most of these genes are associated with the WNT, cell proliferation, apoptosis, fibroblast growth factor, and angiogenesis signaling pathways, of which BAX and WNT were validated by qRT-PCR. Among them, 1451 probes, representing 963 unique genes, were upregulated; however, 2308 probes, representing 1373 unique genes, were downregulated.
CONCLUSION: NOB1 gene silencing by lentivirus-mediated RNA interference can inhibit tumor growth by inducing apoptosis of cancerous human colorectal cells.
Keywords: NOB1, Small RNA interference, Apoptosis, Colorectal cancer, BAX, WNT
Core tip: NOB1, a critically important regulator in yeast, is also required for regulation of cell growth and survival in RKO human colorectal cancer cells. NOB1 knockdown promotes cell apoptosis in both in vitro and in vivo model systems. The gene expression profile suggests the importance of the WNT pathway, cell proliferation, apoptosis, the fibroblast growth factor, and angiogenesis signaling pathways in the function of NOB1.
INTRODUCTION
Colorectal cancer (CRC), one of the most common malignancies worldwide, and is the result of a multi-step and multi-mechanistic process. Abnormalities in apoptotic function have been shown to contribute to both CRC pathogenesis as well as its resistance to chemotherapeutic drugs and radiotherapy[1-3]. Understanding the molecular and cellular mechanisms which contribute to the carcinogenesis and CRC development could facilitate diagnosis and treatment of the disease.
The proteasome, a highly selective proteinase complex, is considered a promising therapeutic target for CRC treatment[4,5]. The proteasome is required for the degradation of many endogenous proteins, including transcriptional factors, cyclins, and tumor suppressors[6-9]. The proteasome 19S regulatory particle (RP) recognizes and degrades ubiquitin-marked proteins[10]. The ubiquitin-proteasome system, one of the most important intracellular degradative pathways, plays a critical role in the regulation of various cellular processes, such as cell cycle progression, differentiation, apoptosis, and angiogenesis[11].
Ribosome biogenesis, a high-energy and essential process, plays a crucial role in cell growth, proliferation, and differentiation[12,13]. The rate of ribosomal processing is highly in tune with extracellular growth signals[14], and is, therefore, tightly coordinated with cell growth and proliferation. An emerging line of evidence suggests that altered ribosome biogenesis may be associated with tumorigenesis[15-17].
The human NOB1 gene encodes a putative protein with a PIN (PilT amino terminus) domain and a zinc ribbon domain[18]. The yeast Nob1p (Nin one binding protein) is required for 26S proteasome function and ribosome biogenesis. Nob1p has an endonuclease-containing PIN domain responsible for cleavage of the 20S pre-rRNA at site D generating the mature 18S-rRNA[19-22]. Granneman et al[22] was able to show the importance of RNA restructuring and protein remodeling in the 3’ region of the 18S rRNA in the Nob1p-dependent cleavage at site D. In addition, using a two-hybrid screen, Nob1p was identified as a protein interacting with Nin1p/Rpn12p (a subunit of the 19S RP of the yeast 26S proteasome)[23,24]. The interaction between Nob1p and 19S RP subunit appears to be crucial for the maturation of the 20S RP[24]. Thus, the human NOB1 might also be involved in ribosome biogenesis and 26S proteasome function in the nucleus[20], and play an important role in cell growth and proliferation.
A recent study indicated that NOB1 RNA interference inhibits human ovarian cancer cell growth through G0/G1 arrest[25]. However, the NOB1 potential role in colorectal cancer has not been demonstrated. A recent study, using immunohistochemistry to determine the expression of NOB1, found that NOB1 was up-regulated in 60 colorectal cancer tissues[26]. RKO, a well-established poorly differentiated human colon carcinoma cell line with wild-type p53, is used as the model for studying NOB1 gene due to the relatively short doubling time and established genetic profile of the cell line. Lentiviral- mediated small interfering RNA (siRNA) was used to inhibit NOB1 expression and investigate the effects of NOB1 knockdown on cell proliferation, cell cycle progression, and apoptosis in RKO. Microarray and qPCR were used to detect and validate NOB1-targeted genes and pathways in colorectal cancer. Herein, a specific downregulation of NOB1 inhibited RKO cell proliferation by inducing cell apoptosis, but not cell cycle arrest. Therefore, NOB1 may serve as a therapeutic target for CRC.
MATERIALS AND METHODS
Reagents and antibodies
RPMI 1640, fetal bovine serum (FBS), Trizol Reagent, and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA, United States). Propidium iodide (PI) was obtained from Sigma-Aldrich (St. Louis, MO, United States). RNase A was from MBI Fermentas (St. Leon-Rot, Germany). The Annexin V-APC Apoptosis Detection Kit was acquired from eBioscience (San Diego, CA, United States). The bicinchoninic acid (BCA) protein assay was purchased from HyClone-Pierce (South Logan, UT, United States). M-MLV Reverse Transcriptase was bought from Promega (Madison, WI, United States). Oligo-dT was procured from Sangon Biotech (Shanghai, China). SYBR Green Master Mixture was purchased from Takara (Otsu, Japan). pGCSIL-green fluorescent protein (GFP), vector and virion-packaging elements (pHelper 1.0 and pHelper 2.0) were obtained from Genechem (Shanghai, China). Rabbit anti-NOB1 polyclonal antibody was bought from either Abcam (Cambridge, MA, United States) or ProteinTech Group (Chicago, IL, United States). Mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), goat anti-rabbit IgG, and goat anti-mouse IgG were from Santa Cruz Biotechnology (Santa Cruz, CA, United States). All other chemicals were of analytical grade.
siRNA construction and lentivirus production
Previously described methods, with minimal modifications, were used to construct siRNA and to produce lentivirus[27]. A 19-nucleotide (CCTGGAGCCAATCTTCAAGAA) siRNA was designed against the human NOB1 mRNA (GenBank accession number NM_014062). In the experiment, siRNA with a scrambled sequence (scr-siRNA; TTCTCCGAACGTGTCACGT) was used as a negative control. siRNAs were synthesized and inserted between the AgeI and EcoRI restriction sites of the pGCSIL-GFP plasmid. The correct siRNA insertion was confirmed by restriction mapping and direct DNA sequencing.
Using Lipofectamine 2000, a recombinant lentiviral vector, pGCSIL-GFP, with pHelper 1.0 [encoding human immunodeficiency virus (HIV) gag, pol, and rev] and pHelper 2.0 (encoding for the VSV-G envelope) was used to co-transfect 293T cells, according to the manufacturer’s instructions. Infectious lentivirus was harvested at 48 h post-transfection, centrifuged to remove cell debris, and then filtered through a 0.45 μm cellulose acetate filter.
Cell culture and infection
The RKO (human colorectal cancer) cell line, purchased from the American Type Culture Collection (ATCC, Manassas, VA, United States), was maintained in RPMI 1640 medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, at 37 °C in a 5% CO2 humidified incubator. RKO cells were subcultured in 6-well tissue culture plates at a density of 5 × 104 cells/well. Lentivirus infection was conducted at 30% cell confluency. Lentivirus was added at different MOIs in serum-free medium at 37 °C in 5% humidified CO2. After 24 h, complete medium was added to the cells. More than 90% of the cells were infected at 4 d post-infection as indicated by the expression of GFP.
Quantitative reverse transcription polymerase chain reaction analysis
The total RNA from scr-siRNA and NOB1-siRNA infected cells was extracted with Trizol reagent according to the manufacturer’s protocol. The RNA quantity and purity were determined by UV absorbance spectroscopy. cDNA was generated by reverse transcription with oligo-dT primer using M-MLV. The quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed using SYBR Green Master Mixture and analyzed on the TAKARA TP800-Thermal Cycler Dice™ Real-Time System. The qRT-PCR primers of each gene are listed in Table 1. The thermal cycling conditions were 15 s at 95 °C, 45 cycles of 5 s at 95 °C, and 30 s at 60 °C. Data were analyzed with TAKARA Thermal Dice Real Time System software Ver3.0. Each reaction was performed with triplicate samples in each group and analyzed individually relative to GAPDH (a normalization control), and differential gene expression was calculated using the 2-ΔΔCt method[28]. Thereafter, data for mRNA expression levels were expressed as a fold difference relative to that of negative control cells.
Table 1.
Primers used in this study
| Genes | Sequence | Length (bp) |
| GAPDH-F | TGACTTCAACAGCGACACCCA | 121 |
| GAPDH-R | CACCCTGTTGCTGTAGCCAAA | |
| NOB1-F | ATCTGCCCTACAAGCCTAAAC | 184 |
| NOB1-R | TCCTCCTCCTCCTCCTCAC | |
| BAX-F | TGCTTCAGGGTTTCATCCA | 296 |
| BAX-R | GGCCTTGAGCACCAGTTT | |
| WNT7B-F | TCCACTGGTGCTGCTTCG | 300 |
| WNT7B-R | GTCACGGGTGCTGTTCTGC |
GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
Western blot analysis
The infected cells were washed twice with PBS, suspended in a lysis buffer (2% mercaptoethanol, 20% glycerol, 4% SDS in 100 mmol/L Tris-HCl buffer, pH 6.8), and placed on ice for 15 min. The suspension was collected after centrifugation at 12000 g for 15 min at 4 °C. Protein concentration was determined by the BCA Protein assay. From each sample, 30 μg of protein was subjected to electrophoresis on 10% SDS-polyacrylamide gel and transferred to a PVDF membrane. TBST (Tris-buffered saline, 0.1% Tween-20) buffer containing 5% non-fat dry milk was used to block non-specific binding for 1 h at room temperature. Membranes were then incubated with the indicated antibodies overnight at 4 °C, and washed three times with TBST. Membranes were incubated with secondary antibodies conjugated to horseradish peroxidase for 2 h at room temperature, and washed three times with TBST. The detected protein signals were visualized by the ECL Plus Western Blotting Detection System (Amersham). GAPDH protein levels were used as a control to verify equal protein loading.
Cell growth
Cell growth was assessed using the Cellomics ArrayScan High Content Screening (HCS) system (Thermo Fisher Scientific, Pittsburgh, PA, United States). Briefly, RKO cells infected with lentivirus-mediated NOB1-siRNA or scr-siRNA were seeded in 96-well plates at a density of 2 × 103 cells/well and cultured at 37 °C in a 5% CO2 humidified incubator. The infected GFP-expressing cells were imaged and counted on the Cellomics ArrayScan HCS Reader once a day for 5 d. The experiment was performed at least three times, independently. The growth curves of the infected cells were constructed.
Colony formation assay
To analyze cell growth, the colony formation analysis was performed using the Cellomics ArrayScan HCS system. Briefly, RKO cells infected with lentivirus-mediated NOB1-siRNA or scr-siRNA were seeded in 96-well plates at a density of 500 cells/well. Cells were cultured for 14 d at 37 °C in a 5% CO2 humidified incubator. Culture medium was changed every 3 d. Cell colonies were imaged and counted using the Cellomics HCS Reader. The experiment was performed in triplicate.
Cell cycle distribution analysis
The cell cycle distribution was determined by DNA staining with propidium iodide (PI) and flow cytometric analysis[29]. In summary, RKO cells seeded in 6 cm culture dishes were infected with lentivirus-mediated NOB1-siRNA or scr-siRNA for 6 d. After infection, the cells were trypsinized, washed with PBS, and fixed in 70% ethanol for at least 1 h at 4 °C. After two washing steps in cold PBS, the cells were resuspended in 1 mL of PBS containing 100 μg/mL RNase A and 50 μg/mL PI, and incubated for 30 min in the dark at room temperature. The percentages of cells in different phases of cell cycle were measured with the Becton Dickinson FACSCalibur flow cytometer using dedicated software. The experiment was repeated three times.
Annexin V apoptosis assay
Annexin V-APC (apoptosis detection kit) was used to detect apoptosis as described by the manufacturer. Briefly, RKO cells seeded in 6-well culture plates were infected with lentivirus-mediated NOB1-siRNA or scr-siRNA for 7 d. After infection, attached cells were trypsinized, washed with PBS, and centrifuged. The cells were washed with 1 × binding buffer, centrifuged, and resuspended in 1 mL 1 × staining buffer. To 100 μL of cell suspension, prepared as described above, 5 μL Annexin V-APC was added, followed by a gentle vortex, and 10 min incubation at room temperature in the dark. Data acquisition and analysis were performed by the Becton Dickinson FACSCalibur flow cytometer using dedicated software. The experiment was repeated three times.
In vivo xenograft tumor model
This animal experiment was approved by the Shanghai Laboratory Animal Ethics Committee. Four to five week old female nude mice were purchased from the Shanghai Laboratory Animal Center of the Chinese Academy of Science and were treated according to the ethics guidelines for animal research. To produce tumors, using a 24-gauge needle, 1 × 106 RKO cells in 0.1 mL of serum-free RPMI 1640 were injected subcutaneously into the right flank of each nude mouse. The mice were randomized into three groups (n = 5 each group). After 2 wk, the first group of nude mice was injected intratumorally with PBS, the second group with lentivirus-mediated scr-siRNA, and the third group with lentivirus-NOB1-siRNA. All of the mice were injected weekly until the completion of experiments. Tumor growth was monitored weekly and measured in two dimensions. Tumor volume was calculated using the V = W2 × L/2 formula, where W and L are the shortest and longest diameters, respectively. After 5 wk, mice were killed and the tumors were immediately fixed in formalin for terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) analysis.
Terminal deoxytransferase-mediated dUTP nick end labeling procedure
Apoptosis was detected using the TUNEL in situ apoptosis detection kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions. After deparaffinization, dehydration, and inactivation of intrinsic peroxidase activity, 20 paraffin sections of each tumor specimen were incubated with 2 μg/mL proteinase K at 37 °C for 15 min. Afterward, the sections were treated with terminal deoxynucleotidyl transferase and biotinylated dUTP. The reaction was stopped with TB buffer (30 mmol/L sodium chloride, 30 mmol/L sodium citrate), followed by microscopic observation of the samples. The controls for the TUNEL procedure were treated in the same manner as the test samples except dH2O was used instead of using TdT enzyme in both kits. No labeling was found in the controls.
Microarray analysis
The scr-siRNA and NOB1-siRNA infected RKO cell gene expression profiles were obtained and compared using the Agilent Human Gene Expression 4 × 44K v2 Microarray kit (Agilent Technologies, Santa Clara, CA, United States). The microarrays were performed following the manufacturer’s protocol. Briefly, the total RNA was extracted using Trizol and further purified with the Qiagen RNeasy kit (Valencia, CA, United States). RNA quantity, quality, and size distribution were checked by the Nanodrop 2000C (A260/A280) and Agilent 2100 Bioanalyzer system (Agilent Technologies, Santa Clara, CA, United States). cDNA was generated by reverse transcription using 0.2 μg of the total RNA. Cy3-CTP-labeled cRNA was synthesized by in vitro transcription (IVT), which was purified by Qiagen RNeasy kit and hybridized to the Agilent Whole Human Genome Oligo Microarrays 4 × 44K. After hybridization, the array was washed and processed using an Agilent DNA microarray scanner (Agilent Technologies). Agilent Feature Extraction Software (FES) was used to read and process the microarray image files. Genespring was employed to determine feature intensities and ratios (including background subtraction and normalization). Genes with ratios > 2 or < 0.5 were considered differentially expressed. The KEGG pathway and Gene Ontology (GO) enrichment analyses, for differentially expressed genes, were performed using the NIH gene annotation software, DAVID[30]. Heat maps were presented using Cluster 3.0 and the Tree View software[31].
Statistical analysis
Statistical analysis was performed using SPSS software version 10.0 (SPSS Inc, Chicago, IL, United States). Data were expressed as the mean of at least three different experiments ± SD. The statistical difference was evaluated using the χ2 test and Student’s t test. Statistically significant differences were defined as P < 0.05.
RESULTS
Downregulation of NOB1 expression by lentivirus-mediated NOB1-siRNA in RKO cells
The efficiency of lentiviral infection of RKO cells was determined through microscopic examination of GFP expression at an MOI of 10 on day 4 after infection (Figure 1A). More than 90% of RKO cells were infected.
Figure 1.
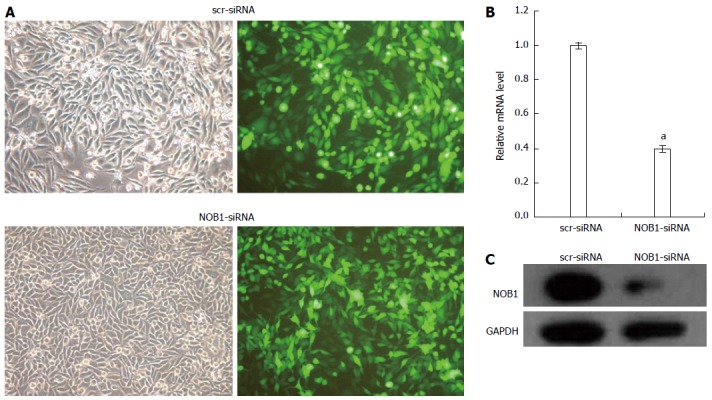
Lentivirus-mediated siRNA decreased NOB1 expression in RKO cells. A: Infection efficiency was estimated 4 d after infection at MOI of 10. Green fluorescent protein (GFP) expression in infected cells was observed under light and fluorescence microscopy, respectively. Light micrograph (left); Fluorescent micrograph (right) (× 200); B: Total RNA was extracted 5 d after infection, and relative NOB1 mRNA expression was determined by qRT-PCR. GAPDH expression was used as a loading control. Data are presented as mean ± SD of three independent experiments. aP < 0.05 vs scr-siRNA; C: Total cellular protein was extracted 7 d after infection and determined by western blot analysis using antibodies against NOB1. GAPDH was used as an internal control. MOI: Multiplicity of infection; GFP: Green fluorescent protein; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
qRT-PCR and Western blot examination of the effect of lentivirus-mediated NOB1-siRNA infection on the silencing of NOB1 expression showed a 60.9% reduction in NOB1 mRNA expression after 5 d of infection compared to scr-siRNA infection (P < 0.05, Figure 1B). Western blot analysis, performed 7 d after infection, showed a significant decrease in NOB1 protein expression in NOB1-siRNA infected RKO cells, as compared to the scr-siRNA infected cells (Figure 1C).
Effects of NOB1 knockdown on cell growth in RKO cells
The effects of NOB1 knockdown on cell function, cell growth, and colony formation were assessed using the Cellomics ArrayScan™ HCS system. As shown in Figure 2A, the GFP-expressing infected cells were counted once a day for 5 d. NOB1-siRNA inhibited cell proliferation in a time-dependent manner. After 5 d of infection, the number of NOB1-siRNA-infected RKO cells was reduced by 92.8% as compared with scr-siRNA infected cells (P < 0.05). Furthermore, NOB1-siRNA infected cells exhibited significant attenuation in the ability to form colonies. After 14 d of infection, the number of cell colonies in NOB1-siRNA infected RKO cells was reduced by 89.7%, as compared with scr-siRNA infected cells (P < 0.05, Figure 2B).
Figure 2.
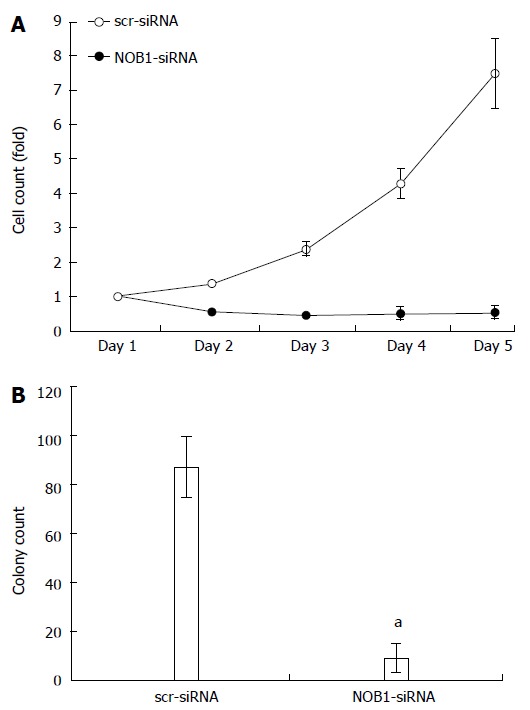
Effect of NOB1 knockdown on cell growth in RKO cells. A: The infected cells expressing GFP were imaged and counted using the Cellomics ArrayScanTM High Content Screening (HCS) Reader once a day for 5 d. Cell growth curves in scr-siRNA and NOB1-siRNA infected RKO cells are shown; B: Cell growth was detected by the colony formation assay in RKO cells 14 d after infection of scr-siRNA and NOB1-siRNA cells. RKO cells were seeded at 500 cells/well and allowed to form colonies. Cell colonies were imaged and counted using the Cellomics ArrayScanTM HCS Reader. Data are presented as mean ± SD of three independent experiments. aP < 0.05 vs scr-siRNA. scr-siRNA: Cells infected with lentivirus-mediated scramble small interfering RNA; NOB1-siRNA: Cells infected with lentivirus-mediated NOB1-siRNA; GFP: Green fluorescent protein; qRT-PCR: Quantitative reverse transcription polymerase chain reaction.
Effects of NOB1 knockdown on cell cycle progression and apoptosis in RKO cells
The effects of NOB1 knockdown on cell cycle progression and apoptosis in RKO cells were examined in an effort to further explore the reason for cell growth reduction. As shown in Figure 3A, there were no significant differences in cell cycle progression, including G0/G1, S, and G2/M phases, between the cells infected with NOB1-siRNA or scr-siRNA.
Figure 3.
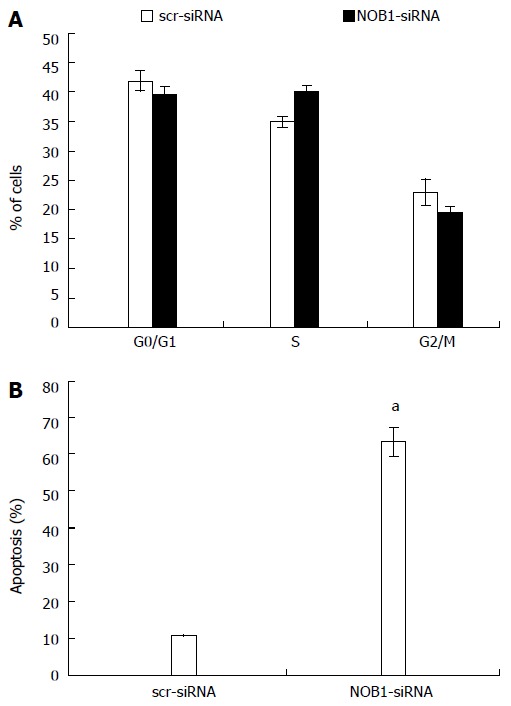
Effects of NOB1 downregulation on cell cycle progression and apoptosis in RKO cells. A: Cell cycle distribution was determined by flow cytometric analysis at day 6 of infection; B: Flow cytometric analysis of cell apoptosis was performed on day 7 of infection. Data are presented as mean ± SD of three independent experiments. aP < 0.05 vs scr-siRNA. scr-siRNA: Cells infected with lentivirus-mediated scramble small interfering RNA; NOB1-siRNA: Cells infected with lentivirus-mediated NOB1-siRNA.
NOB1 knockdown resulted in a marked increase in cell apoptosis (Figure 3B). The percentage of apoptotic cells was 63.2% ± 4.2% in NOB1-siRNA infected RKO cells, and 10.9% ± 0.2% in scr-siRNA infected RKO cells (P < 0.05). To validate the current finding in another colon cancer cell line, HCT116 cells were also infected with NOB1-siRNA and resulted in efficient NOB1 knockdown (Figure 1A). Flow cytometry analysis of cell death revealed that NOB1 knockdown induced significant apoptosis in HCT116 cells (Figure 1B).
Effects of NOB1 knockdown on tumor growth and cell survival in the xenograft mouse model
The effect of lentivirus-mediated Nob1-siRNA on tumor growth in the xenograft mouse model was examined. Western blot showed a reduction in NOB1 expression in xenograft tissue (Figure 4A). Furthermore, NOB1-siRNA also induced extensive cell death as detected by TUNEL assay (Figure 4B, C). After 2 wk the xenograft tumor began to grow; and at week 5 after injection, the xenograft tumor size was as follows (means ± SE): 910 ± 180 mm3 in the PBS control group, 870 ± 1650 mm3 in the scr-siRNA group, and 405 ± 102 mm3 in the NOB1-siRNA group (Figures 4D, E, and F). ANOVA analysis revealed that the inhibitory effect of NOB1-siRNA infection was significant compared with scr-siRNA or PBS injections (P < 0.05). Compared with the starting volume, there was a significant retardation of tumor growth in xenograft mice treated with NOB1-siRNA.
Figure 4.
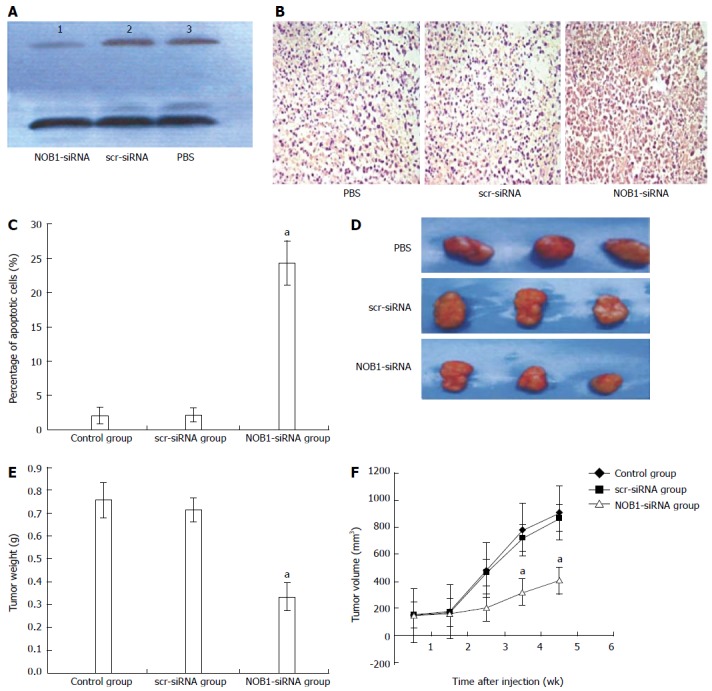
Suppression of tumor growth by lentivirus-mediated NOB1-siRNA in RKO cell xenograft mouse model. A: RKO cell xenograft mice were injected intratumorally with lentivirus-mediated NOB1-siRNA, scr-siRNA, or PBS. The size of the primary tumors was measured every week. Mice were sacrificed after 5 wk. NOB1-siRNA reduced NOB-1 expression in xenografted tumor tissue as determined by Western blot; B: NOB1-siRNA induced significant apoptosis in xenografted tumor tissue detected by TUNEL staining as shown in representative images; C: Plotted percentages of apoptotic cells; D: Typical pictures from RKO cell xenografted tumors infected by PBS, src-RNA and NOB1-siRNA; E: Plotted tumor weight; F: Tumor volume in RKO cell xenograft mouse model after NOB1-siRNA infection. aP < 0.05 vs scr-siRNA. scr-siRNA: Cells infected with lentivirus-mediated scramble small interfering RNA; NOB1-siRNA: Cells infected with lentivirus-mediated NOB1-siRNA; PBS: Phosphate buffered saline.
Microarray analysis identified genes and pathways potentially targeted by NOB1
To determine the NOB1-targeted genes and -pathways in colorectal cancer, the total RNA was extracted from scr-siRNA and NOB1-siRNA infected RKO cells, and was hybridized on the Human Gene Expression 4 × 44K v2 Microarray chip containing 41093 probes, representing 27958 Entrez genes. Among them, 2336 genes with ratio > 2 or < 0.5 were defined as differentially expressed genes and potential NOB1 targets. In an indirect way, NOB1 potentially upregulated 1451 probes representing 963 unique genes. There were also 2308 downregulated probes, representing 1373 unique genes, which may possibly be regulated by NOB1 directly.
NIH DAVID enrichment analysis of 2336 differentially expressed genes demonstrated NOB1 targeting KEGG pathways in cancer [P = 0.001, false discovery rate (FDR) = 1.23%]. With a total of 328 genes in cancer pathways, 56 genes are potentially targeted by NOB1 (Figure 5). Gene Ontology analysis indicated that these 56 genes are found in the WNT, cell proliferation, apoptosis, fibroblast growth factor (FGF), and angiogenesis signaling pathways (Table 2). The expressions of WNT7B and BAX were validated by qRT-PCR (Figure 6), which was consistent with microarray data.
Figure 5.

Heat map showing 56 genes differentially expressed in NOB1-siRNA infected RKO cells and enriched in the cancer pathway.
Table 2.
Gene Ontology enrichment analysis showing NOB1-targeted pathways and genes involved in cancer development
| Gene ontology | Genes | P value | Fold enrichment | FDR |
| WNT pathway | WNT4, WNT7B, WNT5B, MITF, LEF1, WNT9A, TCF7L1, WNT8B, APC | 4.32 × 10-8 | 16.9523 | 7.00 × 10-5 |
| Proliferation | PRKCA, CEBPA, FGFR3, IL-8, FGF9, MITF, TGFBR2, EGLN3, NFKBIA, CDKN1A, CASP3, BAX, VEGFA, FGF1, EGF, APC | 2.03 × 10-7 | 5.0931 | 3.28 × 10-4 |
| Apoptosis | PRKCA, TRAF1, MITF, MLH1, NFKBIA, FADD, BIRC3, DAPK3, CDKN1A, CASP3, NTRK1, BAX, VEGFA, FAS, TRAF4, APC | 2.67 × 10-7 | 4.9854 | 4.33 × 10-4 |
| FGF pathway | FGF5, FGFR3, FGF9, FGF16, FGF1 | 4.64 × 10-6 | 43.1928 | 0.007516 |
| Angiogenesis | IL-8, EPAS1, FGF9, VEGFA, TGFBR2, EGF, FGF1 | 2.33 × 10-5 | 11.8488 | 0.037727 |
FDR: False discovery rate; APC: Adenomatous polyposis coli; IL: Interleukin; VEGFA: Vascular endothelial growth factor A; FGF: Fibroblast growth factor; TGF: Transforming growth factor.
Figure 6.
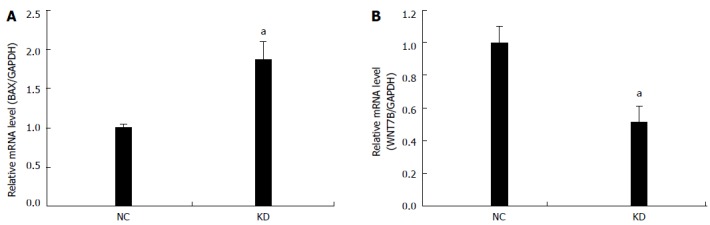
Quantitative reverse transcription polymerase chain reaction demonstrated NOB1-siRNA up-regulating BAX and down-regulating WNT7B mRNA in RKO cells. GAPDH expression was used as a loading control. Data are presented by the mean ± SD of three independent experiments. aP < 0.05 vs scr-siRNA; scr-siRNA: Cells infected with lentivirus-mediated scramble small interfering RNA; NOB1-siRNA: Cells infected with lentivirus-mediated NOB1-siRNA.
DISCUSSION
There has been little research conducted in order to understand the role of human NOB1 gene in colorectal cancer development and treatment. In an effort to expand our understanding, we employed lentiviral-mediated siRNA to inhibit NOB1 expression in RKO human colorectal cancer cells. The efficiency of lentiviral infection in RKO cells was more than 90% at an MOI of 10 on day 4 after infection. The NOB1-siRNA infection effectively reduced the expression of NOB1 in RKO cells, which was confirmed by qRT-PCR and Western blot analysis.
Carcinogenesis upsets the normal balance between cell proliferation and cell death[32]. Our results showed a significant inhibition of RKO cell growth as a result of NOB1 knockdown (Figure 2). The inhibition seemed to be due to induction of cell apoptosis, but not cell cycle arrest, in RKO cells (Figure 3). Furthermore, this phenomenon was validated in HCT116 cells, a p53 null cell line, suggesting that NOB1 is required for the survival of various colon cancer cells and its function is independent of p53. Given that RKO is a p53-positive cell line, our results suggest that apoptosis induced by NOB1-knockdown is p53 independent. The greatest apoptosis-inducing effect occurred 7 d after NOB1-siRNA infection. At 4 d after infection, the cell morphology looked largely normal, which could be explained by the relatively long half-life of NOB1 protein. Although, the most efficient NOB1 knockdown occurred on day 7, mRNA was markedly reduced on day 5 after infection. To explore the underlying mechanisms, microarray analysis was used to identify 2336 genes potentially targeted by NOB1. Further bioinformatic analysis suggested that NOB1 inhibited colorectal cancer through the oncogenic pathways, such as the WNT, cell proliferation, apoptosis, FGF, and angiogenesis signaling pathways. qRT-PCR confirmed that the expression of WNT7B and BAX were altered when NOB1 was knocked down.
Nob1p is required for 26S proteasome function, ribosome biogenesis, and cell viability in yeast[19-24]. Depletion of Nob1p leads to a defect in the processing of 40S ribosome subunits or small subunits[19]. Specifically, it results in the accumulation of 20S rRNA, the precursor of 18S rRNA, which is the primary rRNA component of the small subunit in yeast. Because Nob1p contains a PIN domain, it is believed to be the endonuclease responsible for the site D cleavage producing the mature 18S rRNA[20]. In addition to the PIN domain, Nob1p also contains a zinc ribbon domain, a well-known RNA binding domain. Thus, Nob1p-like proteins may potentially be required for ribosome biogenesis in other eukaryotes. A report showed that Nob1p assists in joining of the 20S core particle with the 19S RP in the nucleus as well as facilitating the 20S core particle maturation. Furthermore, 26S proteasome biogenesis is completed upon Nob1p internalization and degradation by the 26S proteasome[24]. Thus, Nob1p may also be required for ribosome biogenesis and 26S proteasome function in other eukaryotes.
The ubiquitin-proteasome pathway is responsible for most of the intracellular protein degradation[33]. The ribosome is a large protein-RNA complex essential for protein synthesis, a characteristic of cell viability[34]. Because protein synthesis malfunction and protein degradation cause cell apoptosis in tumor cells[35-37], the ubiquitin-proteasome pathway and ribosome biogenesis have become attractive targets in anticancer drug development. Due to the potentially important role of the human NOB1 in 26S proteasome function and ribosome biogenesis, we presumed that NOB1 knockdown might induce cell apoptosis. Considering that genes involved with WNT, apoptosis, FGF and angiogenesis signaling were altered in microarray profiling, further investigations should be focused on deciphering the connection between the role of NOB1 in the ubiquitin-proteasome pathway and ribosome biogenesis with these signaling/pathways.
In conclusion, NOB1 expression can be downregulated specifically and effectively by lentivirus-mediated siRNA. NOB1 knockdown significantly inhibited RKO cell growth by inducing cell apoptosis, but not cell cycle arrest. Furthermore, NOB1 knockdown led to the altered expression of genes involved in multiple pathways or cellular functions, such as the WNT, cell proliferation, apoptosis, FGF, and angiogenesis signaling cascades. Thus, NOB1 should be considered a potential therapeutic target for the treatment of CRC.
COMMENTS
Background
Human NOB1 encodes a protein with a PIN (PilT amino terminus) domain and a zinc ribbon domain. The yeast homolog Nob1p is required for 26S proteasome function and ribosome biogenesis; hence, Nob1p is essential for cell growth. However, the role of NOB1 in human cells remains largely unknown.
Research frontiers
Colorectal cancer (CRC) is one of the most common malignancies worldwide. The molecular events that trigger CRC development remain largely unexplored. Identification of novel therapeutic targets is urgently needed to improve the outcomes of patients suffering from CRC.
Innovations and breakthroughs
The role of NOB1 in CRC is unknown. The results of this study show that NOB1 is required for the survival of CRC cells. siRNA knockdown of NOB1 induced significant cell apoptosis in both in vitro and in vivo model systems. Microarray analysis showed that the WNT, cell proliferation, apoptosis, fibroblast growth factor, and angiogenesis signaling cascades contribute to NOB1 function.
Applications
Given the critical role of NOB1 in CRC, NOB1 may be a novel therapeutic target in the treatment of CRC patients.
Peer review
The role of human NOB1 gene is largely unknown. The authors provide solid evidence to show the importance of NOB1 in the survival of colorectal cancer cells. Inhibition of the NOB1 function by siRNA greatly induces cell apoptosis both in vitro and in vivo. NOB1 seems to be an important and poorly investigated molecular target in colorectal cancer.
Footnotes
Supported by National Natural Science Foundation of China, No. 81272735; Class A Medical and Health Technology Program Project from Zhejiang Province, No. 2010KY178; and the Science and Technology Department of Hunan Province, No. 2010Ck3013.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 5, 2014
First decision: March 27, 2014
Article in press: July 22, 2014
P- Reviewer: Chae SC S- Editor: Ding Y L- Editor: Webster JR E- Editor: Zhang DN
References
- 1.Watson AJ. An overview of apoptosis and the prevention of colorectal cancer. Crit Rev Oncol Hematol. 2006;57:107–121. doi: 10.1016/j.critrevonc.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101–128. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 3.Yang SY, Sales KM, Fuller B, Seifalian AM, Winslet MC. Apoptosis and colorectal cancer: implications for therapy. Trends Mol Med. 2009;15:225–233. doi: 10.1016/j.molmed.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Adams J. Potential for proteasome inhibition in the treatment of cancer. Drug Discov Today. 2003;8:307–315. doi: 10.1016/s1359-6446(03)02647-3. [DOI] [PubMed] [Google Scholar]
- 5.Voorhees PM, Dees EC, O’Neil B, Orlowski RZ. The proteasome as a target for cancer therapy. Clin Cancer Res. 2003;9:6316–6325. [PubMed] [Google Scholar]
- 6.Baumeister W, Walz J, Zühl F, Seemüller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 7.Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- 8.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 9.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 10.Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J Biosci. 2006;31:137–155. doi: 10.1007/BF02705243. [DOI] [PubMed] [Google Scholar]
- 11.Orlowski RZ, Dees EC. The role of the ubiquitination-proteasome pathway in breast cancer: applying drugs that affect the ubiquitin-proteasome pathway to the therapy of breast cancer. Breast Cancer Res. 2003;5:1–7. doi: 10.1186/bcr460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandez-Verdun D, Roussel P. Regulators of nucleolar functions. Prog Cell Cycle Res. 2003;5:301–308. [PubMed] [Google Scholar]
- 13.Thomas G. An encore for ribosome biogenesis in the control of cell proliferation. Nat Cell Biol. 2000;2:E71–E72. doi: 10.1038/35010581. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt EV. The role of c-myc in cellular growth control. Oncogene. 1999;18:2988–2996. doi: 10.1038/sj.onc.1202751. [DOI] [PubMed] [Google Scholar]
- 15.Belin S, Beghin A, Solano-Gonzàlez E, Bezin L, Brunet-Manquat S, Textoris J, Prats AC, Mertani HC, Dumontet C, Diaz JJ. Dysregulation of ribosome biogenesis and translational capacity is associated with tumor progression of human breast cancer cells. PLoS One. 2009;4:e7147. doi: 10.1371/journal.pone.0007147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drygin D, Siddiqui-Jain A, O’Brien S, Schwaebe M, Lin A, Bliesath J, Ho CB, Proffitt C, Trent K, Whitten JP, et al. Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 2009;69:7653–7661. doi: 10.1158/0008-5472.CAN-09-1304. [DOI] [PubMed] [Google Scholar]
- 17.Montanaro L, Treré D, Derenzini M. Nucleolus, ribosomes, and cancer. Am J Pathol. 2008;173:301–310. doi: 10.2353/ajpath.2008.070752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Ni J, Zhou G, Yuan J, Ren W, Shan Y, Tang W, Yu L, Zhao S. Cloning, expression and characterization of the human NOB1 gene. Mol Biol Rep. 2005;32:185–189. doi: 10.1007/s11033-005-3141-7. [DOI] [PubMed] [Google Scholar]
- 19.Fatica A, Oeffinger M, Dlakić M, Tollervey D. Nob1p is required for cleavage of the 3' end of 18S rRNA. Mol Cell Biol. 2003;23:1798–1807. doi: 10.1128/MCB.23.5.1798-1807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fatica A, Tollervey D, Dlakić M. PIN domain of Nob1p is required for D-site cleavage in 20S pre-rRNA. RNA. 2004;10:1698–1701. doi: 10.1261/rna.7123504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamanna AC, Karbstein K. Nob1 binds the single-stranded cleavage site D at the 3’-end of 18S rRNA with its PIN domain. Proc Natl Acad Sci USA. 2009;106:14259–14264. doi: 10.1073/pnas.0905403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Granneman S, Petfalski E, Swiatkowska A, Tollervey D. Cracking pre-40S ribosomal subunit structure by systematic analyses of RNA-protein cross-linking. EMBO J. 2010;29:2026–2036. doi: 10.1038/emboj.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tone Y, Tanahashi N, Tanaka K, Fujimuro M, Yokosawa H, Toh-e A. Nob1p, a new essential protein, associates with the 26S proteasome of growing saccharomyces cerevisiae cells. Gene. 2000;243:37–45. doi: 10.1016/s0378-1119(99)00566-1. [DOI] [PubMed] [Google Scholar]
- 24.Tone Y, Toh-E A. Nob1p is required for biogenesis of the 26S proteasome and degraded upon its maturation in Saccharomyces cerevisiae. Genes Dev. 2002;16:3142–3157. doi: 10.1101/gad.1025602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin Y, Peng S, Yu H, Teng H, Cui M. RNAi-mediated downregulation of NOB1 suppresses the growth and colony-formation ability of human ovarian cancer cells. Med Oncol. 2012;29:311–317. doi: 10.1007/s12032-010-9808-5. [DOI] [PubMed] [Google Scholar]
- 26.Wu DP, He XW. [Expression of NOB1 and its significance in colorectal cancer] Nanfang Yike Daxue Xuebao. 2012;32:420–422. [PubMed] [Google Scholar]
- 27.Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Nunez R. DNA measurement and cell cycle analysis by flow cytometry. Curr Issues Mol Biol. 2001;3:67–70. [PubMed] [Google Scholar]
- 30.Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 31.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1:19–30. doi: 10.1016/s1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 33.Goldberg AL, Stein R, Adams J. New insights into proteasome function: from archaebacteria to drug development. Chem Biol. 1995;2:503–508. doi: 10.1016/1074-5521(95)90182-5. [DOI] [PubMed] [Google Scholar]
- 34.Wilson KS, Noller HF. Molecular movement inside the translational engine. Cell. 1998;92:337–349. doi: 10.1016/s0092-8674(00)80927-7. [DOI] [PubMed] [Google Scholar]
- 35.Wertz IE, Hanley MR. Diverse molecular provocation of programmed cell death. Trends Biochem Sci. 1996;21:359–364. [PubMed] [Google Scholar]
- 36.Nadano D, Sato TA. Caspase-3-dependent and -independent degradation of 28 S ribosomal RNA may be involved in the inhibition of protein synthesis during apoptosis initiated by death receptor engagement. J Biol Chem. 2000;275:13967–13973. doi: 10.1074/jbc.275.18.13967. [DOI] [PubMed] [Google Scholar]
- 37.Almond JB, Cohen GM. The proteasome: a novel target for cancer chemotherapy. Leukemia. 2002;16:433–443. doi: 10.1038/sj.leu.2402417. [DOI] [PubMed] [Google Scholar]