

Abstract
Purpose.
To investigate whether activation of Toll-like receptor 3 (TLR3) promotes the degeneration of retinal ganglion cells (RGCs) by upregulating the protein levels of c-jun N-terminal kinase 3 (JNK3).
Methods.
Toll-like receptor 3-specific activator, Poly(I:C) (polyinosinic-polycytidylic acid), or PBS was injected into the vitreous humor of Thy1-YFP mice. At 24, 48, and 72 hours after treatments, degeneration of RGCs was assessed by using antibodies against brain-specific homeobox/POU domain protein 3a (Brn3a). A TLR3-specific inhibitor was injected into the vitreous humor with or without Poly(I:C). Western blot assays were performed to determine relative levels of TLR3, JNK3, pJNK3, and sterile alpha and HEAT/Armadillo motif-containing 1 (SARM1) proteins in retinal protein extracts, and immunohistochemistry assays were performed to determine their cellular localization in the retina. Mouse eyes were treated with Poly(I:C) or PBS along with MitoTracker Red, and colocalization of MitoTracker Red and JNK3 in the retinas was determined by using antibodies against JNK3.
Results.
Poly(I:C) activated TLR3 and upregulated its downstream target protein JNK3 but not SARM1 in the retina. Poly(I:C) activated TLR3 and upregulated JNK3 specifically in RGCs and promoted a significant degeneration of RGCs over a 72-hour time period. Toll-like receptor 3 upregulated the levels of JNK3 protein in the cytoplasm of RGCs, but not in the mitochondria. Toll-like receptor 3-specific inhibitor downregulated Poly(I:C)-mediated upregulation of JNK3 protein, and, in turn, significantly attenuated TLR3-induced degeneration of RGCs.
Conclusions.
Results presented in this study show that the activation of TLR3 alone promotes the degeneration of RGCs by upregulating the protein levels of JNK3.
Keywords: Toll-like receptor 3, SARM1, JNK3, RGCs
The mechanisms underlying oxidative stress-mediated degeneration of RGCs during glaucoma progression are unclear. This study provides evidence that activation of Toll-like receptor 3 (TLR3) alone promotes the degeneration of RGCs by upregulating the protein levels of JNK3.
Introduction
Toll-like receptors (TLRs) are traditionally known to participate in mammalian innate immune response.1–3 To date, 13 TLRs have been identified.2 These receptors consist of extracellular leucine-rich repeats (LRRs) and cytoplasmic Toll-interleukin 1 receptor (TIR) domains.4 Recent studies have indicated that among the known TLRs, TLR3 plays a significant role in oxidative stress-mediated neuronal damage,2,3,5–8 but it is unclear whether TLR3 plays a role in the degeneration of RGCs.
Toll-like receptors utilize cytoplasmic TIR-containing adaptor molecules, called MyD88 (myeloid differentiation response gene 88), to mediate intracellular signals.1 To date, five members of the MyD88 adaptor protein family have been identified. MyD88 is the first member of the cytosolic adaptor protein family identified that mediates intracellular signaling through a majority of TLRs.9,10 Toll-like receptor 2 mediates intracellular signaling through MyD88-2 and MyD88-3,11–16 and TLR4 mediates intracellular signaling through MyD88-4.17–19 Finally, the role of the fifth member of this family, MyD88-5, also known as sterile alpha and HEAT/Armadillo motif-containing 1 (SARM1), which is conserved to a high degree in flies, worms, and mammals, has remained a mystery for the past few years.20–23 Previous studies have suggested that SARM1 acts as an intracellular adaptor protein for TLR3, and SARM1 promotes stress (oxygen and glucose deprivation)-induced death of cultured HEK-293T and COS-1 cells by translocating c-jun N-terminal kinase 3 (JNK3) into the mitochondria because SARM1 contains a mitochondrial targeting sequence.24,25
Although degeneration of RGCs is a major concern in a number of retinal degenerative conditions including primary open-angle glaucoma (POAG), which affects 60 million people worldwide,26,27 the molecular mechanisms underlying the degeneration of RGCs under glaucomatous conditions are still unclear. Moreover, until now, a role for TLR3 in the degeneration of RGCs in glaucoma has not been investigated. For that matter, the role of TLR3 in intraocular pressure (IOP)-mediated degeneration of RGCs has not been investigated either in human subjects or in animal models related to POAG. Since IOP-mediated tissue stress, including oxidative stress,28,29 may activate TLRs, Tezel's group30 has determined protein levels of TLRs in human POAG donor eyes and found by proteomic analysis that the protein levels of TLR2, -4, -7, -8, and -10, but not TLR3, were upregulated in glaucomatous retinas.30 Immunolocalization studies from the same group reported that TLR2, TLR3, and TLR4 proteins were associated with the astrocytes isolated from glaucomatous retinas.30 However, how TLR3 protein synthesized by astrocytes promotes the degeneration of RGCs under IOP-mediated glaucomatous stress conditions remains unclear.
In addition to TLR3, its downstream target protein JNK3 promotes stress-induced apoptosis of neuronal cells.31 The JNKs, composed of JNK1, JNK2, and JNK3, are a family of serine/threonine protein kinases that become activated by a variety of stimuli; JNK1 and 2 proteins are widely observed, but JNK3 protein is limited to neuronal tissues, testes, and myocytes.32 JNKs regulate the survival of neuronal cells by phosphorylating N-terminal transactivation domain of the c-Jun transcriptional factor.33–39 With respect to POAG, JNK pathway is reported to be activated by many stimuli, including elevated IOP.40–42 JNKs regulate stress-induced neuronal degeneration by phosphorylating JUN, a transcription factor that activates a number of downstream genes involved in neurodegeneration,43,44 and phosphorylated JUN has been reported in RGCs of POAG patients.39,45,46 In addition, earlier studies have reported that phospho-JNK3 (pJNK3) levels were increased in retinas obtained from human glaucoma donor eyes39 and in retinas from rats in which glaucomatous damage was induced experimentally.40,45,47 Although a number of previous studies have reported a detrimental role for JNK signaling in RGC death,48–51 the role of a specific isoform of JNK in IOP-mediated degeneration of RGCs is unclear. Libby's group52 previously reported that activation of JNK2 and JNK3 plays a major role in degeneration of RGCs and their axons. In contrast, Quigley's group53 recently reported that the deletion of Jnk3 does not prevent IOP-induced degeneration of RGCs. Thus, the role of JNK3 in glaucoma pathology is contradictory, and it remains to be determined whether JNK3 plays a significant role in the degeneration of RGCs under glaucomatous conditions.
Based on the above uncertainties regarding the role of TLR3, JNK3, and SARM1 proteins in degeneration of RGCs, in this study, TLR3 was activated in mouse retinas by using a TLR3-specific agonist, Poly(I:C) (polyinosinic-polycytidylic acid),54–56 and experiments were performed to investigate whether TLR3 activation alone promotes the degeneration of RGCs, and, if so, whether TLR3-mediated degeneration of RGCs is mediated by JNK3 and SARM1.
Materials and Methods
Materials
Poly(I:C) was obtained from InvivoGen (San Diego, CA, USA). TLR3/dsRNA complex inhibitor,57 (R)-2-(3-chloro-6-fluorobenzo[b]thiophene-2-carboxamido)-3-phenylpropanoic acid, was obtained from EMD Millipore (Billerica, MA, USA). Toll-like receptor 3 and JNK3 antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA) and Promega (Madison, WI, USA), respectively. SARM1 antibodies were obtained either from Origene (Rockville, MD, USA; for Western blot analysis) or from GenWay Biotech, Inc. (San Diego, CA, USA; for immunohistochemistry). Antibodies against Tuj1 (neuronal class III beta-tubulin) were obtained from Covance (Princeton, NJ, USA), and antibodies against brain-specific homeobox/POU domain protein 3a (Brn3a) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). For immunohistochemical assays, appropriate secondary antibodies conjugated to Alexa Fluor 488 (blue), Alexa Fluor 568 (red), Alexa Fluor 647 (magenta), and MitoTracker Red CMXRos were obtained from Invitrogen (Carlsbad, CA, USA). For Western blot analysis, secondary antibodies conjugated to IRDye 800 or 680 were obtained from LI-COR (Lincoln, NE, USA).
Animals
In this study, all in vivo experiments were performed employing mouse line B6.Cg-Tg (Thy-1-YFPH) 2Jrs/J, in which a subset of retinal ganglion cells (RGCs) express a yellow-fluorescent protein (YFP) under the control of Thy-1 promoter (Jackson Laboratory, Bar Harbor, ME, USA).
Intravitreal Injections
All experiments on mice were performed under general anesthesia, according to Oakland University's Institutional Animal Care and Usage Committee (IACUC) guidelines and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Adult Thy1-YFP mice (8–10 weeks old) were anesthetized by an intraperitoneal injection of ketamine (50 mg/kg body weight) and xylazine (8 mg/kg body weight). After instilling a drop of the topical anesthetic agent proparacaine, Poly(I:C) (2 μg/2 μL volume),58 PBS, or TLR3 inhibitor (5 and 10 μM) was injected into the vitreous humor of right eyes (2 μL volume) of each mouse using a NanoFil syringe equipped with a 36-gauge beveled needle (World Precision Instruments, Sarasota, FL, USA).59 For some experiments, 24 hours after Poly(I:C) or PBS treatment, eyes were treated with an intravitreal injection of MitoTracker Red (500 nM).
Protein Extraction
At 24, 48, and 72 hours after intravitreal injections, mice were euthanized with an overdose of carbon dioxide, and their eyes were enucleated. Retinas were removed carefully and washed three times with PBS. Three or four retinas each were placed in Eppendorf tubes containing 40 μL extraction buffer (1% Nonidet-P40, 20 mM Tris-HCl, 150 mM NaCl, 1 mM Na3VO4, pH 7.4), and the tissues were homogenized. Three separate experiments were performed for each time point to determine the reproducibility of the results. Retinal tissue homogenates were centrifuged at 7840 g for 5 minutes at 4°C and the supernatants were collected. Protein concentration in the supernatants was determined by using Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA).
Western Blot Analysis
Aliquots containing an equal amount of retinal proteins (50 μg) extracted from PBS- or Poly(I:C)-treated eyes were mixed with gel loading buffer and separated by using 10% SDS-PAGE. After electrophoresis, the proteins were transferred onto Immobilon-FL membranes (EMD Millipore, Billerica, MA, USA) and nonspecific binding was blocked with Odyssey blocking buffer containing 0.2% Tween 20 (TBS-T). After incubating with primary antibodies against TLR3, JNK3, pJNK3, SARM1, or actin, membranes were washed with TBS-T and incubated with appropriate secondary antibodies conjugated to IRDye 800 or 680 for 1 hour at room temperature. Finally, the membranes were scanned using the Odyssey two-channel IR-detection scanner (LI-COR, Lincoln, NE, USA).
Immunohistochemistry
After intravitreal injection of PBS or Poly(I:C), eyes were enucleated and fixed in 4% paraformaldehyde for 1 hour at room temperature. Retinas were isolated and processed as whole mounts or embedded in optimum cutting temperature (OCT) compound, and 10-μm-thick radial sections were obtained by using a cryostat according to general methods published from our laboratory.59 Retinal cross sections and whole retinas were then subjected to immunohistochemistry by using antibodies against TLR3, JNK3, and SARM1 with or without Tuj1. Whole retinas and retinal cross sections were observed under a Zeiss Imager Z.2 microscope (Zeiss, Thornwood, NY, USA).
Quantitation of RGCs in the Retina
The number of Brn3a-positive RGCs was assessed by observing whole retinas under the Zeiss Imager Z.2 microscope equipped with epifluorescence as described previously.59 For each retina, Brn3a-positive RGCs in six to eight areas of equal size (450 × 320 μm; ×20 magnification), located at equal distance from the optic disc (850 μm), were photographed using a Zeiss digital camera (Fig. 1A). After digitized images were obtained using the Zeiss camera, images were compiled by using Adobe Photoshop Software 7.0 (Adobe Systems, Inc., San Jose, CA, USA). The number of Brn3a-positive cells in the retinas was quantitated using Nikon Elements AR software (Nikon Instruments, Inc., Melville, NY, USA). Statistical significance was analyzed using a nonparametric Newman-Keuls analog procedure (GB-Stat Software; Dynamic Microsystems, Silver Spring, MD, USA) and expressed as the mean ± SEM.
Figure 1.

Degeneration of RGCs in Poly(I:C)-treated eyes. (A) For each retina, Brn3a-positive RGCs in six to eight areas of equal size (450 × 320 μm; ×20 magnification), located at equal distance from the optic disc (850 μm), were photographed by using a Zeiss digital camera. (B) After obtaining digitized images using a Zeiss camera, images were compiled using Adobe Photoshop Software 7.0. Whole retinas isolated from PBS- or Poly(I:C)-treated eyes were immunostained with Brn3a antibodies. (C) Bar graph shows quantification of the loss of Brn3a-positive RGCs in the retinas isolated from eyes treated with or without Poly(I:C). Images were obtained at ×20 magnification. Scale bar: 50 μm. *P < 0.05 when Brn3a-positive RGCs in the retinas isolated from Poly(I:C)-treated eyes were compared with Brn3a-positive RGCs in the retinas isolated from PBS-treated eyes.
Results
Poly(I:C) Promotes the Degeneration of RGCs
To determine whether activation of TLR3 promotes the degeneration of RGCs, mouse eyes were treated with intravitreal injections of a TLR3-specific agonist, Poly(I:C), or PBS. At 24, 48, and 72 hours after the treatments, retinas were isolated and immunostained with antibodies against Brn3a, a marker for RGCs. Data presented in Figure 1B indicate that Poly(I:C) promoted a progressive degeneration of Brn3a-positive RGCs over a 72-hour time period. Data presented in Figure 1C indicate that Poly(I:C) promoted a significant degeneration of RGCs within the 72-hour time period tested (P < 0.05).
Poly(I:C) Upregulates the Protein Levels of TLR3 and JNK3, but Not SARM1, in the Retina
To determine whether Poly(I:C) promoted the degeneration of RGCs by activating TLR3, JNK3, or SARM1, mouse eyes were treated with intravitreal injections of PBS or Poly(I:C), and Western blot analysis was performed on retinal proteins extracted at 24, 48, and 72 hours after the treatments. Results presented in Figures 2A and 2B indicate that when compared with low protein levels of TLR3 and JNK3 in retinal proteins extracted from PBS-treated eyes, protein levels of both TLR3 and JNK3 were upregulated significantly (P < 0.05) in Poly(I:C)-treated eyes during the 72-hour time period tested. To determine whether TLR3 promotes the degeneration of RGCs by upregulating the protein levels of SARM1, retinal proteins extracted from PBS- or Poly(I:C)-treated eyes were subjected to Western blot analysis by using antibodies against SARM1. Results presented in Figures 2A and 2B indicate that when compared with low levels of SARM1 protein in retinal proteins extracted from PBS-treated eyes, SARM1 protein levels were not increased in retinal proteins extracted from Poly(I:C)-treated eyes.
Figure 2.

Relative levels of TLR3, JNK3, and SARM1 proteins in the retina. (A) Aliquots containing an equal amount of retinal proteins (50 μg) from PBS- or Poly(I:C)-treated eyes were subjected to Western blot analysis using antibodies against TLR3, JNK3, and SARM1. Actin Western bands indicate loading controls for the total proteins. (B) Bar graph shows relative levels of TLR3, JNK3, and SARM1 proteins. *P < 0.05, when TLR3 and JNK3 protein levels in retinal proteins extracted from Poly(I:C)-treated eyes were compared with PBS-treated eyes. SARM1 levels were not increased significantly (NS).
Poly(I:C) Upregulates the Protein Levels of TLR3 and JNK3 in RGCs
To investigate whether activation of TLR3 and upregulation of JNK3 protein promote the degeneration of RGCs in an autocrine fashion or a paracrine fashion through the involvement of other cell types in the ganglion cell layer (GCL), retinas isolated at 24 and 48 hours after Poly(I:C) or PBS treatment were immunostained with antibodies against TLR3 and JNK3. Immunohisotchemical analysis on whole retinas indicated that Poly(I:C) upregulated protein levels of TLR3 (Fig. 3, top) and JNK3 (Fig. 3, bottom) in RGCs, but not in other cell types such as amacrine cells that are located in deeper layers of the retina. Additional immunohistochemical experiments performed on whole retinas indicated that TLR3 (Fig. 4A) and JNK3 protein levels (Fig. 5A) were upregulated in Tuj1-positive RGCs. Experiments performed on retinal cross sections also indicated that TLR3 (Fig. 4B) and JNK3 proteins (Fig. 5B) were localized specifically in RGCs. Although low SARM1 protein levels were localized in RGCs in the retinas isolated from PBS-treated eyes (Fig. 6), consistent with our previously published results,60 Poly(I:C) failed to upregulate the levels of SARM1 protein in RGCs.
Figure 3.

Upregulation of TLR3 and JNK3 proteins in RGCs. Whole retinas isolated from PBS- or Poly(I:C)-treated eyes (at 24 and 48 hours) were immunostained with antibodies against TLR3 and JNK3. Images were obtained at ×20 magnification. Scale bar: 25 μm. Results presented show that the protein levels of TLR3 (top) and JNK3 (bottom) were increased in RGCs in the retinas isolated from Poly(I:C)-treated eyes when compared with the protein levels of TLR3 and JNK3 in RGCs in the retinas isolated from PBS-treated eyes.
Figure 4.

Upregulation of TLR3 protein in Tuj1-positive RGCs. (A) Whole retinas isolated from PBS- or Poly(I:C)-treated eyes (at 48 hours) were immunostained with antibodies against TLR3 and Tuj1. Images were obtained at ×20 magnification. Scale bars: 50 μm. Results presented indicate that Poly(I:C) upregulated the protein levels of TLR3 in Tuj1-positive RGCs. (B) Localization of TLR3 protein in RGCs. Retinal cross section prepared from Poly(I:C)- or PBS-treated eyes (at 48 hours) were immunostained by using antibodies against TLR3. Retinal cross sections were stained with 4′,6-diamidino-2-phenylindole (DAPI) to identify the nuclei. Representative immunohistochemistry results indicate that Poly(I:C) upregulated JNK3 protein in RGCs (white arrows).
Figure 5.

Upregulation of JNK3 protein in Tuj1-positive RGCs. (A) Whole retinas isolated from PBS- or Poly(I:C)-treated eyes (at 48 hours) were immunostained with antibodies against JNK3 and Tuj1. Images were obtained at ×20 magnification. Scale bars: 50 μm. Results presented indicate that Poly(I:C)-mediated upregulation of JNK3 was localized in Tuj1-positive RGCs, but not in other cell types. (B) Localization of JNK3 protein in RGCs. Retinal cross sections prepared from Poly(I:C)- or PBS-treated eyes (at 48 hours) were immunostained by using antibodies against JNK3. Retinal cross sections were stained with DAPI to identify the nuclei. Representative immunohistochemistry results indicate that Poly(I:C)-mediated upregulation of JNK3 protein is localized in RGCs (white arrows). Images were obtained at ×40 magnification.
Figure 6.
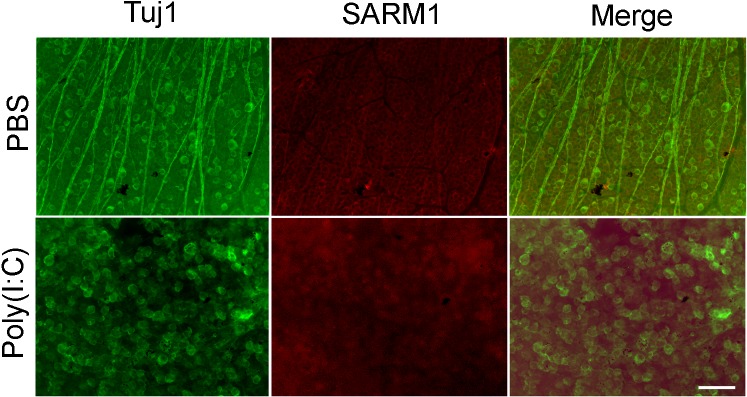
Localization of SARM1 protein in the retina. Whole retinas isolated from PBS- or Poly(I:C)-treated eyes (at 48 hours) were immunostained with antibodies against SARM1 and Tuj1. Images were obtained at ×20 magnification. Scale bar: 50 μm. Results presented indicate that Poly(I:C) failed to upregulate the protein levels of SARM1 in Tuj1-positive RGCs.
Upregulated JNK3 Protein Is Localized in the Cytoplasm, but Not in the Mitochondria
To determine whether Poly(I:C) promotes the degeneration of RGCs by translocating JNK3 into the mitochondria, mouse eyes were treated with PBS or Poly(I:C) for 24 hours. Twenty-four hours after Poly(I:C) treatment, mouse eyes were treated again with an intravitreal injection of MitoTracker Red. Twenty-four hours after MitoTracker Red treatment, retinas were isolated and immunostained with antibodies against JNK3. Results presented in Figure 7 indicate that MitoTracker Red labeled mitochondria in RGCs, as well as in cells in deeper layers of the retinas isolated from PBS-treated eyes, which was expected. Very low JNK3 protein levels were localized in RGCs in the retinas isolated from PBS-treated eyes, but JNK3 protein was not associated with the mitochondria (positive for MitoTracker Red). In addition, even after Poly(I:C) treatment, increased levels of JNK3 protein were not colocalized with the mitochondria in RGCs.
Figure 7.

Localization of JNK3 protein in the cytoplasm of RGCs. Twenty-four hours after intravitreal injection of PBS or Poly(I:C), eyes were treated with an intravitreal injection of MitoTracker Red. Twenty-four hours after MitoTracker Red treatment, retinas isolated from PBS- or Poly(I:C)-treated eyes were immunostained with antibodies against JNK3 and flat mounted. Results presented indicate that MitoTracker Red labeled the mitochondria in RGCs and in cells in the deeper layers of the retina in both PBS- and Poly(I:C)-treated eyes. In PBS-treated eyes, low levels of JNK3 protein were localized in RGCs, but JNK3 protein was not colocalized with MitoTracker Red. In addition, after Poly(I:C) treatment, JNK3 protein levels were upregulated in RGCs, but elevated levels of JNK3 protein was not colocalized with MitoTracker Red. Images were obtained at ×40 magnification. Scale bar: 25 μm.
TLR3 Inhibitor Downregulates JNK3 and pJNK3 Protein Levels in the Retina
Since Poly(I:C) promoted the degeneration of RGCs by upregulating the protein levels of JNK3, to determine whether inhibition of TLR3 activation leads to the downregulation of JNK3 protein, mouse eyes were treated with PBS or Poly(I:C) along with a TLR3-specific inhibitor (5 and 10 μM) for 48 hours, and Western blot analysis was performed on proteins extracted from the retinas. Results presented in Figures 8A and 8B indicate that very low levels of JNK3 and pJNK3 proteins were observed in retinal proteins extracted from PBS-treated eyes, and their protein levels remained unchanged in retinal proteins extracted after treatment of the eyes with PBS along with 5 and 10 μM TLR3 inhibitor (Figs. 8A, 8B). In contrast, JNK3 and pJNK3 protein levels elevated in retinal proteins extracted from Poly(I:C)-treated eyes (Fig. 8C) were downregulated significantly in retinal proteins extracted from mouse eyes treated with Poly(I:C) along with 10 μM TLR3 inhibitor (Fig. 8D) and to a lesser extent in retinal proteins extracted from eyes treated with Poly(I:C) along with 5 μM TLR3 inhibitor.
Figure 8.

Effect of TLR3 inhibitor on JNK3 and pJNK3 protein levels in the retina. Forty-eight hours after mouse eyes were treated with PBS or Poly(I:C) with or without TLR3 inhibitor, total proteins were isolated from the retinas, and aliquots containing an equal amount of retinal proteins (50 μg) were subjected to Western blot analysis using antibodies against JNK3 and pJNK3. Actin Western bands indicate loading controls of total proteins. Bar graphs indicate semiquantitative analysis of JNK3 and pJNK3 protein from the Western blots. Results presented indicate that TLR3 inhibitor did not affect low levels of JNK3 and pJNK3 proteins observed in PBS-treated eyes (A, B). In contrast, Poly(I:C)-mediated upregulation of JNK3 and pJNK3 proteins (C, D) was reduced in eyes treated with Poly(I:C) along with 10 μM concentration of TLR3 inhibitor, but not with 5 μM. *P < 0.05 when JNK3 and pJNK3 protein levels were compared with PBS-treated eyes. **P < 0.05 when JNK3 and pJNK3 protein levels were compared with Poly(I:C)-treated eyes. NS, not significant.
TLR3 Inhibitor Attenuates Poly(I:C)-Induced Degeneration of RGCs
Since TLR3 inhibitor downregulated the protein levels of JNK3, additional experiments were performed to investigate whether decreased levels of JNK3 protein attenuate Poly(I:C)-induced and TLR3-mediated degeneration of RGCs. Mouse eyes were treated with PBS or Poly(I:C) with or without TLR3 inhibitor (5 and 10 μM) for 48 hours, and immunohistochemical analysis was performed on whole retinas by using antibodies against Brn3a. Results presented in Figure 9A indicate that the density of Brn3a-positive RGCs remained unchanged in the retinas isolated from PBS- or PBS- and TLR3 inhibitor-treated eyes. In contrast, Poly(I:C)-induced degeneration of Brn3a-positive RGCs was attenuated significantly (P < 0.05) in the retinas isolated from eyes treated with TLR3 inhibitor (Fig. 9B).
Figure 9.
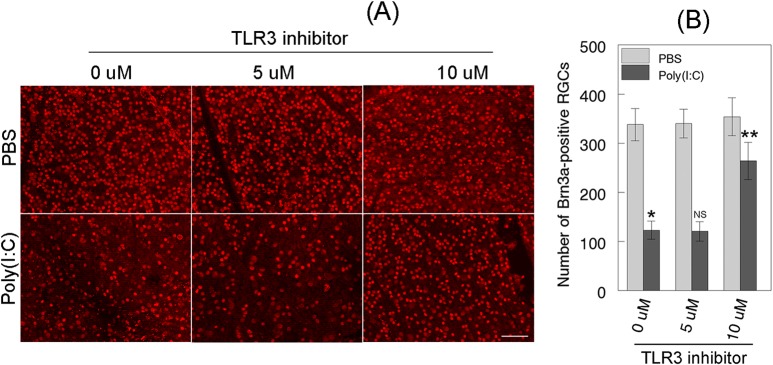
Effect of TLR3 inhibitor on degeneration of RGCs. (A) Forty-eight hours after mouse eyes were treated with PBS or Poly(I:C), with or without TLR3 inhibitor, retinas were isolated and immunostained with Brn3a antibodies. Images were obtained at ×20 magnification. Scale bar: 50 μm. (B) Bar graph indicates a significant reduction (*P < 0.05) in the number of Brn3a-positive RGCs in Poly(I:C)-treated eyes when compared to PBS-treated eyes. Bar graph also indicates that TLR3 inhibitor attenuated Poly(I:C)-induced degeneration of Brn3a-positive RGCs significantly (**P < 0.05). NS, not significant.
Discussion
Primary open-angle glaucoma is a degenerative disease in which irreversible loss of RGCs leads to vision loss.26,27 Despite the fact that elevated IOP has been identified as a major risk factor for the degeneration of RGCs, the mechanisms underpinning the degeneration of RGCs during the progression of POAG are unclear. A few studies have reported that elevated IOP promotes the degeneration of RGCs through oxidative stress.28,29,61–63 But the nature of the oxidative stress and the mediators involved in oxidative stress-induced degeneration of RGCs are unclear.
Previous studies have reported that TLRs may play a role in the degeneration of RGCs in POAG through glial cell-mediated neuroinflammation or IOP-mediated oxidative stress.28,29,64–68 Although the mechanism underlying neuroinflammation-mediated degeneration of RGCs is unclear, a recent study reported that TLR3 is predominantly expressed in astrocytes and that TLR2, -3, and -4 are expressed in microglia in retinas obtained from human donor eyes affected with POAG.30 However, previous studies have reported that glial cells synthesize TLR2 and TLR4 proteins, but not TLR3, which recognizes double-stranded RNA (dsRNA) compared to other TLRs, which recognize single-stranded RNA (ssRNA).69–71 In addition, TLR3 adaptor protein SARM1 is predominantly expressed in neurons, but not in astrocytes or microglia.69–71 Furthermore, even if TLR3 protein is synthesized by astrocytes in the retina, the connection between TLR3 protein in astrocytes, as well as how astrocyte-associated TLR3 protein promotes the degeneration of RGCs under glaucomatous conditions, is unclear. Therefore, to determine whether activation of TLR3 alone induces the degeneration of RGCs by upregulating JNK3 and SARM1 proteins, this study was conducted by activating TLR3 in mouse retinas using a TLR3-specific agonist, Poly(I:C). Results presented in this study show that activation of TLR3 alone upregulates the protein levels of JNK3 but not SARM1 in RGCs and promotes their progressive degeneration over time. In addition, the results show that inhibition of TLR3 activation downregulates the protein levels of JNK3 and attenuates Poly(I:C)-induced degeneration of RGCs. The results presented in this study are significant for several reasons.
First, TLRs recognize molecular patterns of pathogens, for example, bacterial lipopolysaccharide (LPS) and viral double- or single-stranded RNA.72,73 Although microglia are thought to be the primary immune cells that express TLRs in the central nervous system, recent reports indicated that neurons also express the key proteins of the innate immune response including TLRs and their adaptor proteins, such as SARM1, MyD88, and TRIF.69,74 In addition, TLRs have been traditionally known to play important roles during immunological challenge, but recent studies indicate that neuronal TLRs can detect damaging signals and modulate neuronal morphology and function even in the absence of an immunological challenge.4,75 A recent study reported further that subretinal injection of Poly(I:C) activated TLR3 and promoted the degeneration of photoreceptors.58 In concert with these studies, results presented in this study show that activation of TLR3 promotes the degeneration of RGCs by upregulating the protein levels of JNK3.
Second, although a previous study reported that the deletion of Jnk3 did not prevent the death of RGCs under elevated IOP conditions in mice,53 results presented in this study show that activation of TLR3 upregulates the protein levels of JNK3 and pJNK3, and, in turn, promotes the degeneration of RGCs. In addition, results presented in this study show that inhibition of TLR3-mediated upregulation of JNK3 protein attenuates the degeneration of RGCs, indicating that JNK3 does play a causal role in TLR3-mediated degeneration of RGCs. Although the reason(s) for the lack of neuroprotection observed against IOP-mediated degeneration of RGCs in Jnk3-deficient mice is unclear,53 results presented here indicate that JNK3, but not SARM1, was upregulated upon TLR3 activation. Thus, unlike Poly(I:C), which upregulates the protein levels of JNK3 through TLR3, we propose that under glaucomatous conditions, elevated IOP upregulates the protein levels of both JNK3 and SARM1 by two different mechanisms. And even if Jnk3 is deleted, elevated levels of SARM1 alone can promote the degeneration of RGCs in the absence of JNK3, as we have reported previously that upregulation of SARM1 promoted the degeneration of RGCs.60
Third, previous studies using HEK-293T and COS-1 cells indicated that upregulation of SARM1 promoted cell death through SARM1-mediated mitochondrial translocation of JNK3.71,76,77 However, unlike these studies performed on nonneuronal cells, results presented in this study on the retina did not show any evidence that upregulation of JNK3 protein is localized in the mitochondria of RGCs. We believe that JNK3 is not localized in the mitochondria because SARM1, which contains a mitochondrial targeting sequence76 and which mediates the translocation of JNK3 into the mitochondria, was not upregulated in RGCs upon TLR3 activation. Thus, the results presented in this study suggest that JNK3 promotes the degeneration of neuronal cells, in this case RGCs, regardless of whether upregulated levels of JNK3 are localized in the cytoplasm or in the mitochondria. Finally, the results presented in this study do not seem to be specific to Thy1-YFP mice because our unpublished data indicate that Poly(I:C) upregulates the protein levels of TLR3 and JNK3 even in C57BL/6 mice. The only reason for using Thy1-YFP mouse strain is that we routinely use these mice for various other studies and have experience in handling them.
In summary, results presented in this study, for the first time, indicate that as long as TLR3 is activated in RGCs either by oxidative stress or by agents such as Poly(I:C), TLR3 upregulates the protein levels of its downstream target JNK3, and that JNK3, in turn, promotes the degeneration of RGCs. Future studies are warranted to investigate the role of TLR3, JNK3, and SARM1 in the degeneration of RGCs by using appropriate animal models in which IOP can be chronically elevated.
Acknowledgments
Supported by National Eye Institute Project Grant EY017853-01A2 and a grant from Center for Biomedical Research of Oakland University (SKC).
Disclosure: S.K. Chintala, None; N. Putris, None; M. Geno, None
References
- 1. O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007; 7: 353–364. [DOI] [PubMed] [Google Scholar]
- 2. Carty M, Bowie AG. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol. 2011; 81: 825–837. [DOI] [PubMed] [Google Scholar]
- 3. Kenny EF, O'Neill LA. Signalling adaptors used by Toll-like receptors: an update. Cytokine. 2008; 43: 342–349. [DOI] [PubMed] [Google Scholar]
- 4. Liu HY, Chen CY, Hsueh YP. Innate immune responses regulate morphogenesis and degeneration: roles of Toll-like receptors and Sarm1 in neurons. Neurosci Bull. 2014; 30: 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dalod M. Studies of SARM1 uncover similarities between immune and neuronal responses to danger. Sci STKE. 2007; 2007:pe73. [DOI] [PubMed] [Google Scholar]
- 6. Kaiser WJ, Offermann MK. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J Immunol. 2005; 174: 4942–4952. [DOI] [PubMed] [Google Scholar]
- 7. Lin S, Liang Y, Zhang J, et al. Microglial TIR-domain-containing adapter-inducing interferon-beta (TRIF) deficiency promotes retinal ganglion cell survival and axon regeneration via nuclear factor-kappaB. J Neuroinflammation. 2012; 9: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004; 16: 3–9. [DOI] [PubMed] [Google Scholar]
- 9. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001; 413: 732–738. [DOI] [PubMed] [Google Scholar]
- 10. Xu Y, Tao X, Shen B, et al. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000; 408: 111–115. [DOI] [PubMed] [Google Scholar]
- 11. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001; 413: 78–83. [DOI] [PubMed] [Google Scholar]
- 12. Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001; 2: 835–841. [DOI] [PubMed] [Google Scholar]
- 13. Yamamoto M, Sato S, Hemmi H, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002; 420: 324–329. [DOI] [PubMed] [Google Scholar]
- 14. Hoebe K, Du X, Georgel P, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature. 2003; 424: 743–748. [DOI] [PubMed] [Google Scholar]
- 15. Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. 2003; 4: 161–167. [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003; 301: 640–643. [DOI] [PubMed] [Google Scholar]
- 17. Bin LH, Xu LG, Shu HB. TIRP. a novel Toll/interleukin-1 receptor (TIR) domain-containing adapter protein involved in TIR signaling. J Biol Chem. 2003; 278: 24526–24532. [DOI] [PubMed] [Google Scholar]
- 18. Fitzgerald KA, Rowe DC, Barnes BJ, et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med. 2003; 198: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yamamoto M, Sato S, Hemmi H, et al. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003; 4: 1144–1150. [DOI] [PubMed] [Google Scholar]
- 20. Mink M, Fogelgren B, Olszewski K, Maroy P, Csiszar K. A novel human gene (SARM) at chromosome 17q11 encodes a protein with a SAM motif and structural similarity to Armadillo/beta-catenin that is conserved in mouse, Drosophila, and Caenorhabditis elegans. Genomics. 2001; 74: 234–244. [DOI] [PubMed] [Google Scholar]
- 21. Andrade MA, Perez-Iratxeta C, Ponting CP. Protein repeats: structures, functions, and evolution. J Struct Biol. 2001; 134: 117–131. [DOI] [PubMed] [Google Scholar]
- 22. Aviv T, Lin Z, Ben-Ari G, Smibert CA, Sicheri F. Sequence-specific recognition of RNA hairpins by the SAM domain of Vts1p. Nat Struct Mol Biol. 2006; 13: 168–176. [DOI] [PubMed] [Google Scholar]
- 23. Coates JC. Armadillo repeat proteins: beyond the animal kingdom. Trends Cell Biol. 2003; 13: 463–471. [DOI] [PubMed] [Google Scholar]
- 24. Peng J, Yuan Q, Lin B, et al. SARM inhibits both TRIF- and MyD88-mediated AP-1 activation. Eur J Immunol. 2010; 40: 1738–1747. [DOI] [PubMed] [Google Scholar]
- 25. Carty M, Goodbody R, Schroder M, Stack J, Moynagh PN, Bowie AG. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol. 2006; 7: 1074–1081. [DOI] [PubMed] [Google Scholar]
- 26. Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006; 90: 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weinreb RN, Khaw PT. Primary open-angle glaucoma. Lancet. 2004; 363: 1711–1720. [DOI] [PubMed] [Google Scholar]
- 28. Izzotti A, Bagnis A, Sacca SC. The role of oxidative stress in glaucoma. Mutat Res. 2006; 612: 105–114. [DOI] [PubMed] [Google Scholar]
- 29. Zanon-Moreno V, Marco-Ventura P, Lleo-Perez A, et al. Oxidative stress in primary open-angle glaucoma. J Glaucoma. 2008; 17: 263–268. [DOI] [PubMed] [Google Scholar]
- 30. Luo C, Yang X, Kain AD, Powell DW, Kuehn MH, Tezel G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Invest Ophthalmol Vis Sci. 2010; 51: 5697–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000; 103: 239–252. [DOI] [PubMed] [Google Scholar]
- 32. Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013; 13: 679–692. [DOI] [PubMed] [Google Scholar]
- 33. Derijard B, Hibi M, Wu IH, et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994; 76: 1025–1037. [DOI] [PubMed] [Google Scholar]
- 34. Bogoyevitch MA, Arthur PG. Inhibitors of c-Jun N-terminal kinases: JuNK no more? Biochim Biophys Acta. 2008; 1784: 76–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abe N, Cavalli V. Nerve injury signaling. Curr Opin Neurobiol. 2008; 18: 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morishima Y, Gotoh Y, Zieg J, et al. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001; 21: 7551–7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perrin V, Dufour N, Raoul C, et al. Implication of the JNK pathway in a rat model of Huntington's disease. Exp Neurol. 2009; 215: 191–200. [DOI] [PubMed] [Google Scholar]
- 38. Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB. Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001; 98: 10433–10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tezel G, Chauhan BC, LeBlanc RP, Wax MB. Immunohistochemical assessment of the glial mitogen-activated protein kinase activation in glaucoma. Invest Ophthalmol Vis Sci. 2003; 44: 3025–3033. [DOI] [PubMed] [Google Scholar]
- 40. Kwong JM, Caprioli J. Expression of phosphorylated c-Jun N-terminal protein kinase (JNK) in experimental glaucoma in rats. Exp Eye Res. 2006; 82: 576–582. [DOI] [PubMed] [Google Scholar]
- 41. Lukas TJ, Wang AL, Yuan M, Neufeld AH. Early cellular signaling responses to axonal injury. Cell Commun Signal. 2009; 7: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roth S, Shaikh AR, Hennelly MM, Li Q, Bindokas V, Graham CE. Mitogen-activated protein kinases and retinal ischemia. Invest Ophthalmol Vis Sci. 2003; 44: 5383–5395. [DOI] [PubMed] [Google Scholar]
- 43. Ma C, Ying C, Yuan Z, et al. dp5/HRK is a c-Jun target gene and required for apoptosis induced by potassium deprivation in cerebellar granule neurons. J Biol Chem. 2007; 282: 30901–30909. [DOI] [PubMed] [Google Scholar]
- 44. Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001; 29: 629–643. [DOI] [PubMed] [Google Scholar]
- 45. Levkovitch-Verbin H, Harizman N, Dardik R, Nisgav Y, Vander S, Melamed S. Regulation of cell death and survival pathways in experimental glaucoma. Exp Eye Res. 2007; 85: 250–258. [DOI] [PubMed] [Google Scholar]
- 46. Levkovitch-Verbin H, Quigley HA, Martin KR, et al. The transcription factor c-jun is activated in retinal ganglion cells in experimental rat glaucoma. Exp Eye Res. 2005; 80: 663–670. [DOI] [PubMed] [Google Scholar]
- 47. Bessero AC, Chiodini F, Rungger-Brandle E, Bonny C, Clarke PG. Role of the c-Jun N-terminal kinase pathway in retinal excitotoxicity, and neuroprotection by its inhibition. J Neurochem. 2010; 113: 1307–1318. [DOI] [PubMed] [Google Scholar]
- 48. Liu H, Sun H, Liu C. Interference of the apoptotic signaling pathway in RGC stress response by SP600125 in moderate ocular hypertensive rats. Chin J Physiol. 2011; 54: 124–132. [PubMed] [Google Scholar]
- 49. Tezel G, Yang X, Yang J, Wax MB. Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res. 2004; 996: 202–212. [DOI] [PubMed] [Google Scholar]
- 50. Yang X, Luo C, Cai J, Pierce WM, Tezel G. Phosphorylation-dependent interaction with 14-3-3 in the regulation of bad trafficking in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2008; 49: 2483–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee JK, Park J, Lee YD, Lee SH, Han PL. Distinct localization of SAPK isoforms in neurons of adult mouse brain implies multiple signaling modes of SAPK pathway. Brain Res Mol Brain Res. 1999; 70: 116–124. [DOI] [PubMed] [Google Scholar]
- 52. Fernandes KA, Harder JM, Fornarola LB, et al. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis. 2012; 46: 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Quigley HA, Cone FE, Gelman SE, et al. Lack of neuroprotection against experimental glaucoma in c-Jun N-terminal kinase 3 knockout mice. Exp Eye Res. 2011; 92: 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang Z, Stratton C, Francis PJ, et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N Engl J Med. 2008; 359: 1456–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kleinman ME, Yamada K, Takeda A, et al. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature. 2008; 452: 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shiose S, Chen Y, Okano K, et al. Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. J Biol Chem. 2011; 286: 15543–15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Takemura N, Kawasaki T, Kunisawa J, et al. Blockade of TLR3 protects mice from lethal radiation-induced gastrointestinal syndrome. Nat Commun. 2014; 5: 3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murakami Y, Matsumoto H, Roh M, et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 2014; 21: 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ganesh BS, Chintala SK. Inhibition of reactive gliosis attenuates excitotoxicity-mediated death of retinal ganglion cells. PLoS One. 2011; 6: e18305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Massoll C, Mando W, Chintala SK. Excitotoxicity upregulates SARM1 protein expression and promotes Wallerian-like degeneration of retinal ganglion cells and their axons. Invest Ophthalmol Vis Sci. 2013; 54: 2771–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goyal A, Srivastava A, Sihota R, Kaur J. Evaluation of oxidative stress markers in aqueous humor of primary open angle glaucoma and primary angle closure glaucoma patients. Curr Eye Res. 2014; 39: 823–829. [DOI] [PubMed] [Google Scholar]
- 62. Ghanem AA, Arafa LF, El-Baz A. Oxidative stress markers in patients with primary open-angle glaucoma. Curr Eye Res. 2010; 35: 295–301. [DOI] [PubMed] [Google Scholar]
- 63. Hernandez MR, Miao H, Lukas T. Astrocytes in glaucomatous optic neuropathy. Prog Brain Res. 2008; 173: 353–373. [DOI] [PubMed] [Google Scholar]
- 64. Tezel G, Yang X, Luo C, et al. Oxidative stress and the regulation of complement activation in human glaucoma. Invest Ophthalmol Vis Sci. 2010; 51: 5071–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tezel G. Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog Retin Eye Res. 2006; 25: 490–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tezel G, Wax MB. The immune system and glaucoma. Curr Opin Ophthalmol. 2004; 15: 80–84. [DOI] [PubMed] [Google Scholar]
- 67. Tezel G. The role of glia, mitochondria, and the immune system in glaucoma. Invest Ophthalmol Vis Sci. 2009; 50: 1001–1012. [DOI] [PubMed] [Google Scholar]
- 68. Tezel G, Luo C, Yang X. Accelerated aging in glaucoma: immunohistochemical assessment of advanced glycation end products in the human retina and optic nerve head. Invest Ophthalmol Vis Sci. 2007; 48: 1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen CY, Lin CW, Chang CY, Jiang ST, Hsueh YP. Sarm1, a negative regulator of innate immunity, interacts with syndecan-2 and regulates neuronal morphology. J Cell Biol. 2011; 193: 769–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lin CW, Liu HY, Chen CY, Hsueh YP. Neuronally-expressed Sarm1 regulates expression of inflammatory and antiviral cytokines in brains. Innate Immun. 2014; 20: 161–172. [DOI] [PubMed] [Google Scholar]
- 71. Kim Y, Zhou P, Qian L, et al. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J Exp Med. 2007; 204: 2063–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012; 33: 449–458. [DOI] [PubMed] [Google Scholar]
- 73. Liu T, Gao YJ, Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull. 2012; 28: 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kaul D, Habbel P, Derkow K, et al. Expression of Toll-like receptors in the developing brain. PLoS One. 2012; 7: e37767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cameron JS, Alexopoulou L, Sloane JA, et al. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci. 2007; 27: 13033–13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Panneerselvam P, Singh LP, Ho B, Chen J, Ding JL. Targeting of pro-apoptotic TLR adaptor SARM to mitochondria: definition of the critical region and residues in the signal sequence. Biochem J. 2012; 442: 263–271. [DOI] [PubMed] [Google Scholar]
- 77. Panneerselvam P, Singh LP, Selvarajan V, et al. T-cell death following immune activation is mediated by mitochondria-localized SARM. Cell Death Differ. 2013; 20: 478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]