

Abstract
Neuroblastoma is a childhood tumour with heterogeneous characteristics and children with metastatic disease often have a poor outcome. Here we describe the establishment of neuroblastoma patient-derived xenografts (PDXs) by orthotopic implantation of viably cryopreserved or fresh tumour explants of patients with high risk neuroblastoma into immunodeficient mice. In vivo tumour growth was monitored by magnetic resonance imaging and fluorodeoxyglucose–positron emission tomography. Neuroblastoma PDXs retained the undifferentiated histology and proliferative capacity of their corresponding patient tumours. The PDXs expressed neuroblastoma markers neural cell adhesion molecule, chromogranin A, synaptophysin and tyrosine hydroxylase. Whole genome genotyping array analyses demonstrated that PDXs retained patient-specific chromosomal aberrations such as MYCN amplification, deletion of 1p and gain of chromosome 17q. Thus, neuroblastoma PDXs recapitulate the hallmarks of high-risk neuroblastoma in patients. PDX-derived cells were cultured in serum-free medium where they formed free-floating neurospheres, expressed neuroblastoma gene markers MYCN, CHGA, TH, SYP and NPY, and retained tumour-initiating and metastatic capacity in vivo. PDXs showed much higher degree of infiltrative growth and distant metastasis as compared to neuroblastoma SK-N-BE(2)c cell line-derived orthotopic tumours. Importantly, the PDXs presented with bone marrow involvement, a clinical feature of aggressive neuroblastoma. Thus, neuroblastoma PDXs serve as clinically relevant models for studying and targeting high-risk metastatic neuroblastoma.
What's new?
Neuroblastoma is a childhood tumour with heterogeneous characteristics and children with metastatic disease have a poor outcome. Here, the authors established neuroblastoma patient-derived xenografts (PDXs) by orthotopic implantation of viably cryopreserved or fresh tumour explants of patients with high-risk neuroblastoma into immunodeficient mice. The PDXs retained the genotype and phenotype of patient tumours and exhibited substantial infiltrative growth and metastasis to distant organs including bone marrow. PDX-derived neuroblastoma cells were expanded in vitro and retained tumourigenic and metastatic capacity in vivo. The PDXs may thus represent an important tool for investigating neuroblastoma growth and metastasis as well as drug targeting.
Keywords: neuroblastoma, patient-derived xenograft, orthotopic xenograft, neuroblastoma metastasis, bone marrow metastasis, MYCN
Neuroblastoma is a solid childhood tumour, which accounts for 15% of paediatric cancer deaths. The neuroblastoma cell-of-origin is thought to derive from the sympathoadrenal lineage of the neural crest and the primary tumour frequently arises in the adrenal gland, although it can develop at any site of the sympathetic nervous system. Patients with Stage IV tumours display infiltrative growth and metastasis to distant sites such as bone marrow, bone, lymph nodes, liver and lungs. Patients over 18 months of age with metastatic disease or MYCN amplification have a poor prognosis despite intensive treatment with chemotherapy, surgery and radiotherapy.1,2 Relevant neuroblastoma animal models are thus needed to improve the understanding, and ultimately the treatment, of high-risk neuroblastoma.
In current human neuroblastoma xenograft mouse models, tumours have been established subcutaneously and by orthotopic injection of cell lines or short-term cultured primary cells.3–9 Orthotopically growing tumours are more prone to form vascularised xenografts with increased frequency of spontaneous distant metastasis as compared to subcutaneous tumours.5 It has become increasingly clear that tumour cells cultured in vitro can develop aberrant and irreversible genetic and phenotypic changes not consistent with the properties of the original patient tumour.10 In addition, as shown here and previously reported,5,11 cell line–derived orthotopic neuroblastoma xenografts often lack substantial spontaneous metastasis to bone marrow. Although results from cell line-derived studies have been valuable, generation of neuroblastoma patient-derived xenografts (PDXs) by implantation of viable tumour explants from cancer patients is an alternative approach to better maintain patient-derived tumour cell features.12 By implantation of intact patient tumour explants, including human tumour stroma, in vitro induced modifications are bypassed. PDXs have been established by subcutaneous implantation of patient-derived tumour explants from paediatric cancers, including neuroblastoma13 and by implantation of neuroblastoma samples into human pluripotent stem cell-derived experimental teratomas.14 By subcutaneous and/or orthotopic implantation of diverse adult tumours (reviewed in Tentler et al.10), PDX-based tumour models have been increasingly utilised for preclinical drug screening. For this purpose and for mechanistic studies of metastasis and treatment resistance, it would be important to establish orthotopic PDX models of neuroblastoma to faithfully resemble the clinical features of the disease. Establishment of PDXs from rare tumours, like neuroblastoma, is however challenging for investigators with only sporadic access to fresh tumour material. We approached this problem by implantation of viably cryopreserved neuroblastoma samples.
Here we present the establishment, in vivo imaging and detailed characterisation of neuroblastoma patient-derived orthotopic xenografts in immunodeficient mice. Neuroblastoma PDXs retain the undifferentiated histology, neuroblastoma protein marker profile and gene copy number changes of their corresponding patient tumours. PDX-derived cells were cultured as neurospheres where tumour cells expressed typical neuroblastoma gene markers and retained tumour-initiating and metastatic capacity in vivo. Importantly, PDXs exhibit extensive infiltrative growth into surrounding tissues and display widespread metastasis to distant organs, including bone marrow.
Materials and Methods
Cell culture
The human neuroblastoma cell line SK-N-BE(2)c was grown in Minimum Essential Medium at 37°C in 5% CO2, 21% O2 and 95% air. The media was supplemented with 10% fetal bovine serum (FBS), 100 IU/ml penicillin and 100 μg/ml of streptomycin. Media and supplements were obtained from Invitrogen Life Technologies (Carlsbad, CA).
Processing of primary neuroblastoma explants
Primary neuroblastoma samples were obtained from neuroblastoma biopsy or surgery. Intact tumour explants (fragments of approximately 2 mm × 2 mm × 2 mm) were cryopreserved in 90% FBS and 10% dimethyl sulfoxide by stepwise cooling using CoolCell (BioCision, Larkspur, CA) and stored in −80°C freezer. Tumour explants were thawed either immediately prior to implantation into mice or one day prior to implantation and kept in stem cell media [Dulbecco's Modified Eagle's medium (DMEM)/F-12 medium with glutamine, 100 IU/ml penicillin and 100 μg/ml of streptomycin, 2% B27 supplement, 40 ng/ml of basic fibroblast growth factor (FGF) 2 and 20 ng/ml of epidermal growth factor (EGF)] in incubator at 37°C in 5% CO2 and 21% O2. The regional ethical review board at Lund University approved the study (Dnr. 2011/289) and written informed consent was obtained.
Animal procedures
Four- to six-week-old female or male NSG mice were purchased from Charles River (Charles River Laboratories, Wilmington, MA). Mice were housed under pathogen-free conditions and received autoclaved water and food. Mice were anaesthetised using 3% isofluorane inhalation. For the orthotopic adrenal gland injection, the skin and the mesentery membrane were cut and exposure of the left adrenal gland was achieved by displacement of the spleen. 5 × 105 SK-N-BE(2)c cells in 30 µl phosphate buffered saline were injected into the adrenal gland using a 29G syringe. Intact patient-derived tumour explants were incubated in matrigel (BD Biosciences, San Jose) for a minimum of 20 min, then placed on top of the adrenal gland and immediately covered with matrigel. PDX-derived cultured cells were injected either as dissociated single cells (1 × 106 cells) or as spheres (approximately 1 × 106 cells) into the adrenal gland of NSG mice (n = 3; single cells and n = 3; spheres). The mesentery membrane was sown back together and the skin was closed using surgical clips. Mice were sacrificed immediately when exhibiting symptoms of tumour growth. Primary tumour, liver, kidneys, lungs, femur and tibia were fixed in 4% paraformaldehyde for histopathological examination. Pieces of the primary tumour were snap frozen for genomic assays. All animal procedures followed the guidelines set by the Malmö-Lund Ethical Committee for the use of laboratory animals and were conducted in accordance with European Union directive on the subject of animal rights.
Fluorodeoxyglucose–positron emission tomography imaging
FDG-PET scanning was performed to detect orthotopic tumours. The 18F-FDG was produced and provided by Skåne University Hospital Cyclotron Unit. For PET-imaging, mice were intravenously injected with approximately 30 MBq of FDG and were imaged 1 h after administration using a preclinical PET/CT-scanner (Bioscan, USA). Mice were anaesthetised using 2–3% isofluorane and were placed in a temperature regulated animal chamber (Minerve, Bioscan, USA) with the temperature set to 37°C (Minerve multistation controller unit) during the scanning. During the acquisitions, respiration was monitored on PC (SA Instruments, Inc, USA) and anaesthesia levels were adjusted accordingly. CT scanning was performed using the X-ray exposure of 600 ms and 177 μA, and the zoom level set to medium zoom. The CT images were scanned with 180 projections/rotation, pitch 1.0 and a binning of 1:4. The CT reconstruction was performed utilizing a Butterworth filter algorithm. The PET acquisition was performed with the coincidence mode set 1-3. The PET data was acquired in list mode for 30 minutes, and reconstructed dynamically using the OSEM option and rebinning SSRB to 2D LOR with the ring difference set to 8. CT and PET images were evaluated and created using the InVivoScope software (InviCRO).
Magnetic resonance imaging
Mice were anaesthetised using 2–3% isofluorane in 200 ml/min O2 / 200 ml/min NO2. Animals were restrained to an MR bed via head fixation. Breathing was monitored using a pressure sensitive pillow, temperature was monitored using a rectal probe and heating was regulated during MRI data acquisition to keep body temperature at 37°C (SA instruments, Inc, USA). MRI was performed at a field strength of 9.4 T (Agilent Technologies, USA). Respiration triggered fat suppressed fast spin echo images were acquired using a spin echo preparation (10 ms) and an echo spacing of 10 ms (acceleration factor 4, effective echo time: 27 ms, 4 averages). The repetition time was set between 800 and 950 ms depending on the breathing rate of the animal. The acquisition of the data for the 12 slices was spread over 4 respiration phases to achieve a T1-weighting corresponding to a repetition time of about 3.2–3.5 sec. Coronal and sagittal sections were acquired with an in plane resolution of 156×137 μm and a slice thickness of 1 mm, while the transversal direction was acquired with an in plane resolution of 137×137 μm. ImageJ 1.46r with a plugin provided by Agilent to import their proprietary format FDF images was used to analyse the images.
Immunohistochemistry
Xenograft tumours and mice organs were formalin-fixed and bone specimens were decalcified in 10% EDTA (pH 8). Tumours and organs were routinely embedded in paraffin and 4 µm tissue sections were analysed. Haematoxylin & eosin (HistoLab Products AB, Göteborg, Sweden) was used for assessment of histopathology. A list of antibodies and their working dilutions are presented in Supporting information Table 1.
Microscopy and quantification
Images were acquired using Olympus BX51 light microscope with CCD Olympus DP50 camera along with the CellA Analysis Image Processing Program. Ki67 index was calculated by automatic quantification using the Image Pro-Plus software as described previously.15 Neural cell adhesion molecule (NCAM) positive metastatic cells in liver, lungs and bone marrow were manually quantified in 1–2 entire tissue sections/mouse at ×20 magnification.
Single nucleotide polymorphism analysis
Cultured SK-N-BE(2)c cells, fresh tumour explants from SK-N-BE(2)c orthotopic tumours, patient tumours and PDXs were snapfrozen and stored in −80°C freezer for single nucleotide polymorphism (SNP) array analysis. Briefly, DNA was extracted from the cells and tissues using DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) according to the instructions. We used the Affymetrix CytoScan HD platform for SNP array analysis of cultured SK-N-BE(2)c cells, SK-N-BE(2)c tumours, PDXs and patient tumour #3. High resolution microarray analysis was performed by hybridisation of genomic DNA on arrays, using equipment and protocols as recommended by the manufacturer (Affymetrix, Santa Clara, CA). The Affymetrix CytoScan HD chip contains approximately 2.6 million markers distributed throughout all chromosomes; of these almost 750,000 are SNPs enabling detection of copy number neutral loss of heterozygosity. The practical resolution of the method is 10–50 kb for deletions and 30–150 kb for duplications; depending on the probe density of each segment. HumanCytoSNP-12 DNA Analysis BeadChip (299,140 markers ∼10 kb average distance) from Illumina (Illumina, San Diego, CA) was used for SNP array analysis of patient tumour #1 and patient tumour #2. DNA amplification, tagging and hybridisation to the chip were performed according to the manufacturer's protocol (http://www.illumina.com). Data were analysed using GenomeStudio Genotyping Module (Illumina). For exclusion of constitutional copy number polymorphisms, the Database of Genomic Variants (http://projects.tcaq.ca/variation) was used. Genomic position annotations were based on the hg19 build (http://genome.ucsc.edu/) of the human genome sequence. Copy number variation analyses were performed using the Nexus software.
Isolation and in vitro sphere culture of PDX-derived cells
Small tumour pieces were mechanically dissected using sterile scalpels and the minced tissue was collected in a petri dish containing Accutase (Sigma-Aldrich, St. Louis, MO) and incubated for 15–20 min at 37°C, with pipetting every 5 min to dissociate the tissue. The suspension was then transferred to serum-free DMEM/High Glucose (Thermo Scientific, Waltham, MA), excluding nondissociated pieces and centrifuged. The pellet was re-suspended in stem cell medium; DMEM/GlutaMAX™ F-12 (mixed in a 3:1 ratio) supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin, 2% B-27 w/o vitamin A (Gibco-Life Technologies, Grand Island, NY), 40 ng/ml basic FGF (Promega, Madison, WI) and 20 ng/ml EGF (Gibco Life Technologies). Spheres were passaged once a week and cells from early passages (up to the fifth passage) were used for quantitative polymerase chain reaction (qPCR) analysis. PDX-derived cultured cells were injected orthotopically into mice (described in Animal procedures).
Quantitative real-time polymerase chain reaction
Total RNA was extracted using either RNeasy Mini or Micro Kit together with the Qiashredder Kit (Qiagen, Hilden, Germany). An on-column DNAse treatment was included during the extraction. The following complementary DNA synthesis was performed using MultiScribe Reverse Transcriptase enzyme (Applied Biosystems, Foster City, CA) and random primers. Quantitative PCR was carried out in a 7300 Real-Time PCR (RT-PCR) System (Applied Biosystems) with SYBR Green PCR Master mix (Applied Biosystems). Comparative Ct method was used in order to quantify mRNA levels and three reference genes (SDHA, UBC, YWHAZ) were included to normalise gene-expression levels. Experiments were performed in triplicates. Primers are listed in Supporting information Table 2.
Results
In vivo imaging of orthotopic neuroblastoma PDXs
To optimise in vivo imaging techniques for observation of orthotopic neuroblastoma growth, we injected SK-N-BE(2)c neuroblastoma cells into the adrenal gland of NSG mice (n = 9) and tumours were successfully established in all animals. After optimisation of fluorodeoxyglucose–positron emission tomography (FDG-PET) and magnetic resonance imaging (MRI) settings, orthotopic tumour growth was demonstrated as seen from the sagittal, coronal and transversal views. FDG signal was also seen in nontumour regions like brain, heart, kidneys and urinary bladder (Supporting information Fig. 1). For establishment of neuroblastoma PDXs, we implanted orthotopically fresh or viably cryopreserved tumour explants from six patient tumours into NSG mice. Patient characteristics are presented in Table1. Orthotopic PDXs from three patient tumours were established and detected by in vivo imaging. FDG-PET demonstrated heterogeneous uptake of FDG within tumours as seen from the sagittal, coronal and transversal views (Fig. 1a). Orthotopic neuroblastoma PDX growth was confirmed by MRI (Fig. 1b).
Table 1.
Patient tumour characteristics
| No | Age | Origin | Stage | Histology | Patient genomics | PDX | Days | Tissue status |
|---|---|---|---|---|---|---|---|---|
| 1 | 1y4m | primary (AG) | IV | un NB | MYCN amp, 1p- | yes | 70 | Cryopreserved |
| 2 | 2y2m | cerebral met | IV | un NB | MYCN amp, 1p-,+17q | yes | 102 | Cryopreserved |
| 3 | 2y9m | primary (AG) | III | pd NB | MYCN amp, 1p-, +17q | yes | 59 | Fresh |
| 4 | 4y9m | primary (AG) | IV | pd NB | MYCN gain, 1p-, 11q-,+17q | no | NA | Cryopreserved |
| 5 | 2y4m | primary (AG) | IV | pd NB | 1p-,+17q | no | NA | Cryopreserved |
| 6 | 12y | primary | IIb | pd NB | +17q | no | NA | Cryopreserved |
AG = adrenal gland, un NB = undifferentiated neuroblastoma, pd NB = poorly differentiated neuroblastoma. Numbers of days refer to the time period from implantation until PDXs were analysed.
Figure 1.

In vivo imaging and histopathological characterisation of neuroblastoma PDXs. FDG-PET/CT and MRI of neuroblastoma PDXs were performed 3–4 weeks after implantation. Orthotopic PDXs are depicted from the sagittal, coronal and transversal views (a and b). Uptake of FDG is also seen in the brain, heart and urinary bladder (a). The PDX is encircled with a yellow line and located above and below the ipsilateral kidney, encircled in red (b). Gross morphology of neuroblastoma PDX and SK-N-BE(2)c orthotopic tumour (c). PDXs (n = 3) displayed extensive infiltrative growth into ipsilateral kidney, muscle and fat tissue while orthotopic SK-N-BE(2)c tumours (n = 8) exhibited only moderate or no local infiltration (c). PDXs recapitulated the differentiation status of the corresponding patient tumours while SK-N-BE(2)c xenografts showed a uniform undifferentiated histology (d). Ki-67 expression in PDX #1, patient tumour #1 and SK-N-BE(2)c tumours. One section from each patient tumour and four sections from each PDX model were quantified for Ki-67 expression (e). The shown pictures are representative for the mice. Scale bar in C is 100 µm in (c), (d) and (e) and 50 µm in the close-ups of (d) and (e).
Histopathological characterisation of orthotopic neuroblastoma PDXs
Neuroblastoma PDXs developed as large abdominal solid masses with heterogeneous gross morphology, including a haemorrhagic cut surface typical of neuroblastoma (Fig. 1c). The tumours displayed extensive infiltrative growth into surrounding tissues such as the ipsilateral kidney, muscle and fat/connective tissue. In contrast, orthotopic SK-N-BE(2)c tumours only displayed moderate infiltrative growth into surrounding tissues like muscle, pancreas and connective/fat tissue (Fig. 1c), and large parts of the tumours showed expansive growth pattern without obvious signs of infiltration.
Both PDXs and the original patient tumours were stroma-poor tumours consisting of densely packed small round blue tumour cells with scant cytoplasm (Fig. 1d). The PDXs retained the differentiation status of their corresponding patient tumours (undifferentiated neuroblastoma for PDX #1 and #2 and poorly differentiated neuroblastoma for PDX #3). SK-N-BE(2)c orthotopic tumours revealed a uniform undifferentiated histology; small round blue tumour cells with scant cytoplasm and minimal neurofibrillary matrix (Fig. 1d).
Similar to high-stage neuroblastoma in patients, both PDXs and SK-N-BE(2)c tumours were highly proliferating as shown by expression of the cell-cycle marker Ki-67 in a large fraction of tumour cells (Fig. 1e). The Ki-67 labelling index for SK-N-BE(2)c orthotopic tumours was 60%. The Ki-67 index was 74, 76 and 82% for patient tumours #1, #2 and #3, respectively, and 71 ± 7%, 50 ± 2% and 62 ± 5% for the corresponding PDXs.
Expression of neuroblastoma protein markers
Immunohistochemistry revealed expression of NCAM (CD 56) in virtually all tumour cells in patients and the strong expression was retained in all PDXs. Chromogranin A (CHGA) and synaptophysin (SYP) were expressed in almost all tumour cells in patients and heterogeneous expression of these markers was retained in PDXs. Tumour expression of tyrosine hydroxylase (TH) was heterogeneous in both patients and PDXs (Fig. 2). SK-N-BE(2)c orthotopic tumours revealed a more atypical differentiation marker profile, most notably very low number of CHGA and lack of SYP positive cells (Fig. 2).
Figure 2.

Immunohistochemical expression of neuroblastoma markers in neuroblastoma PDXs. NCAM was retained in virtually all tumour cells and CHGA, TH and SYP were heterogenously expressed in PDXs. SK-N-BE(2)c orthotopic tumours exhibited very low numbers of CHGA-positive cells and lacked SYP-positive cells. The shown pictures are representative for the mice. Scale bar is 100 µm in the overviews and 50 µm in the close-ups.
Because previous in vitro culturing of neuroblastoma patient-derived tumour-initiating cells (TICs) resulted in overgrowth by EBV-transformed lymphoblasts,16 EBNA2 expression in PDXs was investigated by immunohistochemistry. We found no evidence of EBNA2 expression in PDXs using EBV-transformed human lymphoblastoid cells as positive control (Supporting information Fig. 2a).
Patient-specific gene copy number changes are retained in neuroblastoma PDXs
Patient tumour #1 and PDX #1 presented with 1p deletion, MYCN amplification, 9p deletion and 17q gain (Fig. 3a). 1p deletion, MYCN amplification and 17q gain were also present in patient tumours #2 and #3 and retained in their corresponding PDXs (Figs. 3b and 3c). Thus, chromosomal aberrations typically found in neuroblastomas were retained in their corresponding PDXs.
Figure 3.
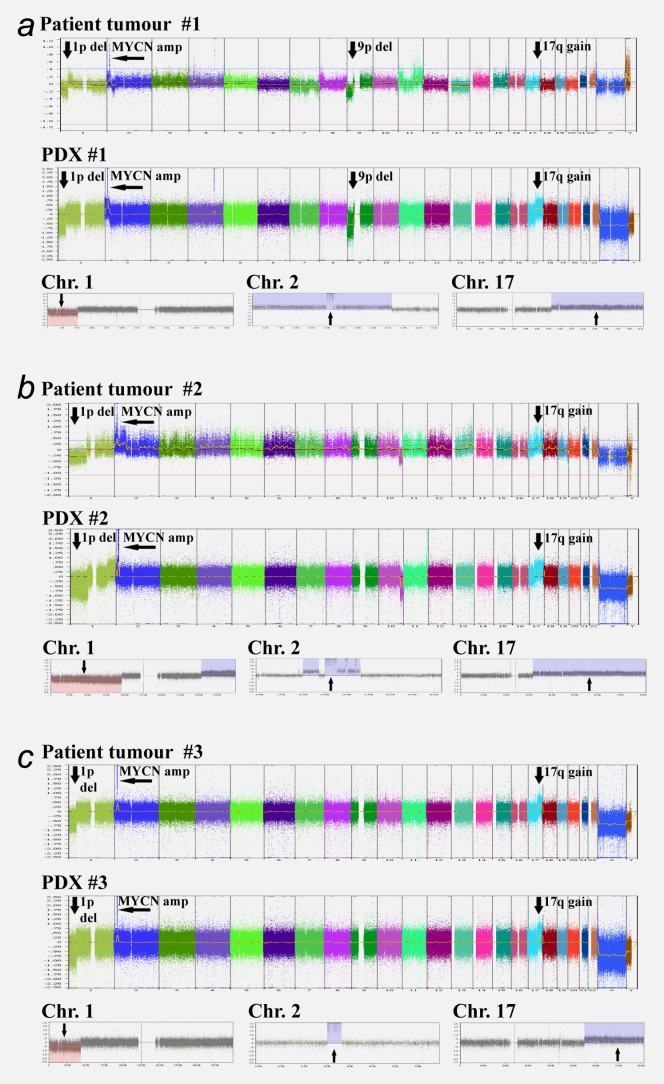
Neuroblastoma PDXs retain patient tumour-specific chromosomal aberrations. Arrows indicate neuroblastoma-typical regions; such as 1p deletion, MYCN amplification, 9p deletion and 17q gain for patient tumours and their corresponding PDXs.
SNP array profiling of SK-N-BE(2)c cell cultures has been performed previously.17,18 Here, SK-N-BE(2)c cell cultures and three SK-N-BE(2)c orthotopic xenografts were analysed by SNP array. MYCN amplification, 1p deletion, 3p deletion and 11q copy number neutral imbalance were retained in orthotopic SK-N-BE(2)c xenografts (Supporting information Fig. 3).
Neuroblastoma PDXs exhibit widespread metastasis to distant organs
Neuroblastoma PDX metastases were demonstrated by immunohistochemistry. In liver, metastatic nodules at subcapsular as well as central locations were present in all PDXs, resembling the appearance and locations of neuroblastoma metastatic growth. Widespread micrometastases were found in the lungs and also, but less frequently, in the ipsilateral kidney (Fig. 4). Importantly, we found substantial metastatic spread to bone marrow in PDXs from all three patients. Bone marrow metastases homogenously expressed NCAM and metastatic tumour cells were dividing as demonstrated by Ki-67 expression (Fig. 4). SK-N-BE(2)c orthotopic tumours metastasised spontaneously to lungs and liver but to a much lesser extent as compared to PDXs (Fig. 4). We found no evidence of SK-N-BE(2)c metastatic cells in bone marrow. Supporting information Table 3 shows the number of metastatic cells in lungs, liver and bone marrow for PDXs (n = 7–9 for each PDX model) and for SK-N-BE(2)c-carrying mice (n = 8).
Figure 4.
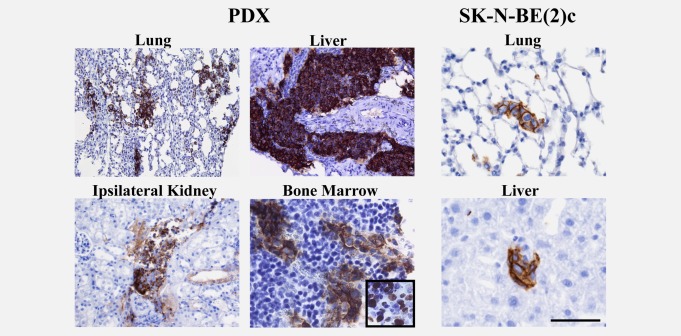
Neuroblastoma PDXs exhibit wide-spread metastasis to distant organs including bone marrow. High numbers of NCAM-positive metastatic cells were found in lungs, liver, ipsilateral kidney and bone marrow of PDXs (7–9 sections from each PDX model). Bone marrow metastases were dividing as shown by Ki-67 expression (inserted field). In contrast, orthotopic SK-N-BE(2)c xenografts (n = 8) exhibited only low numbers of metastatic cells in liver and lungs and no cells in bone marrow. The shown pictures are representative for the mice. Scale bar is 100 µm in PDX lung, liver and ipsilateral kidney, and 50 µm in the PDX bone marrow and SK-N-BE(2)c lung and liver.
Neurosphere-cultured PDX-derived cells express neuroblastoma markers and retain tumourigenic and metastatic capacity in vivo
PDX-derived tumour cells were cultured in serum-free, stem cell-promoting medium, where they formed free-floating neurosphere-like cell structures (Fig. 5a). These cultures were expanded for at least five passages and retained growth and phenotypic properties. RT-qPCR showed that these cultured PDX-derived cells overexpress MYCN (Fig. 5b) and neuroblastoma differentiation markers CHGA, TH, SYP and NPY (Fig. 5c). There was no EBV-specific mRNA expression in any of the PDX-derived cell cultures (Supporting information Fig. 2b). The expression of neuroblastoma gene markers and lack of EBV-specific mRNA expression confirmed the identity of the cultured PDX-derived cells as neuroblastoma cells. The tumour-initiating capacity of the cultured PDX-derived cells was tested by orthotopic injection of dissociated single cells or spheres and all injected animals (n = 3; single cells, and n = 3; spheres) showed tumour take. The tumours exhibited homogenous NCAM expression, heterogenous expression of CHGA and TH, and lacked SYP (Fig. 5d). Using NCAM immunohistochemistry, we found evidence of distant metastases in lungs, liver or bone marrow in all six animals (Fig. 5e). Thus, the in vivo tumourigenic and metastatic capacity of PDX-derived cells were retained after in vitro expansion and orthotopic injections.
Figure 5.

PDX-derived cultured cells express neuroblastoma markers and retain tumourigenic and metastatic capacity in vivo. PDX-derived cells were cultured in serum-free stem cell medium where they formed free-floating spheres (a). MYCN mRNA expression in negative control (the non-MYCN-amplified cell line SH-SY5Y), positive control (the MYCN-amplified cell line SK-N-BE(2)c) and in PDX-derived cells (b). CHGA, TH, SYP and NPY mRNA expression in PDX-derived cell cultures and SK-N-BE(2)c (c). Data are expressed as mean ± SD. Tumours established by orthotopic injection of the PDX-derived cultured cells retained expression of NCAM, CHGA and TH (d). Metastases were found in lungs, liver and bone marrow as shown by NCAM immunohistochemistry (e). Scale bar is 100 µm in A and 50 µm in (d) and (e).
Discussion
Here, we present a pipeline for preclinical modelling of metastatic human neuroblastoma using patient-derived tumour material without passaging the tumour cells in vitro. We show that it is possible to establish orthotopic neuroblastoma PDXs by implantation of fresh and, importantly, viably cryopreserved patient tumour samples, which overcomes the tissue shortage-problem when working with rare tumour forms. Using the clinical imaging modalities FDG-PET and MRI, we further show that orthotopic tumours within the abdominal cavity can be monitored. PDX tumours retained the clinical features of the disease such as the protein marker profile and prognostically significant chromosomal aberrations. Furthermore, PDXs were found to be robust models for invasive neuroblastoma with substantial metastatic spread to distant organs, including bone marrow. Lastly, we show that PDX-derived neuroblastoma cells can be established and passaged in vitro as spheres where they retained their proliferative capacity, neuroblastoma phenotype and tumour-initiating capacity in vivo.
The tumour model presented here is based on orthotopic implantation of tumour tissue on top of the adrenal gland as previous findings have shown that an orthotopic location promotes neuroblastoma growth and behaviour to a large extent mimicking clinical human neuroblastoma growth properties.5 Thus, to monitor tumour growth, we had to use in vivo imaging techniques as abdominal tumours are difficult to detect in early phases and impossible to quantify in terms of tumour spread and tumour volume by using, e.g., calliper. Because tumours are established by implantation of explants we could not utilise imaging modalities that require pre-labelling of single cells for visualisation in vivo. However, as we show here, FDG-PET and MRI are powerful imaging techniques suitable for detection of orthotopic nonmodified neuroblastoma cells. While primary tumours were visualised in all tested tumour-bearing mice, the next and more challenging step, will be to detect metastatic growth in the PDX models. A combination of FDG-PET or MIBG-SPECT, and MRI would probably optimise the conditions for detection of metastases in vivo.
The findings that neuroblastoma PDXs retained clinically important features of the disease make the PDXs more suitable as models for aggressive neuroblastoma as compared to the commonly used cell line-based xenografts. In the SK-N-BE(2)c cell model used here, several aberrant features not typical for the clinical disease were found, most importantly lack of bone marrow involvement. Indeed, one obstacle when studying neuroblastoma metastasis has been the scarcity of xenograft models that retain the metastatic pattern of the disease.11 Here, we show that orthotopic neuroblastoma PDXs are reliable models for human neuroblastoma bone marrow metastasis. The presence of liver and lung metastasis in the PDX models are also consistent with the findings of liver and lung micrometastases in more than half of neuroblastoma patients analysed by immunohistochemistry postmortem.19 Furthermore, we demonstrate that the neuroblastoma protein marker profile and neuroblastoma-associated chromosomal aberrations MYCN amplification, 1p deletion and gain of 17q were retained in PDXs.
Maintaining patient tumour explant viability is a critical parameter for successful establishment of PDXs and the workflow and/or transport of fresh tumour tissue from surgery to implantation into mice can potentially decrease tissue viability and reduce successful in vivo engraftment. Implantation of viably cryopreserved tumour explants can solve this problem as demonstrated here. Cryopreservation of tumour explants further allows for storage of viable primary tumour material in biobanks and can be used for establishment of many diverse PDXs at any lab with animal facilities. This is in particular important for establishment of rare tumours like neuroblastoma. Interestingly, previous studies have shown that the in vivo take rate of cryopreserved and fresh tumour tissue (pancreatic and colorectal carcinoma) is comparable20,21, although a large divergence in tumour take rates between different tumour forms and within a given tumour form can be anticipated. In the case of neuroblastoma it is likely that the tumour take rate drastically drops if low-stage neuroblastomas were implanted. Other parameters that could influence successful in vivo engraftment include tissue viability, time from biopsy to implantation, tumour type, mouse strain, implantation site and the use of co-factors like Matrigel. However, given the scarcity of aggressive neuroblastoma we have not had the possibility to test the effect of all these parameters for engraftment.
Orthotopic neuroblastoma PDXs will be important tools for in vivo drug testing against aggressive neuroblastoma. Although drug testing using subcutaneously implanted neuroblastoma xenografts13 is less time-consuming, it appears, based on the results presented here and by others5,10 that orthotopically established neuroblastoma PDXs is the model of choice for studying and targeting neuroblastoma invasion and metastasis in vivo. For such studies, neuroblastoma PDXs should optimally be used in conjunction with neuroblastoma transgenic mouse models with substantial metastatic capacity.22,23 All established PDXs presented here contain MYCN amplifications and hence, the PDXs are also suitable models for targeting of MYCN in vivo. MYCN is an attractive therapeutic target for patients with high-risk neuroblastoma as MYCN amplification correlates with aggressive disease24 and downregulation of MYCN expression results in decreased tumour cell proliferation, increased cell death and neuronal differentiation of neuroblastoma cells in vitro.25 In addition to orthotopic neuroblastoma PDX models, zebrafish and transgenic mouse models, driven by MYCN alone or in combination with other oncogenes22,26–28 are important complementary preclinical neuroblastoma models.
In conclusion, we present a workflow for preclinical modelling of aggressive human neuroblastoma by FDG/PET and MRI imaging, histopathological examinations and genomic characterisation by use of orthotopic PDXs. Future work will need to expand the pool of neuroblastoma PDXs in order to reflect the large diversity of the disease. For instance up to now, we have only established tumours carrying MYCN amplifications. The PDXs serve as a source for in vivo expansion of patient-derived tumour-initiating cells, which will be valuable for in vitro studies. Furthermore, the recurrent findings of patient tumour-specific features, invasive growth and bone marrow metastasis in the PDX models demonstrate their suitability for mechanistic studies of human neuroblastoma growth and metastasis, and for drug testing against aggressive metastatic neuroblastoma in vivo.
Acknowledgments
Lund University Bioimaging Center (LBIC), Lund University, is gratefully acknowledged for providing experimental resources. We thank Thuy Tran, Gustav Grafström, Adnan Bibic, René in't Zandt and Carina Siversson for technical help with the in vivo imaging studies. This work was supported by grants from the Swedish Childhood Cancer Foundation, the Swedish Cancer Society, the Swedish Research Council, VINNOVA, the SSF Strategic Center for Translational Cancer Research-CREATE Health, the Strategic Cancer Research Program, BioCARE, Crafoord Foundation, The Royal Physiographic Society in Lund, Magnus Bergvall Foundation, Gunnar Nilsson's Cancer Foundation, The Swedish Society of Medicine, Gyllenstiernska Krapperupsstiftelsen, Region Skåne and the research funds of Skåne University Hospital, grants from the FIS (contract PI10/15) and RTICC (Red Tematica de Investigación Cooperativa en Cancer, contracts RD06/0020/0102;RD12/0036/0020), Instituto Carlos III Madrid, and ERDF (European Regional Development Fund).
Glossary
- CHGA
Chromogranin A
- FDG
Fluorodeoxyglucose
- NPY
Neuropeptide Y
- PDX
Patient-derived xenograft
- PET
Positron Emission Tomography
- SNP
Single nucleotide polymorphism
- SYP
Synaptophysin
- TH
Tyrosine Hydroxylase
Supporting Information
Additional Supporting information may be found in the online version of this article
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
References
- Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris JM, Hogarty MD, Bagatell R. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- Coulon A, Flahaut M, Muhlethaler-Mottet A. Functional sphere profiling reveals the complexity of neuroblastoma tumor-initiating cell model. Neoplasia. 2011;13:991–1004. doi: 10.1593/neo.11800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford LM, McKee AE, Zhang L. Neuroblastoma cells isolated from bone marrow metastases contain a naturally enriched tumor-initiating cell. Cancer Res. 2007;67:11234–43. doi: 10.1158/0008-5472.CAN-07-0718. [DOI] [PubMed] [Google Scholar]
- Khanna C, Jaboin JJ, Drakos E. Biologically relevant orthotopic neuroblastoma xenograft models: primary adrenal tumor growth and spontaneous distant metastasis. In Vivo. 2002;16:77–85. [PubMed] [Google Scholar]
- Patterson DM, Shohet JM, Kim ES. Preclinical models of pediatric solid tumours (neuroblastoma) and their use in drug discovery. Curr Protoc Pharmacol. 2011;14 doi: 10.1002/0471141755.ph1417s52. ;Chapter:Unit 14 7. [DOI] [PubMed] [Google Scholar]
- Teitz T, Stanke JJ, Federico S. Preclinical models for neuroblastoma: establishing a baseline for treatment. PLoS One. 2011;6:e19133. doi: 10.1371/journal.pone.0019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate-Eya LT, Ebus ME, Koster J. Newly-derived neuroblastoma cell lines propagated in serum-free media recapitulate the genotype and phenotype of primary neuroblastoma tumours. Eur J Cancer. 2014;50:628–37. doi: 10.1016/j.ejca.2013.11.015. [DOI] [PubMed] [Google Scholar]
- Joseph JM, Gross N, Lassau N. In vivo echographic evidence of tumoral vascularization and microenvironment interactions in metastatic orthotopic human neuroblastoma xenografts. Int J Cancer. 2005;113:881–90. doi: 10.1002/ijc.20681. [DOI] [PubMed] [Google Scholar]
- Tentler JJ, Tan AC, Weekes CD. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–50. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E, Chen L, Viprey VF. Meeting report: 3rd Neuroblastoma Research Symposium, Liverpool, 6–7th November, 2013. Pediatr Blood Cancer. 2014 doi: 10.1002/pbc.25087. [DOI] [PubMed] [Google Scholar]
- Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. 2013;73:5315–9. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton PJ, Morton CL, Tucker C. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49:928–40. doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- Jamil S, Hultman I, Cedervall J. Tropism of the in situ growth from biopsies of childhood neuroectodermal tumours following transplantation into experimental teratoma. Int J Cancer. 2014;134:1630–7. doi: 10.1002/ijc.28498. [DOI] [PubMed] [Google Scholar]
- Tadeo I, Piqueras M, Montaner D. Quantitative modeling of clinical, cellular, and extracellular matrix variables suggest prognostic indicators in cancer: a model in neuroblastoma. Pediatr Res. 2014;75:302–14. doi: 10.1038/pr.2013.217. [DOI] [PubMed] [Google Scholar]
- Mohlin S, Pietras A, Wigerup C. Tumor-initiating cells in childhood neuroblastoma-letter. Cancer Res. 2012;72:821–2. doi: 10.1158/0008-5472.CAN-11-1761. author reply 3. [DOI] [PubMed] [Google Scholar]
- Carr J, Bown NP, Case MC. High-resolution analysis of allelic imbalance in neuroblastoma cell lines by single nucleotide polymorphism arrays. Cancer Genet Cytogenet. 2007;172:127–38. doi: 10.1016/j.cancergencyto.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Kryh H, Caren H, Erichsen J. Comprehensive SNP array study of frequently used neuroblastoma cell lines; copy neutral loss of heterozygosity is common in the cell lines but uncommon in primary tumours. BMC Genomics. 2011;12:443. doi: 10.1186/1471-2164-12-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romansky S, Landing B. Metastatic patterns in childhood tumours. In: Weiss L, Gilbert H, editors. Pulmonary metastasis. Boston: Medical Publications; 1978. pp. 114–7. [Google Scholar]
- Linnebacher M, Maletzki C, Ostwald C. Cryopreservation of human colorectal carcinomas prior to xenografting. BMC Cancer. 2010;10:362. doi: 10.1186/1471-2407-10-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorio C, Bonora A, Orlandini S. Successful xenografting of cryopreserved primary pancreatic cancers. Virchows Arch. 2001;438:154–8. doi: 10.1007/s004280000343. [DOI] [PubMed] [Google Scholar]
- Heukamp LC, Thor T, Schramm A. Targeted expression of mutated ALK induces neuroblastoma in transgenic mice. Sci Transl Med. 2012;4:141ra91. doi: 10.1126/scitranslmed.3003967. [DOI] [PubMed] [Google Scholar]
- Teitz T, Inoue M, Valentine MB. Th-MYCN mice with caspase-8 deficiency develop advanced neuroblastoma with bone marrow metastasis. Cancer Res. 2013;73:4086–97. doi: 10.1158/0008-5472.CAN-12-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–16. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- Westermark UK, Wilhelm M, Frenzel A. The MYCN oncogene and differentiation in neuroblastoma. Semin Cancer Biol. 2011;21:256–66. doi: 10.1016/j.semcancer.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Zhu S, Lee JS, Guo F. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell. 2012;21:362–73. doi: 10.1016/j.ccr.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry T, Luther W, Bhatnagar N. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012;22:117–30. doi: 10.1016/j.ccr.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss WA, Aldape K, Mohapatra G. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985–95. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information