

To the Editor:
Obstructive sleep apnea (OSA) is a highly prevalent condition throughout the lifespan, affecting 2–10% of the general population at any given age. It is associated with an extensive array of cognitive, behavioral, metabolic, and cardiovascular morbidities (1). In recent years, OSA has emerged as an independent risk factor for cardiovascular disease (2) and has been causally associated with a high prevalence of hypertension, atrial fibrillation, congestive heart failure, stroke, and more specifically, coronary heart disease (3). OSA is associated with activation of multiple inflammatory pathways, disruption of lipid metabolism, and endothelial dysfunction through oxidative stress mechanisms (4), all of which predispose to atherogenesis. Long-term chronic intermittent hypoxia during sleep (IH), a prototypic constitutive element of OSA, induces atherosclerosis in murine models and plays a critical role in OSA-associated cardiovascular morbidities (5). However, the molecular mechanisms underlying OSA-induced atherosclerosis are not well understood.
Atherosclerosis is currently viewed as a chronic inflammatory process, in which macrophages play a key pathophysiologic role (6). Monocyte recruitment to the circulation, their entrapment in the vascular wall, and their subsequent differentiation into lipid-laden macrophages are fundamental processes involved in atheromatous plaque formation (7, 8). Shifts across the spectrum of heterogeneous macrophage populations encompassing a continuum between major macrophage phenotypes have been described as a fundamental process in atherogenesis. Generally speaking, proinflammatory macrophages are implicated in foam cell generation, lipid loading, plaque formation, and plaque rupture, whereas anti-inflammatory macrophages play an atheroprotective role (9). In addition to changes in macrophage polarity within the vascular wall, macrophage proliferation has also emerged as a critical determining factor in atherogenesis, although the origin of the expanding macrophage population (i.e., tissue-resident vs. bone-marrow derived) is still controversial (10, 11).
We hypothesized that IH during the sleep period will induce shifts toward a proatherogenic state in the spectrum of macrophages within the vascular wall. To examine our hypothesis, we exposed 8-week-old male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) to IH with alternating 90-second cycles (21% FiO2 followed by 6% FiO2, 20 cycles/h) for 12 hours/day or to room air for 6 weeks. All mice were kept on a regular low-fat chow diet (n = 12–20/group). After exposures, mice were asphyxiated using CO2 and were exsanguinated via cardiac puncture. The vasculature was perfused with PBS containing 20 U/ml heparin. Full-length aortas were dissected, cleaned, and enzymatically digested. Single-cell suspensions were prepared from the digested tissues by cycles of washing and resuspending. Cells were incubated with Fc blocker for 30 minutes to reduce autofluorescence and were subsequently fixed with 1% paraformaldehyde. After two cycles of washing, pelleted cells were incubated with antibodies for macrophage markers previously implicated in atherogenesis and metabolic dysregulation: CD11b-PB, F4/80-PE/Cy7, Ly6c-APC-Cy7, CD36-FITC, and CD64-PE (Biolegend, San Jose, CA). Subsequently, cells were washed and analyzed using BD FACS CANTO II (BD Biosciences, San Jose, CA). Results were analyzed using FlowJo software. Data are expressed as mean ± standard error of the mean. Room air and IH conditions were compared with Student’s t tests or nonparametric testing as appropriate. Statistical significance was assumed at P < 0.05.
Macrophages were defined as CD11b and F4/80 double-positive cells (Figure 1). Significant increases in the percentage of macrophages out of total cell number emerged in IH-exposed mice (6.4% ± 0.3% vs. 8.1% ± 0.3%; P = 0.003). In addition, a trend toward increase in the absolute macrophage number was observed (cells/mg tissue: 247 ± 28 vs. 414 ± 93; P = 0.18). Furthermore, IH also induced shifts in the macrophage population toward a proinflammatory phenotype. Specifically, the IH group showed significantly higher expression (measured by mean fluorescence intensity) of Ly6c (522 ± 23 vs. 699 ± 43; P = 0.004) and increased the percentage of Ly6c(hi) cells (9.2% ± 0.5% vs. 11.7% ± 0.9%; P = 0.03). In contrast, tissue-resident marker CD64 expression in the aortic macrophages was downregulated after IH exposures (mean fluorescence intensity: 3,207 ± 453 vs. 2,967 ± 483; P = 0.03), as well as the percentage of CD64-positive cells (46.4% ± 2.4% vs. 36.9% ± 2.1%; P = 0.006). CD64 expression was threefold higher in Ly6c(lo) compared with Ly6c(hi) cells, thus reinforcing the notion of two distinct macrophage populations. Finally, scavenger receptor CD36 expression was increased in the IH group (mean fluorescence intensity: 1,694 ± 56 vs. 2,129 ± 209; P = 0.005), as was the percentage of CD36+ macrophages (46.7% ± 1.7% vs. 57.3% ± 3.3%; P = 0.03). Of note, despite the decrease in the number of CD64+ cells, the proportion of CD36+ cells out of CD64+ cells was increased in the IH group (14.1% ± 0.9% vs. 20.8% ± 2.2%; P = 0.01).
Figure 1.
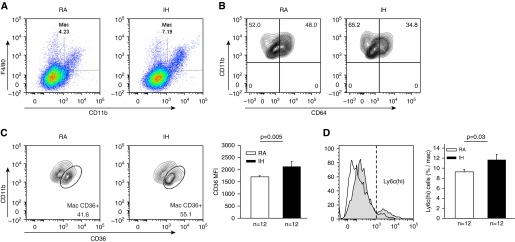
Changes in macrophage populations in the aortic wall of mice exposed to intermittent hypoxia (IH) or room air (RA) during sleep for 6 weeks. (A) Representative example of fluorescence-activated cell sorter analysis of macrophages (CD11b and F4/80 positively labeled cells) and the increase in such population with IH. (B) Representative example of fluorescence-activated cell sorter analysis for CD64+ cells (resident macrophages) illustrating the reduction in resident macrophages after IH exposures. (C) IH is associated with significantly increased expression of CD36 in CD11b and F4/80 positively labeled cells indicating metabolic activation. (D) IH induced significant increases in Ly6C(hi) cell expression, indicating shifts toward proinflammatory macrophages recruited from the bone marrow. MFI = mean fluorescence intensity.
Current findings show, for the first time, an increase in the number of macrophages in the aortic wall after IH exposures. Macrophages have been extensively studied in the context of atherosclerosis, and the presence of CD11b cells is crucial to the initial steps of atherogenesis (6, 8). The expansion of this population at this early stage in the course of IH exposures is suggestive of this being a key step in IH-induced atherosclerotic plaque formation. Furthermore, IH induced several interesting changes in the phenotype of aortic wall macrophages. First, the Ly6c(hi) population was increased, as was the total expression of Ly6c on the aortic wall macrophages. Ly6c is a well-established marker of proinflammatory macrophages recruited from circulating bone marrow-derived monocytes into the arterial wall in the process of atherogenesis (6). Second, increases in Ly6c were accompanied by reciprocal decreases in CD64, a marker of tissue-resident macrophages (12). These findings add to the current debate regarding the source of the expanding macrophage population in the vascular wall during atherogenesis. Indeed, Robbins and colleagues (10) showed that resident macrophages predominate in apolipoprotein E −/− and low-density lipoprotein receptor −/− mice on a high-fat diet. Although our experiments were not designed to resolve this debate (which would require more complex techniques, such as bone marrow depletion and parabiosis), our findings point to the bone marrow as the source of the expanding macrophage population in IH, rather than to resident macrophages, as seen in high-fat diets. Aligned with this interpretation, OSA has been previously shown by our group and others to target and activate specifically the myeloid population in other disease models, such as obesity and cancer (13).
IH also induced increased expression of CD36 surface marker within the macrophage population. CD36 is a membrane glycoprotein participating in oxidized low-density lipoprotein uptake and foam cell formation, the initial critical stage of atherosclerosis (14). Blood monocytes and their lesional progeny also have the potential to emigrate from the plaque (6), and CD36 pathway activation has been shown to inhibit this migration process, which might in turn cause macrophage entrapment in atherosclerotic lesions. Because of its critical role in atherosclerosis, CD36 has also been suggested as a treatment target for atherosclerosis (14). In addition, in line with the proinflammatory interpretation of the changes in our study, CD36 expression has been recently described as identifying a novel metabolically activated macrophage phenotype, which expresses proinflammatory cytokines, but not the classic proinflammatory macrophage surface markers (15).
In summary, IH during the sleep period for 6 weeks leads to expansion of the macrophage population in the aortic wall. In addition to modestly increased cellularity, IH induces changes in macrophage population composition with a shift toward a proinflammatory, metabolically activated phenotype and recruitment of bone marrow-derived macrophages. These changes occur in a relatively resistant animal model for development of atherosclerosis; that is, in the absence of a high-fat diet or of high atherogenic risk predisposing gene knockout mice, thus adding to their potential biological significance. Plans for future studies include the effect of longer IH exposures on macrophage populations within the aortic wall and elucidation of the functional properties associated with the phenotypic switch induced by IH.
Footnotes
D.G. is supported by National Institutes of Health grants HL-65270, HL-086662, and HL-107160.
Author Contributions: A.G.-H. participated in the conceptual framework of the project, performed experiments, analyzed data, and drafted components of the manuscript. I.A., A.K., and S.X.Z. performed experiments. Y.W. served as blinded observer and provided critical input. D.G. conceptualized the project, provided critical input in all phases of the experiments, analyzed data, drafted the ulterior versions of the manuscript, and is responsible for the financial support of the project and the manuscript content. All authors have reviewed and approved the final version of the manuscript.
Author disclosures are available with the text of this letter at www.atsjournals.org.
References
- 1.Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002;165:1217–1239. doi: 10.1164/rccm.2109080. [DOI] [PubMed] [Google Scholar]
- 2.Pack AI, Gislason T. Obstructive sleep apnea and cardiovascular disease: a perspective and future directions. Prog Cardiovasc Dis. 2009;51:434–451. doi: 10.1016/j.pcad.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 3.Loke YK, Brown JW, Kwok CS, Niruban A, Myint PK. Association of obstructive sleep apnea with risk of serious cardiovascular events: a systematic review and meta-analysis. Circ Cardiovasc Qual Outcomes. 2012;5:720–728. doi: 10.1161/CIRCOUTCOMES.111.964783. [DOI] [PubMed] [Google Scholar]
- 4.Gozal D, Kheirandish-Gozal L. Cardiovascular morbidity in obstructive sleep apnea: oxidative stress, inflammation, and much more. Am J Respir Crit Care Med. 2008;177:369–375. doi: 10.1164/rccm.200608-1190PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savransky V, Nanayakkara A, Li J, Bevans S, Smith PL, Rodriguez A, Polotsky VY. Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med. 2007;175:1290–1297. doi: 10.1164/rccm.200612-1771OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schulz C, Massberg S. Atherosclerosis--multiple pathways to lesional macrophages. Sci Transl Med. 2014;6:239ps2. doi: 10.1126/scitranslmed.3008922. [DOI] [PubMed] [Google Scholar]
- 7.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, Bennett M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res. 2007;100:884–893. doi: 10.1161/01.RES.0000260802.75766.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:1120–1126. doi: 10.1161/ATVBAHA.112.300173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, van Rooijen N, et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. Immunological Genome Consortium. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Almendros I, Carreras A, Montserrat JM, Gozal D, Navajas D, Farre R. Potential role of adult stem cells in obstructive sleep apnea. Front Neurol. 2012;3:112. doi: 10.3389/fneur.2012.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park YM. CD36, a scavenger receptor implicated in atherosclerosis. Exp Mol Med. 2014;46:e99. doi: 10.1038/emm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, et al. Metabolic dysfunction drives a mechanistically distinct pro-inflammatory phenotype in adipose tissue macrophages Cell MetabIn press [DOI] [PMC free article] [PubMed] [Google Scholar]