

SUMMARY
Listeria monocytogenes PdxR is a member of the poorly characterized but widespread group of MocR/GabR-type chimeric bacterial proteins that have DNA-binding and aminotransferase-like domains. Using mutational analysis, real-time RT-PCR, transcriptional fusions, gel-shift assays, DNase I footprinting, and in vitro transcription, it was shown that PdxR is a direct activator of the pdxST operon, transcribed divergently from pdxR and responsible for the de novo synthesis of pyridoxal 5′-phosphate (PLP), the major active form of vitamin B6. PLP acts as an anti-activator of PdxR and is the only effector required to reduce the activity of PdxR. PdxR is also a negative autoregulator, and its ability to repress is increased by PLP. A dyad-symmetry sequence, which overlaps the −35 region of the pdxS promoter and lies downstream of the pdxR transcription start point, serves as an important element of the PdxR-binding site. Unexpectedly, some mutations in this activator-binding site, disrupting the dyad-symmetry element, caused constitutive, B6-independent expression from the pdxS promoter. The data suggest that PdxR-like proteins, for which PLP plays just a signaling role, form a separate functional group among the MocR/GabR-type proteins.
Keywords: Listeria monocytogenes, vitamin B6 biosynthesis, pyridoxal 5′-phosphate biosynthesis, MocR/GabR subfamily, GntR family of transcriptional regulators
INTRODUCTION
Chimeric bacterial proteins of the MocR/GabR subfamily belong to the GntR family of transcriptional regulators and are composed of a short N-terminal helix-turn-helix-containing domain (a putative DNA-binding region) of the GntR type and a long C-terminal domain that is similar to full-length aminotransferases (Belitsky and Sonenshein, 2002; Rigali et al., 2002). Aminotransferases are ubiquitous enzymes of nitrogen metabolism that require pyridoxal 5′-phosphate (PLP), the major biologically active form of vitamin B6, as an essential cofactor for catalysis (Eliot and Kirsch, 2004; Schneider et al., 2000).
The MocR/GabR subfamily includes at present thousands of proteins from both Gram-negative and Gram-positive bacteria (Bramucci et al., 2011), but only a handful of its members have been characterized functionally and shown to be DNA-binding proteins that activate transcription of their target genes (Belitsky, 2004a; Belitsky and Sonenshein, 2002; El Qaidi et al., 2013; Jochmann et al., 2011; Wiethaus et al., 2008). One member of the family, Bacillus subtilis GabR, has been described in detail (Belitsky, 2004a), and its crystal structure has been determined (Edayathumangalam et al., 2013). The structural fold of the C-terminal domain of GabR is indeed very similar to the fold of many aminotransferases (fold type I), and PLP appears to be a tightly-bound covalently-linked component of the protein, similar to the case of aminotransferases and other PLP-dependent enzymes (Edayathumangalam et al., 2013; Eliot and Kirsch, 2004; Grishin et al., 1995; Schneider et al., 2000).
GabR is a transcriptional activator of genes for γ-aminobutyrate (GABA) utilization, which requires the presence of GABA, in addition to PLP, for activation of its target genes (Belitsky, 2004a; Belitsky and Sonenshein, 2002). GabR apparently performs an aminotransferase-like partial reaction between GABA and PLP that is required for its activation as a transcriptional regulator (Belitsky, 2004a).
Three MocR/GabR-type proteins from various bacteria, called PdxR proteins, have been shown to be required for the expression of genes of PLP biosynthesis (El Qaidi et al., 2013; Jochmann et al., 2011; Magarvey et al., 2001). PLP is considered to be one of the most versatile cofactors and is essential, in addition to aminotransferases, for activity of numerous other metabolic enzymes, such as amino acid racemases, decarboxylases, aminomutases, and many others (Percudani and Peracchi, 2003; Schneider et al., 2000). Ability of bacterial cells to synthesize PLP is important for virulence of such pathogens as Mycobacterium tuberculosis and Helicobacter pylori (Dick et al., 2010; Grubman et al., 2010).
The functional role and ability of PdxR proteins to respond to vitamin B6 availability in the medium (El Qaidi et al., 2013; Jochmann et al., 2011) suggest that one or more of the B6 vitamers may be a simple effector of these proteins. To characterize further the mechanism by which PdxR proteins regulate expression of their target genes, I studied a putative PdxR-like protein, Lmo2100, of Listeria monocytogenes, a Gram-positive, facultative intracellular bacterium and an important food-borne bacterial pathogen that can cause severe disease in mammals and birds (Vazquez-Boland et al., 2001). In this work, it was shown that L. monocytogenes PdxR is a transcriptional activator of the pdxST genes responsible for de novo PLP biosynthesis. PdxR is inactivated by direct interaction with PLP and, in contrast to GabR, does not require any other effectors for activity.
RESULTS
Phenotype of the pdxST null mutant
The products of the L. monocytogenes lmo2101 and lmo2102 genes (Glaser et al., 2001) are highly similar (82 and 52% identity, respectively) to the products of the pdxS and pdxT genes of B. subtilis (Belitsky, 2004b), which encode two subunits of PLP synthase, the only dedicated enzyme of de novo PLP biosynthesis in many bacteria and other organisms (Fitzpatrick et al., 2007). Hence these genes were renamed as pdxS and pdxT, respectively. The open reading frames of the two listerial genes are separated by only 4 bp, and they form a single transcriptional unit (Toledo-Arana et al., 2009). A deletion-insertion mutation, replacing both the pdxS and pdxT genes with a spectinomycin-resistance cassette, was constructed and introduced into the chromosome of L. monocytogenes strain EGD-e as described in Experimental Procedures. No growth of the resulting strain, BLM1 (ΔpdxST::spc), was observed in a defined medium (see Experimental Procedures) either in liquid culture or on plates. The growth was fully restored by addition of ≥1 μM pyridoxal (PL) or by complementation of the mutation (Fig. 1 and data not shown). It was concluded that the pdxST genes of L. monocytogenes encode a functional PLP synthase, consistent with the known ability of this bacterium to synthesize vitamin B6 (Welshimer, 1963). Apparently, as in all other bacteria, only one de novo pathway of PLP synthesis is present in L. monocytogenes.
Fig. 1. Growth of L. monocytogenes wild-type and mutant strains.

Cells were grown on LDM agar plates without (A, C) or with (B, D) 10 μM PL.
Phenotype of the pdxR null mutant
A putative transcriptional regulator of the MocR/GabR subfamily (Belitsky and Sonenshein, 2002; Rigali et al., 2002) is encoded by the lmo2100 gene that is adjacent to and transcribed divergently from the L. monocytogenes pdxS gene (the two genes are separated by 125 bp) (Glaser et al., 2001). A deletion-insertion mutation, replacing about 0.3 kb of the lmo2100 gene with a spectinomycin-resistance cassette, was constructed and introduced into the chromosome of L. monocytogenes. Strain BLM7 (Δlmo2100::spc) required PL for growth in a manner similar to that of the pdxST null mutant; complementation of the mutation alleviated the PL requirement (Fig. 1). Therefore, the product of the lmo2100 gene appears to be required for pdxST expression, suggesting that it acts as a positive regulator of pdxST transcription. The gene was renamed pdxR; it encodes a 481-amino acid protein, which contains, as do other members of the MocR/GabR subfamily, a N-terminal putative DNA-binding domain and a C-terminal aminotransferase-like domain.
Transcriptional regulation of pdxS and pdxR in L. monocytogenes
As determined by real-time RT-PCR experiments, pdxS expression in the rich Brain Heart Infusion medium (BHI) required the presence of PdxR and was reduced 8-fold in the presence of PL (Table 1). Thus, PdxR behaves indeed as an activator of pdxS expression, and PL or a derivative thereof serves as a likely anti-activator of PdxR. Two other members of the MocR/GabR subfamily, also named PdxR, were shown recently to be B6-responsive activators of the pdxST genes in Corynebacterium glutamicum and Streptococcus pneumoniae cells (El Qaidi et al., 2013; Jochmann et al., 2011).
Table 1.
Expression of the pdxS and pdxR genes in L. monocytogenes
| Genotype | Additions to the medium | pdxS expression | pdxR expression | ||
|---|---|---|---|---|---|
| copies | % | copies | % | ||
| wild-type | none | 1.45 ± 0.32 | 100.0 | 0.141 ± 0.039 | 100.0 |
| PL | 0.18 ± 0.051 | 12.4 | 0.076 ± 0.014 | 53.5 | |
| pdxR | none | 0.013 ± 0.002 | 0.92 | NAa | NA |
| PL | 0.014 ± 0.003 | 0.96 | NA | NA | |
| pdxST | none | NA | NA | 0.43 ± 0.060 | 305.0 |
| PL | NA | NA | 0.078 ± 0.018 | 55.3 | |
Cells were grown in BHI with or without 100 μM PL. In the absence of PL, the mutant strains grew as fast as the wild-type strain or more slowly, depending on the batch of the medium. The data are presented as the number of copies of the pdxS or pdxR transcript per a copy of the rpoB transcript. All values are averages plus/minus standard deviation from at least two experiments.
NA - not applicable. The transcript abundance could not be reliably determined due to the presence of a deletion-insertion in the corresponding gene.
pdxR expression was about 10-fold less efficient than expression of pdxS but was increased 3-fold in the pdxST null mutant in the absence of PL (Table 1), apparently as a result of B6 limitation experienced by the mutant cells. In contrast, it was reduced 2- to 6-fold in the presence of PL both in wild-type and the mutant. Thus, the efficiency of both pdxS and pdxR expression negatively correlated with the abundance of B6. As the pdxR gene was altered by the deletion-insertion mutation, the autoregulation of pdxR in the pdxR mutant could not be faithfully tested by real-time RT-PCR and was analyzed using transcriptional fusions in B. subtilis cells as a surrogate host (see below).
Transcription start points of pdxS and pdxR
Using a RACE approach (Frohman, 1994), two adjacent nucleotides, A and C, located 36 and 37 bp upstream of the pdxS initiation codon, respectively, were identified as apparent 5′ ends of the pdxS mRNA (Fig. 2)(for convenience, the A36 nucleotide was assigned the +1 position). The sequences TTTTTA and TATCAT with three and one mismatches to the consensus −35 and −10 regions of σA-dependent promoters of most bacteria, respectively, and a 17-bp spacer region can be identified upstream of the apparent pdxS transcription start points (Fig. 2). In another RACE experiment, a pyrimidine nucleotide C66, located 66 bp upstream of the pdxR initiation codon, was identified as the apparent 5′ end of the pdxR mRNA. The sequences TTGAAA and TAAGCT with one and three mismatches to the consensus −35 and −10 regions, respectively, and a 17-bp spacer region can be identified upstream of the apparent pdxR transcription start point (Fig. 2). Due to the nature of the RACE experiment, the possibility that A65 serves as an alternative transcription start point for pdxR cannot be excluded. The +1 positions of the pdxR and pdxS genes are separated by only 23 bp, and the −10 promoter regions of the two genes overlap (Fig. 2).
Fig. 2. The double-stranded sequence of the pdxRS regulatory region.
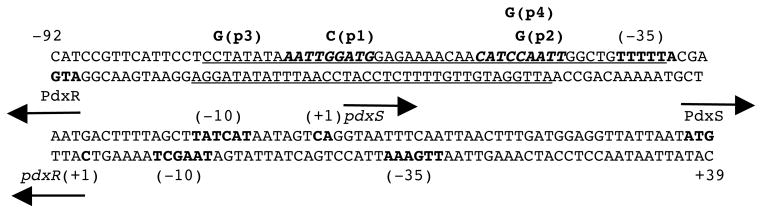
The likely initiation codons, −10 and −35 promoter regions, and the +1 gene positions are in boldface. The directions of transcription and translation are indicated by the arrows. The sequences protected by PdxR on the top and bottom strands in DNase I footprinting experiments are underlined. The dyad-symmetry sequence is in boldface and italicized. The coordinates of the 5′ and 3′ ends of the sequence with respect to the +1 position of the pdxS gene and the locations of the p1 to p4 mutations are indicated.
PdxR is a DNA-binding protein
A C-terminally His6-tagged form of PdxR was purified to near homogeneity (see Experimental Procedures). In a gel-shift experiment, PdxR was able to bind a DNA fragment containing the entire pdxRS regulatory region with an apparent equilibrium dissociation constant (KD) of ~100 nM (Fig. 3A). (KD reflects the PdxR concentration needed to shift 50% of DNA fragments under conditions of vast PdxR excess over DNA). Binding of PdxR appeared to be strongly increased (see below) in the presence of PLP (KD≈1.6 nM), but was not affected by other B6 vitamers, PL, pyridoxine, or pyridoxamine 5′-phosphate (Fig. 3B and data not shown). The apparent ability of PLP to interact with PdxR is consistent with the presence of an aminotransferase-like PLP-binding domain in PdxR and with the in vivo observation that PL, a precursor of PLP, affected the ability of PdxR to activate pdxS expression. PdxR-DNA complexes with the same mobility were formed in the presence and absence of PLP, indicating that PLP does not affect the stoichiometry of PdxR binding (Figs. 3A and 3B). No binding to the regulatory region of L. monocytogenes prfA gene was detected even at 800 nM PdxR, with or without PLP, indicating specificity of PdxR interaction with pdxRS (data not shown).
Fig. 3. Binding of PdxR to the pdxRS regulatory region as detected by a gel-shift assay.
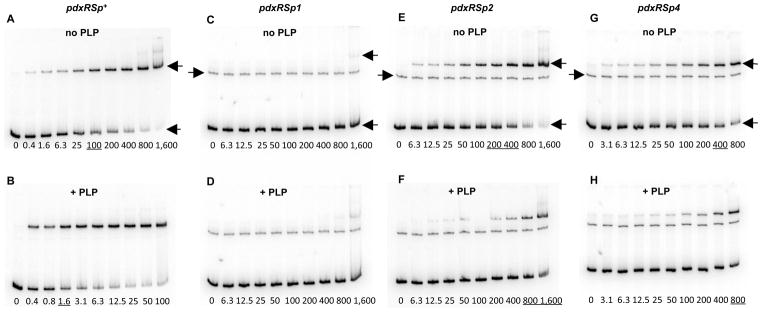
Radioactively labeled pdxRSp+ (A, B), pdxRSp1 (C, D), pdxRSp2 (E, F), and pdxRSp4 (G, H) DNA fragments were incubated with increasing amounts of purified PdxR without (A, C, E, G) or with (B, D, F, H) 100 μM PLP. PdxR monomer concentrations used (nM) are indicated below each lane, and the concentrations, which are needed to shift ~50% of DNA fragments, are underlined. The left-pointing arrows indicate the bands corresponding to unbound DNA and the PdxR-DNA complex, and the right-pointing arrows indicate the bands of unspecific DNA present in some experiments. Each gel-shift assay was repeated at least two times.
DNase I footprinting experiments showed that PdxR protected 30- to 35-nt regions on the bottom and top DNA strands from positions −72 to −43 or −66 to −32, respectively, with respect to the +1 position of the pdxS gene (Figs. 2, 4A, and 4B). The differences in the protected regions on the two strands reflect the known non-uniform and staggered pattern of DNase I cleavage. The protected region overlaps the −35 region of the pdxS promoter and lies downstream of the pdxR transcription start point. Two nucleotides, at positions −64 and −39 of the bottom strand, became hypersensitive to DNase I digestion, indicating an alteration of DNA conformation (often associated with bending) upon PdxR binding (Fig. 4C); the nucleotide at position −52 of the bottom strand, remained sensitive to DNase I digestion, though the flanking nucleotides became protected. Similar changes were detected for the top strand of pdxRS DNA (Fig. 4B).
Fig. 4. DNase I footprinting analysis of PdxR binding to the pdxRS regulatory region.


pdxRSp+ (A, B, C) or pdxRSp2 (C) DNA fragments, radioactively labeled on the bottom (A, C) or top (B) strand, were incubated with increasing concentrations of purified PdxR and in the absence or presence of 100 μM PLP. PdxR monomer concentrations used (nM) are indicated below each lane. The corresponding A + G sequencing ladders are shown in the left and right lanes. The protected areas are indicated by the vertical lines. The positions of some bands (with respect to the +1 position of pdxS) are shown by arrows.
In the presence of PLP, some repositioning of PdxR apparently occurred, as detected by an additional short area of protection around position −78 on the bottom stand (better seen at positions −77 and −74 of the top strand), the disappearance of the hypersensitive band at position −39 and appearance of a new hypersensitive band at position −51 of the bottom strand (Fig. 4). The observed repositioning of PdxR correlates with the strong positive effects of PLP on PdxR- DNA complex formation observed in the gel-shift experiments. Surprisingly, however, using the footprinting approach, it was found that PdxR affinity for DNA was high even in the absence of PLP and was not increased by more than 2-fold in the presence of PLP (Fig. 4, Table 2).
Table 2.
Summary of promoter activities and PdxR affinity for different pdxRS alleles
| Genotype | PL/PLPa | β-gal (pdxS-lacZ) | β-gluc (pdxR-gusA) | PdxR KD | |
|---|---|---|---|---|---|
| gel-shift | footprinting | ||||
| pdxRSp+ | − | 369 | 1.23 | 100 | 3.1–6.3 |
| + | 27.1 | 0.20 | 1.6 | 3.1–6.3 | |
| pdxRSp1 | − | 52.7 | 19.5 | >1,600 | ~1,600 |
| + | 12.4 | 14.0 | >1,600 | ~400 | |
| pdxRSp2 | − | 1,230 | 3.19 | 300 | ~3.1 |
| + | 805 | 12.2 | 1,200 | ~6.3 | |
| pdxRSp4 | − | 1,100 | 2.50 | 400 | ~3.1 |
| + | 605 | 9.16 | 800 | ~3.1 | |
Specific activities of β-galactosidase and β-glucuronidase in TSS medium, enriched with Casamino Acids, were taken from Table 3 and are expressed in Miller units. The values of apparent dissociation constants (KD) were determined from Fig. 3, 4, and 7 and are expressed as nanomolar concentration of monomeric PdxR. For footprinting experiments, KD is approximated as the PdxR concentration needed to decrease the intensity of protected DNA bands two-fold under conditions of vast PdxR excess over DNA.
indicates the presence of PL in the growth medium or PLP in the DNA-binding reactions.
The important difference between footprinting and gel-shift experiments is that free fragments of digested DNA or protein-DNA complexes are loaded on the gel, respectively. Therefore, the gel-shift experiments can reveal the actual affinity of proteins for DNA under tested conditions only if the protein-DNA complexes remain stable during the electrophoresis. Considering that low KD values (i.e., high affinities) determined by the two approaches in the presence of PLP were similar (Table 2), it was concluded that a higher KD value observed in the gel-shift experiments in the absence of PLP reflects not the actual lower affinity of PdxR for DNA, but rather a greater instability of protein-DNA complexes formed by PLP-free PdxR, compared to complexes formed by PLP-bound PdxR, during their loading onto the gel and/or electrophoresis. Despite this rather unusual caveat of the gel-shift experiments, they are fully consistent with the notion, derived from footprinting experiments, that PLP induces a conformational change in PdxR. This conformational change leads to repositioning of PdxR on DNA that correlates with the loss of PdxR’s ability to activate pdxS transcription in vivo and in vitro (see the next section) and also causes stabilization of the PdxR-DNA complex in gel-shift experiments. The repositioning of PdxR is rather subtle, does not involve any significant movement along the DNA strand, and probably affects DNA bending.
No effect of PLP or only a small effect of high concentrations of PLP on DNA binding was found previously in gel-shift experiments for C. glutamicum and S. pneumoniae PdxRs (El Qaidi et al., 2013; Jochmann et al., 2011). However, very low affinities of PdxR for DNA were observed in both cases; no footprinting experiments were reported.
PdxR is a PLP-responsive transcriptional activator of pdxS in vitro
In vitro transcription analysis of pdxS and pdxR expression was performed in run-off experiments using Escherichia coli RNA polymerase and a PCR-generated DNA fragment containing the entire pdxRS intergenic region. No expression from the pdxS promoter was detected in the absence of PdxR (Fig. 5A). Purified PdxR strongly activated transcription from the pdxS promoter. Addition of PLP reduced, in a concentration-dependent manner, formation of the pdxS transcript (Fig. 5B). PdxR also reduced expression from the pdxR promoter either in the presence or absence of PLP, indicating that it serves as a negative autoregulator (Fig. 5)(see also below). Very similar results were obtained with purified B. subtilis RNA polymerase (data not shown). Other B6 vitamers, PL, pyridoxine, pyridoxamine, and pyridoxamine 5′-phosphate, did not affect the activity of PdxR in vitro (data not shown). The addition of any one of 20 proteinogenic amino acids or various other amino group-containing compounds, including non-proteinogenic amino acids, polyamines, and amino sugars, did not affect the ability of PLP to reduce the PdxR-mediated activation of pdxS transcription (data not shown). Thus, PdxR appears to be a direct activator of pdxS and PLP acts as a direct anti-activator of PdxR and appears to be its only effector.
Fig. 5. In vitro transcription from the pdxRS regulatory region.

The pdxRSp+ PCR fragment was transcribed at 37°C for 20 min using E. coli RNA polymerase. (A) The reactions contained increasing amounts of PdxR and no PLP. PdxR monomer concentrations used (nM) are indicated below each lane. (B) The reactions contained no PdxR (first lane) or 200 nM PdxR and increasing concentrations of PLP as indicated below each lane.
Multiple transcripts formed from the pdxR and pdxS promoters apparently reflect RNA polymerase pausing at sites downstream from the corresponding transcription start points. Identity of the transcripts was proved by using pdxRS templates truncated at either the pdxR or pdxS end, which led to the formation of shorter corresponding transcripts (data not shown).
Transcriptional regulation of the pdxS and pdxR promoters in B. subtilis
A bidirectional pdxS-lacZ and pdxR-gusA transcriptional fusion containing the entire L. monocytogenes pdxR-pdxS intergenic region was constructed and integrated at the amyE chromosomal locus of B. subtilis as a prototrophic surrogate host; the B. subtilis cells do not contain a pdxR-like gene upstream of the intrinsic chromosomal pdxST genes, and the latter are not subject to B6-dependent regulation in vivo (unpublished results). Though expression of the pdxR-gusA fusion was readily detected, only extremely low expression of the pdxS-lacZ fusion was observed; no effect of medium composition on the expression of the fusions was observed in the absence of PdxR (Table 3 and data not shown, strain BB2306).
Table 3.
Expression of the bidirectional pdxRS fusion in B. subtilis cells
| Strain | Genotype | pdxRS promoter region | Additions to the medium | β-galactosidase (pdxS-lacZ) | β-glucuronidase (pdxR-gusA) |
|---|---|---|---|---|---|
| BB2306 | wild-type | p+ | Cas. acids | 0.41 ± 0.026 | 7.92 ± 0.16 |
| BB3311 | pdxR+ | none | 35.2 ± 0.35 | 0.62 ± 0.064 | |
| PL | 24.2 ± 1.84 | 0.53 ± 0.021 | |||
| Cas. acids | 369.0 ± 66.8 | 1.23 ± 0.15 | |||
| Cas. acids + PL | 27.1 ± 8.27 | 0.20 ± 0.007 | |||
| BB3630 | pdxT pdxR+ | none | 1260.0 ± 82.1 | 6.14 ± 0.39 | |
| PL | 30.7 ± 1.91 | 0.54 ± 0.021 | |||
| BB3308 | wild-type | p1 | Cas. acids | 0.38 ± 0.007 | 18.8 ± 0.42 |
| BB3312 | pdxR+ | Cas. acids | 52.7 ± 12.2 | 19.5 ± 0.14 | |
| Cas. acids + PL | 12.4 ± 1.32 | 14.0 ± 0.40 | |||
| BB3631 | pdxT pdxR+ | none | 163.0 ± 30.3 | 38.1 ± 3.89 | |
| PL | 19.0 ± 0.57 | 20.9 ± 0.57 | |||
| BB3309 | wild-type | p2 | Cas. acids | 0.47 ± 0.021 | 30.7 ± 0.42 |
| BB3313 | pdxR+ | none | 1090.0 ± 136.0 | 16.1 ± 0.14 | |
| Cas. acids | 1230.0 ± 14.7 | 3.19 ± 0.021 | |||
| Cas. acids + PL | 805.0 ± 35.7 | 12.2 ± 0.42 | |||
| BB3632 | pdxT pdxR+ | none | 1970.0 ± 206.0 | 3.66 ± 0.44 | |
| PL | 999.0 ± 134.0 | 16.6 ± 0.46 | |||
| BB3747 | wild-type | p4 | Cas. acids | 0.52 ± 0.035 | 20.0 ± 0.064 |
| BB3749 | pdxR+ | none | 873.0 ± 164.0 | 12.7 ± 0.21 | |
| Cas. acids | 1,100.0 ± 161.0 | 2.50 ± 0.35 | |||
| Cas. acids + PL | 605.0 ± 180.0 | 9.16 ± 0.69 | |||
| BB3751 | pdxT pdxR+ | none | 1,810.0 ± 30.1 | 3.50 ± 0.31 | |
| PL | 737.0 ± 81.5 | 13.1 ± 0.021 | |||
| BB3748 | wild-type | p1/p2 | Cas. acids | 0.40 ± 0.064 | 13.0 ± 2.13 |
| BB3750 | pdxR+ | Cas. acids | 65.2 ± 0.35 | 15.7 ± 0.85 | |
| Cas. acids + PL | 7.09 ± 1.15 | 13.3 ± 1.41 | |||
| BB3752 | pdxT pdxR+ | none | 220.0 ± 18.3 | 35.4 ± 5.16 | |
| PL | 8.43 ± 1.88 | 26.9 ± 1.74 |
Cells were grown in TSS glucose-ammonium medium with or without 0.5% vitamin-free Casamino Acids (Difco) and 100 μM PL. β-Galactosidase and β-glucuronidase specific activities were assayed and expressed in Miller units. All values are averages plus/minus standard deviation from at least two experiments.
When an intact copy of the L. monocytogenes pdxR gene with its own promoter was integrated at the ectopic sacA locus of the B. subtilis chromosome, much higher expression of the pdxS-lacZ fusion was found in glucose-ammonium minimal medium (Table 3, strain BB3311). This confirms that PdxR is necessary and sufficient for activation of the pdxS promoter and indicates that none of the seven members of the MocR/GabR subfamily (GabR, YdfD, YisV, YdeF, YdeL, YhdI, YcxD) or any other protein present in B. subtilis cells is able to substitute for PdxR. The activity of the pdxS-lacZ fusion increased an additional 36-fold under conditions of B6 limitation created by growing a pdxT null mutant, lacking one of the subunits of PLP synthase, in glucose-ammonium medium (Belitsky, 2004b)(Table 3, strain BB3630). Expression from the pdxS promoter was also elevated about 10-fold in Casamino acids-containing medium in a wild-type strain (Table 3), and that level of expression was increased a further 2.3- to 2.8-fold, to between 866 and 1,030 units, in rich media, such as L-broth, DS medium, or BHI. Increased expression from the pdxS promoter was suppressed when PL was present in the medium (Table 3 and data not shown). These results are fully consistent with the data obtained by real-time RT-PCR in L. monocytogenes and the notion that PdxR is a direct positive regulator of pdxS transcription, whose activity is decreased by PLP, which is formed intracellularly from exogenous PL. Moreover, the level of pdxS-lacZ expression can apparently be used as a reporter for the intracellular concentration of PLP and implies that the latter is reduced in media, containing amino acids and peptides, compared to the PLP concentration in minimal medium. This reduction is likely due to the intensive consumption of PLP during amino acid catabolism.
It should be noted that expression from the pdxS promoter is much higher in the presence, than in the absence of PdxR, even in PL-containing medium (Tables 1 and 3). This apparently reflects the ability of PLP-bound PdxR for low-level activation of the pdxS promoter and is in fact consistent with the in vitro transcription data (Fig. 5B).
Expression of the pdxR-gusA fusion was reduced >10-fold in the presence of PdxR, confirming the in vitro data showing that PdxR is a negative autoregulator. This negative autoregulation was less pronounced, i.e., expression from the pdxR promoter was increased, if the cells were experiencing B6 limitation in the presence of Casamino acids or due to the pdxT mutation, but was restored again in the presence of PL (Table 3), similar to the effects observed in L. monocytogenes cells (Table 1). It was concluded that after repositioning on DNA, PLP-bound PdxR not only loses the ability to activate pdxS but also becomes a stronger repressor of pdxR, i.e., PLP acts as anti-activator of PdxR with respect to pdxS expression and as a co-repressor of PdxR with respect to negative autoregulation. Both roles of PLP make sense because, in the presence of excess PLP, cells need expression of neither the de novo PLP-biosynthetic pathway, nor the activator of this pathway. For unknown reasons, the effect of PLP on the ability of PdxR to autoregulate its expression was observed only in vivo (both in L. monocytogenes and B. subtilis); in the transcription experiments in vitro even free PdxR was able to repress the pdxR promoter (Fig. 5A).
Mutational analysis of the PdxR-binding site
A perfect dyad-symmetry sequence, AATTGGATG-N10-CATCCAATT, was detected within the PdxR-binding site from positions -69 to -42 (with respect to the +1 position of pdxS) (Fig. 2). This dyad-symmetry element is highly conserved in the pdxRS intergenic regions of seven other Listeria spp. (Fig. 6). Single mutations were introduced into each of the 9-bp arms of this dyad symmetry. The p1 mutation, affecting position −65 of the distal arm, significantly reduced the ability of PdxR to activate pdxS expression and almost completely prevented negative autoregulation of pdxR (Table 3). A gel- shift experiment revealed that the apparent ability of PdxR to bind the pdxRSp1 regulatory region was virtually abolished (KD >1.6 μM) both in the absence and presence of PLP (Figs. 3C and 3D). The strong negative effect of the p1 mutation on PdxR binding was confirmed by a footprinting experiment (Fig. 7A, Table 2). It was concluded that the integrity of the distal arm is essential for efficient binding of PdxR and therefore for both activation of the pdxS promoter and repression of the pdxR promoter.
Fig. 6. Multiple alignment of the pdxRS intergenic regions from different Listeria species.

Nucleotide sequences of the entire pdxRS intergenic regions from eight Listeria strains were aligned using the CLUSTAL Omega software (http://www.ebi.ac.uk/Tools/msa/clustalo/). The sequences are flanked by the initiation codons (italicized) of the pdxR and pdxS genes at the 5′ and 3′ ends, respectively. The likely −10 and −35 promoter regions and the +1 position of the L. monocytogenes pdxS are in boldface. The dyad-symmetry sequence (in boldface) and the direct repeat within the PdxR-binding site are shown by arrows. Identical nucleotides in all genomes are indicated by asterisks. The numbering is with respect to the initiation codons of the pdxS genes. The accession numbers for the sequences analyzed are: L. fleischmannii (L.fl.) - NZ_ALWW01000008.1, L. grayi (L.gr.) - NZ_GL538353, L. seeligeri (L.sel.) - ADXK01001019, L. ivanovii (L.iv.) - ADXI01001082, L. innocua (L.in.) - AL596171, L. welshimeri (L.wel.) - NC_008555.1, L. marthii (L.mar.) - ADXF01000815.1, L. monocytogenes (L.mon.) - AL591982.1.
Fig. 7. DNase I footprinting analysis of PdxR binding to the mutant pdxRS regulatory regions.
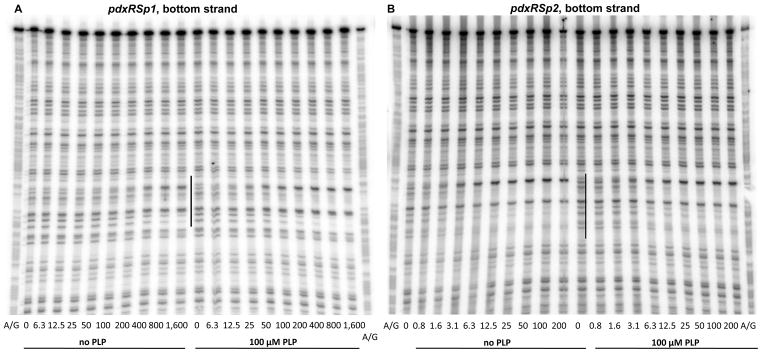
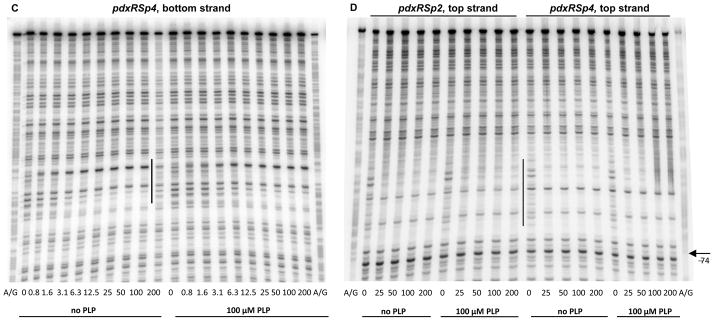
pdxRSp1 (A), pdxRSp2 (B, D), and pdxRSp4 (C, D) DNA fragments, radioactively labeled on the bottom (A, B, C) or top (D) strand, were incubated with increasing concentrations of purified PdxR and in the absence or presence of 100 μM PLP. PdxR monomer concentrations used (nM) are indicated below each lane. The corresponding A + G sequencing ladder is shown in the left and right lanes. The protected areas are indicated by vertical lines.
The p2 mutation at position −46 in the proximal arm of the PdxR-binding site, which is symmetrical to p1, (Fig. 2) had a very different and unusual phenotype. The mutation only slightly affected high pdxS-lacZ expression during B6 limitation (i.e., in the Casamino acids- containing medium or in the pdxT mutant cells), but almost completely prevented the reduction of pdxS expression observed in glucose-ammonium medium or when PL was added (Table 3, strains BB3313 and BB3632). In other words, the p2 mutation almost entirely abolished the ability of PL to inactivate PdxR and led to the B6-independent, constitutive expression of pdxS. This constitutive expression was still fully dependent on PdxR availability (Table 3, strains BB3309 and BB3313).
In a gel-shift experiment, the p2 mutation had only a small effect on the formation of unstable complexes of DNA with PdxR in the absence of PLP but completely prevented the stabilization of such complexes in the presence of PLP; in fact, the apparent KD was reduced 4- fold to ~1.2 μM by PLP addition (Figs. 3E and 3F, Table 2). Importantly, a DNase I protection experiment once again showed that the apparent low affinities of PdxR for p2-containing DNA, both in the absence and presence of PLP, were artifacts of gel-shift experiments, and therefore the p2 mutation did not actually impair PdxR binding (Figs. 7B and 7C, Table 2). However, the inability of PLP to stabilize the pdxRSp2-PdxR complex in a gel-shift assay correlated with the inability of PL to reduce pdxSp2-lacZ expression in vivo (Table 3) and indicated a different mode of PdxR binding to p2-containing DNA. Indeed, the DNase I protection experiment revealed that, in the presence of the p2 mutation, additional residues of the bottom strand, at positions −41 and −40, became protected when PdxR was added. Importantly, the footprinting pattern was not affected by PLP addition and no protection of the −78 region was observed on either strand (Figs. 4C, 7B, and 7D). With regard to positions −39 and −51 and the −78 region, DNA containing the p2 mutation showed the same pattern of protection by free and PLP-bound PdxR as did wild-type DNA in the presence of free PdxR. In all these cases, the resulting protein-DNA complex contained PdxR in a position and conformation that were appropriate for transcription activation from the pdxS promoter. This particular positioning and conformation of PdxR apparently make the complexes with DNA very unstable in a gel-shift experiment.
Considering the unique phenotype of the p2 mutation, another single mutation, p4, was independently introduced at position −47 in the proximal arm of the PdxR-binding site, adjacent to the position of the p2 mutation (Fig. 2). The two single mutations had almost identical phenotypes in vivo (Table 3). The effects of the p4 mutation on PdxR binding were also very similar to the effects of the p2 mutation (Figs. 3G, 3H, 7C, and 7D), consistent with their similar phenotypes in vivo.
To summarize, in the absence of PLP, PdxR molecules interact with the wild-type DNA in a way that allows RNA polymerase to initiate pdxS transcription (Fig. 8). PdxR molecules, which interact with the p2- (or p4)-containing DNA, appear to be locked in a similar position; this positioning of PdxR on the mutant DNA does not change even in the presence of PLP. The constitutive phenotype of the p2- or p4-containing strains indicates that the PLP-induced alteration of PdxR conformation does not prevent transcription activation per se, but rather leads to the altered positioning of PdxR on the wild-type DNA, which is not compatible with the activation of the pdxS promoter (Fig. 8). As far as I know, the p2 and p4 mutations are the first examples of mutations in the activator-binding site, which prevent anti-activation by a low-molecular weight effector and allow constitutive expression of the target genes.
Fig. 8. A model of PdxR interaction with the wild-type and mutant pdxRS regulatory regions.

(A) wild-type DNA, no PdxR; (B) wild-type DNA plus PdxR; (C) wild-type DNA plus PdxR and PLP; (D) p2- or p4-containing DNA plus PdxR and PLP. The model (not to scale) assumes that a PdxR dimer binds to the dyad-symmetry element (indicated by straight arrows) and includes hypothetical PLP-dependent bending induced by PdxR. Bent arrows show the start points and directions of transcription. An asterisk indicates the location of the adjacent p2 and p4 mutations. The position -78 (with respect to pdxS), protected by the PLP-bound form of PdxR on wild-type DNA, is shown.
Interestingly, the ability of the p2 mutation to confer constitutive expression from the pdxS promoter was abolished in the presence of the p1 mutation (i.e., low expression from the pdxSp1/p2 promoter was further strongly reduced in the presence of PL) and is, thus, dependent on the integrity of the distal arm of the PdxR-binding site (Table 3). Incidentally, the combination of the p1 and p2 mutations restored the perfect inverted repeat within the PdxR-binding region (Fig. 2). Because the activity of the p1/p2 promoter remained low, it is the exact sequence of the element and not the dyad symmetry per se that is important for productive interaction with PdxR.
In accord with the observation that PLP-bound PdxR binds to the pdxRSp2 and pdxRSp4 fragments in a manner similar to that of binding of free PdxR to wild-type DNA, both the p2 and p4 mutations abolished the ability of PLP-bound PdxR to repress the pdxR promoter (Table 3). Somewhat paradoxically, some ability to repress the pdxR promoter containing the p2 or p4 mutation was regained under conditions of B6 limitation, indicating that PdxR was still able to interact with PLP and the resulting change in PdxR conformation (or an undetected change in PdxR positioning) affected repression. Additionally, all three mutations, p1, p2, and p4, led to a 2.4- to 3.8-fold increase in the level of pdxR expression in the absence of PdxR, apparently affecting the intrinsic ability of RNA polymerase to initiate transcription from the pdxR promoter (Table 3); the reason for this effect remains unknown.
One more single mutation, p3, was introduced at the conserved −76 position in the region of the PdxR-binding site that is specifically protected on wild-type DNA only in the presence of PLP (Fig. 2). This mutation did not affect the ability of PdxR to regulate expression of the pdxS- lacZ or pdxR-gusA fusions; expression of the latter was reduced ~3-fold under all conditions apparently due to the location of the p3 mutation within the pdxR ribosomal binding site (data not shown).
DISCUSSION
It has been shown that L. monocytogenes PdxR, a member of the MocR/GabR subfamily of the GntR family of transcriptional regulators, is an activator of the pdxST genes responsible for the de novo biosynthesis of PLP. A similar function in other bacteria was previously demonstrated or suggested for three more members of this subfamily, also called PdxR (El Qaidi et al., 2013; Jochmann et al., 2011; Magarvey et al., 2001). It was shown here that L. monocytogenes PdxR responds directly to PLP, which serves as the only effector of the protein. The binding of PLP to L. monocytogenes PdxR is sufficient to cause a likely conformational change and repositioning of the protein on DNA, which affect the ability of the protein both to activate and repress (Fig. 8). As far as I know, the requirement for a B6 vitamer for modulation of activity of a transcriptional regulator is unprecedented among bacteria.
From the mutational analysis, it was concluded that the distal arm of the dyad-symmetry element found within the L. monocytogenes PdxR-binding site is essential for efficient binding of the protein and the proximal arm appears to be required for such an interaction with PdxR that allows the protein to respond to the presence of PLP. Based on their similarity to aminotransferases, the MocR/GabR subfamily members were hypothesized to form head-to-tail dimers and, therefore, bind to direct DNA repeats (Rigali et al., 2002). The head-to-tail dimer configuration of such proteins was confirmed for B. subtilis GabR (Edayathumangalam et al., 2013) and is likely to be true for PdxR. In this respect it is worth noting that a 9-bp sequence, AATTGGcTG, directly repeating the distal arm of the dyad-symmetry element (with one mismatch indicated in lowercase), can be found 15 bp downstream of the first repeat and still within the PdxR-binding site; this sequence overlaps the proximal arm of the dyad symmetry by 4 bp (Fig. 2 and 6). The role of this direct repeat in PdxR binding was not analyzed. Sites similar to these inversely and directly repeated 9-bp sequences were not detected in the binding regions of other characterized proteins of the MocR/GabR subfamily (Belitsky, 2004a; El Qaidi et al., 2013; Jochmann et al., 2011; Wiethaus et al., 2008). Similarly to PdxR, GabR binds to an extended region of DNA, which overlaps the −35 promoter region of the target gene (Belitsky, 2004a).
The signaling role of PLP with respect to PdxR activity is in drastic contrast to the mode of regulation of the activity of GabR, the best characterized member of the MocR/GabR subfamily. In addition to PLP, GabR requires another low-molecular weight compound, GABA, for modulation of its activity (Belitsky, 2004a). Moreover, GabR binds PLP through a covalent Schiff-base linkage, in which the ε-amino group of Lys312 forms an imine bond with the aldehyde group of PLP (Edayathumangalam et al., 2013). PLP appears to act as a tightly bound intrinsic component of GabR, akin to the cofactor role, which PLP plays in aminotransferases and other PLP-dependent enzymes (Eliot and Kirsch, 2004; Schneider et al., 2000). Furthermore, the data strongly indicate that to achieve its active state, GabR performs a transimination reaction between PLP and GABA, in which the imine linkage between Lys312 and PLP is broken and a new imine bond is formed between PLP, which remains bound to GabR non-covalently, and GABA (Belitsky, 2004a). A similar cofactor-like role of PLP is possible for other members of the MocR/GabR-like proteins. For instance, to activate TauR, PLP may interact covalently with taurine, a likely TauR effector (Wiethaus et al., 2008). For such proteins that are not involved in regulation of PLP metabolism, it is hypothesized that the apparent conformational change, which causes their activation, is associated not with PLP binding per se but with binding of another low-molecular compound, such as GABA or taurine, and with the subsequent modification of both this compound and PLP.
It is likely that L. monocytogenes PdxR and other similar proteins, which regulate genes involved in PLP biosynthesis, form a special group within the MocR/GabR subfamily. Members of the PdxR group, like L. monocytogenes PdxR and in contrast to the proteins of the GabR-like group, are hypothesized to respond only to PLP and not to employ additional low-molecular weight compounds for modulation of their activity. In accord with this suggestion, some PdxR proteins do not have the conserved lysine residue that forms a covalent Schiff-base bond with PLP in GabR and aminotransferases (Bramucci et al., 2011; El Qaidi et al., 2013; Jochmann et al., 2011). Assuming that different PdxR proteins interact with PLP in a similar way, it is suggested that for all of them PLP acts as a simple, low-molecular weight effector that causes a conformational change upon non-covalent binding. The aminotransferase-like domain of such proteins, in contrast to GabR-like proteins, is likely to serve only as a PLP-binding domain and does not perform any chemical modification of PLP.
EXPERIMENTAL PROCEDURES
Bacterial strains and culture media
All L. monocytogenes and B. subtilis strains constructed in this study were derivatives of strains EGD-e (Glaser et al., 2001) or SMY (Zeigler et al., 2008), respectively, and are described in Table S1 and in the text. E. coli strain JM107 (Yanisch-Perron et al., 1985) or its pcnB80 derivative (Lopilato et al., 1986) was used for isolation of plasmids. Cells were grown in Brain Heart Infusion medium (BHI) or, for experiment in Fig. 1, in defined medium (LDM), for L. monocytogenes; in DS nutrient broth medium or TSS minimal medium with 0.5% glucose as the carbon source and 0.2% NH4Cl as nitrogen source (Fouet and Sonenshein, 1990), for B. subtilis; and in L broth (Miller, 1972), for E. coli. The same media with addition of agar were used for growth of bacteria on plates. LDM was based on published recipes (Premaratne et al., 1991; Welshimer, 1963) and on the composition of TSS (for K2HPO4, MgSO4 x7H2O, and iron citrate) and DS [for Ca(NO3)2] media and had the following 18 components: MOPS-HCl (pH 7.5), 50 mM; K2HPO4, 2 mM; MgSO4 x7H2O, 0.81 mM (0.02%); a mixture of FeCl3 and Na3-citrate x2H2O, 0.04 g l−1 each (0.004%); Ca(NO3)2, 0.5 mM; glucose, 27.8 mM (0.5%); NH4Cl, 37.4 mM (0.2%); seven amino acids (Leu, Ile, Val, Cys, Met, Arg-HCl, His-HCl) at 100 μg ml−1, each; and four vitamins (in μg ml−1): biotin, 0.5; riboflavin, 0.5; thiamine-HCl, 1; and lipoic acid, 0.005.
The following antibiotics were used when appropriate (in μg ml−1): ampicillin, 50–100; chloramphenicol, 20; tetracycline, 10; spectinomycin, 50; and nalidixic acid, 50, for E. coli strains; chloramphenicol, 5 or 10; and spectinomycin, 100, for L. monocytogenes strains; and chloramphenicol, 2.5; neomycin, 2.5; tetracycline, 15; and spectinomycin, 50, for B. subtilis strains.
General molecular genetic methods
Methods for common DNA manipulations, E. coli electroporation, isolation of B. subtilis chromosomal DNA, transformation of B. subtilis cells, and sequence analysis were as previously described (Belitsky and Sonenshein, 1998; Belitsky and Sonenshein, 2008). Chromosomal DNA of L. monocytogenes was isolated as for B. subtilis, but the cells were disrupted using 0.1 mm silica beads and a Mini-BeadBeater (Biospec Products) for two 30-sec cycles at the maximal setting. All oligonucleotides used in this work are described in Table S2. Chromosomal DNA of L. monocytogenes or plasmids constructed in this work was used as template for PCR. All cloned PCR-generated fragments were verified by sequencing at the Tufts University Core Facility.
Construction of pdxST and pdxR null mutants
A ΔpdxST::spc fragment, in which 97% (1.42 kb) of the pdxS and pdxT open reading frames was replaced by a 1.1-kb spectinomycin-resistance cassette, was constructed by a two-step overlapping PCR method (Wach, 1996). First, 5′- and 3′-flanking fragments were generated by using oBB130 and oBB131 or oBB132 and oBB133 as primers, respectively. Primers oBB131 and oBB132 contain sequences that match the ends of the spc gene from pJL73 (LeDeaux and Grossman, 1995). In the second step, the two PCR products were used as long primers for splicing PCR with pJL73 as template. The spliced product was further amplified using oBB130 and oBB133. The final PCR product was digested with SpeI (a site within pdxR) and SacI and cloned in a temperature-sensitive conjugative plasmid pCON-1 (Smith and Youngman, 1992), digested with XbaI and SacI. The orientation of the spc cassette in the resulting plasmid, pBB1212, was the same as the orientation of the pdxST genes.
To make a ΔpdxR::spc construct, we first deleted a HindIII site from the polylinker of pCON-1 by digesting the plasmid with HindIII, blunt ending the resulting fragment with the DNA polymerase I Klenow fragment, and self-ligating the fragment to obtain pBB1315. The fragment containing the entire pdxR gene was synthesized by PCR using oBB131 and oBB168 as primers. The PCR fragment was digested with PstI and XbaI, cloned in pLG103 (Belitsky and Sonenshein, 2002), and then recloned as an EcoRI-XbaI fragment in pBB1315 to generate pBB1323. To construct pBB1324, a 0.28-kb internal HindIII-SpeI fragment of pdxR was replaced with a 1.1-kb spc cassette (in the orientation opposite to pdxR) that was cut from pJL73 with the same two enzymes.
pBB1212 (cat ΔpdxST::spc) and pBB1324 (cat ΔpdxR::spc) were introduced into an E. coli conjugation donor strain, DW1030/RK231.7 (Tetr) (Thompson and Malamy, 1990) (plasmid RK231.7 is a Kans derivative of RK231 obtained by A. Baughn). The two resulting strains were conjugated at 30°C with L. monocytogenes strain EGD-e, as a plasmid recipient, on BHI agar plates, selecting for chloramphenicol and spectinomycin resistance and using nalidixic acid to counterselect the donor strains. The transconjugants were passaged several times in liquid BHI under selective conditions at 41°C to allow integration of pCON-1 derivatives into the chromosome, and the resulting integrants were passaged several times at non-selective conditions (i.e., in the absence of chloramphenicol but in the presence of spectinomycin and 10 μM of PL) at 30°C to allow for plasmid excision and allelic replacement. Spectinomycin- resistant colonies were isolated at 41°C in the presence of PL and screened for the loss of chloramphenicol-resistance. The replacement of the chromosomal genes by the deletion- insertion-containing alleles in strains BLM1 (pdxST) and BLM7 (pdxR) was searched for by comparing the sizes of PCR fragments from the wild-type and mutant pdxRST chromosomal loci; the loss of the plasmid was confirmed in separate PCR reactions using vector-specific primers.
Complementation of the pdxST and pdxR mutations
A PCR product containing the entire pdxST genes was synthesized by PCR using oBB164 and oBB133 as primers and cut with BclI and HindIII. The resulting 1.88-kb fragment was cloned between the BamHI and HindIII sites of a low copy-number vector pHP13 (Haima et al., 1987). A 1.59-kb PCR product containing the entire pdxR gene was synthesized by PCR using oBB131 and oBB681 as primers, cut with EcoRI and BamHI, and cloned in pHP13. pHP13 and its derivatives were introduced into L. monocytogenes cells by electroporation (Monk et al., 2008).
Isolation of RNA
Samples of 1 to 3 ml of L. monocytogenes cells growing exponentially in BHI with or without 100 μM PL were collected at the OD600≈0.6 by mixing with an equal volume of an 1:1 (vol/vol) mixture of ethanol/acetone (−20°C) and kept at −80°C until further use. The cells were pelleted, washed with 1 ml of 10 mM tris-HCl (pH 8.0) - 1 mM EDTA buffer, and resuspended in 0.8 ml of the TRI Reagent. The cells were disrupted using a beadbeater as described above, and RNA was purified using the Direct-zol RNA kit (Zymo Research). For real-time RT-PCR experiments, purified RNA (~10 μg) was further treated with Turbo DNA- free DNase I (Ambion) according to the manufacturer’s instructions. RNA was quantified using the NanoDrop ND-1000 spectrophotometer (Thermo Scientific).
Real-time RT-PCR
cDNA was synthesized starting from 1 μg of RNA using random hexamer primers and SuperScript II reverse transcriptase (Invitrogen) per the manufacturer’s instructions. The SYBR Green I dye-based PCR reactions were performed according to the manufacturer’s instructions using a LightCycler 480 (Roche Applied Science). The reactions were done in a total volume of 20 μl and contained 4 μl of 6- to 10-fold diluted cDNA or control RNA samples. Primer pairs oBB657-oBB658 or oBB660-oBB661 were used for detection of pdxS or pdxR transcripts, respectively. The rpoB transcript, detected with primers oBB667 and oBB668, was used for normalization. All primers were designed using the PrimerQuest tool (Integrated DNA Technologies). Serial dilutions of L. monocytogenes chromosomal DNA (from 3.2 to 10,000 pg per reaction) were used to create calibration curves for each transcript.
Determination of transcription start points using rapid amplification of cDNA ends (RACE)
cDNA samples were synthesized from 1 μg of RNA in 20-μl reactions using 2 pmol of pdxS- or pdxR-specific primers, oBB154 or oBB130, respectively, and SuperScript II reverse transcriptase (Invitrogen) per the manufacturer’s instructions. 3′ poly(A) tails were added using terminal transferase (New England Biolabs), and cDNA was purified using the PCR Purification kit (Qiagen) and eluted in a 50-μl volume. First-round PCR products were generated using 2 μl of purified cDNA as template, universal anchor primer oKZ69, and primers oBB658 or oBB660 specific for the pdxS and pdxR genes, respectively. The pdxS-specifc product was sequenced directly using primer oBB659. The pdxR-specific product (0.05 μl) was PCR-amplified again using the universal amplification primer oKZ70 and the pdxR-specific primer oBB164 and then sequenced using oBB164. The nucleotide(s) at the junction between the gene-specific sequence and the stretch of A nucleotides, generated from sequencing the poly(A) tail, was assumed to be an apparent 5′ end of mRNA (Frohman, 1994).
Overexpression and purification of PdxR
A PCR product containing the entire pdxR gene with six histidine codons at the 3′ end was synthesized by PCR using oBB162 and oBB163 as primers and cloned downstream of the ara promoter between the EcoRI and SphI sites of the expression vector pBAD18 (Guzman et al., 1995).
The resulting plasmid, pBB1266, was introduced into E. coli strain LMG194 (Δara714) (Guzman et al., 1995), and expression of PdxR was induced in L broth (at OD600≈0.3) by adding L-arabinose to 0.2% and incubation of the cells for an additional 4 hours. PdxR-His6 was purified to almost homogeneity as described previously for GabR (Belitsky, 2004a). Elution from the Ni2+-affinity column (His·Bind resin; Novagen) was with a buffer containing 385 mM imidazole.
In vitro transcription
Assays with E. coli σA-containing RNA polymerase holoenzyme (Epicentre) or a mixture of purified B. subtilis RNA polymerase and σA factor were performed in vitro as described (Belitsky, 2004a; Belitsky and Sonenshein, 2011b). Various amounts of PdxR with or without 100 μM PLP were added to 10-μl reactions as specified in figure legends. A 281-bp pdxRS promoter fragment was synthesized, using pBB1279 as a PCR template and oBB164 and a vector-specific oligonucletide oBB102 as primers, and used as template for in vitro transcription.
Construction of pdxS and pdxR transcriptional fusions
The 0.23-kb fragment, containing the entire 125-bp pdxRS intergenic region, was synthesized by PCR using primers oBB164 and oBB131, digested with BclI and PstI, and cloned between the BglII and SbfI sites of the integrative plasmid pLG103 (Belitsky and Sonenshein, 2002). The resulting plasmid, pBB1279, carries the bidirectional pdxSp+-lacZ and pdxRp+-gusA fusion.
Mutations in the pdxRS regulatory region were introduced by two-step overlapping PCR. In the first step, PCR products containing the 5′ part of the pdxRS regulatory region were synthesized by using oBB164 and mutagenic oligonucleotides, specified in Table S2, as primers. In a similar manner, PCR products containing the 3′ part of the pdxRS regulatory region were synthesized by using mutagenic oligonucleotides, specified in Table S2, and a vector-specific oligonucleotide oBB253 as primers. In the case of the double mutation p1/p2, plasmid pBB1648 (pdxRSp2) was used as template for PCR. The appropriate pairs of PCR products were used in a second, splicing step of PCR mutagenesis as overlapping templates to generate modified fragments containing the entire pdxRS regulatory region; oligonucleotides oBB164 and oBB253 served as primers. The spliced PCR products were cloned in pLG103, as described above, to construct pBB1648 (pdxRSp1), pBB1649 (pdxRSp2), pBB1650 (pdxRSp3), pBB1781 (pdxRSp4), and pBB1782 (pdxRSp1/p2).
B. subtilis strains carrying various lacZ and gusA fusions at the amyE locus were isolated after transforming strain BB1888 (lacA) or BB2511 (amyE::spc lacA) with the appropriate plasmids, by selecting for resistance to chloramphenicol conferred by the plasmids. The transformants were screened for the Amy− phenotype (Belitsky and Sonenshein, 1998) or loss of the spectinomycin-resistance phenotype, which indicated a double crossover, homologous recombination event.
Integration of pdxR into the B. subtilis chromosome
The same pdxR-containing fragment that was used to generate pBB1323 was cloned between the XbaI and EcoRI sites of the integrative plasmid pSac-Kan (Middleton and Hofmeister, 2004) in strain JM107 pcnB80 as a host. The resulting plasmid, pBB1647, was introduced into B. subtilis strain BB3304 (sacA::cat) to create strain BB3305 [sacA::(neo pdxR+)].
Labeling of DNA fragments
The 338-bp PCR products containing the entire pdxRS intergenic region were synthesized using vector-specific oligonucleotides oBB252 and oBB102 as primers and pBB1279 or its mutant derivatives as template. One of the primers for each PCR reaction was labeled using T4 polynucleotide kinase and [γ-32P]-ATP. The labeled PCR products were purified on an 8% nondenaturing polyacrylamide gel or used without purification. oBB252 and oBB102 start 62 bp upstream or 47 bp downstream, respectively, of the BclI and SbfI sites that were used for cloning.
Gel shift assays and DNase I protection experiments
Incubation of CodY with the 32P-labeled promoter fragments was performed in a binding buffer containing 20 mM Tris-Cl (pH 8.0) - 50 mM KCl - 2 mM MgCl2 - 5% glycerol - 0.5 mM EDTA - 1 mM DTT - 0.05% Nonidet P-40 - 25 μg ml−1 sonicated salmon sperm DNA. Samples (11 μl) containing varying amounts of PdxR and less than 1 fmole of DNA were incubated for 16 min at room temperature and separated on 8% nondenaturing 50 mM Tris - 384 mM glycine - 1 mM EDTA polyacrylamide gels in 35 mM Hepes - 43 mM imidazole buffer. In some experiments, B6 vitamers were present in the incubation mixture.
For DNase I protection experiments, samples containing 20–40 fmoles of labeled DNA were incubated with PdxR as described above. One μl of the binding buffer containing 0.1–0.2 U RQ1 DNase I (Promega), 10 mM MgCl2 and 20 mM CaCl2 was then added, followed by addition, after 1 min, of 4 μl of 20 mM EDTA-95% formamide dye solution and subsequent heating of the samples at 80°C for 5 min. The samples were loaded without further purification on 7 M urea - 6% polyacrylamide DNA sequencing gels. The G+A sequencing ladder, generated according to a published procedure by boiling the appropriate samples of labeled DNA for 20 min (Liu and Hong, 1998), served to locate precisely the protected region.
The gels were dried, and the radioactive bands were detected and quantified using storage screens, an Applied Biosystems PhosphorImager, and ImageQuant software (GE Healthcare).
Enzyme assays
β-Galactosidase and β-glucuronidase specific activities were determined as described previously (Belitsky et al., 1995).
Supplementary Material
Acknowledgments
I am grateful to A. L. Sonenshein for encouragement, helpful discussions, and careful reading of the manuscript and to D. E Higgins and A. D. Baughn for the gift of strains and sharing the experimental protocols. This work was supported by research grants from the National Science Foundation (MCB-0110651) to B. R. B. and from the U. S. Public Health Service (GM042219) to A. L. Sonenshein.
Footnotes
No conflict of interest is declared.
References
- Belitsky BR. Bacillus subtilis GabR, a protein with DNA-binding and aminotransferase domains, is a PLP-dependent transcriptional regulator. J Mol Biol. 2004a;340:655–664. doi: 10.1016/j.jmb.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Belitsky BR. Physical and enzymological interaction of Bacillus subtilis proteins required for de novo pyridoxal 5′-phosphate biosynthesis. J Bacteriol. 2004b;186:1191–1196. doi: 10.1128/JB.186.4.1191-1196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky BR, Janssen PJ, Sonenshein AL. Sites required for GltC-dependent regulation of Bacillus subtilis glutamate synthase expression. J Bacteriol. 1995;177:5686–5695. doi: 10.1128/jb.177.19.5686-5695.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky BR, Sonenshein AL. Role and regulation of Bacillus subtilis glutamate dehydrogenase genes. J Bacteriol. 1998;180:6298–6305. doi: 10.1128/jb.180.23.6298-6305.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky BR, Sonenshein AL. GabR, a member of a novel protein family, regulates utilization of γ-aminobutyrate in Bacillus subtilis. Mol Microbiol. 2002;45:569–583. doi: 10.1046/j.1365-2958.2002.03036.x. [DOI] [PubMed] [Google Scholar]
- Belitsky BR, Sonenshein AL. Genetic and biochemical analysis of CodY-binding sites in Bacillus subtilis. J Bacteriol. 2008;190:1224–1236. doi: 10.1128/JB.01780-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belitsky BR, Sonenshein AL. Roadblock repression of transcription by Bacillus subtilis CodY. J Mol Biol. 2011b;411:729–743. doi: 10.1016/j.jmb.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramucci E, Milano T, Pascarella S. Genomic distribution and heterogeneity of MocR-like transcriptional factors containing a domain belonging to the superfamily of the pyridoxal-5′-phosphate dependent enzymes of fold type I. Biochem Biophys Res Commun. 2011;415:88–93. doi: 10.1016/j.bbrc.2011.10.017. [DOI] [PubMed] [Google Scholar]
- Dick T, Manjunatha U, Kappes B, Gengenbacher M. Vitamin B6 biosynthesis is essential for survival and virulence of Mycobacterium tuberculosis. Mol Microbiol. 2010;78:980–988. doi: 10.1111/j.1365-2958.2010.07381.x. [DOI] [PubMed] [Google Scholar]
- Edayathumangalam R, Wu R, Garcia R, Wang Y, Wang W, Kreinbring CA, Bach A, Liao J, Stone TA, Terwilliger TC, Hoang QQ, Belitsky BR, Petsko GA, Ringe D, Liu D. Crystal structure of Bacillus subtilis GabR, an autorepressor and transcriptional activator of gabT. Proc Natl Acad Sci U S A. 2013;110:17820–17825. doi: 10.1073/pnas.1315887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Qaidi S, Yang J, Zhang JR, Metzger DW, Bai G. The vitamin B(6) biosynthesis pathway in Streptococcus pneumoniae is controlled by pyridoxal 5′-phosphate and the transcription factor PdxR and has an impact on ear infection. J Bacteriol. 2013;195:2187–2196. doi: 10.1128/JB.00041-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick TB, Amrhein N, Kappes B, Macheroux P, Tews I, Raschle T. Two independent routes of de novo vitamin B6 biosynthesis: not that different after all. Biochem J. 2007;407:1–13. doi: 10.1042/BJ20070765. [DOI] [PubMed] [Google Scholar]
- Fouet A, Sonenshein AL. A target for carbon source-dependent negative regulation of the citB promoter of Bacillus subtilis. J Bacteriol. 1990;172:835–844. doi: 10.1128/jb.172.2.835-844.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman MA. On beyond classic RACE (rapid amplification of cDNA ends) PCR Methods Appl. 1994;4:S40–58. doi: 10.1101/gr.4.1.s40. [DOI] [PubMed] [Google Scholar]
- Glaser P, Frangeul L, Buchrieser C, Rusniok C, Amend A, Baquero F, Berche P, Bloecker H, Brandt P, Chakraborty T, Charbit A, Chetouani F, Couve E, de Daruvar A, Dehoux P, Domann E, Dominguez-Bernal G, Duchaud E, Durant L, Dussurget O, Entian KD, Fsihi H, Garcia-del Portillo F, Garrido P, Gautier L, Goebel W, Gomez-Lopez N, Hain T, Hauf J, Jackson D, Jones LM, Kaerst U, Kreft J, Kuhn M, Kunst F, Kurapkat G, Madueno E, Maitournam A, Vicente JM, Ng E, Nedjari H, Nordsiek G, Novella S, de Pablos B, Perez-Diaz JC, Purcell R, Remmel B, Rose M, Schlueter T, Simoes N, Tierrez A, Vazquez-Boland JA, Voss H, Wehland J, Cossart P. Comparative genomics of Listeria species. Science. 2001;294:849–852. doi: 10.1126/science.1063447. [DOI] [PubMed] [Google Scholar]
- Grishin NV, Phillips MA, Goldsmith EJ. Modeling of the spatial structure of eukaryotic ornithine decarboxylases. Protein Sci. 1995;4:1291–1304. doi: 10.1002/pro.5560040705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A, Phillips A, Thibonnier M, Kaparakis-Liaskos M, Johnson C, Thiberge JM, Radcliff FJ, Ecobichon C, Labigne A, de Reuse H, Mendz GL, Ferrero RL. Vitamin B6 is required for full motility and virulence in Helicobacter pylori. MBio. 2010:1. doi: 10.1128/mBio.00112-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haima P, Bron S, Venema G. The effect of restriction on shotgun cloning and plasmid stability in Bacillus subtilis Marburg. Mol Gen Genet. 1987;209:335–342. doi: 10.1007/BF00329663. [DOI] [PubMed] [Google Scholar]
- Jochmann N, Gotker S, Tauch A. Positive transcriptional control of the pyridoxal phosphate biosynthesis genes pdxST by the MocR-type regulator PdxR of Corynebacterium glutamicum ATCC 13032. Microbiology. 2011;157:77–88. doi: 10.1099/mic.0.044818-0. [DOI] [PubMed] [Google Scholar]
- LeDeaux JR, Grossman AD. Isolation and characterization of kinC, a gene that encodes a sensor kinase homologous to the sporulation sensor kinases KinA and KinB in Bacillus subtilis. J Bacteriol. 1995;177:166–175. doi: 10.1128/jb.177.1.166-175.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ST, Hong GF. Three-minute G + A specific reaction for DNA sequencing. Anal Biochem. 1998;255:158–159. doi: 10.1006/abio.1997.2457. [DOI] [PubMed] [Google Scholar]
- Lopilato J, Bortner S, Beckwith J. Mutations in a new chromosomal gene of Escherichia coli K-12, pcnB, reduce plasmid copy number of pBR322 and its derivatives. Mol Gen Genet. 1986;205:285–290. doi: 10.1007/BF00430440. [DOI] [PubMed] [Google Scholar]
- Magarvey N, He J, Aidoo KA, Vining LC. The pdx genetic marker adjacent to the chloramphenicol biosynthesis gene cluster in Streptomyces venezuelae ISP5230: functional characterization. Microbiology. 2001;147:2103–2112. doi: 10.1099/00221287-147-8-2103. [DOI] [PubMed] [Google Scholar]
- Middleton R, Hofmeister A. New shuttle vectors for ectopic insertion of genes into Bacillus subtilis. Plasmid. 2004;51:238–245. doi: 10.1016/j.plasmid.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y: 1972. [Google Scholar]
- Monk IR, Gahan CG, Hill C. Tools for functional postgenomic analysis of Listeria monocytogenes. Appl Environ Microbiol. 2008;74:3921–3934. doi: 10.1128/AEM.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percudani R, Peracchi A. A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 2003;4:850–854. doi: 10.1038/sj.embor.embor914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premaratne RJ, Lin WJ, Johnson EA. Development of an improved chemically defined minimal medium for Listeria monocytogenes. Appl Environ Microbiol. 1991;57:3046–3048. doi: 10.1128/aem.57.10.3046-3048.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigali S, Derouaux A, Giannotta F, Dusart J. Subdivision of the helix-turn-helix GntR family of bacterial regulators in the FadR, HutC, MocR, and YtrA subfamilies. J Biol Chem. 2002;277:12507–12515. doi: 10.1074/jbc.M110968200. [DOI] [PubMed] [Google Scholar]
- Schneider G, Kack H, Lindqvist Y. The manifold of vitamin B6 dependent enzymes. Structure Fold Des. 2000;8:R1–6. doi: 10.1016/s0969-2126(00)00085-x. [DOI] [PubMed] [Google Scholar]
- Smith K, Youngman P. Use of a new integrational vector to investigate compartment-specific expression of the Bacillus subtilis spoIIM gene. Biochimie. 1992;74:705–711. doi: 10.1016/0300-9084(92)90143-3. [DOI] [PubMed] [Google Scholar]
- Thompson JS, Malamy MH. Sequencing the gene for an imipenem-cefoxitin-hydrolyzing enzyme (CfiA) from Bacteroides fragilis TAL2480 reveals strong similarity between CfiA and Bacillus cereus beta-lactamase II. J Bacteriol. 1990;172:2584–2593. doi: 10.1128/jb.172.5.2584-2593.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, Barthelemy M, Vergassola M, Nahori MA, Soubigou G, Regnault B, Coppee JY, Lecuit M, Johansson J, Cossart P. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev. 2001;14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast. 1996;12:259–265. doi: 10.1002/(SICI)1097-0061(19960315)12:3%3C259::AID-YEA901%3E3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Welshimer HJ. Vitamin Requirements of Listeria monocytogenes. J Bacteriol. 1963;85:1156–1159. doi: 10.1128/jb.85.5.1156-1159.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiethaus J, Schubert B, Pfander Y, Narberhaus F, Masepohl B. The GntR-like regulator TauR activates expression of taurine utilization genes in Rhodobacter capsulatus. J Bacteriol. 2008;190:487–493. doi: 10.1128/JB.01510-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- Zeigler DR, Pragai Z, Rodriguez S, Chevreux B, Muffler A, Albert T, Bai R, Wyss M, Perkins JB. The origins of 168, W23, and other Bacillus subtilis legacy strains. J Bacteriol. 2008;190:6983–6995. doi: 10.1128/JB.00722-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.