

Abstract
Amyloid deposition has been implicated as the key determinant of Alzheimer’s disease (AD) pathogenesis. Interventions to antagonize amyloid accumulation and mitigate dementia are now under active investigation. We conducted a combined clinical, biochemical and neuropathological assessment of a participant in a clinical trial of the γ-secretase inhibitor, semagacestat. This patient received a daily oral dose of 140 mg of semagacestat for approximately 76 weeks. Levels of brain amyloid-β (Aβ) peptides were quantified using enzyme-linked immunosorbent assays (ELISA). Western blot/scanning densitometry was performed to reveal BACE1, presenilin1, amyloid precursor protein (APP) and its proteolysis-produced C-terminal peptides APP-CT99 and APP-CT83 as well as several γ-secretase substrates. To serve as a frame of reference, the ELISA and Western analyses were performed in parallel on samples from neuropathologically confirmed non-demented control (NDC) and AD subjects who did not receive semagacestat. Neuropathology findings confirmed a diagnosis of AD with frequent amyloid deposits and neurofibrillary tangles in most areas of the cortex and subcortical nuclei as well as cerebellar amyloid plaques. Mean levels of Tris-soluble Aβ40 and glass-distilled formic acid (GDFA)/guanidine hydrochloride (GHCl)-extractable Aβ40 in the frontal lobe and GDFA/GHCl-soluble Aβ40 in the temporal lobe were increased 4.2, 9.5 and 7.7-fold, respectively, in the semagacestat-treated subject compared to those observed in the non-treated AD group. In addition, GDFA/GHCl-extracted Aβ42 was increased 2-fold in the temporal lobe relative to non-treated AD cases. No major changes in APP, β- and γ-secretase and CT99/CT83 were observed between the semagacestat-treated subject compared to either NDC or AD cases. Furthermore, the levels of γ-secretase substrates in the semagacestat-treated subject and the reference groups were also similar. Interestingly, there were significant alterations in the levels of several γ-secretase substrates between the NDC and non-treated AD subjects. This is the first reported case study of an individual enrolled in the semagacestat clinical trial. The subject of this study remained alive for ~7 months after treatment termination, therefore it is difficult to conclude whether the outcomes observed represent a consequence of semagacestat therapy. Additional evaluations of trial participants, including several who expired during the course of treatment, may provide vital clarification regarding the impacts and aftermath of γ-secretase inhibition.
Keywords: Alzheimer’s disease, semagacestat immunotherapy, Alzheimer’s clinical trial, γ-secretase, γ-secretase inhibitors, γ-secretase substrates, amyloid-β
Introduction
The pathophysiology of Alzheimer’s disease (AD) is believed to involve the relentless deposition of amyloid-beta (Aβ) peptides in brain parenchyma and vasculature and the development of intraneuronal neurofibrillary tangles (NFT) composed mainly of the microtubule-associated protein tau. The Aβ peptides are derived from a larger membrane-bound molecule, known as the amyloid precursor protein (APP), by the sequential endoproteolytic cleavage mediated by β- and γ-secretases. It is widely accepted that Aβ peptides can assume either fibrillar (~10 nm) insoluble configurations or soluble dimeric/oligomeric conformations. The amyloid cascade hypothesis postulates amyloid deposition as the critical event in the pathogenesis of AD. Interventions using active and passive anti-Aβ immunotherapies and β- and γ-secretase inhibitors/modulators to prevent or alleviate amyloid deposition and thereby preclude the ravages of dementia are under active investigation.
The γ-secretase inhibitor semagacestat was provided to AD patients exhibiting mild to moderate cognitive impairment in a large randomized, double-blind placebo-controlled clinical trial. A detailed account of the screening, oversight, safety analyses, safety assessments, outcome measures and statistical analysis has been recently published by the Alzheimer’s Disease Cooperative Study Steering Committee and the Semagacestat Study Group [1].
We report clinical and neuropathological data and brain biochemical assessments of a participant in the semagacestat clinical trial (Clinical trials.gov number, NCT00594568). The subject received a daily oral dose of 140 mg of semagacestat for approximately 76 weeks. To provide a frame of reference for the brain neuropathology and biochemical status of the semagacestat-treated patient, we performed parallel analyses of neuropathologically confirmed non-demented control (NDC) subjects and AD subjects who had not received semagacestat treatment. Frontal and temporal brain regions were quantitatively evaluated to determine the levels of soluble and insoluble Aβ peptides using enzyme-linked immunosorbent assays (ELISA). Western blot/scanning densitometry was performed to determine relative amounts of APP and its derived C-terminal peptides (APP-CT99 and APP-CT83), β-site APP-cleaving enzyme-1 (BACE1) and presenilin-1 (PSEN1). To complement this study we examined a battery of γ-secretase substrates represented by amyloid precursor-like protein-1 (APLP1), APLP2, N-cadherin, E-cadherin, Notch-1, Notch-3, Delta-1, Jagged-2, Erb-B4, Eph-A4, neurexin, and neuroligin. In addition, we also quantified the apolipoprotein E (ApoE) protein as well as determined the APOE genotype of each study subject.
Clinical and neuropathological report and treatment synopsis
Clinical history
The semagacestat-treated patient was a 64-year-old female who died with the diagnosis of probable AD. Family history was negative for dementia or other cognitive disorders. According to the family members, symptoms of cognitive impairment had first been noticed about 6 years earlier. Private medical records date from March-April 2005, at which time she was seen by a neurologist for a three- to four-month history of memory problems. The patient was having difficulty performing complicated paperwork and this was giving her considerable anxiety. There was evidence of short term memory failure manifested as iterative speech. In addition, she also started becoming disoriented when driving. Other significant concurrent medical problems included migraine headaches, hypercholesterolemia, osteoporosis, anxiety and cardiac arrhythmia, which was treated by atrial ablation. Upon initial examination the patient was alert and communicative, with normal speech, concentration and attention span. She scored 27/30 on the Mini Mental State Examination (MMSE), losing points for orientation to time and delayed recall. She had normal posture and gait without focal neurological signs. An MRI scan revealed diffuse cortical atrophy and some deep white matter ischemic disease. An EEG showed diffuse slowing consistent with her stage of dementia. A PET scan demonstrated moderately severe hypometabolism bilaterally in frontal, temporal and parietal lobes. A neuropsychological assessment found her performance to be in the average to borderline impaired range. The diagnostic impression was possible mild cognitive impairment and she was started on donepezil.
Over subsequent years the patient exhibited steady cognitive decline documented by MMSE scores of 25/30 in April 2007 and 19/30 in February 2008. She also developed depression, anxiety and oppositional behavior. Memantine was added to her anti-dementia medication regime and she was also treated with anxiolytics and an anti-depressant. She was enrolled in the semagacestat clinical trial (LFAN) in the 140 mg arm, starting in December 2008 and completed the study on August 2010. At her screening visit her Clinical Dementia Rating Scale (CDR) score was 1, MMSE was 18/30, ADAS-Cog score was 26, modified Hachinski score was 0 and Geriatric Depression Scale score was 2. In June 2010, her MMSE was 8/30. In November 2010, her CDR was 3, MMSE was 9/30 and ADAS-Cog was 74. The patient died 4 months later.
Neuropathology report
Gross examination
The brain weighed 1120 grams with normal dura mater and mild midsagittal fibrosis of the leptomeninges. The convexities were symmetrical and showed moderate gyral atrophy of the anterior frontal lobes and parietal lobes, moderate to severe gyral atrophy of the posterior frontal lobes and paracentral regions and mild gyral atrophy of the occipital lobes. The circle of Willis harbored mild patchy atherosclerosis. The mammillary bodies were normal in shape, color and size. The temporal lobes demonstrated moderate atrophy, which was especially prominent at the temporal poles; the unci were grossly unremarkable. The cerebellum and brainstem were externally normal. Cerebral slices exhibited moderate to severe enlargement of the lateral ventricles. A small, circular, brown focal cavitary lesion, measuring 0.1 x 0.1 x 0.3 cm with smooth edges was observed in the white matter immediately adjacent to the primary visual cortex of the left occipital lobe located approximately 1.0 cm rostral to the occipital pole and extending into the occipital pole. The basal ganglia, thalamus and subthalamic nucleus were unremarkable. While the amygdala showed mild atrophy, the head and body of the hippocampus and the parahippocampal gyrus revealed mild to moderate atrophy. The substantia nigra displayed mild bilateral depigmentation. Respective axial and parasagittal slices of the brainstem and cerebellum were normal but friable in texture.
Microscopic examination
Paraffin sections of the left hemibrain sections stained with H&E showed mild to moderate to marked gliosis of upper layers of the cerebral cortex, with evident senile plaques and NFT. A section of the occipital lobe white matter lesion, grossly described above, showed the edge of a cystic cavity within which were small aggregates of hemosiderin-laden macrophages with some bordering gliosis. There was very marked gliosis of entorhinal cortex and mild to moderate gliosis of area CA1 of the hippocampus. The caudate nucleus and putamen demonstrated patchy mild gliosis with occasional tangles in large neurons. The substantia nigra showed mild depletion of pigmented neurons with moderate pigment incontinence. A single Lewy body was present. The locus ceruleus exhibited marked loss of pigmented neurons without Lewy bodies. The remaining sections of the brainstem, cerebellum and upper levels of spinal cord were unremarkable. Large sections stained with H&E showed moderately extensive cerebral white matter rarefaction in the frontal and parietal lobes. Sections stained with Gallyas, Campbell-Switzer and thioflavine-S methods showed frequent senile plaques in all cerebral cortex regions, with frequent diffuse plaques and frequent neuritic and cored plaques. Cerebral amyloid angiopathy was rare to focally sparse. NFT were frequent in all cerebral cortex regions as well as in the amygdala, entorhinal cortex, area CA1 of the hippocampus and hypothalamus. Immunohistochemical staining for phosphorylated α-synuclein demonstrated frequent neuronal perikaryal inclusions and fibers in the olfactory bulb, amygdala and entorhinal/transentorhinal areas. These inclusions were sparse to moderate in the brainstem and cingulate gyrus and rare in the cerebral neocortex.
Neuropathological diagnoses
1) Alzheimer’s disease. 2) Cerebral white matter rarefaction. 3) Microscopic changes of Lewy body disease present in insufficient numbers for diagnosis. 4) Old microscopic hemorrhagic infarct on left occipital lobe white matter.
Materials and methods
Human subjects and neuropathological evaluation
We examined tissue from the frontal and temporal lobes of an AD patient who received infusions of semagacestat (Sema-AD) as well as 6 NDC and 6 non-treated AD cases. These tissues were provided by the Banner Sun Health Research Institute (BSHRI) Brain and Body Donation Program in Sun City, Arizona [2]. All operations of our Institute have been approved by the Banner Health Institutional Review Board. All subjects participating in the Banner Brain and Body Donation Program signed an informed consent approved by the Banner Health Institutional Review Board. A demographic synopsis of each of the subjects includes their age at death, gender, postmortem interval (PMI) and APOE genotype [3] is shown in Table 1. For histopathological analyses, brain coronal sections (~1 cm thick) of the left hemispheres were fixed in formalin and large blocks comprising half coronal sections were sectioned using a frozen microtome at 40 µm thickness. The following regions were investigated: frontal, parietal, occipital, cerebellum, temporal including the amygdala and hippocampus, mid-brain at the level of the substantia nigra, thalamus and striatum. All sections were stained for H&E, Campbell-Switzer, Gallyas and thioflavine-S and scored. A complete list of the sampled regions is given in reference [2]. Coronal sections of the corresponding right hemispheres were immediately frozen in slabs of dry ice, independently packed under vacuum and stored at -86°C. All subjects were neuropathologically appraised for total amyloid plaque, total NFT and cerebral amyloid angiopathy (CAA). These scores were regionally estimated in frontal, temporal, parietal, hippocampal and entorhinal areas by numerical values of 0 (none), 1 (sparse), 2 (moderate) and 3 (frequent) for a maximum additive value of 15. Braak scores were evaluated following the Braak stage method and range from I-VI [4]. The total white matter rarefaction (WMR) score, was evaluated in the frontal, temporal, parietal and occipital white matter areas as none (0), mild (less than 25% affected=1) to moderate (25 to 50% affected=2) to severe (greater than 50% affected=3) for a maximum score of 12. The results of these scores as well as brain weight at autopsy are summarized in Table 1.
Table 1.
Study Subject Demographics and Neuropathology Scores
| NDC | Expired Age (yrs) | Gender | PMI (h) | APOE Genotype | Brain Weight | Total Plaque Score | Total NFT Score | Braak Score | Total WMR Score | Total CAA Score |
|
| ||||||||||
| 1 | 74 | M | 2.5 | 3/3 | 1230 | 0 | 3.5 | II | 1 | n/a |
| 2 | 67 | F | 3.5 | 3/4 | 1150 | 9.9 | 4 | II | 2 | 0 |
| 3 | 69 | M | 2.0 | 3/3 | 1190 | 0 | 0.25 | I | 0 | 0 |
| 4 | 63 | M | 1.5 | 3/3 | 1420 | 0 | 4.3 | II | 4 | 0 |
| 5 | 76 | M | 2.3 | 3/4 | 1375 | 5.5 | 0 | I | 1 | 3 |
| 6 | 68 | F | 2.6 | 3/3 | 1140 | 0 | 3.5 | III | 4 | 0 |
| Mean | 70 | 2.4 | 1251 | 2.6 | 2.6 | 2.0 | 0.6 | |||
|
| ||||||||||
| AD | Expired Age (yrs) | Gender | PMI (h) | APOE Genotype | Brain Weight | Total Plaque Score | Total NFT Score | Braak Score | Total WMR Score | Total CAA Score |
|
| ||||||||||
| 10 | 65 | F | 3.3 | 3/3 | 800 | 15 | 15 | VI | 1 | 0 |
| 12 | 77 | M | 2.3 | 3/4 | 1080 | 14 | 15 | VI | 7 | 2 |
| 13 | 60 | F | 3.5 | 3/3 | 900 | 13.5 | 15 | VI | 3 | 5 |
| 14 | 68 | F | 3.5 | 3/4 | 915 | 14.5 | 15 | VI | 1 | 2 |
| 15 | 79 | F | 5.0 | 3/4 | 1070 | 15 | 15 | VI | 4 | 3 |
| 16 | 68 | F | 4.2 | 3/3 | 1074 | 15 | 15 | VI | 4 | 11 |
| Mean | 70 | 3.6 | 973 | 14.5 | 15.0 | 3.5 | 3.8 | |||
|
| ||||||||||
| Expired Age (yrs) | Gender | PMI (h) | APOE Genotype | Brain Weight | Total Plaque Score | Total NFT Score | Braak Score | Total WMR Score | Total CAA Score | |
|
| ||||||||||
| Sema-AD | 64 | F | 2.2 | 3/4 | 1120 | 14 | 15 | VI | 5 | 4 |
NDC, non-demented control; AD, Alzheimer’s disease; Sema-AD, semagacestat-treated Alzheimer’s disease patient; M, male; F, female; yrs, years; PMI, postmortem interval; h, hours, APOE, apolipoprotein E; NFT, neurofibrillary tangle; WMR, white matter rarefaction; CAA, cerebral amyloid angiopathy.
Prior to the sectioning of the brain, the leptomeninges were gently pulled from the surface of the cerebrum and cerebellum, rinsed with phosphate buffered saline (PBS) and immediately frozen. For the assessment of leptomeningeal CAA, the total leptomeninges were rinsed 9 times in cold distilled water to remove entrapped blood, spread on 14 cm diameter Petri dishes, dried in an oven at 60°C, fixed in absolute ethanol for 1 h, stained by 1% aqueous thioflavine-S for 15 min, rinsed 6 times with 70% ethanol to remove unbound fluorochrome, rinsed with distilled water and immediately observed in an epifluorescent microscope. The numeric scoring method used for the leptomeningeal CAA used the same categories described above for the brain parenchymal CAA.
Chromatographic studies
To determine the chemical nature of the Aβ peptides that are deposited in amyloid plaques in the cerebellar molecular layer, we homogenized this tissue in 90% glass-distilled formic acid (GDFA, 1:10 parts weight/volume) at 4°C, followed by centrifugation at 225,000 x g (Beckman Optima 80LE, SW41 rotor for 1 h). The supernatant was divided into 0.5 ml and submitted to FPLC chromatography on a Superose 12 column (GE Life Sciences, Piscataway, NJ), equilibrated with 80% GDFA. The chromatography was developed at room temperature at a flow rate of 15 ml/h. The fraction containing the peptides ranging from 10-3 kDa, was collected, reduced by vacuum centrifugation and submitted to HPLC on a C8 reverse-phase column using an increasing stepwise gradient of 5% increments of acetonitrile/TFA/water. The chromatography was performed at 80°C. Fractions were collected at acetonitrile levels of 35%, 40%, 45%, 50%, 55%, 60% and 65%, submitted to Western blot characterization and examined using the Aβ42 and 6E10 antibodies (Table 2) as described below.
Table 2.
Antibodies Used in Western Blots
| Primary antibody | Antigen specificity or immunogen | Secondary antibody | Company/Catalog # |
|---|---|---|---|
| 6E10 | Aβ aa 1-16 | M | Covance/SIG-39320 |
| Aβ42 | aa 36-42 of human Aβ | R | Invitrogen 44-344 |
| CT20APP | Last 20 aa of APP | M | Covance/SIG-39152 |
| BACE1 | 3D5 clone, aa 46-460 | M | Kindly provided by Dr. R. Vassar |
| PSEN | C-terminal peptide of human PSEN | R | Cell Signaling Technology/5643 |
| APLP1 | NS0-derived rhAPLP1 extracellular domain | M | R&D Systems/MAB3908 |
| APLP2 | Mouse myeloma cell line NS0-derived recombinant human APLP2 aa 30-680 | M | R&D Systems/MAB49451 |
| N-Cadherin | aa 802-819 of mouse N-Cadherin | M | BD Transduction Laboratories/610920 |
| E-Cadherin | Human E-cadherin C-terminal recombinant protein | M | BD Transduction Laboratories/610182 |
| Notch1 | aa 1946-2209 | R | Proteintech/10062-2-AP |
| Notch3 | Undisclosed peptide | R | Proteintech/55114-1-AP |
| Delta1 | aa 459-723 of human Delta | M | Santa Cruz/sc-377310 |
| Jagged2 | C-terminal synthetic peptide from human jagged2 | R | LifeSpan Biosciences/LS-C100395 |
| Erb-B4 | aa 1258-1308 of human Erb-B4 | R | Santa Cruz/sc-283 |
| EphA4 receptor | C-terminal synthetic peptide from human EphA4 receptor | M | Life Technologies/37-1600 |
| Neurexin 1, 2, 3 | CT tails of α/β neurexins 1,2,3 | R | Synaptic Systems/175003 |
| Neuroligin | aa 718-843 of rat neuroligin 1 | R | Synaptic Systems/129013 |
| ApoE | Recombinant ApoE | G | Millipore/AB947 |
| GAPDH | Full-length human GAPDH protein | M | Life Technologies/39-8600 |
| Actin Ab-5 | Clone C4 | M | BD Transduction Laboratories/A65020 |
| Actin | N-terminus of human α-actin | R | Abcam/Ab37063 |
CT, C-terminal; APP, amyloid-β precursor protein; BACE, β-site APP-cleaving enzyme; aa, amino acid; APLP, amyloid precursor-like protein; PSEN, preseniln; ApoE, apolipoprotein E; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; M, HRP conjugated AffiniPure goat-anti mouse IgG (catalog # 115-035-146, Jackson Laboratory); R, HRP conjugated AffiniPure goat-anti rabbit IgG, (catalog # 111-035-144, Jackson Laboratory); G, HRP conjugated AffiniPure bovine-anti goat IgG (catalog # 805-0.35-180, Jackson Laboratory).
Aβ40 and Aβ42 ELISA
For a complete protocol, please see reference [5]. In brief, gray matter samples from the frontal and temporal lobe (200 mg) were each homogenized in 1600 µl of 20 mM Tris, 5 mM EDTA, pH 7.8 supplemented with a protease inhibitor cocktail (PIC, Roche Diagnostics, Mannheim, Germany). The homogenate was centrifuged at 500,000 x g (Beckman Optima TLX, 120.2 TLA rotor, Brea, CA) for 20 min and the supernatant collected and saved for total protein assays (Micro BCA, Pierce, Rockford, IL) and Aβ40 and Aβ42 ELISA. The remaining pellet was suspended in 1200 µl ml of 90% GDFA and centrifuged as described above. The supernatant was harvested and dialyzed in deionized H2O followed by 0.1 M ammonium bicarbonate. The samples were lyophilized then re-suspended in 1000 µl 5 M guanidine-hydrochloride, 50 mM Tris, pH 8.0 with PIC. Samples were centrifuged (500,000 x g 20 min) and the total protein levels of the supernatant quantified with a Pierce Micro BCA protein assay kit. Tris-soluble and GDFA/GHCl-soluble Aβ40 and Aβ42 levels were measured with ELISA kits from Life Technologies Corp. (Carlsbad, CA) following the manufacturer’s instructions.
Western blots
Frontal and temporal gray matter (200 mg) samples were each homogenized in 2 ml of 5% SDS, 5 mM EDTA, 20 mM Tris-HCl, pH 7.8 and centrifuged for 30 min in a Beckman 22R centrifuge at 20°C. The Micro BCA protein assay kit (Pierce) was used to measure total protein. A complete Western blot protocol is available in reference [5]. In summary, 40 µg or 100 µg of total protein from each case was loaded onto 4-12% Bis-Tris gels (Life Technologies Corp). After electrophoretic separation, proteins were transferred onto 0.45 µm nitrocellulose membranes (Life Technologies Corp). The APLP2 blots were run under non-reducing conditions (without DTT or antioxidant). To provide a total protein loading control, all membranes were stripped and re-probed with anti-mouse actin, anti-rabbit actin antibodies or a GAPDH antibody (APLP2). Details of each antibody used in Western blotting are provided in Table 2. The trace quantity of each band (defi-ned as the measured area under each band’s intensity profile in units of optical density (OD) x mm) was measured using Quantity One software (Bio-Rad, Hercules, CA, USA). The OD x mm results for each primary antibody were adjusted by the OD x mm for actin or GAPDH.
Statistical analyses
GraphPad Prism 5 software (La Jolla, CA) was used for data analyses. Significance was set at P ≤ 0.05. Only AD and NDC cases were compared using an unpaired, 2-tailed t-test.
Results
Neuropathology results and cerebellar Aβ analysis>
Data from NDC and AD individuals were used as a frame of reference for the biochemical comparative analyses (Table 1). The 6 NDC were represented by 4 males and 2 females (mean age of death: 70 years; range 63-76) and the 6 AD cases were represented by 1 male and 5 females (mean age of death: 70; range 60-79). The overall mean PMI was less than 3.6 h. As expected the brain weights of the NDC had, on average, about 20% more mass (1,251 g) than those of the AD cohort (973 g), while the Sema-AD case was intermediate (1,120 g). The APOE genotype of the Sema-AD treated patient was ε3/4 which was well represented in the reference groups. The APOE ε4 frequency was 17% in the NDC and 25% in the AD cohorts. The total plaque score was zero in 4 of the 6 NDC cases while in the remaining 2 cases, it averaged 7.7. By contrast, the average value in the AD group was 14.5 (out of a maximum of 15) similar to that of the Sema-AD case. In reference to the total NFT score, the mean total NFT scores for the NDC and AD were 2.6 and 15, respectively, while the Sema-AD case had a score of 15. These scores were in accord with the Braak staging being I-III and VI for the NDC and AD, respectively. The total WMR ranged from 0 to 4 in the NDC and 1 to 7 in the AD group, while in the Sema-AD case it was 5. The total CAA score in the NDC ranged from 0 to 3 while in the AD group it ranged from 1 to 11. The total CAA score in the Sema-AD treated patient was 4.
Representative histological sections shown in Figures 1 and 2, reveal the frontal, parietal, occipital, temporal, striatum, amygdala and hippocampus regions of the Sema-AD patient which display abundant AD amyloid plaques and NFT which reached maximum neuropathological scores (Table 1). Parenchymal and leptomeningeal vessels revealed areas of patchy aggregated CAA (Figure 3).
Figure 1.
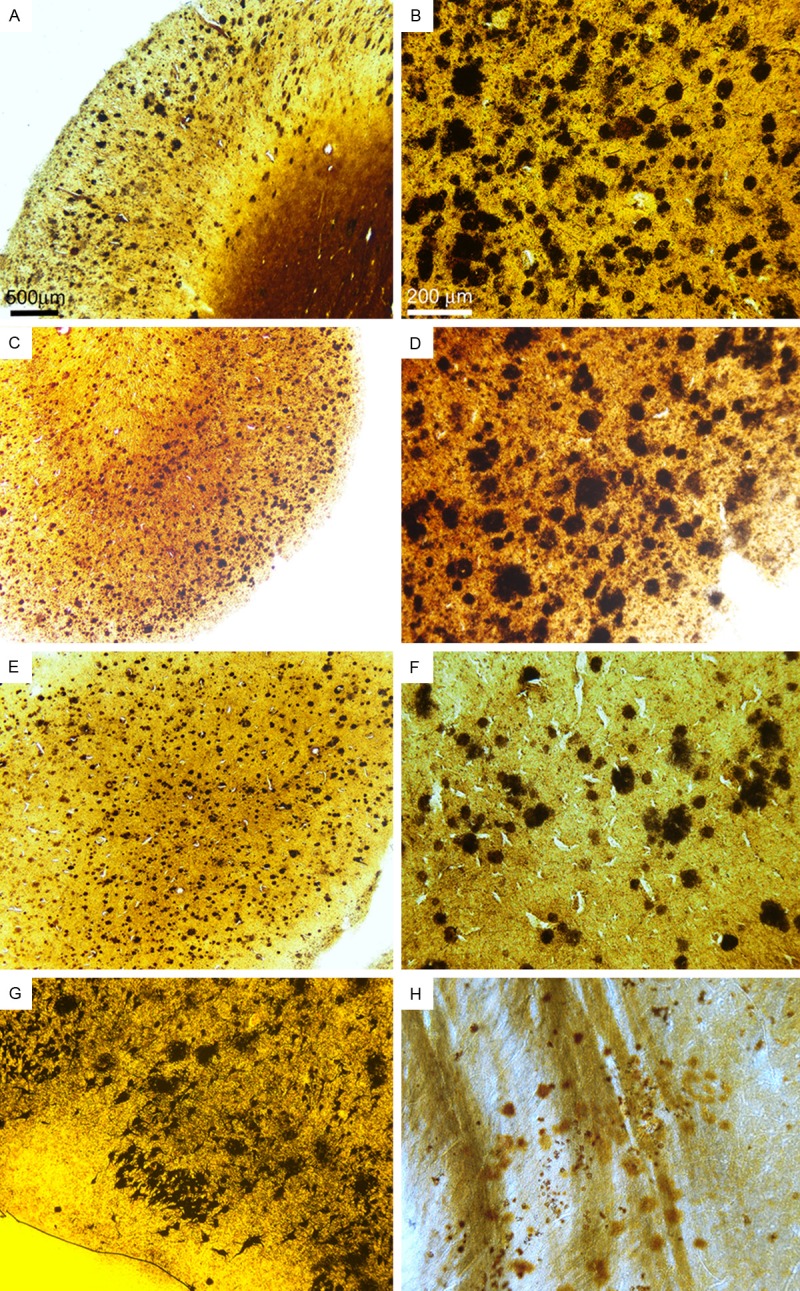
Amyloid plaque deposition in the cerebral cortex, amygdala and striatum in the Sema-AD patient (Campbell-Switzer stain). A and B: Frontal cortex. C and D: Parietal cortex. E and F: Occipital cortex. G: Amygdala. H: Striatum. All sections demonstrated abundant amyloid plaques in densities observed in non-semagacestat treated individuals. Magnifications: A, C and E, 25X. B, D and F-H - 100X.
Figure 2.
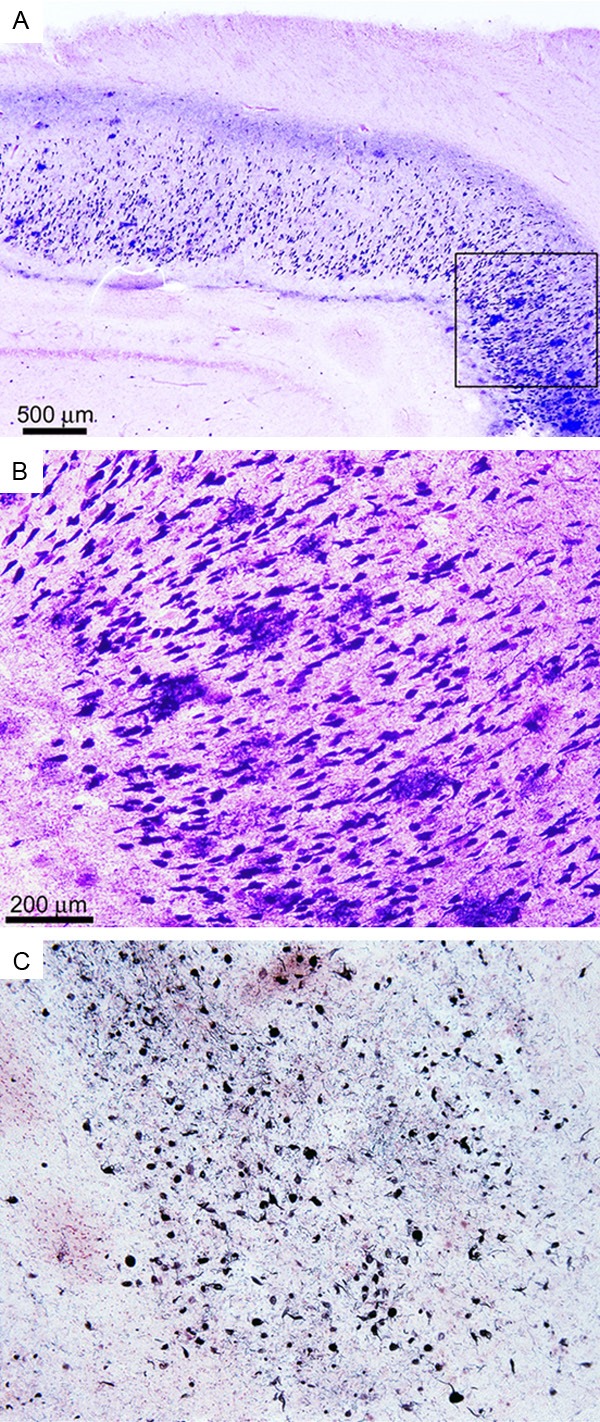
Neurofibrillary tangle pathology in the Sema-AD patient (Gallyas stain). (A) Hippocampus sh-owing a high density of NFT with a cluster of amyloid plaques (far right). Magnification 25X. (B) A higher magnification of the box in (A) demonstrates abundant NFT surrounding amyloid plaques. Magnification 100X. (C) Abundant distribution of NFT in the striatum. Magnification 100X.
Figure 3.
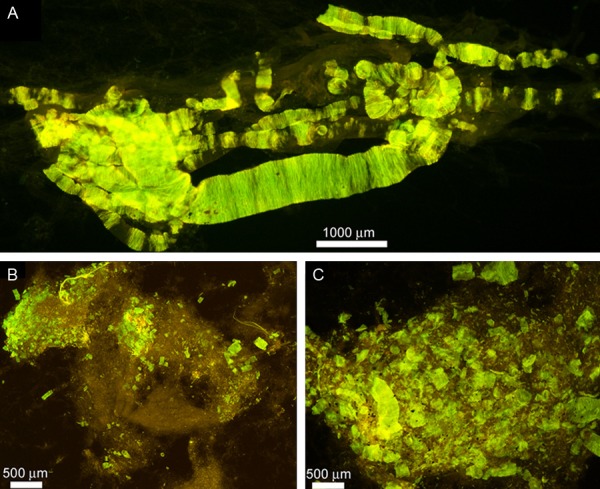
Whole mounts of cerebral vessels exhibiting amyloid angiopathy in the Semagacestat-treated patient. (A) Cortical vessels stained by thioflavine-S. To isolate the vessels small cubes of cerebral cortex, approximately 0.5 cm, were stirred in 50 mM Tris-HCl buffer pH 7.5 containing 5% SDS. After 72 h the only remaining structures are the insoluble extracellular matrix of the vasculature and associated amyloid deposits. Magnification 100X. (B and C) Leptomeninges showing abundant clusters of vascular amyloid deposits stained by thioflavine-S. Magnifications: (B) 25X and (C) 100X.
Of special interest was the cerebellum, which in the molecular layer demonstrated well defined amyloid plaque conglomerates of parallel filamentous bundles almost perpendicularly oriented to the surface of the granular layer (Figure 4). To determine the composition of these filaments, cerebellum from the Sema-AD case was homogenized in 90% GDFA, separated by FPLC (Figure 5A), followed by HPLC (Figure 5B) and analyzed by Western blot (Figure 5B, inset). Positive reactions of cerebellar samples with both 6E10 (epitope Aβ1-16) and Aβ42 (epitope Aβ36-42) antibodies suggests detection of a peptide with the N-terminal sequence of Aβ and a C-terminus at position Aβ42.
Figure 4.
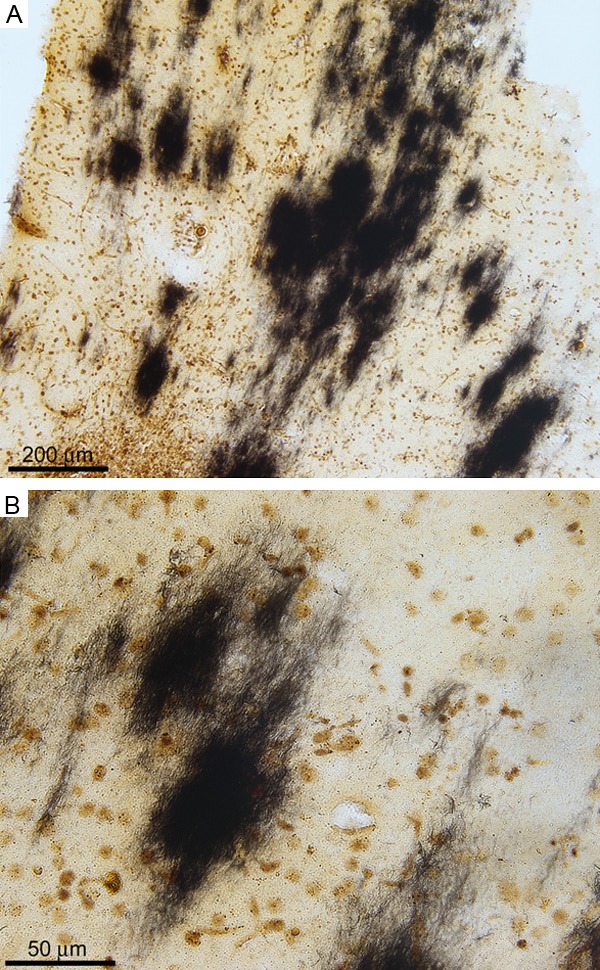
Cerebellar amyloid plaques in the Sema-AD patient (Campbell-Switzer stain). A. In some areas of the cerebellum, the deposits of amyloid are abundant. The plaques apparently have an elongated shape with the major axis oriented perpendicular to the surface of the cerebellar convolutions. Magnification 100X. B. At higher magnification, the plaques appear to be composed of discrete bundles of amyloid fibrils which are clearly observed on the peripheral areas. Magnification 400X.
Figure 5.
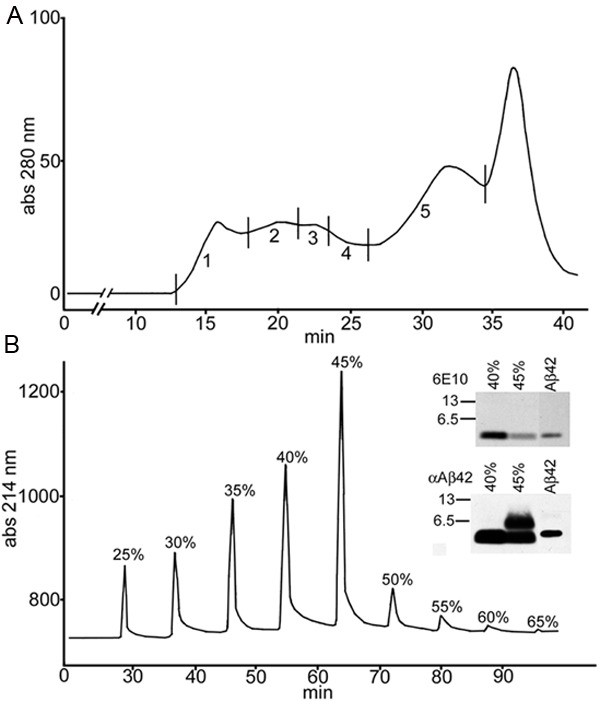
Chromatographic and Western blot analyses of cerebellum from the Sema-AD patient. (A) FPLC (size-exclusion Superose 12 column) profile of formic acid solubilized cerebellar tissue. Five fractions were collected. Fraction number 5 contained the Aβ peptides. (B) HPLC (C8 reverse-phase column) separation of fraction 5 from the FPLC shown in (A). Western blots detected Aβ peptides in fractions 40% and 45% acetonitrile concentration. The 6E10 antibody, against the N-terminal region of Aβ, was stripped from the blot and the same blot was reprobed with Aβ42 antibody, raised against the C-terminal region of the Aβ peptide. Molecular weight markers are shown in the left of each blot and reported in kDa. Aβ1-42 standard was loaded into the third lane. The Aβ dimer is shown in the 45% peak using the Aβ42 antibody.
Aβ eLISA quantifications
Quantitative assessment of the Aβ peptides in the frontal lobe by ELISA (see Table 3) revealed that the Sema-AD case total Tris-soluble Aβ40 and Aβ42 levels were 464 pg/mg of total protein and 51 pg/mg, respectively. On the other hand, the GDFA/GHCl solubilized pools amounted to 162 ng/mg of total protein and 200 ng/mg of total protein for the Aβ40 and Aβ42, respectively. In the frontal cortex of the reference group, the NDC Tris-soluble fraction contained an average 26 pg of Aβ40 per mg of total protein and 11 pg of Aβ42 per mg of total protein while the GDFA/GHCl-soluble fraction yielded an average of 0.38 ng of Aβ40 per mg of total protein and 2.78 ng of Aβ42 per mg of total protein. These yields were moderately elevated in the AD cohort where the Tris-soluble fraction contained 111 pg/mg of total protein for Aβ40 and 61 pg/mg of total protein for Aβ42 with GDFA/GHCl solubilization yielding 17 ng/mg of protein for Aβ40 and 232 ng/mg of protein for Aβ42.
Table 3.
Aβ ELISA Quantifications
| FRONTAL | ||||
| NDC (n=5)* | Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GDFA/GHCl Aβ40 ng/mg total protein | GDFA/GHCl Aβ42 ng/mg total protein |
| Mean | 26 (0-44) | 11 (0-57) | 0.38 (0-0.68) | 2.78 (0-7.50) |
| AD (n=5)* | Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GDFA/GHCl Aβ40 ng/mg total protein | GDFA/GHCl Aβ42 ng/mg total protein |
| Mean | 111 (0-408) | 61 (0-109) | 17 (2.22-68.36) | 232 (105-383) |
| Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GDFA/GHCl Aβ40 ng/mg total protein | GDFA/GHCl Aβ42 ng/mg total protein | |
| Sema-AD | 464 | 51 | 162 | 200 |
|
| ||||
| TEMPORAL | ||||
| NDC (n=5)* | Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GDFA/GHCl Aβ40 ng/mg total protein | GDFA/GHCl Aβ42 ng/mg total protein |
| Mean | 15 (0-51) | 6 (0-32) | 0.41 (0.09-1.04) | 1.54 (0-5.742) |
| AD (n=5)* | Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GDFA/GHCl Aβ40 ng/mg total protein | GDFA/GHCl Aβ42 ng/mg total protein |
| Mean | 32 (0-99) | 60 (54-65) | 4.82 (2.44-7.96) | 287 (90-597) |
| Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GDFA/GHCl Aβ40 ng/mg total protein | GDFA/GHCl Aβ42 ng/mg total protein | |
| Sema-AD | 32 | 56 | 37 | 562 |
One case from the AD group and one case from the NDC group were eliminated because they were extreme outliers.
NDC, non-demented control; AD, Alzheimer’s disease; Sema-AD, semagacestat-treated Alzheimer’s disease patient; Tris, 20 mM Tris, 5 mM EDTA, pH 7.8; GDFA/GHCl, glass-distilled formic acid/5 M guanidine-hydrochloride, 50 mM Tris, pH 8.0 (see Materials and Methods for details). The range is shown in parentheses next to the mean.
Quantitative evaluation of the Aβ peptides in the temporal lobe by ELISA (see Table 3) demonstrated that in the Sema-AD case the Tris-soluble levels of Aβ40 and Aβ42 were 32 pg/mg and 56 pg/mg of total protein, respectively. In the same lobe, the GDFA/GHCl solubilized extracts contained 37 ng/ng total protein and 562 ng/mg total protein of Aβ40 and Aβ42, respectively. The NDC group temporal cortex Tris-soluble fraction yielded an average of 15 pg of Aβ40 per mg of total protein and 6 pg of Aβ42 per mg of total protein while the GDFA/GHCl-soluble fraction had an average of 0.41 ng of Aβ40 per mg of total protein and 1.54 ng of Aβ42 per mg of total protein. These yields were increased in the AD group where the Tris-soluble fraction contained 32 pg/mg of total protein for Aβ40 and 60 pg/mg of total protein for Aβ42 and, as expected, significantly elevated after GDFA/GHCl solubilization yielding 4.82 ng/mg of protein for Aβ40 and 287 ng/mg of protein for Aβ42.
APP, APP-CTF, BACE1 and PSEN1-CTF
In spite of these substantial differences in Aβ content, there were no quantitative differences in the levels of holoAPP and its proteolytic derivatives CT99 and CT83 between Sema-AD patient and the NDC and AD reference cases in both the frontal and temporal regions (Figure 6A and 6B). However, in the temporal lobe, APP-CT83 was significantly lower in the AD cohort than in the NDC; P=0.0138 (Figure 6B). Likewise, there were no major deviations in the amounts of BACE1 (Figure 6C and 6D) and PSEN1-C-terminal fragment (Figure 6E and 6F) when the Sema-AD patient was compared to NDC and AD cases. Interestingly, the NDC group had statistically significant higher levels of PSEN1-C-terminal fragment than the AD cohort in the temporal lobe (Figure 6F, P=0.004). Increased amounts of the C-terminal fragment of PSEN1 in the NDC frontal lobe relative to the AD group were also observed, but the difference was not statistically significant (Figure 6E).
Figure 6.

Western blot analyses of APP, APP-CTF, BACE1 and PSEN-CTF. Western blotting was used to detect APP, APP-CT99 and APP-CT83 in the frontal cortex (A) and temporal cortex (B), BACE1 in the frontal cortex (C) and temporal cortex (D) and a CTF of PSEN in the frontal cortex (E) and temporal cortex (F). Data are reported in optical density units. Actin reprobes are shown below each primary antibody blot. Molecular weights, reported in kDa, are shown to the left of each blot. Abbreviations: NDC, non-demented controls, Se-AD, semagacestat-treated AD patient; AD, Alzheimer’s disease; APP, amyloid precursor protein; CT, C-terminal; BACE1, β-site amyloid precursor protein-cleaving enzyme-1; PSEN, presenilin; CTF, C-terminal fragment. For statistical treatment, AD and NDC cases were compared using an unpaired, 2-tailed t-test.
Substrates of γ-secretase and ApoE
In reference to the 12 γ-secretase substrates that were investigated (APLP1, APLP2, N-cadherin, E-cadherin, Notch-1, Notch-3, Delta-1, Jagged-2, Erb-B4, Eph-A4, neurexin and neuroligin), no major differences were observed when the Sema-AD case was compared to the NDC and AD cohorts in both the frontal and temporal regions (see Figures 7, 8, 9 and 10). However, a comparison between the reference group levels of NDC versus AD revealed significant statistical differences in temporal N-cadherin (Figure 7F, P=0.0006), frontal Jagged-2 (Figure 8G, P=0.0004), temporal Jagged-2 (Figure 8H, P=0.0002), frontal neurexin-α (Figure 10A, P=0.0446) and temporal neurexin-β (Figure 10B, P=0.0181). The ApoE values did not differ between NDC and AD or between these two groups and the Sema-AD patient (Figure 10E and 10F).
Figure 7.

Western blot analyses of APLP1, APLP2, N-cadherin and E-cadherin. Western blotting was used to detect APLP1 in the frontal cortex (A) and temporal cortex (B), APLP2 in the frontal cortex (C) and temporal cortex (D), N-cadherin in the frontal cortex (E) and temporal cortex (F) and E-cadherin in the frontal cortex (G) and temporal cortex (H). The APLP2 blot was run under non-reducing conditions. Data are reported in optical density units. Actin reprobes are shown below each primary antibody blot. Molecular weights, reported in kDa, are shown to the left of each blot. Abbreviations: NDC, non-demented controls, Se-AD, semagacestat-treated AD patient; AD, Alzheimer’s disease; APLP, amyloid precursor-like protein. For statistical treatment, AD and NDC cases were compared using an unpaired, 2-tailed t-test.
Figure 8.

Western blot analyses of Notch1, Notch3, Delta1 and Jagged2. Western blotting was used to detect Notch1 in the frontal cortex (A) and temporal cortex (B) cortex, Notch3 in the frontal cortex (C) and temporal cortex (D), Delta1 in the frontal cortex (E) and temporal cortex (F) and Jagged in the frontal cortex (G) and temporal cortex (H). Data are reported in optical density units. Actin reprobes are shown below each primary antibody blot. Molecular weights, reported in kDa, are shown to the left of each blot. Abbreviations: NDC, non-demented controls, Se-AD, semagacestat-treated AD patient; AD, Alzheimer’s disease; NICD, Notch intracellular domain. For statistical treatment, AD and NDC cases were compared using an unpaired, 2-tailed t-test.
Figure 9.

Western blot analyses of ErbB4 and Eph-A4. Western blotting was used to detect ErbB4 in the frontal cortex (A) and temporal cortex (B) and Eph-A4 in the frontal cortex (C) and temporal cortex (D). Data are reported in optical density units. Actin reprobes are shown below each primary antibody blot. Molecular weights, reported in kDa, are shown to the left of each blot. Abbreviations: NDC, non-demented controls, Se-AD, semagacestat-treated AD patient; AD, Alzheimer’s disease. For statistical treatment, AD and NDC cases were compared using an unpaired, 2-tailed t-test.
Figure 10.

Western blot analyses of neurexin 1/2/3, neuroligin-1 and ApoE. Western blotting was used to detect the α and β chains of neurexin 1/2/3 in the frontal cortex (A) and temporal cortex (B), neuroligin-1 in the frontal cortex (C) and temporal cortex (D) and ApoE in the frontal cortex (E) and temporal cortex (F). Data are reported in optical density units. Actin reprobes are shown below each primary antibody blot. Molecular weights, reported in kDa, are shown to the left of each blot. Abbreviations: NDC, non-demented controls, Se-AD, semagacestat-treated AD patient; AD, Alzheimer’s disease. For statistical treatment, AD and NDC cases were compared using an unpaired, 2-tailed t-test.
Discussion
Semagacestat is a γ-secretase inhibitor that was developed by Eli Lilly and Company for the treatment of mild-to-moderate AD. Phase 3 was a randomized, double-blind, placebo-controlled clinical trial. The study was initially designed to randomize 501 patients to the placebo arm, 507 patients to the 100 mg/day dose of semagacestat and 529 patients to the 140 mg/day dose of semagacestat. The patient presented here was randomized to the higher dose and completed the study, receiving treatment for approximately 76 weeks. The study was terminated due to unanticipated adverse events [1]. The subject lived approximately 7 months after semagacestat treatment ceased and was evaluated once after that event.
To our knowledge, this is the first report of neuropathological and biochemical data from a patient who completed the semagacestat clinical trial. Between December of 2008 to November of 2010 the Sema-AD patient’s ADAS-Cog score increased significantly, from 26 to 74. Her MMSE score dropped from 18 to 9, and her CDR increased from 1 to 3. Taken together, these data suggest that the patient declined rapidly within a period of less than 2 years which coincided with her semagacestat treatment.
In the semagacestat phase III clinical trial, patients receiving 140 mg of the drug had a mean increase in their ADAS-Cog of 7.8 points at week 76 of treatment, while the Sema-AD patient in this study demonstrated a 48-point difference. The clinical data on this patient indicated a rapid cognitive decline. Several studies have raised the possibility that low concentrations of γ-secretase inhibitors may increase Aβ levels [6-8]. In addition, many critical molecules that are involved in multiple metabolic pathways are γ-secretase substrates [9]. The possibility exists that essential processing activities of these molecules could be inhibited at far lower levels of semagacestat than required to suppress Aβ production [6]. However, it is important to note that several studies in humans established that semagacestat, at a dose of 140 mg, is quickly absorbed (T-max=0.5 h) with a maximum (t-time) concentration in plasma of ~1 h and a half-life in plasma of ~2.5 h [10,11]. Since this patient remained alive for ~7 months after the termination of treatment, it is difficult to ascertain whether any toxic effects or neuropathological changes could be adjudicated to semagacestat treatment.
The postmortem report confirmed the final diagnosis of AD with frequent amyloid deposits and NFT in most areas of the cerebral cortex and subcortical nuclei as well as cerebellar amyloid plaques. Amyloid plaques in the cerebellum are commonly observed in AD and Down’s syndrome cases, and have been mostly classified as diffuse plaques [12-18]. We were intrigued by the homogeneous morphology of the cerebellar plaques which exhibited an array of compact parallel bundles with clearly discernible fibrillar structures similar to the subpial cortical Aβ deposits [19]. Chemical analysis of the cerebellar plaques in Down’s syndrome demonstrated the presence of P3 peptide composed of residues Aβ17-42 [20], similar to that characterized as the main component of AD parenchymal diffuse plaques [21], which are associated with quiescent microglia [22,23]. However, in the Sema-AD case we examined, the Aβ deposited in the cerebellum was mainly composed of Aβ n-42, although the possible presence of Aβ17-42 in these deposits cannot be definitively ruled out. If precipitation of the highly insoluble and aggregation-prone P3 peptides occurred during the biochemical manipulations, these molecules would have been undetectable in our Western blots.
The abundance of amyloid and NFT lesions and the early age of onset and duration of the disease were consistent with the APOE e3/4 genotype of the subject. The age and fact that the subject had reported memory issues and exhibited cognitive function test score declines at the outset of the study suggests amyloid deposition processes were already well advanced at the inception of treatment. Semagacestat was not anticipated to impact the distribution or persistence of already deposited amyloid; rather the drug was intended to suppress Aβ synthesis with the hope of modifying the course of dementia. In our postmortem analyses, we detected major differences in the average levels of Tris-soluble Aβ40 and GDFA/GHCl-soluble Aβ40 in the frontal lobe and GDFA/GHCl-soluble Aβ40 in the temporal lobe which were increased 4.2, 9.5 and 7.7-fold, respectively, in the Sema-AD patient when compared to the average Aβ peptides observed in the non-treated AD group. These discrepancies could be interpreted as semagacestat favoring cleavage of the γ-secretase site at position Aβ40 which may explain the increased levels of the shorter Aβ peptide. However, CAA in the semagacestat-treated subject was classified as ‘mild’ and did not seem to reflect this increase in Aβ40. In addition, the Sema-AD GDFA/GHCl-extracted Aβ42 was 2-fold increased in the temporal lobe relative to non-treated AD cases. Semagacestat was expected to block production of Aβ which could have resulted in the accumulation of CT99/CT83 in response to the blocking of γ-secretase activity. Yet, we found no significant changes in the levels of APP, β- and γ-secretases and APP-CT99/CT83 in the frontal and temporal lobes of the Sema-AD patient when compared to the NDC and AD reference groups. Perhaps this reflects the fact that drug therapy had ceased 7 months prior to death and subsequent autopsy. Interestingly, in the temporal lobe, we observed that the levels of PSEN-CTF and APP-CT83 peptides were significantly lower in the AD group relative to the NDC, an observation that deserves a wider investigation on the potential role of these molecules in the pathogenesis and pathophysiology of AD.
The γ-secretase exerts its proteolytic activity on over 90 substrates [9]. This transmembrane aspartate proteolytic enzyme is composed of four subunits: PSEN1/2, APH1, Pen2 and nicastrin. In all likelihood, the proteolytic activity of presenilin is modulated by important allosteric interactions with the other 3 members of the γ-secretase complex [24]. The γ-secretase has a high spectrum of biological functions, which in most cases results in the generation of signaling mediated pathways and transcription factors that intervene in embryonic/fetal organ development, maintenance of homeostasis and have a role in large number of diseases including neurodegeneration and cancer [25]. The unfortunate accelerated cognitive decline of semagacestat-treated subjects suggests that neurodegeneration may involve more than amyloid deposition and raises the possibility that direct modulation or suppression of presenilin functions actually intensified dementia progression [26]. Therefore, special emphasis was dedicated to the evaluation of molecules known to be substrates of the PSEN/γ-secretase. Although our data revealed no trend differences between our solitary Sema-treated subject and the NDC and AD cases, we nevertheless observed some intriguing statistically significant differences between NDC and AD. For example, in the frontal lobe, Jagged2 and neurexin-α were different while in the temporal lobe these differences were manifested in APP-CT83, C-terminal fragment of PSEN1, N-cadherin, Jagged2, and neurexin-β. With the exception of N-cadherin, all of these γ-secretases substrates were significantly lower in the AD group relative to the NDC. If confirmed in a larger set of samples, these gray matter protein differences may suggest that substrates other than APP are also affected in sporadic AD implying an altered PSEN/γ-secretase activity upon these substrates. In a recent investigation, we determined ErbB4 levels in the white matter of 10 familial AD cases due to PSEN mutations in which 6 out of 10 cases the ErbB4 was decreased relative to the statistical confidence limits established by the NDC [27].
Global inhibition of γ-secretase is likely to have deleterious repercussions on a large number of important cellular and tissue functions. However, new generations of more selective γ-secretase modulators, specifically targeting APP and Aβ production and sparing other substrates, are presently under development [28,29]. The abundance of neuropathological lesions and the faster course of the disease signs and symptoms as well as the general toxicity shown by semagacestat and other γ-secretase inhibitors such as avagacestat, begacestat and ELND006 [30,31] suggest that a detailed pathological and chemical investigation in the aftermath of these treatments is an urgent necessity. This may yield invaluable information regarding the fundamental mechanisms promoting dementia and key insights for the design of future therapeutic agents and clinical trials to mitigate or prevent Alzheimer’s disease.
Acknowledgements
This study was supported by the National Institute on Aging grants R01 AG019795 and R21 AG035078 (AER). The Brain Donation Program at Banner Sun Health Research Institute (TGB) is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. The funders had no role in study design, data collection, analysis or interpretation of data, decision to publish or preparation of the manuscript.
Disclosure of conflict of interest
Roher AE, Maarouf CL, Kokjohn TA, Whiteside CM, Kalback WM, and Serrano G have no conflicts of interest to declare. Jacobson SA receives royalties from American Psychiatric Publishing, Inc. for published works. Sabbagh MN receives grants/contracts from Pfizer, Eisai, Neuronix, Lilly, Avid, Piramal, GE, Avanir, Elan, Takeda, DART, Navidea and Functional Neuromodulation, is an advisor for Biogen, Lilly, Piramal, Eisai and Avid and receives royalties from Wiley and Tenspeed (Random House).
References
- 1.Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Siemers E, Sethuraman G, Mohs R. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013;369:341–350. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 2.Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L, Connor DJ, Sabbagh MN, Rogers J. The Sun Health Research Institute Brain Donation Program: description and experience, 1987-2007. Cell Tissue Bank. 2008;9:229–245. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 5.Maarouf CL, Daugs ID, Kokjohn TA, Walker DG, Hunter JM, Kruchowsky JC, Woltjer R, Kaye J, Castano EM, Sabbagh MN, Beach TG, Roher AE. Alzheimer's disease and non-demented high pathology control nonagenarians: comparing and contrasting the biochemistry of cognitively successful aging. PLoS One. 2011;6:e27291. doi: 10.1371/journal.pone.0027291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barthet G, Shioi J, Shao Z, Ren Y, Georgakopoulos A, Robakis NK. Inhibitors of gamma-secretase stabilize the complex and differentially affect processing of amyloid precursor protein and other substrates. FASEB J. 2011;25:2937–2946. doi: 10.1096/fj.11-183806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lanz TA, Karmilowicz MJ, Wood KM, Pozdnyakov N, Du P, Piotrowski MA, Brown TM, Nolan CE, Richter KE, Finley JE, Fei Q, Ebbinghaus CF, Chen YL, Spracklin DK, Tate B, Geoghegan KF, Lau LF, Auperin DD, Schachter JB. Concentration-dependent modulation of amyloid-beta in vivo and in vitro using the gamma-secretase inhibitor, LY-450139. J Pharmacol Exp Ther. 2006;319:924–933. doi: 10.1124/jpet.106.110700. [DOI] [PubMed] [Google Scholar]
- 8.Burton CR, Meredith JE, Barten DM, Goldstein ME, Krause CM, Kieras CJ, Sisk L, Iben LG, Polson C, Thompson MW, Lin XA, Corsa J, Fiedler T, Pierdomenico M, Cao Y, Roach AH, Cantone JL, Ford MJ, Drexler DM, Olson RE, Yang MG, Bergstrom CP, McElhone KE, Bronson JJ, Macor JE, Blat Y, Grafstrom RH, Stern AM, Seiffert DA, Zaczek R, Albright CF, Toyn JH. The amyloid-beta rise and gamma-secretase inhibitor potency depend on the level of substrate expression. J Biol Chem. 2008;283:22992–23003. doi: 10.1074/jbc.M804175200. [DOI] [PubMed] [Google Scholar]
- 9.Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J Alzheimers Dis. 2011;25:3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yi P, Hadden C, Kulanthaivel P, Calvert N, Annes W, Brown T, Barbuch RJ, Chaudhary A, Ayan-Oshodi MA, Ring BJ. Disposition and metabolism of semagacestat, a {gamma}-secretase inhibitor, in humans. Drug Metab Dispos. 2010;38:554–565. doi: 10.1124/dmd.109.030841. [DOI] [PubMed] [Google Scholar]
- 11.Willis BA, Zhang W, Ayan-Oshodi M, Lowe SL, Annes WF, Sirois PJ, Friedrich S, de la Pena A. Semagacestat pharmacokinetics are not significantly affected by formulation, food, or time of dosing in healthy participants. J Clin Pharmacol. 2012;52:904–913. doi: 10.1177/0091270011407195. [DOI] [PubMed] [Google Scholar]
- 12.Joachim CL, Morris JH, Selkoe DJ. Diffuse senile plaques occur commonly in the cerebellum in Alzheimer's disease. Am J Pathol. 1989;135:309–319. [PMC free article] [PubMed] [Google Scholar]
- 13.Mavroudis IA, Fotiou DF, Adipepe LF, Manani MG, Njau SD, Psaroulis D, Costa VG, Baloyannis SJ. Morphological changes of the human purkinje cells and deposition of neuritic plaques and neurofibrillary tangles on the cerebellar cortex of Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2010;25:585–591. doi: 10.1177/1533317510382892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolf DS, Gearing M, Snowdon DA, Mori H, Markesbery WR, Mirra SS. Progression of regional neuropathology in Alzheimer disease and normal elderly: findings from the Nun study. Alzheimer Dis Assoc Disord. 1999;13:226–231. doi: 10.1097/00002093-199910000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Larner AJ. The cerebellum in Alzheimer's disease. Dement Geriatr Cogn Disord. 1997;8:203–209. doi: 10.1159/000106632. [DOI] [PubMed] [Google Scholar]
- 16.Li YT, Woodruff-Pak DS, Trojanowski JQ. Amyloid plaques in cerebellar cortex and the integrity of Purkinje cell dendrites. Neurobiol Aging. 1994;15:1–9. doi: 10.1016/0197-4580(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 17.Wisniewski HM, Bancher C, Barcikowska M, Wen GY, Currie J. Spectrum of morphological appearance of amyloid deposits in Alzheimer’s disease. Acta Neuropathol. 1989;78:337–347. doi: 10.1007/BF00688170. [DOI] [PubMed] [Google Scholar]
- 18.Braak H, Braak E, Bohl J, Lang W. Alzheimer's disease: amyloid plaques in the cerebellum. J Neurol Sci. 1989;93:277–287. doi: 10.1016/0022-510x(89)90197-4. [DOI] [PubMed] [Google Scholar]
- 19.Yamaguchi H, Nakazato Y, Yamazaki T, Shoji M, Kawarabayashi T, Hirai S. Subpial beta/A4 amyloid deposition occurs between astroglial processes in Alzheimer-type dementia. Neurosci Lett. 1991;123:217–220. doi: 10.1016/0304-3940(91)90934-l. [DOI] [PubMed] [Google Scholar]
- 20.Lalowski M, Golabek A, Lemere CA, Selkoe DJ, Wisniewski HM, Beavis RC, Frangione B, Wisniewski T. The “nonamyloidogenic” p3 fragment (amyloid beta17-42) is a major constituent of Down’s syndrome cerebellar preamyloid. J Biol Chem. 1996;271:33623–33631. doi: 10.1074/jbc.271.52.33623. [DOI] [PubMed] [Google Scholar]
- 21.Gowing E, Roher AE, Woods AS, Cotter RJ, Chaney M, Little SP, Ball MJ. Chemical characterization of A beta 17-42 peptide, a component of diffuse amyloid deposits of Alzheimer disease. J Biol Chem. 1994;269:10987–10990. [PubMed] [Google Scholar]
- 22.Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol. 1997;56:321–339. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Mattiace LA, Davies P, Yen SH, Dickson DW. Microglia in cerebellar plaques in Alzheimer’s disease. Acta Neuropathol. 1990;80:493–498. doi: 10.1007/BF00294609. [DOI] [PubMed] [Google Scholar]
- 24.Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, Shi Y. Three-dimensional structure of human gamma-secretase. Nature. 2014;512:166–70. doi: 10.1038/nature13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jurisch-Yaksi N, Sannerud R, Annaert W. A fast growing spectrum of biological functions of gamma-secretase in development and disease. Biochim Biophys Acta. 2013;1828:2815–2827. doi: 10.1016/j.bbamem.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 26.Shen J, Kelleher RJ III. The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roher AE, Maarouf CL, Malek-Ahmadi M, Wilson J, Kokjohn TA, Daugs ID, Whiteside CM, Kalback WM, Macias MP, Jacobson SA, Sabbagh MN, Ghetti B, Beach TG. Subjects harboring presenilin familial Alzheimer’s disease mutations exhibit diverse white matter biochemistry alterations. Am J Neurodegener Dis. 2013;2:187–207. [PMC free article] [PubMed] [Google Scholar]
- 28.Hall A, Patel TR. gamma-Secretase modulators: current status and future directions. Prog Med Chem. 2014;53:101–145. doi: 10.1016/B978-0-444-63380-4.00003-2. [DOI] [PubMed] [Google Scholar]
- 29.Pettersson M, Stepan AF, Kauffman GW, Johnson DS. Novel gamma-secretase modulators for the treatment of Alzheimer’s disease: a review focusing on patents from 2010 to 2012. Expert Opin Ther Pat. 2013;23:1349–1366. doi: 10.1517/13543776.2013.821465. [DOI] [PubMed] [Google Scholar]
- 30.Mikulca JA, Nguyen V, Gajdosik DA, Teklu SG, Giunta EA, Lessa EA, Tran CH, Terak EC, Raffa RB. Potential novel targets for Alzheimer pharmacotherapy: II. Update on secretase inhibitors and related approaches. J Clin Pharm Ther. 2014;39:25–37. doi: 10.1111/jcpt.12112. [DOI] [PubMed] [Google Scholar]
- 31.Hopkins CR. ACS chemical neuroscience molecule spotlight on ELND006: another gamma-secretase inhibitor fails in the clinic. ACS Chem Neurosci. 2011;2:279–280. doi: 10.1021/cn2000469. [DOI] [PMC free article] [PubMed] [Google Scholar]