

Abstract
Rationale
MicroRNA miR145 has been implicated in vascular smooth muscle cell differentiation, but its mechanisms of action and downstream targets have not been fully defined.
Objective
Here, we sought to explore and define the mechanisms of miR145 function in smooth muscle cells.
Methods and Results
Using a combination of cell culture assays and in vivo mouse models to modulate miR145, we characterized its downstream actions on smooth muscle phenotypes. Our results show that the miR-143/145 gene cluster is induced in smooth muscle cells by coculture with endothelial cells. Endothelial cell-induced expression of miR-143/145 is augmented by Notch signaling and accordingly expression is reduced in Notch receptor-deficient cells. Screens to identify miR145-regulated genes revealed that the TGFβ pathway has a significantly high number of putative target genes, and we show that TGFβ receptor II (TGFBR2) is a direct target of miR145. Extracellular matrix (ECM) genes that are regulated by TGFBR2 were attenuated by miR145 overexpression, and miR145 mutant mice exhibit an increase in ECM synthesis. Furthermore, activation of TGFβ signaling via angiotensin II infusion revealed a pronounced fibrotic response in the absence of miR145.
Conclusions
These data demonstrate a specific role for miR145 in the regulation of matrix gene expression in smooth muscle cells, and suggest that miR145 acts to suppress TGFβ-dependent ECM accumulation and fibrosis, while promoting TGFβ-induced smooth muscle cell differentiation. Our findings offer evidence to explain how TGFβ signaling exhibits distinct downstream actions via its regulation by a specific microRNA.
Keywords: microRNA, smooth muscle cells, TGFβ, differentiation, matrix, growth factor
INTRODUCTION
MicroRNAs have been cast as modulators of gene expression, whose fundamental function is to fine-tune cellular phenotypes in response to intrinsic signals or environmental stress.1 In the vasculature, the ability of cells within the vessel wall to adjust to a range of cues is critically important for maintaining proper flow and pressure. While the endothelial cells serve as the primary sensor of blood vessels, smooth muscle cells act as the essential workhorse, by providing stability and contraction as needed. Smooth muscle cells are dynamic cells that can exist in a range of phenotypes. In addition to being contractile, these cells can be proliferative and/or exist in a synthetic state, where they secrete extracellular matrix (ECM) that is needed for vessel wall stability.2 The ability of smooth muscle cells to undergo phenotypic transitions is essential for vascular development and remodeling associated with changes in blood flow; however in vascular disease, phenotypic modulation can have a detrimental impact by contributing to disease pathology.
A host of mediators have been implicated in the control of phenotypic modulation.2 The transcription factor tandem, serum response factor (SRF) and Myocardin are master regulators of smooth muscle differentiation that drive cells towards a contractile phenotype.3, 4 Notch signaling has been shown to be important for smooth muscle development and differentiation, and plays a specific role in endothelial cell-dependent maturation.5, 6 Two opposing growth factor signaling pathways, platelet-derived growth factor (PDGF) and transforming growth factor (TGF)-β have been shown to drive cells towards a proliferative and differentiated phenotype, respectively.2 Additionally, the TGFβ pathway can induce matrix synthesis under certain conditions, suggesting that this pathway might have a dual role in smooth muscle cell programming.7 With the discovery of microRNAs that are enriched in smooth muscle cells, an obvious question is how they might contribute to phenotypic modulation by intervening with these established regulatory pathways. Not surprisingly, several microRNAs have been associated with the regulation of smooth muscle phenotypes.8 For example, miR-1 was shown to be induced by Myocardin, can inhibit proliferation and is reduced in a carotid artery ligation model.9 miR-21 also exhibits a pro-differentiation profile by being regulated by BMP and TGFβ and is important for TGFβ-mediated smooth muscle maturation.10, 11 In contrast, miR-221 is induced by PDGF signaling and can drive vascular smooth muscle cells towards a dedifferentiated state, partially through downregulation of Myocardin.12, 13
In 2009, a series of publications highlighted the importance of the miR-143/145 microRNA cluster in the regulation of smooth muscle cell phenotypes.14–18 The results showed that microRNA-143/145 are highly expressed in contractile smooth muscle and are reduced in proliferative conditions. Data indicated that genetic loss of these microRNAs in mice, while not lethal, caused a decrease in smooth muscle stress fiber formation and an increase in rough endoplasmic reticulum, both indicators of a less differentiated and more synthetic phenotype. Attempts to identify targets of this miR cluster revealed they had a hand in the regulation of proliferation, actin remodeling, and contractility genes.19, 20 Despite the consensus that miR-143/145 contribute to a differentiated phenotype, inconsistencies in the data using different experimental models strongly suggests that miR-143/145 function is context-dependent.
In this study we show that miR145 is induced in smooth muscle cells by endothelial cell signaling. Endothelial cells promote the increase in miR145 expression through Notch signaling, consistent with a differentiated phenotype. Examination of putative miR145 target genes revealed that miR145 regulates TGFβ receptor II (TGFBR2) expression and governs the expression of downstream matrix genes in smooth muscle cells. Our results suggest that miR145 functions to modulate TGFβ signaling in smooth muscle cells as a mechanism to suppress matrix gene expression, while sparing smooth muscle-specific differentiation genes. These actions of miR145 may have implications in disease progression, where suppression of detrimental matrix synthesis by miR145 could be used to alleviate fibrosis in a range of tissues.
METHODS
Cell culture
Primary cultures of human aortic smooth muscle cells (HAoSMCs) were purchased from Vasculife and grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Mediatech, Inc.) supplemented with 10% fetal bovine serum (FBS) (Hyclone), 2mM glutamine, 1mM sodium pyruvate and 100U/ml penicillin-streptomycin. Human mesenchymal stem cells (HMSCs) were purchased from Sciencell, and cultured in DMEM supplemented as above with 5% FBS. Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza, and grown in EBM-2 supplemented with the bullet kit as recommended (Lonza). Primary cells between passages 7–8 were used for all experiments. For virus production, TN-293 cells were purchased from Stratagene and cultured in DMEM supplemented as above with 10% FBS. Mouse embryo fibroblasts (MEFs) were isolated from embryonic day 10.5 mouse embryos and cultured in DMEM supplemented as above with 5% FBS.21 HEK293 cells and PAC122 cells were cultured in DMEM supplemented as above with 5% FBS. All cultures were maintained in humidified 5% CO2 at 37°C. For coculture, 3x104 mural cells were seeded in 12-well plates, and after adhesion, 3x104 HUVECs were added. To separate endothelial cells from HAoSMCs and HMSCs, anti-PECAM1-conjugated Dynabeads (Invitrogen) were used according to manufacturer’s instructions. We have demonstrated efficacy of this purification procedure previously.23 The purity of the smooth muscle cells was verified by costaining the separated cells for PECAM1 and ACTA2 and counting cell number. The separated smooth muscle cell population was greater than 99% pure. All cell coculture experiments, unless indicated, were performed in media consisting of EBM-2 supplemented with the bullet kit. NOTCH inhibitor, DAPT (anyl-2-phenyl]glycine-1,1-dimethylethyl ester, Calbiochem) was added to specified wells at the time of plating at 10μM. BMP inhibitor, LDN193189 (Reagentsdirect) was added at 100nM, and TGFβ inhibitor, SB431542 (Reagentsdirect) was added at 1μM. For TGFβ1 treatment, cells were serum starved for 24 hours before TGFβ1 (Peprotech) was added at a 10ng/ml concentration. For conditioned media assays, after 24 hours conditioning, media from HUVECs or HAoSMCs was transferred to HAoSMCs. For transwell assays, 4x104 HAoSMCs were plated on 12-well plates, and 0.4μm pore-size transwell inserts (Corning Costar) were inserted containing 2x104 HUVECs or HAoSMCs.
Primary mouse aorta smooth muscle cell culture
Mice were euthanized at 4–5 weeks of age and a midsternal thoracotomy was performed. The thoracic aorta was isolated and adventitia was carefully removed in cold PBS (Phosphate Buffered Saline). Aorta was digested with 1mg/ml Collagenase II (Sigma, C6885) and 100μg/ml Elastase (Sigma, E0127) at 37°C for 40 minutes. After digestion, cells were pelleted and plated in DMEM with 10% FBS. The next morning, cells were washed with PBS 3–4 times, followed by media refresh every 48 hours. Primary cells at passage 2 were used for experiments.
Quantitative Real-Time PCR (qPCR)
Total RNA was isolated using TRIzol reagent following manufactures’ instructions (Invitrogen). Mouse tissue was first homogenized using TissueLyzer II (Qiagen). RNA was reverse transcribed with M-MLV reverse transcriptase (Promega) to generate cDNA. Real-time PCR was performed using a StepOne PCR system (Applied Biosystems) with SYBR Green. Taqman assays were performed to detect mature microRNAs using a Taqman microRNA assay kit (Life technologies # 4427975).
Immunoblotting
Equivalent amounts of protein were run on 10% SDS-PAGE gels, transferred to nitrocellulose membranes (Millipore), and subjected to incubation using primary antibodies to TGFBR2 (Cell Signaling, 3713), SERPINE1 (BD Transduction, 612025), Fibronectin (FN1) (BD, 610078), Collagen I (Col1A1) (Abcam, ab292), Elastin (ELN) (Abcam, ab77804), Calponin1 (CNN1) (Sigma, C2687), SM α-ACTIN (ACTA2) (Sigma, 1A4), Tubulin (TUBB2A) (Sigma, T7816) and GAPDH (Novus Bio, NB300-221), Phospho-SMAD2 (Ser465/467)(Cell Signaling, 3101), SMAD2 (Cell Signaling, 5339), Phospho-p38 MAPK (Cell Signaling, 9211), p38 MAPK (Cell Signaling, 9219).
RNA mimic, miRNA inhibitor, and siRNA transfection
HAoSMCs were plated in a 12-well plate at 3x104 cells/well. After 12 hours, the cells were transfected with miR145 or control RNA mimic at 40nM using Lipofectamine RNAiMAX (Invitrogen). For miR145 inhibition, 200nM of miRVana inhibitor MH11480 (Life Technologies) was transfected into 6x104 cells/well HAoSMCs using RNAiMAX as directed. For coculture experiments, HUVECs were added 24 hours after transfection. For siRNA knockdown, HAoSMCs were plated in a 12-well plate at 6x104 cells/well. Cells were transfected with 40 nM of control, Notch2 or Notch3 siRNA using RNAiMAX (Invitrogen). After 24 hours, cells were cocultured with 6 x104 HUVEC for additional 96 hours and collected for qPCR analysis. siRNA to knockdown JAG1 in endothelial cells (Dharmacon M-011060-02) was used at 80 nM with RNAiMAX transfection reagent prior to coculture. Knockdown was verified by qPCR and Western blot analysis (Online Figure II and 24).
Lentivirus expression
Mouse NICD1 cDNA was cloned into pCDF1-MCS2-EF1-copGFP (System Biosciences) in front of the CMV promoter using BamHI and EcoRI sites. NICD2, NICD3 and DN-MAML constructs were made as described previously.25 The lentiviral plasmids were transfected into TN-293 cells using Lipofectamine 2000 (Invitrogen), and the viral particles were amplified and purified as described.26
Plasmid transfection and Luciferase assays
psi-CHECK2-TGFBR2 3′UTR plasmid was obtained from Addgene plasmid #31882.27 HEK293 or PAC1 cells were plated in a 12 well plate and transfected with 500ng plasmid and RNA mimics at 100nM concentration. 24 hours later, Dual luciferase assay was performed to measure the firefly luciferase conjugated to the 3′UTR normalized to Renilla luciferase activity followed the instructions of manufacturer (Promega). The miR145 target site in the TGFBR2 3′UTR was mutated from AACTGGAA to AAAAAAAA by PCR mutagenesis.
Collagen secretion assay
Cell culture medium was incubated with 25% (NH4)2SO4 at 4°C overnight. The secreted collagen was pelleted by centrifugation at maximal speed and resuspended in 950μl of 50μM Sirius Red at room temperature. The stained collagen was centrifuged down and dissolved in 0.1M KOH. The absorbance was determined in spectrophotometer of 540nm wavelength.
miR145 knockout animals and angiotensin II infusion
The mouse studies were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee at the Research Institute at Nationwide Children’s Hospital. miR145 knockout mice, referred to here as miR145−/− were generated and generously provided by Dr. Eric Olson,18 and maintained in C57Bl/6 background. miR145+/− mice were crossed to generate wildtype and miR145−/− mice. For Angiotensin II (Ang II) infusion, wild-type and miR145−/− mice (20 to 24 weeks old) were randomly divided into two treatment groups: one group (n = 6 per group) received vehicle (0.9% saline) and the other group was administered Ang II (1.4 mg/kg/day, Sigma, St. Louis, MO) via Alzet mini osmotic pumps (Durect Corporation, Model 2004, Cupertino, CA). Briefly, mini-pumps were filled with either vehicle or Ang II and allowed to prime for 48-hours prior to surgical implantation according to the manufacturer’s instructions. Pumps were implanted subcutaneously under 2% isoflurane anesthesia using aseptic technique, after which they were given buprenorphine for pain in drinking water and monitored until ambulation. A subset of AngII-infused mice were injected daily with TGFβ receptor inhibitor, SB431542 (Selleckchem) at 10 mg/kg/day in DMSO for 14 days. All other mice received DMSO vehicle injections as controls. After 14 days of treatment, mice were sacrificed and tissues were harvested for RNA isolation or histological analysis.
Ex vivo culture of mouse aorta
Thoracic aortas were dissected from 4-week old mice, and the endothelial layer was carefully removed by scraping with scalpel. After cutting into two equal halves, aorta pieces were cultured in EBM-2 with 10% FBS for 24 hours and then serum starved in DMEM with 0.25% FBS for additional 24 hours. After starvation, ex vivo cultured aortas were treated with or without TGFβ1 for 24 hours.
Immunohistochemistry and histology
After fixation in 4% paraformaldehyde, tissues were processed, embedded in paraffin, and sectioned at 8 μm. Sections were then incubated with primary antibodies, ACTA2 (1:1000, SIGMA, Cat: A2547), TGFBR2 (1:100, Santa Cruz, sc-400) overnight at 4 °C. Primary smooth muscle cells were cultured on chamber slides and fixed with 4% PFA at room temperature for 1 hour. Fluorescence from same area was quantified and normalized to DAPI intensity. Masson’s trichrome staining was performed on sections using a kit purchased from Sigma following kit instructions. Quantification of trichrome staining was performed using Image-Pro Plus software.
Statistical analysis
Data analyses were performed using GraphPad Prism and comparisons between data sets were made using a Student’s t test or ANOVA. Differences were considered significant if P < 0.05, and data are presented as mean ± standard deviation (SD). Data shown are representative of at least three independent experiments.
RESULTS
miR-143/145 is induced in smooth muscle cells by endothelial cells
Endothelial cells can regulate the phenotype of smooth muscle cells and our lab previously demonstrated that cocultured endothelial cells promote vascular smooth muscle cell differentiation.26, 28 To examine the extent of this modulation by endothelial cells, we measured the expression of the microRNA gene cluster miR-143/145, which has been linked to governing smooth muscle differentiation.14, 16, 18 Coculture of human umbilical vein endothelial cells (HUVECs) with human aortic smooth muscle cells (HAoSMCs) caused an increase in precursor microRNA pri-miR-143/145 transcript as well as the individual mature microRNAs (Figure 1A–C). The expression of both the precursor and mature microRNAs was observed at high levels after 48 hours of incubation and was sustained, albeit at reduced levels up to six days in coculture. Smooth muscle α-actin RNA and protein (ACTA2) showed a similar expression profile in cocultured aortic smooth muscle cells, compared to miR-143/145 (Figure 1D,E). Analysis of other smooth muscle-enriched microRNAs,8 miR-21, miR-26a, and miR-221 revealed that miR-143/145 were uniquely upregulated by cocultured endothelial cells (Online Figure IA). Expression of miR-143/145 could also be induced in smooth muscle cells cocultured with microvascular endothelial cells, and was also upregulated in cocultured smooth muscle precursors, mesenchymal stem cells (Online Figure IB–F). Thus, miR-143/145 shows an increase in expression consistent with the smooth muscle differentiation phenotype induced by endothelial cells.
Figure 1. Endothelial cells induce miR-143/145 expression in vascular smooth muscle cells.
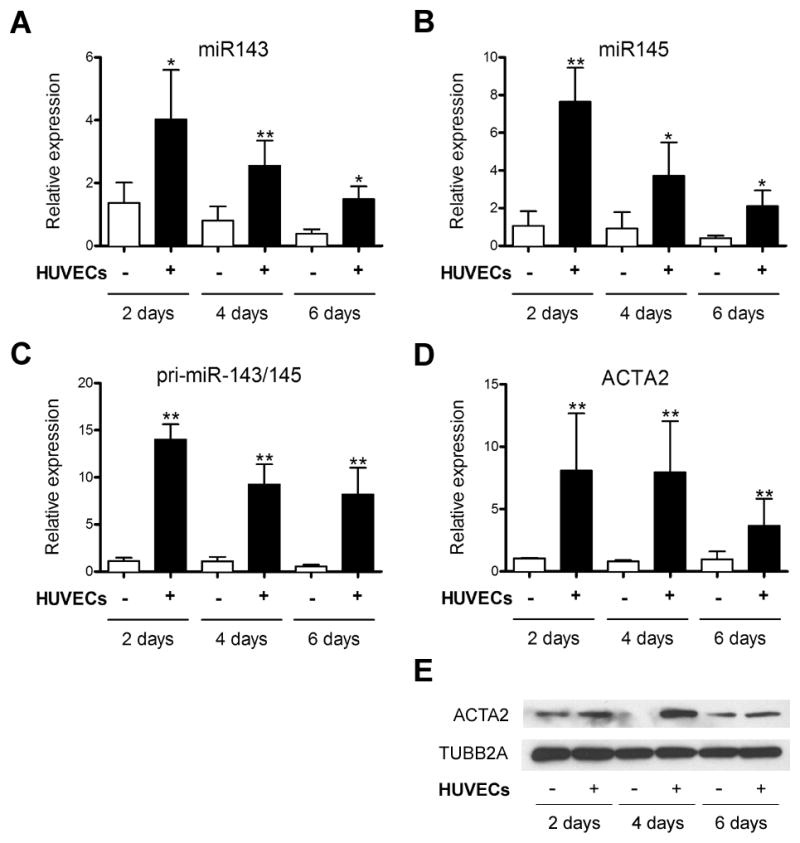
Human aortic smooth muscle cells (HAoSMCs) were cocultured with human umbilical vein endothelial cells (HUVECs) for 2, 4 and 6 days, separated using anti-PECAM1-conjugated dynabeads, and smooth muscle-derived RNA was subjected to qPCR analysis for (A) mature miR143, (B) mature miR145, (C) pri-miR-143/145 transcript and (D) ACTA2 expression. (E) Western blot of ACTA2 protein samples isolated from alone and cocultured smooth muscle cells following separation. Tubulin (TUBB2A) was used as loading control. * P < 0.05, **P < 0.01 relative to control without endothelial cells.
miR-143/145 is regulated by Notch signaling
To determine whether the induction of miR-143/145 required cell-cell contact between endothelial cells and smooth muscle cells, we first treated smooth muscle cells with conditioned media. miR-143/145 levels remained low in smooth muscle cells treated with HUVEC-conditioned media, similar to those treated with HAoSMC-conditioned media, whereas the control cocultured cells showed robust induction (Figure 2A). We also utilized a transwell assay to physically separate cocultured cells. Like the conditioned media experiments, smooth muscle expression of miR-143/145 was not increased by endothelial cells separated by a transwell membrane (Figure 2B). Our data suggested that miR-143/145 expression required cell-cell contact, and given that membrane-bound Notch signaling is known to be critical for endothelial cell-induced smooth muscle differentiation,26 we tested the role of Notch signaling in miR-143/145 expression.
Figure 2. Examination of miR-143/145 transcript expression in relation to Notch signaling.

(A) HAoSMCs were treated with conditioned media collected from HAoSMCs or HUVECs for 48 hours. (B) HAoSMCs were cultured with HUVECs seeded in a transwell insert as indicated for 48 hours. Cocultured cells were used as a positive control. (C) HAoSMCs were cultured with HUVECs in the presence or absence of Notch inhibitor DAPT for 48 hours. (D) HAoSMCs were lenti-virally transduced with GFP, as control, or dnMAML for 48 hours, and then cultured alone or with HUVECs for an additional 48 hours. (E) HAoSMCs cultured alone or with HUVECs, which were transfected with JAG1 siRNA. (F) HAoSMCs were cultured with HUVECs in the presence or absence of BMP receptor inhibitor (ALK2/3), LDN193189 and TGFβ receptor inhibitor (ALK4/5/7), SB431542 for 48 hours. (G) The Notch1 (NICD1), Notch2 (NICD2) or Notch3 (NICD3) intracellular domains were overexpressed in HAoSMCs by lenti-viral infection for 96 hours. A GFP-expressing virus was used as control. (H) Ascending aorta and descending aorta were isolated from Notch3+/− (heterozygous) and Notch3−/− (null) mice to isolate RNA for miR-143/145 analysis. (I) RNA was collected from MEFs isolated from embryos with mutations in the Notch2 and Notch3 genes. In all panels, expression of the pri-miR-143/145 transcript was analyzed by qPCR. For coculture experiments, HAoSMCs were separated from endothelial cells prior to analysis. * P < 0.05, **P < 0.01, n.s. not significant.
Using the chemical inhibitor, DAPT and a lentiviral-delivered dominant-negative-mastermind-like1 (dn-MAML) protein29 to block Notch signaling, we measured the induction of miR-143/145 in smooth muscle cells. Both strategies to block Notch signaling resulted in an almost complete absence of miR-143/145 induction by endothelial cells (Figure 2C,D and Online Figure II). Moreover, knockdown of the Notch ligand, JAGGED-1 (JAG1) on endothelial cells also caused a loss of miR-143/145 induction in smooth muscle cells (Figure 2E, Online Figure II). In contrast, receptor inhibitors to prevent TGFβ (ALK4/5/7) and BMP (ALK2/3) signaling, both which have been previously shown to induce miR-143/145 expression, did not block the upregulation (Figure 2F, Online Figure II).30, 31 We additionally targeted the NOTCH2 and NOTCH3 receptors by siRNA,24 in smooth muscle cells and demonstrated that loss of either receptor attenuated the endothelial cell-induced upregulation of miR-143/145 (Online Figure II). Further, overexpression of the intracellular domains of Notch1, Notch2, or Notch3 promoted robust expression of miR-143/145 in smooth muscle cells (Figure 2G). To test if Notch signaling regulates miR-143/145 expression in vivo, we isolated ascending and descending aortas from Notch3-deficient mice32 and examined expression. The data show that miR-143/145 levels are decreased by approximately 25% in the Notch3 null mice compared to heterozygous controls (Figure 2H). This was further confirmed using mouse embryonic fibroblasts (MEFs) deficient in both Notch2 and Notch3. Homozygous null MEF cells exhibited greater than 40% decrease in miR-143/145 expression compared to Notch2/Notch3 double heterozygotes (Figure 2I).21, 33 These data are consistent with previous published in vitro findings,34 and show that Notch signaling regulates miR-143/145 expression in vivo. The modest reduction of expression in the absence of Notch2 and Notch3 suggest that other mediators, such as Notch1 or Myocardin might contribute to its expression as previously shown.16, 18, 34
miR145 targets the TGFβ signaling pathway
Expression and deletion analysis of miR-143/145 have suggested a role in the modulation of smooth muscle cell phenotypes, and although targets for these microRNAs have been identified, the analysis is incomplete. In an attempt to identify key signaling pathways that miR-143/145 may influence we performed a computational screen with the PANTHER classification system 35 using data derived from TargetScan.36 TargetScan, identified 406 putative target genes for miR-143 and 717 possible targets for miR145. These target genes were screened as a group using PANTHER to identify pathways, which were preferentially targeted by these individual miRs. miR-143 targeted pathways unrelated to miR145 (not shown), Interestingly, miR145 preferentially targeted two opposing signaling pathways in smooth muscle, PDGF and TGFβ (Online Figure IIIA). The PDGF pathway had been previously shown to be a miR145 target and suppressor of miR145 expression,37 while TGFβ was shown to be upstream activator of miR145 expression,30, 31 suggesting that miR145 may exist in a negative feedback loop to dampen TGFβ signaling. Overexpression of a miR145 mimic in smooth muscle cells caused a decrease in TGFβ readout genes, SERPINE1 (PAI-1) and SMAD7, 38, 39 suggesting a decrease in TGFβ signaling (Online Figure IIIB). A cursory examination of the 16 putative target genes within the TGFβ pathway showed that 7 of the 16 exhibited a significant decrease in transcript expression in the presence of a miR145 mimic in smooth muscle cells (Online Figure IIIC). The most significantly downregulated of these genes was the TGFβ receptor II (TGFBR2). Examination of the 3′ untranslated region (UTR) of the TGFBR2 gene revealed a highly conserved miR145 target sequence (Figure 3A). To test if this element was a functional seed sequence, the 3′UTR was cloned into a luciferase expression vector to evaluate its response to miR145.27 Cotransfection of the luciferase reporter with the miR145 mimic into HEK 293 or PAC1 smooth muscle cells 22 showed the 3′UTR conveyed decreased expression, whereas a mutated version of the 3′UTR that could not be recognized by miR145 was not affected (Figure 3B). The ability of miR145 to regulate TGFBR2 was further confirmed by Western blot, where overexpression of the miR145 mimic reduced TGFBR2 protein in smooth muscle cells (Figure 3C). Furthermore, we saw a reduction of TGFBR2 RNA and protein in smooth muscle cells cocultured with endothelial cells (Online Figure IVA, B), consistent with an increase in endogenous miR145 (Figure 1). Inhibition of miR145 by an antagomir revealed that TGFBR2 expression is attenuated by miR145 in smooth muscle cells cultured alone and additionally the downregulation of TGFBR2 by cocultured endothelial cells is dependent upon miR145 function (Online Figure IVC).
Figure 3. TGFβ receptor II (TGFBR2) is a direct target of miR145.
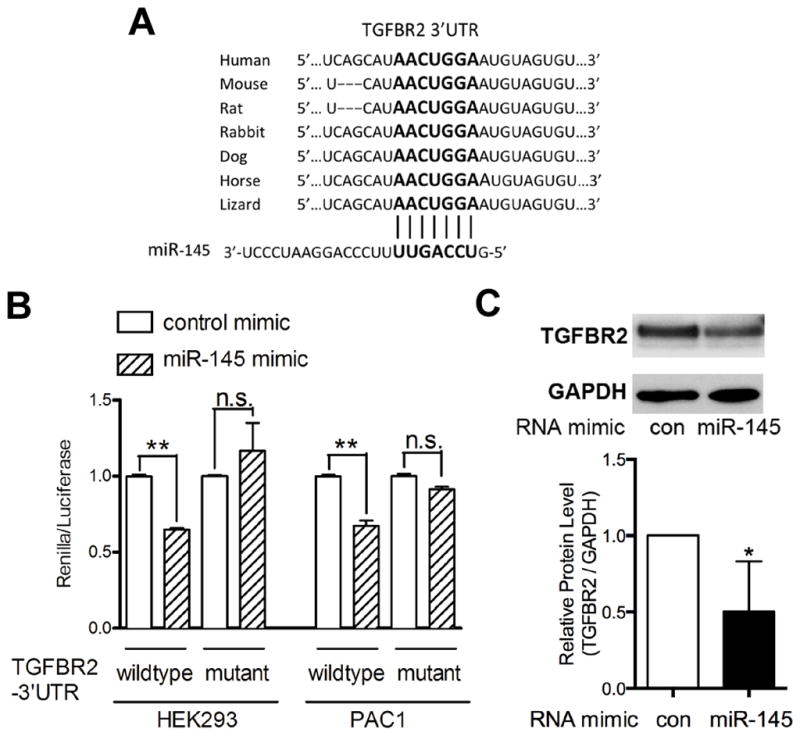
(A) The seed sequence (bold) of miR145 on TGFBR2 3′UTR is highly conserved across species. (B) Wildtype or mutant 3′UTR of TGFBR2 gene was cloned into the Renilla reporter plasmid. The TGFBR2-3′UTR constructs were transfected into HEK293 and PAC1 cells with control or miR-145 mimic, followed by Renilla luciferase assays. (C) Western blot analysis of TGFBR2 protein level in HAoSMCs transfected with a miR-145 or control (con) mimic. Graph shows quantification from 3 separate experiments. * P < 0.05, **P < 0.01, n.s. not significant.
miR145 regulates TGFβ-pathway genes and matrix gene expression in smooth muscle cells
In smooth muscle cells, TGFβ signaling and miR145 are pro-differentiation mediators; 14, 16, 18, 40 yet, our data indicate that miR145 suppresses the expression of certain TGFβ pathway genes. Therefore, we sought to explore how miR145 would influence TGFβ-dependent smooth muscle gene expression. Overexpression of the miR145 mimic in smooth muscle cells in the presence and absence of TGFβ1 ligand showed, as before, a decrease in TGFBR2 and SERPINE1 RNA and protein expression (Figure 4A, D). As expected, the smooth muscle marker genes, ACTA2, CNN1 (h1-calponin), and TAGLN (SM22α) all were increased with TGFβ stimulation and miR145 mimic alone, and together appeared to exhibit an additive response, suggesting independent mechanisms of action (Figure 4B,D). Together these data indicate that miR145 has a selective effect on TGFβ signaling and may facilitate unique downstream events. Deletion analysis of TGFBR2 in smooth muscle cells of mice revealed an important role in the regulation ECM genes, like elastin, collagens, and the matrix crosslinking genes, Lox and Lox1.41, 42 We examined expression of these matrix genes in the presence of the miR145 mimic, and similar to TGFBR2 expression, we observed a decrease in expression at both RNA and protein level (Figure 4C, D). Interestingly, while Fibronection (FN1) transcripts were decreased by miR145, we saw no significant effect on protein expression. This data demonstrate that in smooth muscle cells, miR145 selectively regulates TGFβ signaling and blocks matrix synthesis, while permitting expression of smooth muscle differentiation genes.
Figure 4. miR145 regulates TGFβ-dependent matrix gene synthesis.

HAoSMCs were transfected with miR-145 mimic in the presence or absence of TGFβ1. (A) qPCR was performed to measure the expression of TGFBR2 and SERPINE1, (B) smooth muscle differentiation genes and (C) matrix genes. (D) Western blot to analyze the protein expression of smooth muscle and matrix genes. * P < 0.05, **P < 0.01, n.s. not significant.
miR145 deficiency increases TGFβ signaling in smooth muscle cells in vivo
To investigate whether miR145 regulates TGFβ-dependent matrix gene expression in vivo, we utilized miR145-deficient (miR145−/−) mice.18 miR145 null mice are viable with deficits in smooth muscle function.14–18 We isolated aortas from adult wild-type and miR145−/− mice and performed immunostaining and Western analysis to detect Tgfbr2 and Acta2 expression (Figure 5A–D). Expression of Tgfbr2 in the miR145−/− aortas was increased compared to wild-type, while Acta2 expression showed slight but insignificant difference between wild-type and miR145-deficient mice. Aorta tissue was isolated and cultured ex vivo with or without TGFβ1 and matrix gene expression was measured by qPCR (Figure 5E). While there were no significant differences in the basal level of expression between wild-type and miR145 null mice, after TGFβ1 challenge there was a greater induction in the absence of miR145. We additionally isolated aortic smooth muscle cells from wild-type and miR145 null mice and measured matrix gene expression in cultured cells. Expression of some matrix and matrix synthesis genes was increased at basal levels in the absence of miR145, and all showed an increased level of expression in response to TGFβ1 (Online Figure VA,B). Collagen secretion was also measured from the culture media and showed an increase in the miR145 null mice (Online Figure VC). Western blot on extracts from cultured cells to detect Tgfbr2 and Acta2 showed consistent results indicative of miR145 suppression (Online Figure VD). These findings demonstrate that miR145 functions to repress matrix gene expression and might likely act to govern aberrant ECM deposition. Given that downstream targets of the TGFβ pathway were altered by the loss of miR145, we measured if TGFβ signaling mediators were changed. Analysis of phosphorylated Smad2 and p38 MAP kinase in aorta extracts from wild-type and miR145-deficient mice revealed a trending but not significant increase in SMAD2 phosphorylation, while we observed a significant increase in phosphorylated p38 MAP kinase (Figure 5F). Thus, these data indicate that miR145 attenuates TGFβ signaling and matrix synthesis, and the direct targeting of Tgfbr2 by miR145 likely contributes to these suppressive abilities.
Figure 5. miR145 null mice have elevated Tgfbr2.

(A) Ascending aortas were isolated from wild-type and miR145 mutant mice and stained for Tgfbr2 (green), Acta2 (red) and DAPI (blue). (B, C) Intensity of Tgfbr2 and Acta2 were quantified and normalized to DAPI. (D) Western blot to analyze the protein expression of Tgfbr2 and Acta2 in aortas, and quantified relative to Tubb2a levels. (E) Aortas were isolated from wild-type and miR145 mutant mice, and cultured in the presence or absence of TGFβ1. RNA was extracted after 24 hours and subjected to qPCR analysis. (F) Western blot to analyze levels of phosphorylated-Smad2 and phosphorylated-P38 MAP kinase in aortas. Amounts were quantified relative to total protein levels.. * P < 0.05, **P < 0.01, n.s. not significant.
Loss of miR145 exacerbates angiotensin II-induced fibrosis
The data indicate that miR145 regulates TGFBR2 and matrix synthesis and this is most evident when the TGFβ pathway is robustly activated. Fibrotic diseases are associated with excess TGFβ signaling in activated fibroblasts, and given that miR145 also is expressed in cardiac fibroblasts,43–45 we hypothesized that it might act as a suppressor of pathological fibrosis. To test this, we infused angiotensin II (AngII) into mice to induce TGFβ-dependent cardiovascular fibrosis.46, 47 Infusion of AngII (1.4 mg/kg/day)48 by osmotic pumps for 14 days in wild-type and miR145-deficient mice revealed data consistent with our previous results. AngII caused interstitial and perivascular fibrosis within the hearts of wild-type mice, and this fibrosis was significantly increased in the miR145 knockout animals. Examination of the hearts of these mice by Masson’s trichrome staining revealed a much more pronounced collagen accumulation in the miR145 null mice (Figure 6). To determine if the enhanced fibrosis observed in the miR145-deficient mice was dependent upon TGFβ receptor signaling we treated a subset of AngII-infused mice with a TGFβ receptor inhibitor. Tgfbr2 requires Tgfbr1 to function,49 so we utilized a well-characterized Tgfbr1 inhibitor (ALK4/5/7) (SB431562) to block receptor activity. The data show that blocking TGFβ receptor activity reversed the AngII-induced fibrosis (Figure 6). Furthermore, examining vascular fibrosis of the coronary arteries by trichrome staining revealed a significant increase in collagen deposition in miR145 null mice compared to wild-type mice, which was attenuated by addition of the TGFβ inhibitor (Figure 7A–C). Analysis of the aorta revealed no overt difference in collagen content by trichrome staining (not shown), and there was no significant difference in Col1A1 transcript expression in wild-type compared to miR145 null mice infused with AngII (Figure 7D). However, we did observe significant increases in the expression levels of Serpine1 and Ctgf between AngII-infused miR145-deficient animals compared to control groups (Figure 7E, F). To assess if the increase in fibrosis markers could be correlated to Tgfbr2 expression, we examined Tgfbr2 transcripts. Consistent with our previous data (Figure 5), Tgfbr2 expression in miR145-deficient aortas was higher than in wild-type mice, and there was a significant increase of expression in both wild-type and mutant mice infused with AngII compared to the saline-treated wild-type control (Online Figure VI). However, we failed to see a greater increase of Tgfbr2 expression in the miR145 null mice in the presence of AngII, indicating that other factor likely contribute to the enhanced fibrosis phenotype in the miR145 mutant mice. Interestingly, the RNA expression of these TGFβ-dependent genes was not attenuated by the TGFβ receptor inhibitor, suggesting additional mediators contribute to their regulation. Together, these data demonstrate that miR145 has the capacity to regulate TGFβ-dependent responses in pathological conditions, and acts to selectively regulate matrix synthesis independent of smooth muscle differentiation genes.
Figure 6. Loss of miR145 exacerbates angiotensin II-induced fibrosis.

(A) Masson’s trichrome staining of representative heart sections of wild-type or miR145-deficient mice infused with angiotensin II (AngII) or control saline for 14 days, highlighting collagen deposition in blue. Subset of mice injected with TGFβ receptor inhibitor SB431542 (SB). (B) Quantification of trichrome staining to measure collagen in the different groups. **P < 0.01.
Figure 7. Loss of miR145 promotes angiotensin II-induced collagen deposition in coronary arteries.

Masson’s trichrome staining of (A) left anterior descending artery (LAD) and (B) septal artery (SA) of wild-type or miR145-deficient mice infused with angiotensin II (AngII) or control saline for 14 days, highlighting collagen deposition in blue. TGFβ receptor inhibitor (SB)431542 was injected in a subset of mice. (C) Quantification of collagen from trichrome stained sections of the different groups. (D–F) qPCR was performed to measure the expression of Cola1, Serpine1 and Ctgf in RNA from aorta. * P < 0.05, **P < 0.01, n.s. not significant.
DISCUSSION
Previous studies demonstrated a role for miR145 in regulating smooth muscle differentiation.14–18 Collectively the data indicate that miR145 acts to drive smooth muscle-specific gene expression in a vast regulatory loop that includes activation of Myocardin and inactivation of dedifferentiation mediator KLF4.8 Our data support this pro-differentiation notion, as we show that miR-143/145 transcript is induced by endothelial cells during Notch-regulated smooth muscle differentiation. Previously, smooth muscle cells were shown to take up miR145 from exosomes secreted by endothelial cells, which was demonstrated to have an atheroprotective role.50 Thus, endothelial cells provide miR-143/145 to smooth muscle cells via multiple strategies that might be linked to a distinct function. TGFβ is a well-described smooth muscle differentiation inducer, and studies have shown that TGFβ activates miR145 expression.30, 31 Our initial finding that miR145 preferentially targeted TGFβ signaling genes suggested it might exist in a negative feedback loop. Though this may be the case, further analysis in this study revealed that miR145 targeted distinct subsets of TGFβ-dependent genes. We show that in smooth muscle cells TGFβ activates both matrix genes and smooth muscle-specific genes, but miR145 specifically attenuated the expression of the matrix genes, leaving the smooth muscle-specific genes unaffected. This selective effect on TGFβ target genes implies that miR145 functions to control the actions of TGFβ and define smooth muscle cell phenotypes. A proposed model is illustrated in Figure 8, where an increase in TGFβ causes both matrix genes and smooth muscle differentiation genes. Under conditions that drive a contractile phenotype, miR145 expression is high causing reduced TGFβ to preferentially activate smooth muscle differentiation genes and suppress matrix. Under conditions in which a synthetic phenotype is warranted, miR145 levels are reduced that allows for TGFβ-dependent matrix gene expression to ensue.
Figure 8.
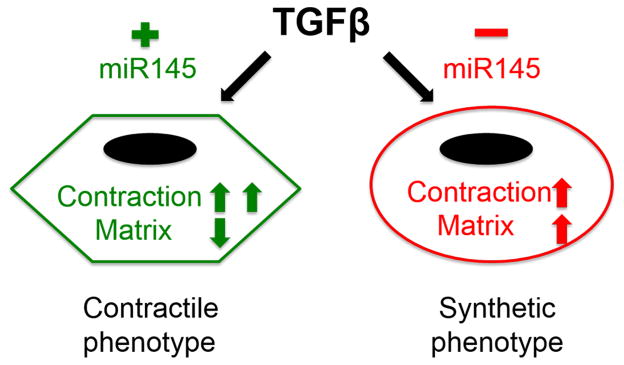
Loss of miR145 switches TGFβ-regulated contractile phenotype to synthetic phenotype in smooth muscle cells.
Our data demonstrate that TGFBR2 is a direct target of miR145 and this finding led us to examine matrix gene expression because of the established link to this receptor’s activity. Tgfbr2 is essential for normal vascular development and mouse knockouts have shown a role in the regulation of matrix synthesis, elastogenesis, and aortic wall homeostasis.41, 42 Human mutations found in this receptor cause Loeys-Dietz and Marfan syndrome type 2.51 The ability of miR145 to decrease TGFBR2 levels in smooth muscle cells is one mechanism through which it regulates matrix, but we expect other direct targets of the TGFβ pathway and matrix synthesis also contribute to this outcome. Indeed, we demonstrated that additional genes in the TGFβ pathway are decreased by miR145, but whether these are direct targets is currently under investigation. TGFβ signaling is a complex pathway that involves autoregulation of both a positive and negative nature and mutations in TGFβ components cause paradoxical increases in TGFβ signaling.7, 52 Our attempts to quantify changes in TGFβ signaling under differing miR145 levels by measuring SMAD and p38 MAP kinase phosphorylation did reveal small increases in their activity in the absence of miR145. Additionally, examination of promoter regions for SMAD binding elements (SBE) of five matrix genes that were downregulated by miR145 mimic only found a potential SBE in the Col1A1 promoter (not shown). We are presently unable to explain the exact mechanism through which miR145 suppresses matrix gene expression. Furthermore it is unclear if suppression of matrix genes by miR145 is solely through the TGFβ pathway and TGFBR2.
Finally, data presented here indicate that miR145 might serve as a critical checkpoint in the development of TGFβ-associated diseases. TGFβ signaling is linked to a range of cardiovascular diseases, many of which are based on inappropriate ECM deposition.52 Our results demonstrate that cardiac and perivascular fibrosis is increased in the absence of miR145, thus finding ways to increase or maintain miR145 in fibrotic diseases may have beneficial consequences. A recent report analyzing miR145 in cardiac fibroblasts indicated that just as in smooth muscle cells, miR145 promoted a contractile phenotype, and consistent with our results, loss of miR145 increased scarring in response to injury.43 Given that miR145 can preferentially down regulate TGFβ-dependent matrix synthesis while leaving other TGFβ-responsive pathways unaffected could serve as a valuable treatment strategy.
Supplementary Material
Novelty and Significance.
What Is Known?
miR145 functions to modulate smooth muscle cell and fibroblast differentiation.
miR145 favors a differentiated rather than a synthetic phenotype.
TGFβ signaling has been linked to promoting both synthetic (fibrotic) and differentiated phenotypes.
What New Information Does This Article Contribute?
The TGFβ signaling pathway is a target of miR145.
miR145 selectively regulates downstream targets of TGFβ signaling.
Loss of miR145 in mice increases cardiovascular fibrosis.
In this study, we found that microRNA miR145 can selectively suppress TGFβ-dependent matrix synthesis while promoting smooth muscle-specific gene expression. In the absence of miR145 there was an increase in a synthetic or fibrotic phenotype. These findings suggest that miR145 acts to discriminate TGFβ targets and this effect facilitates a differentiated phenotype. Thus, in the presence of miR145, TGFβ functions as a mediator of differentiation, and in the absence of miR145, TGFβ exhibits a synthetic and matrix-expressing phenotype. These results offer insight into how microRNAs modulate smooth muscle cells and fibroblasts through their unique regulation of TGFβ signaling and provide evidence for a mechanism in which TGFβ induces different downstream actions via regulation by miR145
Acknowledgments
SOURCES OF FUNDING
American Heart Association to BL (GIA), NZ (predoc), and National Institutes of Health to AJT (K99 HL116769). STK was supported by Award Number Grant TL1TR001069 from the National Center For Advancing Translational Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Advancing Translational Sciences or the National Institutes of Health.
Nonstandard Abbreviations and Acronyms
- ECM
extracellular matrix
- TGFβ
transforming growth factor β
- PDGF
platelet-derived growth factor
- BMP
bone morphogenic protein
- HAoSMCs
human aortic smooth muscle cells
- HUVECs
human umbilical vein endothelial cells
- UTR
untranslated region
- qPCR
quantitative polymerase chain reaction
- Ang II
angiotensin II
Footnotes
DISCLOSURES
None
References
- 1.Sevignani C, Calin GA, Siracusa LD, Croce CM. Mammalian microRNAs: a small world for fine-tuning gene expression. Mammalian genome: official journal of the International Mammalian Genome Society. 2006;17:189–202. doi: 10.1007/s00335-005-0066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 3.Miano JM. Serum response factor: toggling between disparate programs of gene expression. Journal of molecular and cellular cardiology. 2003;35:577–93. doi: 10.1016/s0022-2828(03)00110-x. [DOI] [PubMed] [Google Scholar]
- 4.Parmacek MS. Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation. Circulation research. 2007;100:633–44. doi: 10.1161/01.RES.0000259563.61091.e8. [DOI] [PubMed] [Google Scholar]
- 5.Boucher J, Gridley T, Liaw L. Molecular pathways of notch signaling in vascular smooth muscle cells. Frontiers in physiology. 2012;3:81. doi: 10.3389/fphys.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A. 2008;105:1955–9. doi: 10.1073/pnas.0709663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends in cell biology. 2010;20:556–67. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Davis-Dusenbery BN, Wu C, Hata A. Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:2370–7. doi: 10.1161/ATVBAHA.111.226670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Yin H, Jiang Y, Radhakrishnan SK, Huang ZP, Li J, Shi Z, Kilsdonk EP, Gui Y, Wang DZ, Zheng XL. Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:368–75. doi: 10.1161/ATVBAHA.110.218149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circulation research. 2007;100:1579–88. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 12.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. The Journal of biological chemistry. 2009;284:3728–38. doi: 10.1074/jbc.M808788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circulation research. 2009;104:476–87. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. The Journal of clinical investigation. 2009;119:2634–47. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circulation research. 2009;105:158–66. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–10. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell death and differentiation. 2009;16:1590–8. doi: 10.1038/cdd.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23:2166–78. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albinsson S, Sessa WC. Can microRNAs control vascular smooth muscle phenotypic modulation and the response to injury? Physiological genomics. 2011;43:529–33. doi: 10.1152/physiolgenomics.00146.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rangrez AY, Massy ZA, Metzinger-Le Meuth V, Metzinger L. miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circulation Cardiovascular genetics. 2011;4:197–205. doi: 10.1161/CIRCGENETICS.110.958702. [DOI] [PubMed] [Google Scholar]
- 21.Wang Q, Zhao N, Kennard S, Lilly B. Notch2 and Notch3 function together to regulate vascular smooth muscle development. PLoS One. 2012;7:e37365. doi: 10.1371/journal.pone.0037365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothman A, Kulik TJ, Taubman MB, Berk BC, Smith CW, Nadal-Ginard B. Development and characterization of a cloned rat pulmonary arterial smooth muscle cell line that maintains differentiated properties through multiple subcultures. Circulation. 1992;86:1977–86. doi: 10.1161/01.cir.86.6.1977. [DOI] [PubMed] [Google Scholar]
- 23.Lilly B, Kennard S. Differential gene expression in a coculture model of angiogenesis reveals modulation of select pathways and a role for Notch signaling. Physiological genomics. 2009;36:69–78. doi: 10.1152/physiolgenomics.90318.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin CH, Lilly B. Notch signaling governs phenotypic modulation of smooth muscle cells. Vascul Pharmacology. 2014 doi: 10.1016/j.vph.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Zhao N, Liu H, Lilly B. Reciprocal regulation of syndecan-2 and Notch signaling in vascular smooth muscle cells. The Journal of biological chemistry. 2012;287:16111–20. doi: 10.1074/jbc.M111.322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu H, Kennard S, Lilly B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circulation research. 2009;104:466–75. doi: 10.1161/CIRCRESAHA.108.184846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanyam D, Lamouille S, Judson RL, Liu JY, Bucay N, Derynck R, Blelloch R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat Biotechnol. 2011;29:443–8. doi: 10.1038/nbt.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin CH, Lilly B. Endothelial Cells Direct Mesenchymal Stem Cells Toward a Smooth Muscle Cell Fate. Stem cells and development. 2014 doi: 10.1089/scd.2014.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weng AP, Nam Y, Wolfe MS, Pear WS, Griffin JD, Blacklow SC, Aster JC. Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol Cell Biol. 2003;23:655–64. doi: 10.1128/MCB.23.2.655-664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long X, Miano JM. Transforming growth factor-beta1 (TGF-beta1) utilizes distinct pathways for the transcriptional activation of microRNA 143/145 in human coronary artery smooth muscle cells. The Journal of biological chemistry. 2011;286:30119–29. doi: 10.1074/jbc.M111.258814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G, Hata A. down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. The Journal of biological chemistry. 2011;286:28097–110. doi: 10.1074/jbc.M111.236950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krebs LT, Xue Y, Norton CR, Sundberg JP, Beatus P, Lendahl U, Joutel A, Gridley T. Characterization of Notch3-deficient mice: normal embryonic development and absence of genetic interactions with a Notch1 mutation. Genesis. 2003;37:139–43. doi: 10.1002/gene.10241. [DOI] [PubMed] [Google Scholar]
- 33.McCright B, Gao X, Shen L, Lozier J, Lan Y, Maguire M, Herzlinger D, Weinmaster G, Jiang R, Gridley T. Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development. 2001;128:491–502. doi: 10.1242/dev.128.4.491. [DOI] [PubMed] [Google Scholar]
- 34.Boucher JM, Peterson SM, Urs S, Zhang C, Liaw L. The miR-143/145 cluster is a novel transcriptional target of Jagged-1/Notch signaling in vascular smooth muscle cells. The Journal of biological chemistry. 2011;286:28312–21. doi: 10.1074/jbc.M111.221945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–86. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18:1139–46. doi: 10.1038/nsmb.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quintavalle M, Elia L, Condorelli G, Courtneidge SA. MicroRNA control of podosome formation in vascular smooth muscle cells in vivo and in vitro. J Cell Biol. 2010;189:13–22. doi: 10.1083/jcb.200912096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lund LR, Riccio A, Andreasen PA, Nielsen LS, Kristensen P, Laiho M, Saksela O, Blasi F, Dano K. Transforming growth factor-beta is a strong and fast acting positive regulator of the level of type-1 plasminogen activator inhibitor mRNA in WI-38 human lung fibroblasts. EMBO J. 1987;6:1281–6. doi: 10.1002/j.1460-2075.1987.tb02365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–5. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 40.Grainger DJ, Metcalfe JC, Grace AA, Mosedale DE. Transforming growth factor-beta dynamically regulates vascular smooth muscle differentiation in vivo. J Cell Sci. 1998;111 (Pt 19):2977–88. doi: 10.1242/jcs.111.19.2977. [DOI] [PubMed] [Google Scholar]
- 41.Jaffe M, Sesti C, Washington IM, Du L, Dronadula N, Chin MT, Stolz DB, Davis EC, Dichek DA. Transforming growth factor-beta signaling in myogenic cells regulates vascular morphogenesis, differentiation, and matrix synthesis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:e1–11. doi: 10.1161/ATVBAHA.111.238410. [DOI] [PubMed] [Google Scholar]
- 42.Choudhary B, Zhou J, Li P, Thomas S, Kaartinen V, Sucov HM. Absence of TGFbeta signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis. 2009;47:115–21. doi: 10.1002/dvg.20466. [DOI] [PubMed] [Google Scholar]
- 43.Wang YS, Li SH, Guo J, Mihic A, Wu J, Sun L, Davis K, Weisel RD, Li RK. Role of miR-145 in cardiac myofibroblast differentiation. Journal of molecular and cellular cardiology. 2014;66:94–105. doi: 10.1016/j.yjmcc.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 44.Yang S, Cui H, Xie N, Icyuz M, Banerjee S, Antony VB, Abraham E, Thannickal VJ, Liu G. miR-145 regulates myofibroblast differentiation and lung fibrosis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:2382–91. doi: 10.1096/fj.12-219493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghosh AK, Quaggin SE, Vaughan DE. Molecular basis of organ fibrosis: potential therapeutic approaches. Experimental biology and medicine (Maywood, NJ) 2013;238:461–81. doi: 10.1177/1535370213489441. [DOI] [PubMed] [Google Scholar]
- 46.Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-beta signaling in vascular fibrosis. Cardiovascular research. 2007;74:196–206. doi: 10.1016/j.cardiores.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 47.Williams B. Angiotensin II and the pathophysiology of cardiovascular remodeling. The American journal of cardiology. 2001;87:10C–17C. doi: 10.1016/s0002-9149(01)01507-7. [DOI] [PubMed] [Google Scholar]
- 48.Bouzeghrane F, Mercure C, Reudelhuber TL, Thibault G. Alpha8beta1 integrin is upregulated in myofibroblasts of fibrotic and scarring myocardium. Journal of molecular and cellular cardiology. 2004;36:343–53. doi: 10.1016/j.yjmcc.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 49.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 50.Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M, Braun T, Urbich C, Boon RA, Dimmeler S. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nature cell biology. 2012;14:249–56. doi: 10.1038/ncb2441. [DOI] [PubMed] [Google Scholar]
- 51.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature genetics. 2005;37:275–81. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 52.Doyle JJ, Gerber EE, Dietz HC. Matrix-dependent perturbation of TGFbeta signaling and disease. FEBS letters. 2012;586:2003–15. doi: 10.1016/j.febslet.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.