

Abstract
Background
Synthetic L-ascorbic acid (vitamin C) is widely used as a preservative and nutrient in food and pharmaceutical industries. In the current production method, D-glucose is converted to L-ascorbic acid via several biochemical and chemical steps. The main source of L-ascorbic acid in human nutrition is plants. Several alternative metabolic pathways for L-ascorbic acid biosynthesis are known in plants. In one of them, D-galacturonic acid is the precursor. D-Galacturonic acid is also the main monomer in pectin, a plant cell wall polysaccharide. Pectin is abundant in biomass and is readily available from several waste streams from fruit and sugar processing industries.
Results
In the present work, we engineered the filamentous fungus Aspergillus niger for the conversion of D-galacturonic acid to L-ascorbic acid. In the generated pathway, the native D-galacturonate reductase activity was utilized while the gene coding for the second enzyme in the fungal D-galacturonic acid pathway, an L-galactonate consuming dehydratase, was deleted. Two heterologous genes coding for enzymes from the plant L-ascorbic acid pathway – L-galactono-1,4-lactone lactonase from Euglena gracilis (EgALase) and L-galactono-1,4-lactone dehydrogenase from Malpighia glabra (MgGALDH) – were introduced into the A. niger strain. Alternatively, an unspecific L-gulono-1,4-lactone lactonase (smp30) from the animal L-ascorbic acid pathway was introduced in the fungal strain instead of the plant L-galactono-1,4-lactone lactonase. In addition, a strain with the production pathway inducible with D-galacturonic acid was generated by using a bidirectional and D-galacturonic acid inducible promoter from the fungus. Even though, the lactonase enzyme activity was not observed in the resulting strains, they were capable of producing L-ascorbic acid from pure D-galacturonic acid or pectin-rich biomass in a consolidated bioprocess. Product titers up to 170 mg/l were achieved.
Conclusions
In the current study, an L-ascorbic acid pathway using D-galacturonic acid as a precursor was introduced to a microorganism for the first time. This is also the first report on an engineered filamentous fungus for L-ascorbic acid production and a proof-of-concept of consolidated bioprocess for the production.
Keywords: L-ascorbic acid, D-galacturonic acid, L-galactonic acid, Citrus peel, Aspergillus niger, Metabolic engineering
Background
L-Ascorbic acid (L-AA), also known as vitamin C, is a six-carbon organic compound with reducing agent properties. It occurs naturally in many animal and plant cells having biological functions, such as being an antioxidant and enzyme cofactor [1,2]. Synthetic L-AA is commercially used for several purposes in food, beverage, feed and pharmaceutical industries. The annual production of synthetic L-AA is about 110 000 tonnes with the fluctuating market price of about 10 USD per kg [3].
Industrial L-AA production has been traditionally based on the Reichstein process which is an efficient multi-step and mostly chemical manufacturing method converting D-glucose to L-AA [4]. In the first step, D-glucose is hydrogenated to D-sorbitol followed by the oxidation of D-sorbitol to L-sorbose that is commonly carried out using a bacterial fermentation [3]. The resulting L-sorbose is then chemically oxidized to 2-keto-L-gulonate and lactonized to L-AA. Other proposed and reported biotechnological steps have focused on the microbial conversion of D-sorbitol, L-sorbose or D-glucose to 2-keto-L-gulonic acid that is the last intermediate in the Reichstein process [5]. Currently, the predominant industrial process for L-AA production is a so called two-step fermentation process [3]. In the first step, D-sorbitol is oxidized to L-sorbose using Gluconobacter oxydans. The second step is a mixed fermentation with Ketogulonicigenium vulgare and Bacillus megaterium, converting L-sorbose to 2-keto-L-gulonic acid which is then chemically converted to L-AA.
In addition to the partially biotechnological processes, some approaches using wild type or engineered microbes for the direct L-AA production have been reported in the literature. A one-step process from biomass sugar to L-AA would be advantageous when compared to the currently used two-step fermentation process. Yeast have a native biosynthetic pathway for D-erythroascorbic acid (D-EA), a 5-carbon analogue of L-AA, where D-arabinose is converted to D-EA via D-arabino-1,4-lactone [6]. The D-EA pathway seems to be relatively unspecific converting also L-galactose and L-galactono-1,4-lactone (L-galL) to L-AA [7,8]. The reactions are similar to the reactions of L-AA synthesis in plants via the Smirnoff-Wheeler (S-W) pathway. In addition to yeast, a strain of K. vulgare, the bacterium that is used in the two-step fermentation process, is capable of converting D-sorbitol, L-sorbose, L-sorbosone and L-gulose directly to L-AA [9]. Also several strains of algae producing small concentrations of extracellular L-AA have been reported in the literature [10].
In the field of metabolic engineering, there are a few attempts to introduce the plant S-W pathway to yeast either by overexpressing the native substrate-unspecific D-EA pathway genes or expressing the S-W pathway genes from Arabidopsis thaliana. The resulting strains were capable of converting L-galactose, D-glucose and D-galactose to L-AA [11–13]. The product titers were reported to be about 100 mg l−1 from L-galactose [11], 0.1 mg l−1 OD−1 from D-glucose [12] and 30 mg l−1 from D-galactose [13].
In plants, L-AA is considered to be synthesized predominantly through the S-W pathway where D-glucose is converted via GPD-D-mannose and L-galactose to L-galL which is oxidized to L-AA in the final step by the L-galL dehydrogenase (GALDH) [14]. In addition to S-W, a few alternative pathways for L-AA synthesis have been observed in plants. One of them originates from D-galacturonic acid (D-galUA) which is reduced to L-galactonic acid (L-galA) and further lactonized to L-galL, which is the last intermediate in the S-W pathway [1]. Even though the function of a D-galUA reductase for L-AA biosynthesis was shown in higher plants [15], the evidence of the pathway converting D-galUA to L-AA is still incomplete – the gene coding for an L-galL aldonolactonase (ALase) catalysing the lactonization of L-galA to L-galL has been described only in the photosynthetic organism Euglina gracilis [16]. This activity was however found in animals as part of the biosynthetic L-AA pathway. In the animal pathway, L-gulonic acid is lactonized to its corresponding 1,4-lactone by an ALase that is encoded by the smp30 gene. The enzyme is unspecific and can also catalyse the lactonization of L-galA [17].
In the present study, instead of using the plant S-W pathway for L-AA synthesis, we have focused on the alternative pathway converting D-galUA to L-AA. D-GalUA is the main monomer in pectin, a heteropolysaccharide found from plant cell walls, which is especially abundant in some non-woody biomass types, such as citrus fruit peels and sugar beet pulp. Both of these materials are abundantly available as agro-industrial side streams. The enzymes for degradation of pectin to D-galUA the catabolic pathways for its utilization are known in many microbial organisms. In fungi, such as Aspergillus niger, the pathway for D-galUA catabolism is reductive having a similar reduction step from D-galUA to L-galA as in the L-AA synthesis pathway originating from D-galUA in plants (Figure 1) [18]. Previously, we reported engineered A. niger strains capable of converting pure D-galUA and citrus processing waste (CPW) to L-galA that can be chemically converted to L-AA [19,20]. In the present study, we have engineered A. niger strains that are capable of direct production of L-AA from D-galUA and CPW.
Figure 1.
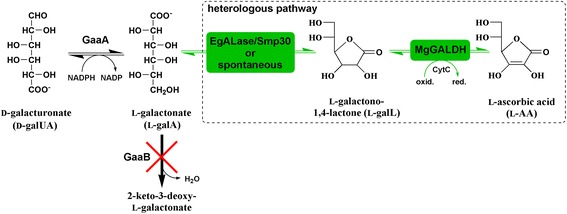
Schematic figure of the disrupted fungal D-galUA pathway and the heterologous pathway introduced in A. niger for L-AA biosynthesis: native GaaA (D-galUA reductase), native GaaB (L-galA dehydratase), EgALase (L-galL aldonolactonase from Euglena gracilis ), Smp30 (aldonolactonase from rat functioning in the animal L-AA pathway) and MgGALDH (L-galL dehydrogenase from Malpighia glabra).
Results
Pathway assembly and transcription
The fungal catabolic D-galUA pathway and the proposed D-galUA originating biosynthetic L-AA pathway in plants share the same reduction step yielding L-galA in the first reaction (Figure 1). Thus, the A. niger strain ΔgaaB with the disrupted L-galA dehydratase gene, which is known to accumulate L-galA, was used as a platform strain. In addition, two heterologous genes from the plant or animal L-AA pathway, an ALase and a GALDH, were introduced to the strain. Several different A. niger ΔgaaB based strains with an ALase (EgALase or smp30) and a GALDH (MgGALDH) or only the GALDH were generated and tested (Table 1). The MgGALDH gene that was used contained its native mitochondrial signal peptide region at the beginning of the ORF. In addition, a constitutive (PgpdA) and a D-galUA inducible (PgaaA/C) promoter were compared.
Table 1.
Plasmids and A. niger strains used in this work
| Plasmid | Genes expressed | Promoter |
|---|---|---|
| JKp1-EgALase | The codon optimized Euglina gracilis Alase (EgALase) [GenBank: AB306917] | gpdA |
| JKp1-Smp30 | The codon optimized rat Smp30 [GenBank: CAA48786] | gpdA |
| JKp1-MgGALDH | The codon optimized Malpighia glabra GALDH (MgGALDH) [GenBank: ACG75919] ORF | gpdA |
| Bidir-EgALase-MgGALDH | EgALase and MgGALDH | gaaA/C |
| Strain | Genes expressed | Description |
| ∆gaaB | none | ATCC 1015 with the deleted L-GalA dehydratase gene gaaB |
| ∆gaaB-Mg | MgGALDH | ∆gaaB with MgGALDH expressed under gpdA promoter |
| ∆gaaB-Eg-Mg (PgpdA) | EgALase and MgGALDH | ∆gaaB with EgALase and MgGALDH, both expressed under gpdA promoter |
| ∆gaaB-Smp30-Mg | Smp30 and MgGALDH | ∆gaaB with Smp30 and MgGALDH, both expressed under gpdA promoter |
| ∆gaaB-Eg-Mg (PgaaA/C) | EgALase and MgGALDH | ∆gaaB with EgALase and MgGALDH, both expressed under bidirectional gaaA/C promoter |
To avoid unnecessary and energy wasting expression of the L-AA pathway genes in non-producing conditions, the strain ΔgaaB-Eg-Mg (PgaaA/C) with D-galUA inducible EgALase and MgGALDH expression was generated using the bidirectional gaaA/C promoter. Since there are no reports on the use of gaaA/C promoter in heterologous expression, the functionality of the D-galUA inducible transcription of EgALase and MgGALDH from the gaaA/C promoter was tested using RT-qPCR (Figure 2). The transcription of both of the heterologous genes was clearly induced at 20 hours after the shift to D-galUA containing medium. The pattern of MgGALDH transcription was similar to the transcription of the native gaaA gene, which was expected since MgGALDH was orientated in the same way behind the bidirectional gaaA/C promoter as the gaaA gene is. The transcription of EgALase was higher when compared to gaaA and MgGALDH after the D-galUA induction. A similar observation was reported earlier in the case of native gaaC gene (2-keto-3-deoxy-L-galactonate aldolase) which had a higher transcription when compared to gaaA growing in D-galUA medium [21]. EgALase had the same orientation at gaaA/C promoter as gaaC gene in the wild type strain.
Figure 2.
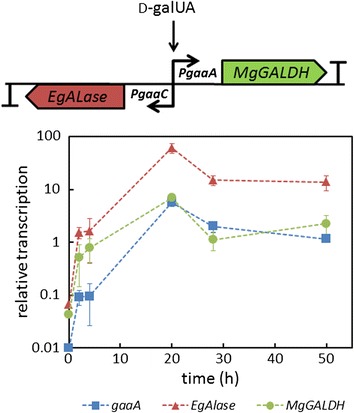
The D-galUA inducible expression cassette and relative transcription of gaaA (blue squares), EgALase (red triangles) and MgGALDH (green circles) in the strain Δ gaaB - Eg - Mg ( PgaaA/C ) after the shift from pre-cultures to medium supplemented with D-galUA and D-xylose. Values were normalized to the expression of actin. Error bars represent ± SEM (n = 3).
Enzyme activities
Enzyme activities of the introduced L-AA pathway genes were measured from crude extracts. The GALDH activity assay was based on the measurement of reduced cytochrome C as a result from oxidation of L-galL. The activity was tested from the parental strain ΔgaaB and from the further engineered strains that were cultured 20 h in the medium supplemented with 20 g l−1 D-galUA and 5 g l−1 D-xylose (Figure 3). All the strains with MgGALDH under the constitutive gpdA promoter showed the activity for L-galL while only residual activity was found from the parental strain ΔgaaB. In the case of ΔgaaB-Eg-Mg (PgaaA/C) strain, the GALDH activity was lower; however, it was still significantly higher if compared to the values from ΔgaaB (P < 0.05, Student’s t-test).
Figure 3.
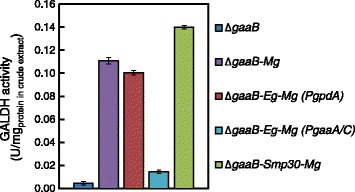
GALDH activity for L-galL from the crude extracts of Δ gaaB , Δ gaaB - Mg Δ gaaB - Eg - Mg ( PgpdA ), Δ gaaB - Eg - Mg ( PgaaA/C ) and Δ gaaB - Smp30 - Mg cultured in minimal medium supplemented with D-galUA and D-xylose for 20 h. All of the values from the strains with introduced MgGALDH differed significantly from the value from ΔgaaB (P < 0.05, Student’s t-test). Error bars represent ± SEM (n = 3).
In the lactonization of L-galA to L-galL, which is catalysed by an ALase, protons are absorbed in an equimolar manner increasing the pH in the reaction. The relation between absorption at 405 nm and proton concentration was quantified being ∆A405 = 0.003 per 10 μM of protons which should provide high enough sensitivity for the ALase assay. We tested all the engineered strains for ALase activity in the presence of L-galA; however, we could not detect this activity.
Production of L-AA
The parental strain ΔgaaB and the different engineered strains were tested for L-AA production even though the lactonase activities were not detectable. We used flask cultures on defined medium supplemented with D-galUA and D-xylose and with the initial pH adjusted to 3.0. With the strain ΔgaaB only L-AA concentrations of below 3.5 mg l−1 were observed in the cultures (Figure 4). All the engineered strains having MgGALDH under gpdA or gaaA/C promoter produced L-AA at concentrations of around 55–83 mg l−1. Also the strain ΔgaaB-Mg without an ALase produced similar L-AA concentrations as the strain ΔgaaB-Eg-Mg (PgpdA) with the introduced EgALase gene. In fact, the L-AA production corresponds to the GALDH activity in the strains having MgGALDH under PgpdA (Figure 3). The clearest difference between the engineered strains was the timing of the highest L-AA concentration in the medium – all the strains with MgGALDH under gpdA promoter had the highest titer at 48 h whereas ΔgaaB-Eg-Mg (PgaaA/C) had the highest titer at 96 h. The decreasing L-AA concentrations during the time course may be due to the instability of the product causing by the exposure to air. In addition, the production turned out to be pH dependent since L-AA production was not observed when the initial pH was 5.0 (data not shown).
Figure 4.
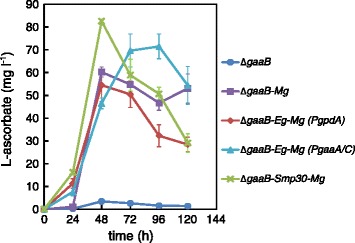
L-Ascorbate production by engineered A. niger strains in minimal medium supplemented with D-galUA and D-xylose. Error bars represent ± SEM (n = 3).
Even though, D-xylose was used as a co-substrate, one of the reasons for limited L-AA production may be the lack of NADPH for the D-galUA reductase gaaA in the first reaction in the pathway. In addition, energy and carbon sources are needed for the expression of heterologous L-AA pathway genes. To overcome these possible limitations and to test L-AA production from a pectin-rich residue, we used CPW as substrate containing a complex mix of different sugars and sugar acids in the form of polysaccharides. We decided to use ΔgaaB-Eg-Mg (PgaaA/C) strain in order to avoid unnecessary and energy wasting expression of the heterologous genes at the beginning of fermentation when free D-galUA is not yet available. In addition, we used higher fungal biomass in the inoculation in order to ensure CPW hydrolysis and speed up the process. L-AA production from CPW turned out to be more efficient than in defined media conditions with respect to the final concentrations (Figure 5). A lag phase of about 48 h was observed before the production started at a higher rate, which is probably due to rather slow pectin hydrolysis to D-galUA and utilization of the more preferred carbon sources, such as D-glucose present in CPW. The D-galUA content in CPW that was prepared as the CPW used in this study was reported to be 27% on a dry mass basis [20]. Other detected main components after pectinase hydrolysis were L-arabinose, D-galactose, D-glucose and L-rhamnose. Even though, L-AA production started later than when compared to the defined conditions with the ΔgaaB-Eg-Mg (PgaaA/C) strain the highest titer (around 170 mg l−1) was achieved at 96 h as was the case of the cultures in the defined conditions. As in the case of defined conditions, a decrease in L-AA concentration was observed after 96 h probably due to the instability of the product. The CPW used as a substrate did not contain detectable amounts of L-AA. In the control fermentations with the parental strain ΔgaaB, the observed L-AA concentrations were below 15 mg l−1.
Figure 5.
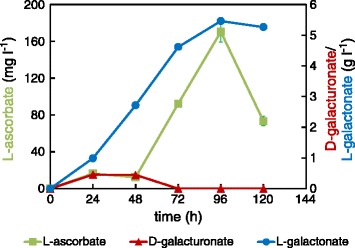
Concentration of extracellular L-ascorbate (green squares), D-galUA (red triangles) and L-galA (blue circles) from a culture of the strain Δ gaaB - Eg - Mg ( PgaaA/C ) in minimal medium supplemented with CPW. Error bars represent ± SEM (n = 3) and where not visible, are smaller than the symbol.
Discussion
Synthetic L-AA is a widely used compound in several industries. Currently it is produced in a multistep process including chemical and biochemical steps. This study is the first report on an engineered filamentous fungus for direct L-AA production. To the best of our knowledge, the L-AA titer of 170 mg l−1 that was achieved in this work is the highest among engineered fungal strains. In addition, the pathway for L-AA biosynthesis originating from D-galUA was for the first time introduced to a heterologous host. In the previous literature, L-AA production was reported to be achievable from D-glucuronic acid and D-galUA using wild type strains of A. niger [22] or yeast [23]. However, the detection method for L-AA that was used in these studies (2,6-dichlorophenolindophenol method) could not distinguish between L-AA and other similar compounds, such as D-EA [24].
We decided to use the GALDH from M. glabra (acerola), which is known for its vitamin C rich fruits, including the native N-terminal mitochondrial signal peptide. The kinetic parameters of MgGALDH are not known but the expression level of MgGALDH is known to be higher in M. glabra tissue when compared to the corresponding GALDH gene (AtGALDH) expression in Arabidopsis thaliana [25]. AtGALDH gene codes for a protein having 75.25% homology with MgGALDH and kinetic parameters of Km 0.17 mM and kcat 134 s−1 for L-galL [26]. We have also tested the expression of AtGALDH gene in the A. niger ΔgaaB strain, however, L-AA was not observed in the resulting strains (data not shown). In plants, the GALDH locates on the inner membrane of mitochondria binding non-covalently FAD as a cofactor [26]. During the oxidation of L-galL to L-AA, electrons are shuttled to cytochrome c and further to the electron transport chain. The recombinant AtGALDH without mitochondrial targeting signal has been produced in E. coli as a cytoplasmic protein [26]. The resulting enzyme was active with L-galL when cytochrome c was added to the reaction mixture. However, in a living cell the functionality of a GALDH is probably dependent on its correct localization enabling interaction with mitochondrial cytochrome c. Thus it is crucial that the mitochondrial targeting signal in a plant GALDH is also functional in A. niger. Mitochondrial inner membrane proteins are most often targeted to their location by an N-terminal cleavable peptide [27]. The targeting peptide is typically positively charged among different organisms. However, in yeast, mitochondrial signal peptides are generally shorter, less alpha-helix forming and less hydrophobic when compared with the plant signal peptides [27]. The cleavage site for the signal peptides seem to be relatively conserved between yeast and plants consisting of tyrosine, leucine, phenylalanine and arginine residues [27]. In the case of AtGALDH and MgGALDH, the estimated signal peptides differ being 102 and 84 residues long, respectively [28]. The shorter signal peptide in MgGALDH may be more suitable for mitochondrial targeting in A. niger, however, the localization of MgGALDH in the engineered A. niger strains was not investigated in this study.
Despite several attempts, the ALase activity for L-galL could not be detected from any of EgALase or smp30 expressing strains. In addition to A. niger strains, we have also tried to express both of the ALases in yeast or E. coli strains, however, without detectable activity. The lactonase assay that was used in this work is based on a pH change and a pH indicator in the reaction mixture. The assay should provide high enough sensitivity for the activity and it has been routinely and successfully used in many studies. Consequently, the most likely explanations for the non-detectable ALase activity are inactivity of the protein in heterologous hosts or inactivation during cell disruption. In the literature, EgALase was produced at low temperature (15°C) in an E. coli strain co-expressing the Trigger Factor chaperone protein [16]. In the case of Smp30 from animal L-AA pathway, the recombinant protein was produced in E. coli co-expressing two chaperones GroEL and GroES [17]. In addition, it has been reported that production of human Smp30 protein in E. coli without chaperones led up to the insoluble and inactive proteins and formation of inclusion bodies [29,30]. Thus poor folding of the ALase proteins in a heterologous host is a possible explanation for the unsuccessful expression.
Regardless of the non-detectable ALase activity, several A. niger strains with a functional GALDH were capable of L-AA production. The lactonization reaction might be spontaneous and may take place extracellularly. In the lactonization reaction, protons are assimilated, which mean that a low pH shifts the equilibrium towards the lactone form. This would explain the fact that only low pH enabled L-AA production. Thus it is possible that L-galA is first secreted out from the cells and spontaneously formed L-galL is then transported back to the cells and oxidized to L-AA. On the other hand, the parental strain ∆gaaB is known for its higher capacity to produce L-galA at low pH [19,20] which may provide more substrate for the L-AA pathway also in the engineered strains when cultured at low pH. The production improved when CPW was used as substrate. In addition to the higher fungal biomass in the inoculation, the improvement was possibly due to the additional carbon sources that were constantly released from the substrate during the fermentation providing energy such as reducing power for the strain. The product titer was still relatively low and possible limited by the poor lactonization reaction from L-galA to L-galL. In addition, L-AA concentrations started to decrease during the fermentation. This is likely due to the oxidation that occurs by the exposure to air. Thus it would be beneficial for the process to optimize factors, such as, aeration and timing of the harvesting. Nevertheless, this is the first demonstration of a consolidated bioprocess for L-AA production.
Conclusions
We have engineered A. niger strains to redirect the D-galUA pathway to L-AA synthesis. The native catabolic D-galUA pathway was disrupted and the biosynthetic L-AA pathway from plants originating from D-galUA was introduced. In addition, we built a strain having the L-AA pathway under D-galUA inducible expression. The resulting strains were capable of L-AA production from pure D-galUA but also from CPW that is a pectin-rich biomass residue. Final L-AA titers up to around 170 mg l−1 were achieved with the engineered strains.
Methods
Strains
The Aspergillus niger strain ATCC 1015 (CBS 113.46) was used as a wild type. The engineered strain of ATCC 1015 with the deleted L-galA dehydratase gaaB [19,20] was used as a host strain for the L-AA production strains that are listed in Table 1. All the plasmids were produced in E. coli TOP10 cells and the homologous recombination for the plasmid construction was carried out in the S. cerevisiae strain ATCC 90845.
Plasmid construction
For introducing the biosynthetic L-AA pathway in A. niger, the heterologous genes coding for a GALDH or an ALase, listed in Table 1, were custom synthetized as codon optimized ORFs (Genscript) and inserted to the JKp1-hph plasmid [19] under the gpdA promoter. Alternatively, the GALDH from Malpighia glabra (MgGALDH) and the ALase from Euglena gracilis (EgALase) were both expressed under the native bidirectional promoter (657 bp) of the A. niger D-galUA reductase (gaaA) and the 2-keto-3-deoxy-L-galactonate aldolase (gaaC) genes that are clustered in the genome. In the expression cassette, MgGALDH was orientated as gaaA and EgALase as gaaC in A. niger genome. Terminators that were used in the bidirectional cassette for MgGALDH and EgALase were the native gaaA and gaaC terminators, respectively, both 505 bp downstream from the stop codon. All of the expression cassettes contained hygromycin B phosphotransferase (hph) gene under the gpdA promoter for the selection of transformants.
The codon optimized ORFs were released with SacI and XmaI (both NEB) and ligated to the JKp1 plasmid. The resulting plasmids JKp1-EgALase/Smp30/MgGALDH were linearized with SpeI (NEB). For constructing the cassette with D-galUA inducible expression, the bidirectional gaaA/C promoter and terminators of gaaA and gaaC genes were amplified from A. niger ATCC1015 genomic DNA with the primers P1-P6 as described in Table 2. The EgALase, MgGALDH and the hph gene (from JKp1) were amplified with the primers P7-P12 (Table 2). All the six amplified fragments, containing 40 bp flanks for homologous recombination, and an EcoRI and BamHI digested platform plasmid pRS426 were transformed to yeast using the Gietz method [31] and the transformants were selected on SCD-URA plates. Several yeast colonies were collected and the resulting plasmid Bidir-EgALase-MgGALDH was rescued and sequenced with the primers P13-P25 (Table 2). The resulting expression cassette was linearized with EcoRI and BamHI (both NEB) before A. niger was transformed.
Table 2.
Primers used in this work
| Primer | Sequence | Description |
|---|---|---|
| P1 | AGGCATCTGTCTGAGAGGCAACCGTGGCGA | Amplification of gaaA/C promoter, forward |
| GAGTCCGCATTCTTTGATCTGCTGTTAGTT | ||
| P2 | GGTGACGAAGTGTGCGATTGAGCGTGATAA | Amplification of gaaA/C promoter, reverse |
| AACGAAACATTGTGATTGCTGTGGTGTAAA | ||
| P3 | TAAGTTGGAGAAGTTGTTTCCGTCGCTCGAT | Amplification of gaaA terminator, forward |
| GCCATTTGAATACCTTAGAGAAGCTTGTATG | ||
| P4 | CGTCTCTCCGCATGCCAGAAAGAGTCACCGG | Amplification of gaaA terminator, reverse |
| TCACTGTACCATCTCCATCTCCTTCCCG | ||
| P5 | GCCCCCCCTCGAGGTCGACGGTATCGATAAGC | Amplification of gaaC terminator, forward |
| TTGATATCGAATTCCTGTTGGAGAGAGGGTGTGT | ||
| P6 | CCCAGCCCCAGGTCCGCCACCCGCAGAGTTCCGT | Amplification of gaaC terminator, reverse |
| TTGTGATCCATTGTATCATATAGATTATGAC | ||
| P7 | ATGCGGACTCTCGCCACG | Amplification of Eg ALase, forward |
| P8 | TCACAAACGGAACTCTGCGG | Amplification of Eg ALase, reverse |
| P9 | ATGTTTCGTTTTATCACGCT | Amplification of Mg GALDH, forward |
| P10 | TCAAATGGCATCGAGCGAC | Amplification of Mg GALDH, reverse |
| P11 | GTACAGTGACCGGTGACTCT | Amplification of hph, forward |
| P12 | GCTGGAGCTCCACCGCGGTGGCGGCCGCTCTAGA | Amplification of hph, reverse |
| ACTAGTGGATCCTTGGAGATTTCAGTAACGTT | ||
| P13 | GAGGTCGACGGTATCGATAAGC | Sequencing of the bidirectional Eg/Mg cassette |
| P14 | TGATACAATGGATCACAAACGG | " |
| P15 | CAACAGAGAACAGACCGCCA | " |
| P16 | GTGTTGCGAAGCTGTAGTTGG | " |
| P17 | ATTTACACCACAGCAATCAC | " |
| P18 | AAAGAAGCGTGTTCGAGTCC | " |
| P19 | ATACGGAGGATGAAGCCCTC | " |
| P20 | GCCAGCGGAAGGAGATTACG | " |
| P21 | GGCAGTGATTGAGGCTGTGG | " |
| P22 | AGTAAGCGAAGGAGAATGTG | " |
| P23 | AGTACTTTGCTACATCCATACTCC | " |
| P24 | ATTCGGACCGCAAGGAATCG | " |
| P25 | TGTCGGGCGTACACAAATCG | " |
| P26 | AGCCGTGTTTCAATGTCGTG | " |
| P27 | CGCTCTAGAACTAGTGGATC | " |
| P28 | CAACATTGTCATGTCTGGTGG | qPCR of actin, forward |
| P29 | GGAGGAGCAATGATCTTGAC | qPCR of actin, reverse |
| P30 | AGGACACGATTACTCTACTTG | qPCR of gaaA, forward |
| P31 | GAGCCCATATAATGGAAGTAC | qPCR of gaaA, reverse |
| P32 | TCCGGGTGGACCCCGCTAAG | qPCR of EgALase, forward |
| P33 | TGAAACACGGCTCCGGCGTC | qPCR of EgALase, reverse |
| P34 | TAGCAAGTGGCGCGGTGTCC | qPCR of MgGALDH, forward |
| P35 | TCGTGATCTCACCGCCCCGA | qPCR of MgGALDH, reverse |
All the resulting expression cassettes were introduced into the A. niger ΔgaaB strain by protoplast transformation method [32]. Transformants were screened for integration of the cassette by growth in the presence of 400 μg ml−1 hygromycin B (Calbiochem). Strains having both genes (smp30 or EgALase + MgGALDH) expressed in JKp1 under gpdA promoter were generated by co-transformation. Integration of the transformed cassettes into the genome was confirmed with colony PCR using Phire direct PCR kit (Thermo Scientific) and the primers P26 and P27 (Table 2).
Media and culture conditions
Luria Broth (LB) culture medium supplemented with 100 μg ml−1 of ampicillin and cultural conditions of 37°C and 250 rpm were used for E. coli cultures. For yeast pre-cultures, YPD medium (10 g yeast extract l−1, 20 g peptone l−1 and 20 g D-glucose l−1) was used and after transformation SCD-URA plates (uracil deficient synthetic complete media supplemented with 20 g D-glucose l−1) were used for uracil auxotrophy selection. Yeast cultures were carried out at 30°C and all the liquid cultures at 250 rpm. For the A. niger submerged fermentations, spores were generated on potato-dextrose plates and 0.9*108 spores were inoculated into 50 ml of YP medium (10 g yeast extract l−1, 20 g peptone l−1) containing 30 g gelatin l−1. Mycelia were pre-grown in 250-ml Erlenmeyer flasks by incubating overnight at 28°C, 200 rpm and harvested by vacuum filtration, rinsed with sterile water and weighted. In the A. niger transformations and fermentations, A. nidulans defined minimal medium [33], containing (in g l−1) 6 NaNO3, 0.52 KCl, 0.52 MgCl2 and 1.52 KH2PO4 was used. In the transformations the minimal medium was supplemented with 1.2 M D-sorbitol, 10 g l−1 of D-glucose and 20 g l−1 of agar and the pH was adjusted to 6.5. The minimal medium used in the fermentations for L-AA production, enzymatic assays and transcriptional analysis was supplemented with 20 g l−1 of D-galUA and 5 g l−1 of D-xylose or with 40 g l−1 of dried citrus processing waste (CPW). The pH was adjusted to 3. The CPW contained peel and pulp from the processed oranges and was received from the Federal University of Paraná in Curitiba, Brazil. The submerged cultures supplemented with pure D-galUA and D-xylose were inoculated with 1 g l−1 (cdw) and the submerged cultures supplemented with CPW were inoculated with 3 g l−1 (cdw) of pre-grown mycelia.
Enzymatic assays
For enzymatic assays, A. niger mycelia were harvested by vacuum filtration. Crude extracts were prepared in 100 mM sodium phosphate buffer pH 7 with protease inhibitor (Complete, Roche). The cells were disrupted with 0.4 mm diameter glass beads using a bead beater (Precellys 24, Bertin Technologies) and solid residues were removed by centrifugation. The GALDH assay was based on the cytochrome c reduction at 550 nm as described by Leferink et al. [26]. Crude extracts from the each strain were supplemented with 500 μM of FAD (flavin adenine dinucleotide), which is an essential cofactor for MgGALDH, and were tested for the activity by following the cytochrome c reduction after addition of 5 mM L-galL. The enzymatic assay for the lactonase activity (from EgALase and Smp30) was based on a pH change in the reaction from L-galA to L-galL that absorbs protons and was assayed as described by Ishikawa et al. [16]. The pH was followed at 405 nm using p-nitrophenol as a pH indicator and the correspondence between ∆405 nm and the absorption of protons were quantified using the titration with HCl.
Transcriptional analysis
Samples of 2 ml were collected from the cultures and the mycelium was harvested by vacuum filtration. The filtered mycelium was frozen with liquid nitrogen and stored at −80°C. RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) and ~1 μg of total RNA was used for cDNA synthesis with the Transcriptor High Fidelity cDNA Synthesis Kit (Roche), following the manufacturer’s instructions. cDNA samples were diluted with RNase free water (Roche) and were used for RT-qPCR using a LightCycler II with the LightCycler SYBR green I Master mix (both Roche). The expression of gaaA, EgALase, MgGALDH and actin (ATCC 1015 200483-mRNA) were quantified using the primers P28-P35 (Table 2). The signal from each of the target genes was normalized to actin using the accompanying software (Advance Relative Quantification tool).
Chemical analyses
Samples of 2 ml were removed at intervals and mycelium was separated from the supernatant by centrifugation. The concentration of D-galUA and L-galA was determined by HPLC using a Fast Acid Analysis Column (100 mm × 7.8 mm, BioRad Laboratories, Hercules, CA) linked to an Aminex HPX-87H organic acid analysis column (300 mm × 7.8 mm, BioRad Laboratories) with 5.0 mM H2SO4 as eluent and a flow rate of 0.5 ml min−1. The column was maintained at 55°C. Peaks were detected using a Waters 410 differential refractometer and a Waters 2487 dual wavelength UV (210 nm) detector. The L-AA content was measured using the commercial Ascorbic Acid Assay Kit II (Sigma-Aldrich) following the manufacturer’s instructions.
Acknowledgements
This work was supported by the Academy of Finland through the Sustainable Energy (SusEn) program (Grant 271025) and by the CNPq of Brazil in the form of a cooperation project.
Abbreviations
- L-AA
L-ascorbic acid
- D-EA
D-erythroascorbic acid
- D-galUA
D-galacturonic acid
- L-galA
L-galactonic acid
- L-galL
L-galactono-1,4-lactone
- GALDH
L-galactono-1,4-lactone dehydrogenase
- ALase
Aldonolactonase
- CPW
Citrus processing waste
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JK and PR designed and JK carried out all the experimental work and analyzed the data. JK and PR drafted the manuscript. PR and MP designed the fundamental concept and participated in the coordination of the study. All the authors read and approved the final manuscript.
Contributor Information
Joosu Kuivanen, Email: joosu.kuivanen@vtt.fi.
Merja Penttilä, Email: merja.penttila@vtt.fi.
Peter Richard, Email: peter.richard@vtt.fi.
References
- 1.Valpuesta V, Botella MA. Biosynthesis of L-ascorbic acid in plants: new pathways for an old antioxidant. Trends Plant Sci. 2004;9(12):573–7. doi: 10.1016/j.tplants.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Linster CL, Van Schaftigen E. Vitamin C biosynthesis, recycling and degredation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 3.Pappenberger G, Hohmann H. Industrial production of L-ascorbic acid (Vitamin C) and D-isoascorbic acid. Adv Biochem Eng Biotechnol. 2014;143:143–88. doi: 10.1007/10_2013_243. [DOI] [PubMed] [Google Scholar]
- 4.Reichstein T, Grüssner A. Eine ergiebige synthese der l-ascorbinsäure (C-Vitamin) Helv Chim Acta. 1934;17(1):311–28. doi: 10.1002/hlca.19340170136. [DOI] [Google Scholar]
- 5.Bremus C, Herrmann U, Bringer-Meyer S, Sahm H. The use of microorganisms in L-ascorbic acid production. J Biotechnol. 2006;124(1):196–205. doi: 10.1016/j.jbiotec.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 6.Kim S, Huh W, Lee B, Kang S. D-Arabinose dehydrogenase and its gene from Saccharomyces cerevisiae. Biochimica et Biophysica Acta. 1998;1429(1):29–39. doi: 10.1016/S0167-4838(98)00217-9. [DOI] [PubMed] [Google Scholar]
- 7.Onofri S, Poerio E, Serangeli P, Tosi S, Garuccio I, Arrigoni O. Influence of L-galactonic acid γ-lactone on ascorbate production in some yeasts. Antonie Van Leeuwenhoek. 1997;71(3):277–80. doi: 10.1023/A:1000161921389. [DOI] [PubMed] [Google Scholar]
- 8.Hancock RD, Galpin JR, Viola R. Biosynthesis of L-ascorbic acid (vitamin C) by Saccharomyces cerevisiae. FEMS Microbiol Lett. 2000;186(2):245–50. doi: 10.1111/j.1574-6968.2000.tb09112.x. [DOI] [PubMed] [Google Scholar]
- 9.Sugisawa T, Miyazaki T, Hoshino T. Microbial production of L-ascorbic acid from D-sorbitol, L-sorbose, L-gulose, and L-sorbosone by Ketogulonicigenium vulgare DSM 4025. Biosci Biotechnol Biochem. 2005;69(3):659–62. doi: 10.1271/bbb.69.659. [DOI] [PubMed] [Google Scholar]
- 10.Running J, Severson D, Schneider K. Extracellular production of L-ascorbic acid by Chlorella protothecoides, Prototheca species, and mutants of P. moriformis during aerobic culturing at low pH. J Ind Microbiol Biotechnol. 2002;29(2):93–8. doi: 10.1038/sj.jim.7000275. [DOI] [PubMed] [Google Scholar]
- 11.Sauer M, Branduardi P, Valli M, Porro D. Production of L-ascorbic acid by metabolically engineered Saccharomyces cerevisiae and Zygosaccharomyces bailii. Appl Environ Microbiol. 2004;70(10):6086–91. doi: 10.1128/AEM.70.10.6086-6091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Branduardi P, Fossati T, Sauer M, Pagani R, Mattanovich D, Porro D. Biosynthesis of vitamin C by yeast leads to increased stress resistance. PLoS One. 2007;2(10):e1092. doi: 10.1371/journal.pone.0001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosa JC, Colombo LT, Alvim MC, Avonce N, Van Dijck P, Passos FM. Metabolic engineering of Kluyveromyces lactis for L-ascorbic acid (vitamin C) biosynthesis. Microb Cell Fact. 2013;12(1):59. doi: 10.1186/1475-2859-12-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wheeler GL, Jones MA, Smirnoff N. The biosynthetic pathway of vitamin C in higher plants. Nature. 1998;393(6683):365–9. doi: 10.1038/30728. [DOI] [PubMed] [Google Scholar]
- 15.Agius F, González-Lamothe R, Caballero JL, Muñoz-Blanco J, Botella MA, Valpuesta V. Engineering increased vitamin C levels in plants by overexpression of a D-galacturonic acid reductase. Nat Biotechnol. 2003;21(2):177–81. doi: 10.1038/nbt777. [DOI] [PubMed] [Google Scholar]
- 16.Ishikawa T, Nishikawa H, Gao Y, Sawa Y, Shibata H, Yabuta Y, et al. The pathway via D-galacturonate/L-galactonate is significant for ascorbate biosynthesis in Euglena gracilis: identification and functional characterization of aldonolactonase. J Biol Chem. 2008;283(45):31133–41. doi: 10.1074/jbc.M803930200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondo Y, Inai Y, Sato Y, Handa S, Kubo S, Shimokado K, et al. Senescence marker protein 30 functions as gluconolactonase in L-ascorbic acid biosynthesis, and its knockout mice are prone to scurvy. Proc Natl Acad Sci U S A. 2006;103(15):5723–8. doi: 10.1073/pnas.0511225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richard P, Hilditch S. D-galacturonic acid catabolism in microorganisms and its biotechnological relevance. Appl Microbiol Biotechnol. 2009;82(4):597–604. doi: 10.1007/s00253-009-1870-6. [DOI] [PubMed] [Google Scholar]
- 19.Kuivanen J, Mojzita D, Wang Y, Hilditch S, Penttila M, Richard P, et al. Engineering filamentous fungi for conversion of D-galacturonic acid to L-galactonic acid. Appl Environ Microbiol. 2012;78(24):8676–83. doi: 10.1128/AEM.02171-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuivanen J, Dantas H, Mojzita D, Mallmann E, Biz A, Krieger N, et al. Conversion of orange peel to L-galactonic acid in a consolidated process using engineered strains of Aspergillus niger. AMB Express. 2014;4(1):1–8. doi: 10.1186/s13568-014-0033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mojzita D, Wiebe M, Hilditch S, Boer H, Penttilä M, Richard P. Metabolic engineering of fungal strains for conversion of D-galacturonate to meso-galactarate. Appl Environ Microbiol. 2010;76(1):169–75. doi: 10.1128/AEM.02273-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sastry KS, Sarma P. Glucuronic acid, a precursor of ascorbic acid in Aspergillus niger. Nature. 1957;179:44–45. doi: 10.1038/179044a0. [DOI] [PubMed] [Google Scholar]
- 23.Petrescu S, Hulea SA, Stan R, Avram D, Herlea V. A yeast strain that uses D-galacturonic acid as a substrate for L-ascorbic acid biosynthesis. Biotechnol Lett. 1992;14(1):1–6. doi: 10.1007/BF01030904. [DOI] [Google Scholar]
- 24.Smirnoff N, Running JA, Gatzek S. Introduction. In: Asard H, May JM, Smirnoff N, editors. Vitamin C: its Functions and Biochemistry in Animals and Plants. New York: Garland Science/BIOS Scientific Publishers; 2004. p. 19. [Google Scholar]
- 25.Badejo AA, Fujikawa Y, Esaka M. Gene expression of ascorbic acid biosynthesis related enzymes of the Smirnoff-Wheeler pathway in acerola (Malpighia glabra) J Plant Physiol. 2009;166(6):652–60. doi: 10.1016/j.jplph.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Leferink NG, van den Berg WA, van Berkel WJ. l-Galactono-γ-lactone dehydrogenase from Arabidopsis thaliana, a flavoprotein involved in vitamin C biosynthesis. FEBS J. 2008;275(4):713–26. doi: 10.1111/j.1742-4658.2007.06233.x. [DOI] [PubMed] [Google Scholar]
- 27.Mossmann D, Meisinger C, Vögtle F. Processing of mitochondrial presequences. Biochimica et Biophysica Acta. 2012;1819(9):1098–106. doi: 10.1016/j.bbagrm.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241(3):779–86. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 29.Choi MS, Saxena A, Chilukuri N. A strategy for the production of soluble human senescence marker protein-30 in Escherichia coli. Biochem Biophys Res Commun. 2010;393(3):509–13. doi: 10.1016/j.bbrc.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 30.He XX, Luo G, Zhou S. Soluble expression of recombinant human Smp30 for detecting serum Smp30 antibody levels in hepatocellular carcinoma patients. Asian Pac J Cancer Prev. 2013;14(4):2383–6. doi: 10.7314/APJCP.2013.14.4.2383. [DOI] [PubMed] [Google Scholar]
- 31.Gietz D, St Jean A, Woods RA, Schiestl RH. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20(6):1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yelton MM, Hamer JE, Timberlake WE. Transformation of Aspergillus nidulans by using a trpC plasmid. Proc Natl Acad Sci U S A. 1984;81(5):1470–4. doi: 10.1073/pnas.81.5.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barratt RW, Johnson GB, Ogata WN. Wild-type and mutant stocks of Aspergillus nidulans. Genetics. 1965;52(1):233. doi: 10.1093/genetics/52.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]