

Abstract
Mitochondrial dysfunction (primary or secondary) is detrimental to intermediary metabolism. Therapeutic strategies to treat/prevent mitochondrial dysfunction could be valuable for managing metabolic and age-related disorders. Here, we review strategies proposed to treat mitochondrial impairment. We then concentrate on redox-active agents, with mild-redox potential, who shuttle electrons among specific cytosolic or mitochondrial redox-centers. We propose that specific redox agents with mild redox potential (−0.1 V; 0.1 V) improve mitochondrial function because they can readily donate or accept electrons in biological systems, thus they enhance metabolic activity and prevent reactive oxygen species (ROS) production. These agents are likely to lack toxic effects because they lack the risk of inhibiting electron transfer in redox centers. This is different from redox agents with strong negative (−0.4 V; −0.2 V) or positive (0.2 V; 0.4 V) redox potentials who alter the redox status of redox-centers (i.e., become permanently reduced or oxidized). This view has been demonstrated by testing the effect of several redox active agents on cellular senescence. Methylene blue (MB, redox potential ≅10 mV) appears to readily cycle between the oxidized and reduced forms using specific mitochondrial and cytosolic redox centers. MB is most effective in delaying cell senescence and enhancing mitochondrial function in vivo and in vitro. Mild-redox agents can alter the biochemical activity of specific mitochondrial components, which then in response alters the expression of nuclear and mitochondrial genes. We present the concept of mitochondrial electron-carrier bypass as a potential result of mild-redox agents, a method to prevent ROS production, improve mitochondrial function, and delay cellular aging. Thus, mild-redox agents may prevent/delay mitochondria-driven disorders.
Keywords: mitochondria, diaminopheniothiazines, methylene blue, redox-centers, redox potential
1. Overview
Among the cellular metabolic network, mitochondrial metabolism occupies a unique position for its vital role in cellular energy metabolism, intracellular signaling, and regulating cell death and survival. Thus, compromised mitochondrial function puts the cellular metabolic network in disarray. Additionally, oxidative and nitrosative stresses covalently alter key components of the cellular network. Furthermore, free radicals and oxidants (e.g., superoxide radical and hydrogen peroxide, respectively) can alter the redox status of various redox centers due to their ability to interact with iron or copper in redox centers. Mitochondria are enriched with such redox centers. Therefore, it is no surprise that mitochondrial dysfunction plays a central role in the pathogenesis of numerous human diseases (e.g., neurodegeneration, diabetes). In fact, mitochondrial dysfunction is as detrimental to the cellular metabolic network as genetic mutations are when they happen in a gene that plays a central role in cellular metabolism [1–3].
In this review, we examine the emerging concept of mitochondrial medicine and review current efforts that may set the course for future drug development. We are particularly interested in the role that mild redox agents may play in designing new mitochondria targeted drugs. We discuss the significance of the redox potential in regard to determining the specificity of the interaction with physiologic redox centers (Scheme 1). We also discuss the chemical structure, which is essential for the compartmentalization of the redox agent within the cell (e.g., mitochondria vs. cytosol). We are using our research on methylene blue (MB), where we set the concept of electron bypass by mild redox agent, as a tool to prevent free radicals production and enhance cellular metabolic activity. MB seems to possess the basic features for both chemical structure and being a redox agent with the potential that it will not excessively accumulate in mitochondria and will not compromise the oxidation state of the physiologic redox centers.
Scheme 1.

The effect of the various categories of redox active agents on the oxidation state of physiologic redox agents. Three proposed cases of redox agents varying in redox activity and their potential outcomes in physiologic redox centers. Strong oxidizing redox agent (case A): represents the outcome of a strong oxidizing redox (redox potential ⋙ 0) agent being introduced to the physiologic center. Because of the strong oxidizing potential, the redox status of the physiologic redox center would be compromised. A strong oxidizing redox agent would have a high redox potential, which would in turn remove electrons from other reactants. Thus, taking electrons from the physiologic redox centers and inhibiting metabolic output. Strong reducing redox agent (case B): represents the outcome of a strong reducing agent (redox potential ⋘ 0) being introduced. A strong reducing agent would have a strong ability to donate electrons to another reactant, in turn creating the toxic effect of inhibiting metabolic input to the redox center. Mild redox agent (case C) represents a redox agent with a mild redox potential (e.g., MB). A redox agent with a mild redox potential (close to 0) will enter the mitochondria in its oxidized form. Select physiologic redox centers (e.g., NADH-dehydronase of complex I) will reduce the redox agent (e.g., MB is reduced to MBH2). It is proposed that mild redox agents will be reoxidized by a second physiologic center (e.g., cyt c). The significance of this is shown by the bypassing of the production of superoxide radicals and the cycling between the reduced and oxidized form of MB (thus, low concentration is needed) while preventing extreme transition in the redox states on physiologic redox centers. We propose this to be, in part, the mechanism by which MB serves as an electron carrier, inhibiting the production of superoxide radicals (x in bold refers to inhibition, dashed arrows in case C refers to the reversibility of the electron transfer to secondary redox center). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Impaired mitochondrial function is often linked to physical and cognitive impairments in age-related disorders [4]. Impaired mitochondrial function interferes with energy and intermediary metabolism, increases the production of oxidants, and increases the risk for tissue dysfunction. These declines are linked, in part, to age-associated changes in mitochondria in neuronal and muscle cells leading to impaired hippocampal or muscular functions, respectively. For example, the decline in the activity of mitochondrial function, energy hypometabolism, and increased oxidative stress are associated with the early signs of various age-related dementias. The risk for mitochondrial dysfunction increases as a result of nutritional deficiencies, exposure to environmental toxins, aging, or due to genetic disorders. Therefore, mitochondria-protecting agents may be potential drugs to prevent or delay age-related neurodegenerations such as Alzheimer’s disease (AD), the most common age-related dementia. Adequate mitochondrial function for preventing age-related disorders is accepted by most scientists as a promising therapeutic strategy. This view led to the search for strategies to delay or protect mitochondria, which involves different disciplines such as pharmacological, nutriceuticals, and DNA manipulation.
Mitochondria-targeted treatments could also prevent mitochondrial dysfunction that may result from drug toxicity and its side effects. Certain therapeutic drugs can involve mitochondrial toxicity (e.g., ifosfamide, doxorubicin) [5–10] and may cause cell death and organ malfunctioning [11]. Thus, protecting mitochondria may preserve normal organ function and minimize the side effects of important drugs.
Genetic manipulation of mitochondrial dysfunction as a therapeutic strategy is very challenging, particularly when it involves genes specific for mitochondrial DNA (mtDNA). In light of the absence of a suitable technology to manipulate mtDNA in a clinical setting, as is the case in Nuclear DNA (nDNA), pharmacology is the only resort left for mitochondria-based therapy. Preventative strategies could be a key strategy for delaying the age-related health disorders.
In addition to therapeutic strategies, diagnostic markers are also needed as they will facilitate the cause-effect relation between mitochondrial dysfunction and a disorder (e.g., the current debate about the role of mitochondria in autism, diabetes, and obesity). In this regard, clinical practice will tremendously benefit from such markers and it will make it easy for clinicians to identify the metabolic abnormalities linked to or caused by mitochondrial dysfunction. Based on our previous research, we propose that specific redox active agents have the potential to improve mitochondrial function.
2. Mitochondria and cellular metabolism
Many of the cellular biochemical activities are confined to the mitochondria, including reactions of energy transduction, calcium homoeostasis, iron assimilation into heme or iron-sulfur clusters, steroid synthesis, redox-homeostasis, control of apoptosis, in addition to the synthesis of specific metabolites (e.g., tetrahydrobiopterin). Although some of these functions are assigned to specific types of cells (e.g., steroid synthesis), others are more common (e.g., heme synthesis). However, impairment to any of these mitochondrial functions would compromise cellular intermediary metabolism, which impairs cellular function. Additionally, in most cases, mitochondrial impairment is accompanied by an increase in the production of reactive oxygen species (ROS). Oxidative damage to macromolecules, DNA in particular, is a second key contributor to cellular dysfunction. Oxidative stress and loss of cellular Ca2+ homeostasis are closely linked. They are both key outcomes of mitochondrial bioenergetic failure and are common denominators in the pathophysiology of many neurodegenerative diseases such as AD. Mitochondria themselves are also major targets for oxidative stress and abnormal intracellular Ca2+, as both synergistically activate the mitochondrial apoptotic pathway. Exposure to environmental toxins, nutritional deficiencies, aging, or genetic factors can increase the risk for mitochondrial dysfunction. However, the molecular mechanisms that contribute to mitochondrial impairment in aging or age-related disorders are not always clear. Importantly, impairment to mitochondrial metabolism in a disorder will have detrimental consequences regardless if it plays a primary or secondary role in the mechanism of the disease.
3. Mitochondrial medicine
Regardless if mitochondrial impairment is the primary or secondary cause for a health condition, preventing mitochondrial dysfunction is presumed to have clinical benefits. This theme drives the emerging field of research on mitochondria-targeted medicine [4,12–14]. Mitochondrial medicine is aimed at taking the research on mitochondria from basic concepts to translational research and clinical practice. However, the search for mitochondria-targeted pharmaceutical or nutriceutical agents with mitochondria-protecting activity is quite a challenging task. Our understanding of mitochondrial biochemistry, physiology, and genetics in addition to the mitochondrial role in health and disease are instrumental for designing new and specific drugs and strategies to prevent or treat specific mitochondria-based disorders [15,16]. Although we know a lot about mitochondria, the challenge is bridging between these disciplines to design therapeutic strategies to improve mitochondrial function or specifically target specific mitochondrial pathways. Several methods were proposed to prevent mitochondrial dysfunction and improve mitochondrial metabolism. These methods rely on the development of pharmaceutical [17] or nutriceutical agents [18].
3.1. Mitochondrial micronutrients
Micronutrients (e.g., vitamins and minerals) are part of a regular diet and are naturally directed to the mitochondria or other specific localization in the cells, guided by their biological properties. As for their biological efficacy, it may vary and will depend on the existence of previous deficiencies. Nutriceutical agents other than micronutrients (e.g., polyphenols) can be promising mitochondria protective agents; however, more research is needed to establish this view [19–21]. Usually, no concern is associated with the consumption of nutriceuticals, unless it is exaggerated. Other agents that are mitochondrial metabolites or effectors (e.g., Krebs cycle intermediates, Lipoic acid, melatonin, sex hormones) are intended to compensate for a presumed shortage in one or more specific micronutrients and support mitochondrial metabolism [19,21–23].
3.2. Mitochondria-targeted pharmaceuticals
There are two types of mitochondria-targeted molecules, those that are dependent upon the mitochondrial membrane potential and those that are not. The nonmembrane potential-dependent molecules make use of small, cell-permeable peptides that use a structural motif of alternating aromatic residues and basic amino acids. Pharmaceutical agents, on the other hand, are believed to be targeted to the mitochondrial matrix, using their biophysical properties as lipophylic cations or by conjugating them with mitochondria-targeting effective molecules [24]. There has been more work done with membrane potential-dependent molecules, which use delocalized lipophobic cations as the conjugated mitochondrial-directing agent. One molecule that has been demonstrated to have a high oral bioavailability and to concentrate in the mitochondria, and is thus frequently used with promising results, is the lipophilic cation mitoQ [24–26]. However, the clinical implication of this approach is awaiting the final results of current clinical trials.
A third approach to targeting the mitochondria are agents with mild redox (Oxidation-reduction) activity. Mitochondria are rich in the redox centers iron-sulfur, heme (heme-a, –b, and –c), Flavin mononucleotide (FMN), Flavin adenine dinucleotide (FAD), copper, and natural coenzyme Q. These are confined mostly to the innermembrane.
Because the physiology of mitochondria depends on major redox activities, we propose to use redox active agents to target the enzyme-complexes of the mitochondria that depend on redox centers for activity. These centers are the major source of production of free radicals by the mitochondria. Our research in this direction has provided promising results using diaminophenothiazines. We will describe our proposed mechanism of “electron bypass” activity of mild redox active agents and its role in enhancing mitochondrial activity and preventing free radical production.
4. Challenges to mitochondria-targeted drugs
Several challenges that face the search for mitochondria targeted drugs can be listed. The membrane permeability of the designed drug is a double fold challenge. The double membranes that surround the mitochondrial matrix are a major challenge. The drug first has to cross the cellular membrane, followed by the mitochondrial inner membrane, which exhibits a low permeability factor. Drugs that fail to selectively reach the mitochondria may accumulate in unintended targets [13,27].
Delocalized lipophylic cations possess the biophysical features that make them ideal for the purpose of targeting drugs to the mitochondria. However, this category of drugs tends to accumulate to very high concentrations in the mitochondrial compartments due to the high membrane potential (negative inside) of the mitochondria, thus mitochondria become a sink for delocalized lipophylic cation compounds [28]. This, in some cases, could be associated with serious concern regardless of chemical or biological activity of the drug under discussion. Although it is a powerful principle (especially when successful as in the case of mitoQ), extreme caution should be exercised because such drugs can readily poison the mitochondria (e.g., Rhodamine, JC1), especially in cases of chronic extended use. Additionally, some of the delocalized lipophylic cations accumulation may exceed the concentration of its intramitochondrial target, which lowers the efficacy of the drug and enhances its side effects, leading to mitochondrial poisoning. Such high intra-mitochondrial concentration may mask the benefit of the drug. Mitochondrial toxicity can also originate from the high-matrix concentrations of the drug, which may drive reactions not seen at much lower concentrations. The high concentration of the drug may also affect the matrix pH. Furthermore, the matrix of mitochondria is very concentrated and rich in proteins. Adding additional chemical at high concentration to this rich milieu may compromise the solubility of the matrix proteins and may lead to precipitation and structural changes. Furthermore, if a drug tends to accumulate in the membrane, it may interfere with electron transport complexes (ETC) activities and lead to uncoupling of the mitochondria. The innermembrane of the mitochondria plays a key role in energy transduction. This aspect is also particularly unique because the metabolic activity of the mitochondria varies depending on the physical activity of the body, which in turn may alter pharmacokinetics and efficacy of the drug. Additionally, parameters established based on experiments in vitro may not always apply to in vivo conditions, such as fold of accumulation, degree of interfering with redox status, or pH of the mitochondria. In conclusion, lipophylic cations that excessively accumulate in the mitochondria should be avoided, particularly if chronic use is intended (such as aging).
Pharmacological studies for candidate drugs are needed. It is presumed that clearance as well as the turnover rate will be slow due to the fact that the drug has to diffuse through three membranes to reach the extracellular milieu. Therefore, we believe that the selection criteria for mitochondrial drugs should be very stringent and exceed that of other nonmitochondrial drugs. Given the significance of mitochondria for energy and intermediary metabolism, the therapeutic index of these drugs should be very high. Several drugs (or agents) that are currently used for treating disorders not related to the mitochondria can cause mitochondrial toxicity [8–10]. Structural and functional examination of these drugs can provide insight into the design of newly planned mitochondrial drugs. Designing drugs that allow intermittent rather than continuous treatment may improve the therapeutic outcome of lipophylic cation-based drugs. This is quite an attractive approach because it may minimize the risk of side effects.
5. The search for mitochondria-protecting agents
Several natural compounds, mitochondrial metabolites, and pharmaceuticals were reported to provide some protection to the mitochondria. Natural compounds could be a promising source for exploring new molecules with the potential of mitochondria-protective activity. Antioxidant and neuroprotective agents are a primary lead to designing mitochondria-based therapeutic strategies for neurodegenerative disorders. Functional impairment of nerve endings occurs in the early stages of neurotoxicity, especially in those diseases with excitotoxic and depleted energy metabolism components as is the case in AD. Here, we bring several examples of the research investigation into mitochondria-protecting drugs or nutriceuticals as related to the various health conditions, emphasizing the significance of mitochondrial impairment in health and disease [9,29].
5.1. Mitochondria-protecting natural agents
Nakao et al. have recently shown that astaxanthin (a red carotenoid from sea food) leads to higher heart mitochondrial membrane potential and contractility index when compared with the control group. These findings suggest the possible cardioprotection of dietary astaxanthin and mitochondria may play a mechanistic role in astaxanthin activity [30]. Oxidation of lipids and proteins secondary to brain mitochondrial dysfunction contributes to neuronal injury following traumatic brain injury (TBI). Mustafa et al. described mitochondria protection using vitamin E derivative antioxidant (U-83836E). This treatment significantly preserved Ca+2 buffering capacity and attenuated the reduction in respiratory control ratio values after TBI. U-83836E also significantly lowers the levels of oxidative damage markers (e.g., 4-hydroxynonenal and 3-nitrotyrosine) in both cortical homogenates and mitochondria post injury [31]. This effect of U-83836E is probably due to protecting the mitochondrial membranes from oxidative damage (Camins A, 1998). Coenzyme Q10 significantly protects against oxidative stress and mitochondrial dysfunction in neurodegenerative disorders, especially Parkinson’s disease [32,33]. Clinical trials using Coenzyme Q10 have shown neuroprotective potential in Parkinson patients. This suggests a correlation between the activity of mitochondria protection and the ability to improve clinical outcome of a disorder [34].
Martins et al. have recently shown that low-molecular-mass peptide fractions (Ba-V) from Bothrops atrox snake venom is as effective as cyclosporin A (CsA) in its ability to inhibit the calcium/phosphate-induced mitochondrial swelling. This indicates the potential of Ba-V to prevent mitochondrial permeability transition (MPT) and probably apoptosis. Ba-V did not generate side effects as determined by the production of ROS. The authors conclude [35] that Ba-V may be useful in the future strategies for the treatment of mitochondria-based diseases due to the important role of the mitochondrial dysfunction and, especially MPT, in the development of neuropathies [35].
S-allylcysteine (SAC, a natural constituent of fresh garlic) is a well-known antioxidant that protected the mitochondria in in vivo models using 3-nitropropionic acid (3-NP). 3-NP is a specific inhibitor of mitochondrial complex II, thus it impairs both electron transport through ETC and alters the activity of Krebs cycle. Chronically administered 3-NP produces selective lesions in the striatum. 3-NP-striatally impaired rats received SAC for 7 days. SAC protected nerve ending within the first hours (1 and 3) after the toxic insult with 3-NP. SAC protected nerve endings from oxidative damage and energy depletion caused by mitochondrial dysfunction by 3-NP [36]. Tetramethylpyrazine (TMP) is a principal ingredient of the plant Ligusticum wallichi Franchat, used for treatment of cardiovascular and cerebrovascular ischemic disorders (used in Chinese medicine). TMP protects against kainate-induced excitotoxicity in vitro and in vivo. TMP partially alleviated kainate-induced neuronal loss in the hippocampus. This neuronal protection by TMP was attributed to preserving the integrity of the mitochondrial membrane potential, ATP production, and complex I and III activities. Stabilization of mitochondrial function was linked to the observation that TMP could function as a reductant/antioxidant (redox agent) to quench ROS, block lipid peroxidation, and protect enzymatic antioxidants such as glutathione peroxidase and glutathione reductase. These results suggest that TMP may protect mitochondria quenching of free radicals [37].
Ionizing radiation causes damage to mitochondria and leads to impaired function. Exposure of cells to γ-radiation induces the production of ROS, which are the chemical tool by which ionizing radiation damages macromolecules. The radioprotective effect of phloroglucinol (1,3,5-trihydroxybenzene), a phlorotannin compound isolated from Ecklonia cava, against γ-radiation-induced oxidative damage (e.g., lipid, DNA, and protein) has been shown in vitro and in vivo. Phloroglucinol significantly decreased the level of radiation-induced intracellular ROS and damage to cellular components. Phloroglucinol prevents the loss of cell viability caused after exposure to γ-radiation. Phloroglucinol reduced the radiation-induced loss of the mitochondrial membrane potential, reduced the levels of the active forms of caspase 9 and 3, and elevated the expression of bcl-2. Thus, phloroglucinol reduces the radiation-induced mitochondrial dysfunction and prevents apoptosis. Furthermore, the inhibition of radiation-induced apoptosis by phloroglucinol was also exerted by inhibiting mitogen-activated protein kinase kinase-4 (MKK4/SEK1), c-Jun NH(2)-terminal kinase and activator protein-1. Phloroglucinol restored the level of reduced glutathione and the active subunit of glutamate-cysteine ligase, which is a rate-limiting enzyme in glutathione biosynthesis. In an in vivo study, phloroglucinol administration in mice provided substantial protection against death and oxidative damage following whole-body irradiation [38].
5.2. Mitochondria-protecting pharmaceuticals
Recent understandings for mitochondrial biology have led to the proposal for potential selective targeting of drugs to manipulate mitochondrial function, such as the use of infrared light therapy to alter mitochondrial function. Although modifications to the mitochondrial genome are challenging, they carry promising potential for therapy if successful. The current intensive research to rationally design and target drugs to mitochondria could lead to more effective interventions in treating diseases with mitochondrial dysfunctions.
Carvedilol, an antihypertensive with strong antioxidant properties, was able to counteract the renal damage by Cisplatin by preventing the mitochondrial dysfunction [39]. Cisplatin-induced nephrotoxicity includes mitochondrial dysfunction. Rodrigues et al. have shown that Cisplatin induces the collapse of mitochondrial membrane potential, decline in Ca uptake, and impaired respiration. The Oxidative phosphorylation (OXPHOS) capacity was preserved by carvedilol [39]. This study suggests that carvedilol is a potential drug for the adjuvant nephroprotective therapy during cisplatin chemotherapy. Carvedilol intrinsic antioxidant activity, which has been described to protect cardiac mitochondria from oxidative injury, also protects cardiac mitochondria from doxorebucin toxicity [5].
Pinacidil (and diazoxide), a potassium channel opener, significantly improved heart preservation and functional parameters during organ preservation in cold. The mitochondrial respiratory function and energy metabolism were also preserved in pinacidil as compared with the control group without pinacidil. This effect of pinacidil appears to depend on both mitochondrial and sarcolemmal adenosine triphosphate sensitive potassium channel [40,41]. Protecting mitochondrial function can help prolong donor organ preservation. This is important for organ transplant surgeries.
The protective effect of the 4-anilinoquinazoline derivative (PD153035) on mitochondria was demonstrated in cardiac ischemia/reperfusion model. Nanomolar concentrations of PD153035 strongly protect against heart and cardiomyocyte damage induced by ischemia/reperfusion and cyanide/aglycemia. Interestingly, PD153035 activated K+ transport (mitoK-ATP) channel in isolated mitochondria, demonstrating that this protection is dependent on mitoK(ATP) activation. The authors conclude that PD153035 is a potent cardioprotective compound and acts in a mechanism involving mito-K(ATP) activation [42].
Ralph et al. propose that mitochondrial complex II could serve as a pharmaceutical target for modulating oxidant formation and thus preventing (or increasing) oxidative stress. He described diazoxide to prevent oxidant production and protect normal cells [43].
Low molecular weight drugs are likely to cross the plasma membrane and reach the mitochondria. However, the Szeto-Schiller (SS) peptide [44,45] antioxidants represent a novel approach that uses the targeted delivery of antioxidants to the inner mitochondrial membrane. SS peptides were shown to scavenge hydrogen peroxide and peroxynitrite, block lipid peroxidation, and inhibit apoptosis. Rocha et al. propose the use of SS peptides to treat ischemia-reperfusion injury in addition to neurodegenerative disorders [46].
Mildronate, an aza-butyrpbetaine, is an anti-ischemic agent which is cardioprotective and prevents dysfunction of mitochondrial complex I. Mildronate also acts as a neuroprotective agent. Mildronate reduced the mitochondrial toxicity of azidothymidine (anti-HIV drug) [47]. Mildronate prevented the changes in the expression of mitochondrial complex IV. The authors propose anti-neurodegenerative (anti-apoptotic) and anti-inflammatory action of mildronate [48]. The tertiary nitrogen group of mildronate may be mostly charged in the low pH of the intermembrane space of mitochondria while the carboxylic group may be negatively charged, thus modulating its permeability through the inner mitochondria preventing its excessive accumulation.
6. Redox active agents
Mitochondrial metabolic activity depends on various redox centers, including iron-sulfur, heme (heme-a, –b, and –c), FMN, FAD, copper, and coenzyme Q. The redox activity is fundamental for redox metabolism (e.g., iron homeostasis or NADH-NADPH), energy metabolism, intermediary metabolism (e.g., Ca2+ homeostasis, gluconeogensis, TCA intermediates), and oxidants and antioxidants balance (e.g., GPX). However, redox activity of mitochondria provides the source of free radicals in living cells.
Methylene blue (3,7-Bis-dimethylamino-phenazathionium) is a redox active agent with mild redox potential, which is close to zero. MB is a redox indicator that has a mild redox potential of 10 mV [49]. The low redox potential allows MB to cycle readily between oxidized and reduced forms using various redox centers such as those present in mitochondria. Several NAD(P)H-dependent dehydrogenases also reduce MB to form the colorless leucomethylene blue (MBH2). MBH2 can be reoxidized to MB by O2 in the absence of suitable electron acceptors such as cytochrome c [50,51] or other heme-proteins [52].
MB is soluble in both water and organic solvents; the lipid solubility of MBH2 is higher than that of MB. Electron delocalization in MB results in a partial positive charge being located on both nitrogen and sulfur atoms, which may increase the permeability of MB through membranes. Thus, MBH2 and MB can enter the mitochondria [53] and other intracellular compartments such as lysosomes [54]. MB is the most effective agent reported so far to delay cellular senescence and enhance specific biochemical activities of the mitochondria. Functional implication of these changes suggests that mitochondrial function and activity are also improved by MB. Indeed, at nanomolar concentrations, MB increased the activity of mitochondrial cytochrome c oxidase (complex IV), heme synthesis, cell resistance to oxidants, and oxygen consumption. The in vitro effect of MB was also maintained in vivo when administered to old mice at low dose [55]. MB prevented the age-related decline in spatial memory and grip strength in old mice. In addition, MB increased mitochondrial complex IV activity by 100% and 50% in the brains and hearts of old mice, respectively. MB prevented the age-related decline in protein content of the brain. In the aging human brain (ages from 30 to 90 years old), a decrease of 5–15% in protein content has been previously described [56]. Thus, MB has the potential to improve mitochondrial function in fibroblasts, brain, and heart. Although the mechanism of action of MB is still under investigation, we believe that the mild redox potential of MB plays an important role in its effect on mitochondria.
7. Proposed mechanism for mild redox active agents that improve mitochondrial function
Redox active agents can be categorized in three categories: strong oxidizing agents (e.g., ferricynide), strong reducing agents (e.g., dithionite), and mild redox agents (e.g., MB). The three categories of redox agents varying in redox activity can vary in their potential outcomes in physiologic redox centers (Scheme 1). In the case of strong oxidizing redox agent (proposed redox potential range 0.2–0.4 V) interacting with physiologic redox centers, the redox status of the physiologic redox center would be compromised. A strong oxidizing redox agent would have a high redox potential, which would in turn remove electrons from other reactants. Thus, taking electrons from the physiologic redox centers and inhibiting metabolic output. In the case of strong reducing redox agent (proposed redox potential in the range −0.4 to −0.4 V) interacting with physiologic redox centers, the agent would have a strong ability to donate electrons to another reactant, in turn creating the toxic effect of inhibiting metabolic input to the redox center. The third category is when a redox agent with a mild redox potential (proposed redox potential in the range −0.1 to 0.1 V) interacts with physiologic redox centers. Such agents will enter the cell in its oxidized form and select physiologic redox centers (e.g., NADH-dehydronase of complex I) will reduce it (e.g., MB is reduced to MBH2). It is proposed that mild redox agents will be reoxidized by a second physiologic center (e.g., cyt c). The significance of this is the potential of bypassing the production of superoxide radicals and the cycling between the reduced and oxidized form of MB (thus, only low concentrations required) while preventing extreme transition in the redox states on physiologic redox centers [51]. Because MBs redox potential (10 mV) is considered mild as compared with the redox potential of the ETC components (Scheme 2, [57]), we propose this to be, in part, the mechanism by which MB serves as an electron carrier inhibiting the production of superoxide radicals [51].
Scheme 2.
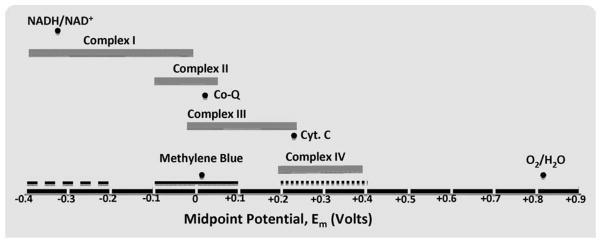
The proposed potential ranges for mild and strong reducing or oxidizing agents relative to midpoint potential ranges and the redox potentials for specific mitochondrial electron carriers. The midpoints potential ranges for each of the four ETC groups (complexes I, II, III, and IV) are shown as solid gray bars. Dark circles show the redox potentials for the labeled specific electron carriers important for ETC, or for the redox potential of MB. The solid black bars show the proposed range of positive redox potential for a strong oxidizing agent (dotted bar), a strong negative redox potential for a reducing agent (dashed bar), and a mild redox potential mild redox agent (solid bar) [57]. The statements of strong and mild relates to the redox potentials usually found in physiological settings.
Our findings, in conjunction with previous studies, suggest that intracellular MB is a typical mild redox agent. MB is likely to cycle between the oxidized (MB) and the reduced (MBH2) forms [50,52]. Some NADH-Reduced form of flavin mononucleotide(FMNH)-dependent enzymes can reduce MB to MBH2 (e.g., NADH-dehydrogenase of complex I), whereas mitochondrial cytochrome c (cyt c) can reoxidize MBH2 to MB [50,52]. The hydrophobicity of MB increases upon reduction, which facilitates it crossing the inner membrane from the matrix to reach cyt c [51]. This cycling property of MB probably explains the low concentrations of MB needed to affect mitochondrial functions. cyt c carries electrons from complex III to complex IV. We suspect that as MBH2 increases the rate of the reduction of cyt c over and above the normal enzymatic reduction by complex III, it triggers the induction of complex IV. Complex IV catalyzes the electron transfer from the reduced cyt c to O2 to form H2O. Thus, we hypothesize that the increase in the rate of reduction of cyt c by MBH2 [50,52] could explain, in part, the drive for the induction of complex IV that occurs in the presence of 100 nM MB. The signaling mechanism that communicates between the nDNA and mtDNA to increase the biogenesis of complex IV in response to MB is under investigation. We speculate that the mild redox agent alters the cell redox signaling. Thus, possible molecular factors important for the mechanism of mitochondria protection by redox agents may involve NO production, PGC1a, NRF1/2, AMPK, or Sirtuins (through the effect of MB on NADH/NAD ratio). The effect of MB on NADH may also prevent a back up of electrons on certain redox centers such as FMNH of complex I that can increase ROS [51].
We also suggest that the mild redox potential of MB allows it to cycle between its oxidized and reduced forms using physiologic redox centers as we proposed earlier [51]. For example, if this cycling takes place between FMNH of complex I (reducing center) and cyt c as the oxidizing center, it may decrease superoxide radical production by the mitochondria, particularly from the FMNH of complex I [51]. In a case that MB is replaced by a strong oxidizing or reducing agent, it may totally oxidize FNMH and prevent electron flow through complex I, inhibiting ETC. In the case that MB is replaced by strong reducing agent, it may leave most of the ETC redox centers reduced which may increase free radical production and alter enzymatic reactions.
The chemistry of the redox agent also plays a key role in regard to determining the redox potential and defining the specificity of the interaction with redox centers. The chemical structure is also essential for the compartmentalization of the redox agent within the cells (e.g., mitochondria vs. cytosol). MB seems to possess the ideal conditions for both chemical structures so it does not excessively accumulate in the cellular compartments and the mild redox potential so it does not compromise the oxidation state of the physiologic redox centers. Scheme 1 depicts this concept of superiority of mild redox agents over strong oxidizing or reducing redox agents for minimal side effects and toxicity.
8. Conclusions
The risk for mitochondrial dysfunction increases as a result of nutritional deficiencies, exposure to environmental toxins, aging, or genetic disorders. We described the potential of specific mild redox active agents to protect mitochondria and enhance their function. We propose that specific mild redox active agents may optimize mitochondrial function and prevent the production of oxidants due to their ability to redox cycle using mitochondrial redox centers [51]. We used our understanding of the beneficial effect of diaminopheniothiazines (e.g., MB) on mitochondrial activity to demonstrate this hypothesis. Accumulation of these agents in the mitochondria is not necessary, thus eliminating potential side effects. The most intriguing finding is that a low chronic dose of MB increases complex IV in brain and heart and in vitro [51,55]. Complex IV is found in ≈5-fold excess over the other ETC of the mitochondria [58], which may indicate the physiological significance of excess complex IV [59]. We believe that each of the mitochondrial complexes (e.g., complex I) can also be induced using the proper redox active agent. MB has the potential to prevent the decline in complex IV, which is a key cytopathology of AD. Thus, by increasing the brain reserve of complex IV, we propose that MB could delay or slow the age-related decline in complex IV, thus preserving mitochondrial function, energy metabolism, and memory retention in AD. Similarly, other mitochondrial ETC could be manipulated using specific redox agents that match their redox potential.
Acknowledgments
Supported by NIH grant R15AG041414 from the National Institute On Aging and AFAR grants to H.A. H. Atamna has filed a patent on MB, aging and cellular senescence.
References
- 1.Boveris A, Navarro A. Brain mitochondrial dysfunction in aging. IUBMB Life. 2008;60:308–314. doi: 10.1002/iub.46. [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC. A mitochondrial paradigm for degenerative diseases and ageing. Novartis Found Symp. 2001;235:247–263. doi: 10.1002/0470868694.ch20. discussion 263–246. [DOI] [PubMed] [Google Scholar]
- 3.Yap LP, Garcia JV, Han D, Cadenas E. The energy-redox axis in aging and age-related neurodegeneration. Adv Drug Deliv Rev. 2009;61:1283–1298. doi: 10.1016/j.addr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pereira GC, Silva AM, Diogo CV, Carvalho FS, Monteiro P, Oliveiraalo PJ. Drug-induced cardiac mitochondrial toxicity and protection: from doxorubicin to carvedilol. Curr Pharm Des. 2011;17:2113–2129. doi: 10.2174/138161211796904812. [DOI] [PubMed] [Google Scholar]
- 6.Hrushesky WJ, Olshefski R, Wood P, Meshnick S, Eaton JW. Modifying intracellular redox balance: an approach to improving therapeutic index. Lancet. 1985;1:565–567. doi: 10.1016/s0140-6736(85)91218-8. [DOI] [PubMed] [Google Scholar]
- 7.Park IS, Lee HJ, Lee YS, Hwang JS, Lee MS. Ifosfamide-induced encephalopathy with or without using methylene blue. Int J Gynecol Cancer. 2005;15:807–810. doi: 10.1111/j.1525-1438.2005.00140.x. [DOI] [PubMed] [Google Scholar]
- 8.Wallace KB. Mitochondrial off targets of drug therapy. Trends Pharmacol Sci. 2008;29:361–366. doi: 10.1016/j.tips.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Neustadt J, Pieczenik SR. Medication-induced mitochondrial damage and disease. Mol Nutr Food Res. 2008;52:780–788. doi: 10.1002/mnfr.200700075. [DOI] [PubMed] [Google Scholar]
- 10.Boelsterli UA, Lim PL. Mitochondrial abnormalities—a link to idiosyncratic drug hepatotoxicity? Toxicol Appl Pharmacol. 2007;220:92–107. doi: 10.1016/j.taap.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 11.Morales AI, Detaille D, Prieto M, Puente A, Briones E, Arevalo M, Leverve X, Lopez-Novoa JM, El-Mir MY. Metformin prevents experimental gentamicin-induced nephropathy by a mitochondria-dependent pathway. Kidney Int. 2010;77:861–869. doi: 10.1038/ki.2010.11. [DOI] [PubMed] [Google Scholar]
- 12.Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weissig V. Targeted drug delivery to mammalian mitochondria in living cells. Expert Opin Drug Deliv. 2005;2:89–102. doi: 10.1517/17425247.2.1.89. [DOI] [PubMed] [Google Scholar]
- 14.Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev. 2002;54:101–127. doi: 10.1124/pr.54.1.101. [DOI] [PubMed] [Google Scholar]
- 15.Koponen T, Cerrada-Gimenez M, Pirinen E, Hohtola E, Paananen J, Vuohelainen S, Tusa M, Pirnes-Karhu S, Heikkinen S, Virkamaki A, Uimari A, Alhonen L, Laakso M. The activation of hepatic and muscle polyamine catabolism improves glucose homeostasis. Amino Acids. doi: 10.1007/s00726-011-1013-0. in press. [DOI] [PubMed] [Google Scholar]
- 16.Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamm-Alvarez S, Cadenas E. Mitochondrial medicine and therapeutics. II Preface. Adv Drug Deliv Rev. 2009;61:1233. doi: 10.1016/j.addr.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Ames BN. Prevention of mutation, cancer, and other age-associated diseases by optimizing micronutrient intake. J Nucleic Acids. 2010;2010:1–10. doi: 10.4061/2010/725071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borras C, Gambini J, Lopez-Grueso R, Pallardo FV, Vina J. Direct antioxidant and protective effect of estradiol on isolated mitochondria. Biochim Biophys Acta. 2010;1802:205–211. doi: 10.1016/j.bbadis.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Carrasco-Pozo C, Gotteland M, Speisky H. Protection by apple peel polyphenols against indometacin-induced oxidative stress, mitochondrial damage and cytotoxicity in Caco-2 cells. J Pharm Pharmacol. 2011;62:943–950. doi: 10.1211/jpp.62.06.0017. [DOI] [PubMed] [Google Scholar]
- 21.Dessolin J, Schuler M, Quinart A, De Giorgi F, Ghosez L, Ichas F. Selective targeting of synthetic antioxidants to mitochondria: towards a mitochondrial medicine for neurodegenerative diseases? Eur J Pharmacol. 2002;447:155–161. doi: 10.1016/s0014-2999(02)01839-3. [DOI] [PubMed] [Google Scholar]
- 22.Inarrea P, Casanova A, Alava MA, Iturralde M, Cadenas E. Melatonin and steroid hormones activate intermembrane Cu,Zn-superoxide dismutase by means of mitochondrial cytochrome P450. Free Radic Biol Med. 2011;50:1575–1581. doi: 10.1016/j.freeradbiomed.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ames BN. Optimal micronutrients delay mitochondrial decay and age-associated diseases. Mech Ageing Dev. 2010;131:473–479. doi: 10.1016/j.mad.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 25.Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 26.Porteous CM, Logan A, Evans C, Ledgerwood EC, Menon DK, Aigbirhio F, Smith RA, Murphy MP. Rapid uptake of lipophilic triphenylphosphonium cations by mitochondria in vivo following intravenous injection: implications for mitochondria-specific therapies and probes. Biochim Biophys Acta. 2010;1800:1009–1017. doi: 10.1016/j.bbagen.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Yousif LF, Stewart KM, Kelley SO. Targeting mitochondria with organelle-specific compounds: strategies and applications. Chembiochem. 2009;10:1939–1950. doi: 10.1002/cbic.200900185. [DOI] [PubMed] [Google Scholar]
- 28.Rottenberg H. Membrane potential and surface potential in mitochondria: uptake and binding of lipophilic cations. J Membr Biol. 1984;81:127–138. doi: 10.1007/BF01868977. [DOI] [PubMed] [Google Scholar]
- 29.Reddy VD, Padmavathi P, Kavitha G, Gopi S, Varadacharyulu N. Emblica officinalis ameliorates alcohol-induced brain mitochondrial dysfunction in rats. J Med Food. 2011;14:62–68. doi: 10.1089/jmf.2010.1122. [DOI] [PubMed] [Google Scholar]
- 30.Nakao R, Nelson OL, Park JS, Mathison BD, Thompson PA, Chew BP. Effect of astaxanthin supplementation on inflammation and cardiac function in BALB/c mice. Anticancer Res. 2011;30:2721–2725. [PubMed] [Google Scholar]
- 31.Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem. 2011;114:271–280. doi: 10.1111/j.1471-4159.2010.06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas B, Beal MF. Mitochondrial therapies for Parkinson’s disease. Mov Disord. 2010;25 (Suppl 1):S155–S160. doi: 10.1002/mds.22781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spindler M, Beal MF, Henchcliffe C. Coenzyme Q10 effects in neurodegenerative disease. Neuropsychiatr Dis Treat. 2009;5:597–610. doi: 10.2147/ndt.s5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yong-Kee CJ, Salomonczyk D, Nash JE. Development and validation of a screening assay for the evaluation of putative neuroprotective agents in the treatment of Parkinson’s disease. Neurotox Res. 19:519–26. doi: 10.1007/s12640-010-9174-2. [DOI] [PubMed] [Google Scholar]
- 35.Martins NM, Ferreira DA, Rodrigues MAC, Cintra AC, Santos NA, Sampaio SV, Santos AC. Low-molecular-mass peptides from the venom of the Amazonian viper Bothrops atrox protect against brain mitochondrial swelling in rat: potential for neuroprotection. Toxicon. 2011;56:86–92. doi: 10.1016/j.toxicon.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 36.Elinos-Calderon D, Robledo-Arratia Y, Perez-De La Cruz V, Maldonado PD, Galvan-Arzate S, Pedraza-Chaverri J, Santamaria A. Antioxidant strategy to rescue synaptosomes from oxidative damage and energy failure in neurotoxic models in rats: protective role of S-allylcysteine. J Neural Transm. 2011;117:35–44. doi: 10.1007/s00702-009-0299-5. [DOI] [PubMed] [Google Scholar]
- 37.Li SY, Jia YH, Sun WG, Tang Y, An GS, Ni JH, Jia HT. Stabilization of mitochondrial function by tetramethylpyrazine protects against kainate-induced oxidative lesions in the rat hippocampus. Free Radic Biol Med. 2011;48:597–608. doi: 10.1016/j.freeradbiomed.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Kang KA, Zhang R, Chae S, Lee SJ, Kim J, Jeong J, Lee J, Shin T, Lee NH, Hyun JW. Phloroglucinol (1,3,5-trihydroxybenzene) protects against ionizing radiation-induced cell damage through inhibition of oxidative stress in vitro and in vivo. Chem Biol Interact. 2011;185:215–226. doi: 10.1016/j.cbi.2010.02.031. [DOI] [PubMed] [Google Scholar]
- 39.Rodrigues MA, Rodrigues JL, Martins NM, Barbosa F, Curti C, Santos NA, Santos AC. Carvedilol protects against the renal mitochondrial toxicity induced by cisplatin in rats. Mitochondrion. 2011;10:46–53. doi: 10.1016/j.mito.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Yang L, Yu T. Prolonged donor heart preservation with pinacidil: the role of mitochondria and the mitochondrial adenosine triphosphate-sensitive potassium channel. J Thorac Cardiovasc Surg. 2011;139:1057–1063. doi: 10.1016/j.jtcvs.2009.10.042. [DOI] [PubMed] [Google Scholar]
- 41.Kopustinskiene DM, Liobikas J, Skemiene K, Malinauskas F, Toleikis A. Direct effects of K(ATP) channel openers pinacidil and diazoxide on oxidative phosphorylation of mitochondria in situ. Cell Physiol Biochem. 2011;25:181–186. doi: 10.1159/000276552. [DOI] [PubMed] [Google Scholar]
- 42.Cavalheiro RA, Marin RM, Rocco SA, Cerqueira FM, da Silva CC, Rittner R, Kowaltowski AJ, Vercesi AE, Franchini KG, Castilho RF. Potent cardioprotective effect of the 4-anilinoquinazoline derivative PD153035: involvement of mitochondrial K(ATP) channel activation. PLoS One. 2011;5:e10666-1–8. doi: 10.1371/journal.pone.0010666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ralph SJ, Moreno-Sanchez R, Neuzil J, Rodriguez-Enriquez S. Inhibitors of succinate: quinone reductase/complex ii regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm Res. 2011;28:2695–2730. doi: 10.1007/s11095-011-0566-7. [DOI] [PubMed] [Google Scholar]
- 44.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8:E521–E531. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cho J, Won K, Wu D, Soong Y, Liu S, Szeto HH, Hong MK. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron Artery Dis. 2007;18:215–220. doi: 10.1097/01.mca.0000236285.71683.b6. [DOI] [PubMed] [Google Scholar]
- 46.Rocha M, Hernandez-Mijares A, Garcia-Malpartida K, Banuls C, Bellod L, Victor VM. Mitochondria-targeted antioxidant peptides. Curr Pharm Des. doi: 10.2174/138161210793292519. in press. [DOI] [PubMed] [Google Scholar]
- 47.Klusa V, Pupure J, Isajevs S, Rumaks J, Gordjushina V, Kratovska A, Taivans I, Svirskis S, Viksna L, Kalvinsh I. Protection of azidothymidine-induced cardiopathology in mice by mildronate, a mitochondria-targeted drug. Basic Clin Pharmacol Toxicol. 2006;99:323–328. doi: 10.1111/j.1742-7843.2006.pto_543.x. [DOI] [PubMed] [Google Scholar]
- 48.Pupure J, Isajevs S, Skapare E, Rumaks J, Svirskis S, Svirina D, Kalvinsh I, Klusa V. Neuroprotective properties of mildronate, a mitochondria-targeted small molecule. Neurosci Lett. 2010;470:100–105. doi: 10.1016/j.neulet.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 49.Kamat P, Mimitijevic N, Fessenden R. Photoelectrochemistry in particulate systems: electron-transfer reactions of small CdS colloids in acetonitrile. J Phys Chem. 1987;91:396–401. [Google Scholar]
- 50.McCord JM, Fridovich I. The utility of superoxide dismutase in studying free radical reactions. II The mechanism of the mediation of cytochrome c reduction by a variety of electron carriers. J Biol Chem. 1970;245:1374–1377. [PubMed] [Google Scholar]
- 51.Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H, Ames BN. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. Faseb J. 2008;22:703–712. doi: 10.1096/fj.07-9610com. [DOI] [PubMed] [Google Scholar]
- 52.Kelner MJ, Bagnell R, Hale B, Alexander NM. Methylene blue competes with paraquat for reduction by flavo-enzymes resulting in decreased superoxide production in the presence of heme proteins. Arch Biochem Biophys. 1988;262:422–426. doi: 10.1016/0003-9861(88)90393-1. [DOI] [PubMed] [Google Scholar]
- 53.Gabrielli D, Belisle E, Severino D, Kowaltowski AJ, Baptista MS. Binding, aggregation and photochemical properties of methylene blue in mitochondrial suspensions. Photochem Photobiol. 2004;79:227–232. doi: 10.1562/be-03-27.1. [DOI] [PubMed] [Google Scholar]
- 54.Mellish KJ, Cox RD, Vernon DI, Griffiths J, Brown SB. In vitro photodynamic activity of a series of methylene blue analogues. Photochem Photobiol. 2002;75:392–397. doi: 10.1562/0031-8655(2002)075<0392:ivpaoa>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 55.Atamna H, Gharib A. Methylene Blue Induces Mitochondrial Complex IV and Improves Cognitive Function and Grip Strength in Old Mice. In: McNeill Alexander S., editor. Neurodegeneration: Theory, Disorders and Treatments. Chapter 3. Nova Science Publishers; 2011. pp. 63–86. [Google Scholar]
- 56.Naber D, Dahnke HG. Protein and nucleic acid content in the aging human brain. Neuropathol Appl Neurobiol. 1979;5:17–24. doi: 10.1111/j.1365-2990.1979.tb00610.x. [DOI] [PubMed] [Google Scholar]
- 57.Tyler DD. The Mitochondrion in Health and Disease. VHC Publishers, Inc; New York: 1992. [Google Scholar]
- 58.Capaldi RA. Arrangement of proteins in the mitochondrial inner membrane. Biochim Biophys Acta. 1982;694:291–306. doi: 10.1016/0304-4157(82)90009-0. [DOI] [PubMed] [Google Scholar]
- 59.Atamna H, Kumar R. Protective role of methylene blue in Alzheimer’s disease via mitochondria and cytochrome c oxidase. J Alzheimers Dis. 2010;20 (Suppl 2):S439–S452. doi: 10.3233/JAD-2010-100414. [DOI] [PubMed] [Google Scholar]