

Abstract
Purpose of review
The mutational patterns of cancer genomes allow conclusions or generation of hypotheses as to what mechanisms or environmental, dietary or occupational exposures might have created the mutations and therefore will have contributed to the formation of the cancer. The arguments for cancer causation are particularly convincing when epidemiological evidence can support the theory that a particular exposure is linked to the cancer and when the mutational process can be recapitulated in experimental systems. In this review, I will summarize recent evidence from cancer genome sequencing studies to exemplify how the environment can modulate tumor genomes.
Recent findings
Mutation data from cancer genomes clearly implicate the UVB component of sunlight in melanoma skin cancers, tobacco carcinogen-induced DNA damage in lung cancers and aristolochic acid, a chemical compound found in certain herbal medicines, in urothelial carcinomas of exposed populations. However, large-scale sequencing is beginning to unveil other unique mutational spectra in particular cancers, such as A to C mutations at 5′AA dinucleotides in esophageal adenocarcinomas and complex mutational patterns in liver cancer. These data sets can form the basis for future studies aimed at identifying the carcinogens at work.
Summary
The findings have substantial implications for our understanding of cancer etiology and cancer prevention.
Keywords: mutations, melanoma, lung cancer, esophageal cancer, liver cancer
INTRODUCTION
Environmental factors have long been associated with development of cancer in humans. As early as 1775, it was observed that squamous cell carcinoma of the scrotum was prevalent in chimney sweeps as a results of exposure to soot representing the first type of tumor linked to occupational and environmental exposure [1]. In the 20th century, experimental exposure of animals, mostly rodents, to various carcinogens provided evidence that ultraviolet light and chemical constituents of tobacco smoke can promote effective formation of tumors [2–4]. Since humans are also exposed to these agents, this indirect evidence suggested that at least some types of human cancer are due to environmental factors. However, more definitive proof was missing and was obtained only when DNA sequencing was used to characterize genes and genomes in human tumors in search of fingerprints that can be traced back to environmental factors. In this review, I will discuss recent progress mostly derived from high-throughput sequencing of cancer genomes that has solidified our knowledge of mutational patterns in exposure-associated cancers but has also unveiled the existence of previously unrecognized mutational signatures suggesting that human populations are exposed to carcinogens of unknown identity.
Mutational patterns deduced from TP53 mutations in human cancer
The application of Sanger sequencing to candidate oncogenes or tumor suppressor genes showed that the gene TP53, coding for the tumor suppressor p53, was frequently mutated in a variety of different cancer types [5]. Interestingly, even today, after numerous entire tumor genomes or exomes have been sequenced, TP53 still stands at the top of the list as the most frequently mutated gene in human cancer, and this holds true for many if not most types of malignancies. Two major TP53 mutational databases containing tens of thousands of mutations have been established that summarize the types of mutations reported in the literature [6, 7].
Sequence analysis of the TP53 gene in non-melanoma skin cancers (basal and squamous cell carcinoma), a type of tumor strongly linked epidemiologically to sun exposure of the fair-skinned population, revealed convincing evidence that the mutations found in these tumors were caused by ultraviolet light from the sun [8]. A large percentage of skin cancer-associated TP53 mutations were C to T transitions at dipyrimidine sites and often included CC to TT tandem mutations. It was known from previous laboratory-based studies that ultraviolet light induces exactly these types of mutations. The tandem CC to TT mutations are extremely rare among any other carcinogen-induced mutations and are almost never found in tumors of internal body sites. This work provided solid evidence for the existence of environmentally induced mutations in human cancer.
Analysis of the TP53 gene in liver cancers revealed a unique type of mutation at codon 249, at which the sequence AGG was frequently converted to AGT [9, 10]. This mutation was particularly common in liver tumors from specific geographic areas in which food contamination with the mycotoxin aflatoxin is a major problem. Since aflatoxin is known to induce G to T transversions in many experimental systems, this exposure remains a strong candidate for the mutagenic process observed in liver cancer. However, the specific targeting of a single guanine at codon 249 by this compound is still unexplained and may involve interaction with other mechanisms including, for example, co-existence of hepatitis B virus [11]. If the codon 249 mutations are removed from the TP53 mutation database [12], a more heterogenous pattern is apparent that includes frequent mutations at A:T base pairs and is very different from that, for example found in colon cancer.
Lung cancers are characterized by TP53 mutations that are dominated by G to T transversions. This mutation class is more prevalent in smokers than in nonsmokers suggesting the involvement of tobacco smoke carcinogens [13]. A major class of such carcinogens is present in the tar fraction of cigarette smoke condensate and consists of the group of polycyclic aromatic hydrocarbons (PAHs). Bezo[a]pyrene is a major and well-studied member of this group of chemicals. When entering cells or tissues, these compounds are metabolically activated as part of detoxification processes. This metabolic activation leads to the formation of diol epoxide derivatives, intermediates that can strongly bind to DNA leading to the formation of covalent DNA adducts that are mutagenic and can produce G to T transversions [1]. In the TP53 gene, the G to T transversions are very frequent at six specific mutational hotspot sites [14]. Exposure of human bronchial cells to the diol epoxide of benzo[a]pyrene led to the formation of strong DNA damage hotspots exactly at the same positions that are frequently mutated (as G to T) in lung cancer from tobacco smokers [15]. This data provided a specific molecular link between smoking and lung cancer.
Whole cancer genome sequencing studies
With the invention of high-throughput DNA sequencing technology, it has become possible to analyze all genes of a cancer genome, extending the analysis beyond TP53 and a few other genes studied in isolation previously. The focus has often been on exome sequencing because genes with so-called driver mutations that alter the coding sequence need to be identified. Even at this level of analysis, it became clear very soon that most mutations found in tumors are unlikely to have an effect on cancer initiation or progression [16]. For example, mutations affecting the third base of a codon are often silent and their frequencies in cancers are almost the same as would be expected as the result of a random process. Nonetheless, the large number and specific types of mutations (silent or non-silent) have provided a rich source of information for understanding mutational patterns in tumors [17]. Some examples of such mutational spectra are shown in Figure 1. Sequencing entire cancer genomes reveals an even more unbiased view on the mutational landscape since only a small number of the mutations are selected during tumorigenesis. Large-scale cancer genome sequencing studies have uncovered mutational signatures for many types of human cancer. Many of these signatures are similar to those previously established from TP53 sequencing data but several new ones have also been found [18–20]. Perhaps with the exception of melanoma and certain urothelial cancers (see below), it is rare that a cancer genome is dominated by only one single specific mutational signature [18]. During the lifetime of a patient, and then during the development and progression of the tumor itself, many potentially mutagenic processes are at work. These include, but are not limited to, inherent DNA polymerase errors, spontaneous decay of DNA, for example in the form of 5-methylcytosine deamination at CpG sites, spontaneous, e.g. oxidative DNA damage-induced erroneous polymerase bypass during replication, and exogenous exposure-induced DNA damage leading to mutations during DNA replication. A computational approach was developed to dissect the different kinds of mutational processes that may have operated to shape a cancer genome [21]. Interestingly, this approach has revealed tumor-specific mutational signatures with most types of tumors having two or more of such signatures. Several signatures are most likely due to endogenous mutagenic mechanisms. One of them includes the aforementioned C to T transition mutations at CpG sites. Since most CpGs are methylated in mammalian genomes, the mechanism seems to involve 5-methylcytosine (5mC) bases. The dogma is that such mutations are due to spontaneous deamination of 5mC leading to thymine followed by inefficient repair of the resulting mismatch. However, other mechanisms are also possible, as discussed previously [22], including poorly studied pathways that may involve enzymatic oxidation products of 5mC such as 5-hydroxymethylcytosine, 5-formylcytosine or 5-carboxylcytosine, which are recently recognized components of the epigenetic code [23]. Another endogenous pathway that seems to produce a mutational signature in several types of cancer involves cytosine deaminases of the AID/APOBEC family. This pathway may be responsible for the frequent mutations observed at the cytosine of 5′TpC dinucleotides where the cytosine often gets converted into thymine [22, 24]. These endogenous processes, although important, are not the focus of this review, but have been reviewed recently [25]. In the following paragraphs, I will discuss several interesting examples of mutational signatures that are likely caused by environmental exposures.
Figure 1. Examples of mutation spectra for human lung cancer, liver cancer and colorectal cancer.
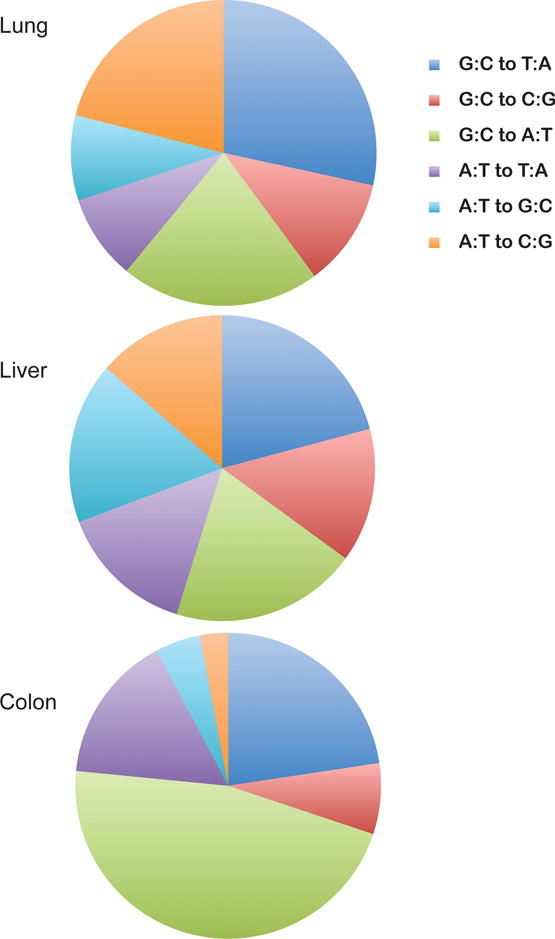
Data for base substitution mutations were obtained from the COSMIC database. Note that the data set may be biased to some extent because single gene-specific datasets (e.g. KRAS, CTNNB1) are included, which may be affected by base composition and/or selection for specific mutations.
Region-specific mutation frequencies
Mutation frequencies along the genome depend on many variables. In general, important parameters are the potency of the DNA damaging agent, the type of DNA lesion formed, DNA repair, and the structure and mutagenicity of the damage, which is dictating error-prone versus error-free DNA polymerase bypass. Moreover, local mutation frequencies in sub-compartments of the genome are strongly influenced by sequence- or gene-specific DNA repair, chromatin structure of the locus and by DNA replication. For example, DNA damage associated with crosslinked, dimerized or bulky DNA lesions such as adducts of polycyclic hydrocarbons or UV-induced pyrimidine dimers are subject to transcription-coupled DNA repair [26]. This repair pathway operates preferentially on the transcribed DNA strand leading to a preponderance of the typical exposure-related mutation on the non-transcribed strand, e.g. as seen for G to T mutations linked to PAH compounds [27]. In fact, this gene-associated repair and strand bias can clearly be observed for genome-wide mutations in UV light-associated melanomas [28] and in smoking-associated lung cancers [29].
A recent analysis further revealed that DNA sequences associated with regulatory regions of the genome, which are collectively reflected as DNAseI hypersensitive sites, have a substantially reduced local mutation density [30]. This reduced mutation density was associated with nucleotide excision repair, which may be preferentially targeted to such regions of open chromatin. Earlier studies had revealed a similar phenomenon at domains near transcription start sites of genes, which were repaired preferentially [31] suggesting that transcription-coupled and regulatory domain-specific DNA repair play important roles in shaping the mutational landscapes of cancer genomes.
Mutational heterogeneity across the cancer genome is also affected by replication timing. Genomic regions can replicate early or late during the S phase of the cell cycle. It was reported that the average mutation density is 2.1-fold higher in the late replicating 10% of genes versus the early replicating 10% of genes [19]. Mechanistic explanations are currently lacking but could possibly be linked to a depletion of the nucleotide pool in late S phase.
Mutations in melanoma genomes are linked to UVB exposure
Melanoma is a lethal type of skin cancer with rising incidence in many parts of the world. Although factors other than sunlight exposure play a role in transformation of melanocytes to invasive melanoma, exposure to ultraviolet light from the sun plays a very significant role, in particular for melanomas occurring on sun-exposed body sites [32]. Different wavelengths of the ultraviolet spectrum reach the earth’s surface and might be involved in tumor formation, both for melanoma and non-melanoma skin cancers [33]. However, the efficacy of ultraviolet B wavelengths (280 nm to 320 nm) in inducing tumors in mice is much stronger than that of UVA (320 to 400 nm) [34]. The first sequencing of an entire melanoma genome indeed revealed a striking predominance of UVB-specific mutations [28]. The mutations were characterized by an abundance of C to T and CC to TT transitions at dipyrimidine sites and this same pattern was also observed in subsequent melanoma sequencing studies [35–38]. These data make a convincing case for UVB irradiation from the sun being a strong mutational driving force in melanoma formation.
Mutations in lung cancer genomes are due to tobacco smoking
Lung cancer causes well over one million deaths each year. Most lung cancers are caused by chemicals in tobacco smoke [39]. Similar to UV-associated melanomas, the genomes of lung cancers (adenocarcinomas, squamous cell and small cell lung cancers) are characterized by some of the highest mutation frequencies among all cancers [19, 20, 40]. A prevalent type of mutation in lung cancers is the G to T transversion, which occurs with a strand bias towards the non-transcribed DNA strand [18]. Importantly, lung cancer genomes from smokers contain up to 10 times more mutations than those from nonsmokers [41, 42] attesting to the overwhelming mutational power of cigarette smoke exposure. The G to T mutational preference is reminiscent of the data obtained earlier with the TP53 gene reinforcing the notion that polycyclic aromatic hydrocarbons are likely involved in their etiology [13]. Even at the level of neighboring base sequence analysis, the G to T transversions are often targeted to methylated CpG dinucleotides, similar as in TP53 [14, 29]. This same mutational specificity was previously derived also experimentally for benzo[a]pyrene-induced DNA damage [43] strongly supporting a role for this class of tobacco smoke carcinogens in lung cancer etiology.
Esophageal adenocarcinoma: signature of an unidentified carcinogenic process operating at 5′AA sequences
The incidence of esophageal adenocarcinoma (EAC) has increased significantly in recent decades. Gastrointestinal reflux disease, Barrett’s esophagus (intestinal metaplasia), smoking and obesity are major factors in disease etiology. Recent exome sequencing of EACs revealed an A to C transversion signature, which was not present in esophageal squamous cell carcinomas [44]. This signature was also seen in a much larger collection of samples with 149 samples of EACs sequenced at all exons and 15 samples processed by whole genome sequencing [45]. In the whole genome data set, A to C mutations comprised an average of 34% of all mutations, which is much larger than that observed in any other tumor type. As shown by nearest neighbor base analysis, 84% of the A to C mutations were flanked at the 5′ side by an adenine, i.e. the mutational target sequence is 5′AA [45]. Higher levels of gene expression were correlated with lower levels of AA to AC mutation frequency and a small strand bias towards the nontranscribed strand was apparent. This data suggest that transcription-associated DNA repair processes reduce A to C mutations at AA sequences. Although the molecular pathway involved in this very unique mutational signature is unknown, the nature of the dinucleotide target and the bias towards non-transcribed genomic regions suggest that perhaps a bulky or crosslinked type of DNA damage may be involved in this process. Future mechanistic work identifying this signature in experimental mutation detection systems will be critically important towards finding an etiological agent or pathway for esophageal adenocarcinoma.
Urothelial carcinoma and aristolochic acid
Herbal remedies including Aristolochia plants have been implicated in the development of nephrotoxicity and urothelial cell carcinoma of the upper urinary tract [46]. These plant extracts contain aristolochic acid, a compound that can form DNA adducts at adenines after metabolic activation [47]. Studies on TP53 mutations in these tumors had shown an enrichment of mutations at A/T base pairs mostly in the form of A to T transversions [48]. This type of mutation is a known mutagenic signature of aristolochic acid in experimental systems [49]. Recently, two studies were conducted to analyze the genomes of upper urinary tract urothelial cell carcinomas from a Taiwanese population with suspected aristolochic acid exposure [50, 51]. Mutation frequencies were extremely high (about 150 mutations per megabase) and a similar mutational signature was found in both studies that corresponded to the earlier TP53 data. A to T mutations comprised over 70% of all mutations. These mutations were frequent at a particular sequence motif (T/CAG) and occurred predominantly on the nontranscribed DNA strand. Interestingly, the authors identified a similar A to T transversion-dominated mutation pattern in several hepatocellular carcinoma genomes suggesting that exposure to aristolochic acid may perhaps also be linked to liver cancer [51].
Complex mutations in liver cancer genomes
There have been several sequencing studies reporting genomic mutations in liver cancers, mostly hepatocellular carcinomas (HCC) [52–56]. In the HCC mutation data sets, two prominent signatures were identified: G to T transversions and mutations at A/T base pairs. The G to T transversions, with strand bias towards the nontranscribed strand, are also frequent in lung cancers from cigarette smokers and may therefore originate from exposure to carcinogens forming bulky DNA adducts that are only repaired efficiently on the transcribed DNA strand [13]. Up to now, genome-sequencing data for liver cancer in aflatoxin-exposed populations have not yet been reported.
The origin of mutations at A/T base pairs, which are otherwise rare in most cancer types, is currently unknown. Some of the cases of dominant A to T transversions may be due to consumption of herbal preparations that contain aristolochic acid as described above. There are only a few other mutagens that are known to selectively cause mutations at adenines, for example certain PAH compounds [57] and vinyl chloride [58]. However, the man-made vinyl chloride is more likely to be relevant in occupational settings rather than for the general population and induces angiosarcomas rather than HCC. The molecular origin of the common A to G transition mutations in liver cancer (and in other tumors), where adenine is located on the nontranscribed DNA strand, has so far remained enigmatic. Further research is required to improve our understanding of the molecular origins of the complex mutational patterns seen in hepatocellular carcinomas.
CONCLUSIONS
Recent genome sequencing data are beginning to reveal several unique mutational signatures in cancer genomes that are unexplained. Examples include liver cancer and espophageal cancer. As more and more tumor genomes are sequenced, we can expect that the mutational signatures will become more refined. This type of work could also help understand rare types of cancer that are linked to particular environmental factors or the effects of unique exposures in different parts of the world. There is a large gap, however, between recognizing these specific mutational signatures and actually identifying the causative agent. To close this gap, it would be desirable to develop experimental systems that can screen candidate carcinogens or mutagens to delineate their specific mutational characteristics in order to ultimately find matches with the signatures present in tumor genomes (Figure 2). At a medium to high throughput level, this may be achieved, for example, by shotgun sequencing of exposed cell populations or organisms [59], perhaps in combination with ultrasensitive DNA sequencing technologies [60].
Figure 2. Use of mutational spectra to deduce the origins of cancer.
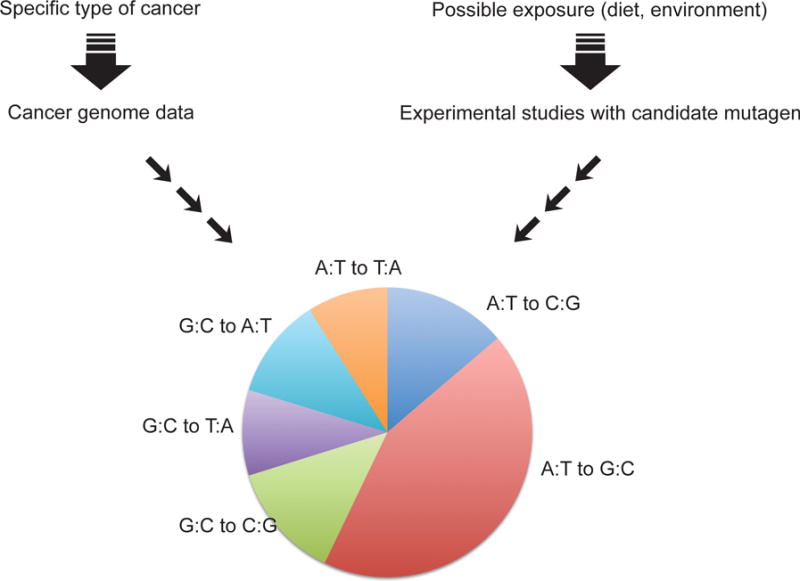
Cancer genome sequencing provides large datasets that can be used to derive mutational spectra (left). Candidate mutagens can then be tested in experimental systems (right) to determine if their mutational specificity is similar to the spectra observed in human cancer. The spectrum shown is hypothetical.
Key points.
Cancer genome sequencing data support strong effects of the environment on cancer mutations in UVB-associated melanomas, tobacco smoking-induced lung cancers and in urothelial cell carcinomas induced by aristolochic acid found in herbal medicines.
Sequencing of esophageal adenocarcinomas revealed an unusual mutation signature of A to C mutations at 5′AA dinucleotides suggesting that an unknown carcinogen is causing these mutations.
Mutational patterns in hepatocellular carcinoma are complex and often involve G to T transversions or mutations at A/T base pairs likely dependent on the type of exposure.
Acknowledgments
Work of the author was supported by NIH grant ES006070. This manuscript is dedicated to the fond memory of Victor Fung, Ph.D., former Program Officer at NCI and former Scientific Review Officer of the Cancer Etiology study section of CSR, NIH, for his wisdom, compassion, integrity, and his love of science.
Footnotes
Conflicts of interest: G.P.P. declares no conflict of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest.
** of outstanding interest.
- 1.Luch A. Nature and nurture – lessons from chemical carcinogenesis. Nat Rev Cancer. 2005;5:113–125. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- 2.Rusch HP, Baumann CA. Tumor production in mice with ultraviolet radiation. Am J Cancer. 1939;35:55–62. [Google Scholar]
- 3.Croninger AB, Graham EA, Wynder EL. Experimental production of carcinoma with tobacco products. V. Carcinoma induction in mice with cigar, pipe, and all-tobacco cigarette tar. Cancer Res. 1958;18:1263–1271. [PubMed] [Google Scholar]
- 4.Winkelmann RK, Baldes EJ, Zollman PE. Squamous cell tumors induced in hairless mice with ultraviolet light. The Journal of investigative dermatology. 1960;34:131–138. [PubMed] [Google Scholar]
- 5.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 6.Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 7.Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Hum Mutat. 2014;35:672–688. doi: 10.1002/humu.22552. [DOI] [PubMed] [Google Scholar]
- 8.Brash DE, Rudolph JA, Simon JA, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350:429–431. doi: 10.1038/350429a0. [DOI] [PubMed] [Google Scholar]
- 10.Hsu IC, Metcalf RA, Sun T, et al. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350:427–428. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- 11.Ortiz-Cuaran S, Villar S, Gouas D, et al. Association between HBX status, aflatoxin-induced R249S TP53 mutation and risk of hepatocellular carcinoma in a case-control study from Thailand. Cancer Lett. 2013;331:46–51. doi: 10.1016/j.canlet.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 12.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfeifer GP, Denissenko MF, Olivier M, et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–7451. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 14.Hainaut P, Pfeifer GP. Patterns of p53 G–>T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22:367–374. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 15.Denissenko MF, Pao A, Tang M-s, Pfeifer GP. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 16.Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014 doi: 10.1038/nrg3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev. 2014;24:52–60. doi: 10.1016/j.gde.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19*.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. This article shows that mutation frequency across the genome is strongly correlated with replication timing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20**.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. This article extracted 20 mutational signatures from over 7,000 human cancers and identified several of them as cancer-specific signatures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–259. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfeifer GP. Mutagenesis at methylated CpG sequences. Curr Top Microbiol Immunol. 2006;301:259–281. doi: 10.1007/3-540-31390-7_10. [DOI] [PubMed] [Google Scholar]
- 23.Pfeifer GP, Kadam S, Jin SG. 5-hydroxymethylcytosine and its potential roles in development and cancer. Epigenetics & chromatin. 2013;6:10. doi: 10.1186/1756-8935-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nik-Zainal S, Alexandrov LB, Wedge DC, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuong KJ, Loeb LA. APOBEC3B mutagenesis in cancer. Nat Genet. 2013;45:964–965. doi: 10.1038/ng.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 27.Denissenko MF, Pao A, Pfeifer GP, Tang M-s. Slow repair of bulky DNA adducts along the nontranscribed strand of the human p53 gene may explain the strand bias of transversion mutations in cancers. Oncogene. 1998;16:1241–1247. doi: 10.1038/sj.onc.1201647. [DOI] [PubMed] [Google Scholar]
- 28.Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pleasance ED, Stephens PJ, O’Meara S, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30*.Polak P, Lawrence MS, Haugen E, et al. Reduced local mutation density in regulatory DNA of cancer genomes is linked to DNA repair. Nat Biotechnol. 2014;32:71–75. doi: 10.1038/nbt.2778. The article identified regulatory elements of the genome as having lower mutation density. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tu Y, Tornaletti S, Pfeifer GP. DNA repair domains within a human gene: selective repair of sequences near the transcription initiation site. EMBO J. 1996;15:675–683. [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell D, Fernandez A. The photobiology of melanocytes modulates the impact of UVA on sunlight-induced melanoma. Photochemical & photobiological sciences: Official journal of the European Photochemistry Association and the European Society for Photobiology. 2012;11:69–73. doi: 10.1039/c1pp05146f. [DOI] [PubMed] [Google Scholar]
- 33.Pfeifer GP, Besaratinia A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem Photobiol Sci. 2012;11:90–97. doi: 10.1039/c1pp05144j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Fabo EC, Noonan FP, Fears T, Merlino G. Ultraviolet B but not ultraviolet A radiation initiates melanoma. Cancer Res. 2004;64:6372–6376. doi: 10.1158/0008-5472.CAN-04-1454. [DOI] [PubMed] [Google Scholar]
- 35.Berger MF, Hodis E, Heffernan TP, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikolaev SI, Rimoldi D, Iseli C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44:133–139. doi: 10.1038/ng.1026. [DOI] [PubMed] [Google Scholar]
- 37.Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gartner JJ, Parker SC, Prickett TD, et al. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc Natl Acad Sci USA. 2013;110:13481–13486. doi: 10.1073/pnas.1304227110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hecht SS. Lung carcinogenesis by tobacco smoke. International journal of cancer Journal international du cancer. 2012;131:2724–2732. doi: 10.1002/ijc.27816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–1134. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoon JH, Smith LE, Feng Z, et al. Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Res. 2001;61:7110–7117. [PubMed] [Google Scholar]
- 44.Agrawal N, Jiao Y, Bettegowda C, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012;2:899–905. doi: 10.1158/2159-8290.CD-12-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45**.Dulak AM, Stojanov P, Peng S, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45:478–486. doi: 10.1038/ng.2591. This article identified A to C transversions at AA dinucleotide sequences as a type of frequent mutation in esophageal adenocarcinomas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmeiser HH, Nortier JL, Singh R, et al. Exceptionally long-term persistence of DNA adducts formed by carcinogenic aristolochic acid I in renal tissue from patients with aristolochic acid nephropathy. Int J Cancer. 2014;135:502–507. doi: 10.1002/ijc.28681. [DOI] [PubMed] [Google Scholar]
- 47.Arlt VM, Schmeiser HH, Pfeifer GP. Sequence-specific detection of aristolochic acid-DNA adducts in the human p53 gene by terminal transferase-dependent PCR. Carcinogenesis. 2001;22:133–140. doi: 10.1093/carcin/22.1.133. [DOI] [PubMed] [Google Scholar]
- 48.Grollman AP, Shibutani S, Moriya M, et al. Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc Natl Acad Sci USA. 2007;104:12129–12134. doi: 10.1073/pnas.0701248104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Z, Hergenhahn M, Schmeiser HH, et al. Human tumor p53 mutations are selected for in mouse embryonic fibroblasts harboring a humanized p53 gene. Proc Natl Acad Sci USA. 2004;101:2963–2968. doi: 10.1073/pnas.0308607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50**.Hoang ML, Chen CH, Sidorenko VS, et al. Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci Transl Med. 2013;5:197ra102. doi: 10.1126/scitranslmed.3006200. This article, along with ref. 51, was the first to show genome-wide A to T mutations as a characteristic signature in urothelial cell carcinomas of a population exposed to the carcinogen aristolochic acid. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51**.Poon SL, Pang ST, McPherson JR, et al. Genome-wide mutational signatures of aristolochic acid and its application as a screening tool. Sci Transl Med. 2013;5:197ra101. doi: 10.1126/scitranslmed.3006086. This publication, along with ref. 50, was the first to show genome-wide A to T mutations as a characteristic signature in urothelial cell carcinomas of a population exposed to the carcinogen aristolochic acid. [DOI] [PubMed] [Google Scholar]
- 52.Fujimoto A, Totoki Y, Abe T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 53.Totoki Y, Tatsuno K, Yamamoto S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43:464–469. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- 54.Huang J, Deng Q, Wang Q, et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44:1117–1121. doi: 10.1038/ng.2391. [DOI] [PubMed] [Google Scholar]
- 55.Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shibata T, Aburatani H. Exploration of liver cancer genomes. Nat Rev Gastroenterol Hepatol. 2014;11:340–349. doi: 10.1038/nrgastro.2014.6. [DOI] [PubMed] [Google Scholar]
- 57.Yoon JH, Besaratinia A, Feng Z, et al. DNA damage, repair, and mutation induction by (+)-Syn and (−)-anti-dibenzo[a,l]pyrene-11,12-diol-13,14-epoxides in mouse cells. Cancer Res. 2004;64:7321–7328. doi: 10.1158/0008-5472.CAN-04-1094. [DOI] [PubMed] [Google Scholar]
- 58.Jackson MA, Lea I, Rashid A, et al. Genetic alterations in cancer knowledge system: analysis of gene mutations in mouse and human liver and lung tumors. Toxicol Sci. 2006;90:400–418. doi: 10.1093/toxsci/kfj101. [DOI] [PubMed] [Google Scholar]
- 59.Meier B, Cooke SL, Weiss J, et al. C. elegans whole genome sequencing reveals mutational signatures related to carcinogens and DNA repair deficiency. Genome Res. 2014 doi: 10.1101/gr.175547.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kennedy SR, Salk JJ, Schmitt MW, Loeb LA. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013;9:e1003794. doi: 10.1371/journal.pgen.1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]