

Abstract
Complex mechanisms involving genomic aberrations in numerous proteins and pathways are believed to be a key cause of many diseases such as cancer. With recent advances in genomics, elucidating the molecular basis of cancer at a patient level is now feasible, and has led to personalized treatment strategies whereby a patient is treated according to his or her genomic profile. However, there is growing recognition that existing treatment modalities are overly simplistic, and do not fully account for the deep genomic complexity associated with sensitivity or resistance to cancer therapies. To overcome these limitations, large-scale pharmacogenomic screens of cancer cell lines – in conjunction with modern statistical learning approaches - have been used to explore the genetic underpinnings of drug response. While these analyses have demonstrated the ability to infer genetic predictors of compound sensitivity, to date most modeling approaches have been data-driven, i.e. they do not explicitly incorporate domain-specific knowledge (priors) in the process of learning a model. While a purely data-driven approach offers an unbiased perspective of the data – and may yield unexpected or novel insights - this strategy introduces challenges for both model interpretability and accuracy. In this study, we propose a novel prior-incorporated sparse regression model in which the choice of informative predictor sets is carried out by knowledge-driven priors (gene sets) in a stepwise fashion. Under regularization in a linear regression model, our algorithm is able to incorporate prior biological knowledge across the predictive variables thereby improving the interpretability of the final model with no loss – and often an improvement - in predictive performance. We evaluate the performance of our algorithm compared to well-known regularization methods such as LASSO, Ridge and Elastic net regression in the Cancer Cell Line Encyclopedia (CCLE) and Genomics of Drug Sensitivity in Cancer (Sanger) pharmacogenomics datasets, demonstrating that incorporation of the biological priors selected by our model confers improved predictability and interpretability, despite much fewer predictors, over existing state-of-the-art methods.
1. Introduction
High-throughput technologies such as microarray and deep sequencing have been extensively used to reveal that cancer subtypes can be molecularly defined based on their corresponding genomic alterations [1-4]. Moreover, two large-scale pharmacogenomics cell line screens have become available with genomic profiles and drug response of hundreds of clinical and preclinical anti-cancer compounds: the Cancer Cell Line Encyclopedia (CCLE) [5, 6] and the Genomics of Drug Sensitivity (Sanger) projects [7-9]. Both studies demonstrated that genomic features identified by modern machine learning algorithm could be a viable preclinical tool for identifying potential drug sensitivity or resistance markers, with the potential for guiding precision medicine applications and clinical trial design.
In contrast to data-driven pharmacogenomic modeling, decades of experimental molecular biology has produced a detailed (albeit incomplete) knowledge of gene-gene regulatory networks and pathways. The Kyoto Encyclopedia for Genes and Genomes (KEGG), for example, is a collection of comprehensive pathway information derived from experimental analyses and literature curation [10]. Pathway Commons is another rich resource that integrates biological pathway and molecular interaction information from many publicly available databases [11]. Importantly, pathway databases represent only the static regulatory relationships between genes or gene products and are typically context independent [12]. In addition, it is well known that pathways are not functionally independent but are highly coupled processes, with constitutive pathway genes playing multiple roles within different biological processes.
As computational approaches for modeling therapeutic response are being increasingly used in research and translational applications, systematic analyses and best practices recommendations have been recently published [13, 14]. However, these studies have primarily focused on computational or algorithmic improvements. Integrating prior knowledge in predictive algorithms may increase the biological interpretability of these models, and potentially mitigate issues of data over-fitting. Several analytical studies have already incorporated pathways or network information in the variable selection framework [15-21] or used network knowledge to identify differentially expressed genes [22, 23]. However, most of these studies considered only pre-selected pathways as “prior knowledge”, impeding an unbiased assessment of how each pathway is individually associated with model performance. In addition, group lasso algorithms [24-26] were proposed for solving the group sparsity problem. However, biological priors such as pathways are highly coupled and overlapping, and therefore do not optimally match the conditions required for group lasso.
In this study, we present the Stepwise Group Sparse Regression (SGSR) model, developed to leverage prior knowledge in order to improve predictive power and interpretability in the context of modeling drug response with genomic data. Specifically, we embedded a prior selection procedure into sparse regression, such that it could specify preferences for particular combination of priors in the model. The rationale is derived from forward stepwise selection, in which selection of gene-set-coherent features are encouraged through regularization, while the best combination of feature sets is determined by forward stepwise method. We first explored the effectiveness of the SGSR as compared to LASSO, Ridge and Elastic Net regression on the CCLE and Sanger cell line studies [5-7, 9, 13, 14], and then analyzed whether informative pathway priors improved the selection of previously validated drug-targets in our model, e.g. MAPK pathway genes for MEK inhibitors. We also demonstrated and compared the effectiveness and power of SGSR using different genomic features as input variables, e.g. gene-expression (EXP) vs. copy-number alterations (COPY). For the public accessibility, we provide an R package at https://github.com/Sage-Bionetworks/SGSR, and share all results through https://www.synapse.org/#!Synapse:syn2600070.
2. Material and Methods
2.1. Materials: Datasets and Prior knowledge databases Datasets
The CCLE and Sanger datasets contain anti-cancer compound screening data performed on large panels of molecularly characterized cancer cell lines. Both datasets contain high-throughput gene expression and copy number alterations, as well as mutation status on a subset of genes, summarized to gene-level features. Here, we utilize either EXP or COPY dataset to predict drug responses.
In Sanger we have 664 cell lines with EXP measurements on 12,024 genes (643 cell lines with COPY data on 12,082 genes), whereas CCLE has 491 cell lines with EXP measurements on 18,897 genes (488 cell lines with COPY data on 21,217 genes). All data was normalized as described in the original papers [5, 7]. Both studies provided multiple drug dose statistics such as IC50 and ActArea (or AUC) to summarize dose-response curves to compound sensitivity values for each cell line. We chose ActArea with CCLE and IC50 with Sanger, respectively, based on our previous analyses showing their predictive benefit [13]. In addition, we chose 28 out of 138 compounds in Sanger and all 24 compounds in CCLE: 14 overlapping drugs in both cell line studies, selected for cross-comparison. One of the main objectives of the proposed model is to improve interpretability by taking advantage of prior knowledge on pathways that may be implicated in sensitivity/resistance patterns to anti-cancer compounds. Sanger has drug response data to many agents that are not being investigated as anti-neoplastic drugs or that have multiple - and overlapping - targets, making interpretation of the results difficult. We decided to select for downstream analyses Sanger compounds for which there is substantial level of evidence in the literature in terms of preclinical or clinical oncological translation, making sure that we had at least one drug that inhibits relevant targets (known cancer drivers) included in the final list.
Prior knowledge databases
Curated pathway databases represent a valuable resource for scientists studying biological processes in cancer. We take advantage of this information accumulated over years of biomedical research and define a knowledge-driven prior as a set of genes that are mapped to a curated pathway. We anticipate that our model selects a set of pathways – and corresponding genes – that are most likely functionally important for drug sensitivity patterns, therefore increasing biological interpretability of the final set of features. Thus, our prior incorporated predictive model goes beyond traditional analyses by learning the complex structure of input variables and their functional relationships with response. As input to the SGSR model we used the GRAPH Interaction from pathway Topological Environment (graphite: R package built in Bioconductor [27]), providing access to publicly available canonical pathway databases such as KEGG (n=232), Biocarta (n=254), NCI/Nature (n=177) and gene ontology (GO) Biological Processes (n=825) and Molecular Functions (n=396) in MSigDB 3.0 [28]).
2.2. Baseline regularized regression methods
A major challenge in the development of predictive models utilizing high-dimensional, genomic data is finding the optimal trade-off between predictive performance and model sparsity (often associated with model interpretability). In the context of drug sensitivity modeling, this trade-off is particularly acute as the selection of biomarkers for patient stratification is a primary goal. Simultaneously, model performance is used to evaluate the ultimate feasibility of drug prediction, and robustness of the biomarkers. Moreover, the incorporation of prior knowledge into data-driven models is a non-trivial task. Biological priors are highly coupled and oftentimes redundant, thereby complicating the process by which they might be included in model building.
To resolve these problems, we have implemented a predictive modeling framework that systematically incorporates prior biological knowledge. Here we present the prior incorporated sparse regression model and its internal prior selection procedure in terms of forward-stepwise selection. Throughout the text we consider the linear regression model y = Xβ + ε, where y represents the (n × 1) vector of responses X, corresponds to the (n × p) matrix of features, β corresponds to the (p × 1) vector of regression coefficients, and ε represents a (n×1) noise vector. The original problem is to estimate vector of coefficients with least square criteria. In the “large p (features), small n (samples)” paradigm, the solution to the least-squared problem is undetermined and requires constraining the model space. Recent studies have shown that regularized regression can lead to practical solutions for modeling high-dimensional genomic data [13, 29-33]. Specifically, the LASSO model imposes an L1 penalty on and typically results in sparse solutions where most coefficients are exactly zero. Conversely, the Ridge model imposes an L2 penalty on its model parameters and often produces a model where most coefficients are non-zero. However, in practice the use of these penalty functions have several limitations: the LASSO selects at most n variables before it saturates and if there is a group of highly correlated variables the LASSO tends to select one representative from a group and ignore the other components in the group. Meanwhile, models based on Ridge regression tend to perform well [13], but are hard to interpret due to lack of feature selection. To address these problems, Elastic Net regression linearly combines the L1 and L2 penalties of the LASSO and Ridge methods and optimizes two hyper-parameters ( λ1 and λ2) . Even though the Elastic Net regression method is able to select features that are not identified by LASSO because of high pairwise correlation – while still remaining parsimonious – there are intrinsic limitations of data-driven models: biological insights of model features can only be extracted after extensive post processing steps, including pathway enrichment analyses.
2.3. Stepwise Group Sparse Regression (SGSR)
The SGSR model is based on a stepwise forward “prior” selection procedure (see Figure 1 describing the workflow of the SGSR algorithm). We first define the following terms: , Gi, and correspond to the set of model features, genes in a gene set, and Mean Squared Error (MSE), respectively, corresponding to the i-th gene set and k-th round of the algorithm. We define L as the total number of pathways (gene sets) in the database. In the initialization step, we train a standard LASSO model without utilization of gene set priors, optimizing the LASSO's single hyper-parameter using 5-fold cross-validation. We define the set of selected features in this model as M0 and its MSE as MSE0. The stepwise forward prior selection process begins by evaluating the addition of each gene set to the previous model and the model that results in the largest reduction of MSE is selected as the model input for the next round (see Figure 1). Of note, the newly added genes from each gene set are unpenalized in the LASSO model, allowing them to enter into the model as a group. If none of the L models produces a lower MSE than the previous optimal model, then the iteration terminates and the previous Mk−1 is returned.
Figure 1.
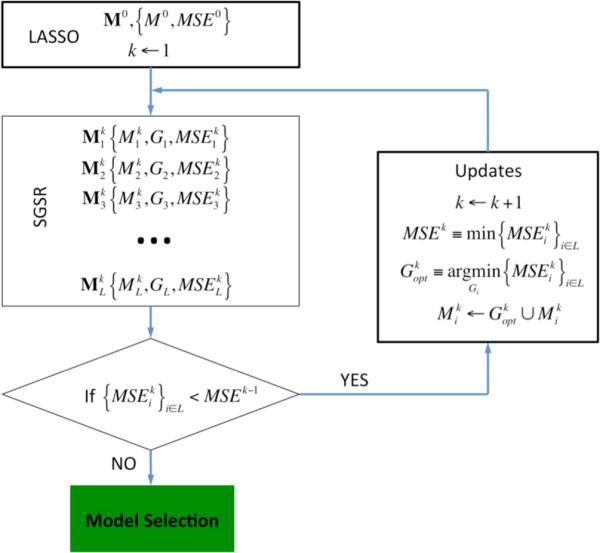
Workflow describing the SGSR algorithm. We define , Gi, and as the set of model features, genes in a gene set, and Mean Squared Error (MSE), respectively, corresponding to the i-th gene set and k-th round of the algorithm.
More specifically, we have and in the k-th round, where , are the coefficients trained by incorporating i-th prior's genes in its LASSO model, β∉i are the coefficients for the predictors which do not belong to i-th prior's genes so that they should be penalized, β∈i are the coefficients that correspond to i-th prior's genes and should be always unpenalized in the model training, and is the set of all L models in the k-th round of the stepwise selection procedure. When is satisfied, we select the k-th best prior by Finally, the algorithm's iteration is terminated either when no further MSE gain is achieved or when all pathways of given database are selected.
2.4. Assessment of model performance
For SGSR model running, we randomly split the input dataset into five non-overlapping sample groups: 4/5ths of the samples are used for training, whereas 1/5th of the samples are used for testing. The 5-fold cross validation scheme is once again applied within the 4/5ths training samples so that we can tune the parameters and have an optimized set of priors. Afterwards, we apply the model in the remaining 1/5th test samples and assess the final performance by summarizing the 5 sets of predicted drug responses with the Weighted Root Mean Squared Error (WRMSE) metric. The key reason for dividing the RMSE by the average of variance from observed and predicted values is that we can give proper weights to check whether or not the training procedure is successful. In the present analysis we discarded genomic features that have missing data in samples or that have a variance smaller than 0.02. At each split we obtained a prediction vector , where j ∈ {1,2,...,5}, and we computed a single WRMSE between the concatenated predicted vector, , and the full observed response,
3. Results
3.1. Model assessment with fixed sparsity
Using the SGSR framework, we are interested in generating models that are simultaneously sparse (i.e. have a minimal set of features in the model) and optimally predictive. As the Elastic Net regression framework was developed to optimize this tradeoff, we compare the SGSR method with the Elastic Net model to determine whether incorporation of pathway knowledge can improve performance. Specifically, we compared overall model performance of SGSR and Elastic Net at comparable levels of model sparsity. Results using the CCLE and Sanger data sets are shown in Figure 2.
Figure 2.

Comparison of model performance (weighted RMSE) between ElasticNet and the Ridge, LASSO, and SGSR algorithms. ElasticNet models are constrained to have a comparable number of features to the SGSR model. Each point corresponds to a single drug model. (A) CCLE with EXP (B) CCLE with COPY (C) Sanger with EXP (D) Sanger with COPY are applied for SGSR with 5 distinctive available pathway databases such as KEGG, BIOCARTA, Nature/NCI, GO_BP and GO_MF.
In general, we observed an overall improvement in predictive performance using the SGSR model over Elastic Net regression, in which the latter is constrained to have the same number of features as SGSR. This pattern is consistent, regardless of the pathway database selected, with the exception of the GO_BP pathways applied on the Sanger data set. Consistent with our previous work [13], we observed that models utilizing EXP data are more accurate. Interestingly, knowledge-driven priors significantly improved model performance when using COPY as input data, particularly in CCLE (P<0.0001 for all models, Wilcox rank sum test with all 5 corresponding pathway databases) while the performance improvement in Sanger with COPY depended on the type of pathway database that was utilized (see Table 1 (A)). Due to marginal gains of predictive performance with EXP, not all SGSR models were statistically significant. Overall, SGSR improved predictive accuracy over Elastic Net in the majority of comparisons (see “performance gain ratio” in Table 1(A)).
Table 1.
Performance assessment with Wilcox rank sum test and performance percentage. Orange are depicted for CCLE while light green are for Sanger (A) pairwise assessment table for fixed sparsity in Figure 2, (B) Pairwise assessment table for Figure 3 and 4 when LASSO is used for benchmarked model (C) Pairwise assessment table for Figure 3 and 4 when Ridge is used for benchmarked model. Performance percentage is computed by counting how many drug models of SGSR ourperform the benchmark model. Red and green are depicted when SGSR shows better performance (>50%) than the benchmark model.
| Benchmarked with Elastic Net regression | ||||||||
|---|---|---|---|---|---|---|---|---|
| (A) | CCLE | Sanger | ||||||
| EXP | COPY | EXP | COPY | |||||
| wilcox test p-value |
performance percentage |
wilcox test p-value |
performance percentage |
wilcox test p-value |
performance percentage |
wilcox test p-value |
performance percentage |
|
| SGSR_KEGG | 0.19 | 79.20% | 2.35E-05 | 100% | 0.10 | 82.10% | 0.23 | 57.10% |
| SGSR_BIOCARTA | 0.04 | 95.80% | 3.88E-06 | 95.80% | 0.05 | 82.10% | 0.02 | 75.00% |
| SGSR_NCI | 0.14 | 95.80% | 1.26E-06 | 95.80% | 0.18 | 82.10% | 0.07 | 78.60% |
| SGSR_GO_BP | 0.08 | 91.70% | 3.55E-05 | 95.80% | 0.64 | 32.10% | 0.25 | 57.10% |
| SGSR_GO_MF | 0.25 | 83.30% | 6.46H-05 | 91.70% | 0.05 | 92.90% | 0.31 | 64.30% |
| Benchmarked with Lasso | ||||||||
|---|---|---|---|---|---|---|---|---|
| (B) | EXP | COPY | EXP | COPY | ||||
| wilcox test p-value |
performance percentage |
wilcox test p-value |
performance percentage |
wilcox test p-value |
performance percentage |
wilcox test p-value |
performance percentage |
|
| Ridge | 0.14 | 83.30% | 1.76E-01 | 62.50% | 0.27 | 67.50% | 0.57 | 46.40% |
| SGSR_KEGG | 0.13 | 83.30% | 2.61E-05 | 100% | 0.05 | 92.90% | 0.06 | 78.60% |
| SGSR_BIOCARTA | 0.02 | 95.80% | 1.50E-04 | 91.70% | 0.02 | 89.30% | 0.01 | 85.70% |
| SGSR_NCI | 0.10 | 91.70% | 7.12E-05 | 95.80% | 0.09 | 89.30% | 0.07 | 78.60% |
| SGSR_GO_BP | 0.02 | 95.80% | 1.10E-05 | 91.70% | 0.21 | 78.60% | 0.02 | 71.40% |
| SGSR_GO_MF | 0.10 | 91.70% | 2.61E-05 | 91.70% | 0.008 | 89.30% | 0.07 | 82.10% |
| Benchmarked with Ridge | ||||||||
|---|---|---|---|---|---|---|---|---|
| (C) | EXP | COPY | EXP | COPY | ||||
| wilcox test p-value |
performance percentage |
wilcox test p-value |
performance percentage |
wilcox test p-value |
performance percentage |
wilcox test p-value |
performance percentage |
|
| Lasso | 0.86 | 16.70% | 0.83 | 37.50% | 0.73 | 28.60% | 0.43 | 50% |
| SGSR_KEGG | 0.44 | 45.80% | 0.002 | 95.80% | 0.21 | 75.00% | 0.10 | 64.30% |
| SGSR_BIOCARTA | 0.12 | 83.30% | 0.002 | 87.50% | 0.08 | 82.10% | 0.03 | 82.10% |
| SGSR_NCI | 0.48 | 45.80% | 0.001 | 87.50% | 0.25 | 75.00% | 0.06 | 78.60% |
| SGSR_GO_BP | 0.17 | 83.30% | 0.0001 | 95.80% | 0.43 | 53.60% | 0.02 | 75.00% |
| SGSR_GO_MF | 0.36 | 66.70% | 0.0003 | 87.50% | 0.04 | 85.70% | 0.05 | 78.60% |
3.2 Assessment of data-driven model vs. knowledge-driven model
We also investigated the performance of the data-driven models and the SGSR knowledge-driven model, independent of sparsity constraints. Figures 3 and 4 summarize the results of the two data-driven models (Ridge & LASSO) with SGSR using several pathway databases. In general, we observed that Ridge outperforms LASSO, consistent with previous work [13]. The improvement of SGSR over LASSO was generally higher than what we observed with Ridge over LASSO. Using the CCLE data set, SGSR with COPY markedly outperformed the data-driven models while SGSR with EXP produced marginally better performance results (see Figure 3 and Table 1 (left orange panels of B and C)). Similarly, with the Sanger data, differences in favor of the SGSR algorithm showed consistent trends for both the COPY and EXP models (see Figure 4 and Table 1 (right light green panels of B and C)). Of note, the final number of predictors in SGSR models was on average only marginally increased as compared with the LASSO models (91.7%, 94.2%, 87.9% and 79.3% in CCLE EXP, CCLE COPY, Sanger EXP and Sanger COPY, respectively).
Figure 3.

Performance comparison for CCLE pharmacogenomics data. (A) Predictability score with WRMSE metric of LASSO, Ridge, SGSR with KEGG, Biocarta, NCI/Nature, GO-BP and GO-MF pathways using EXP data across the 24 CCLE drugs, (B) Performance discrepancy between benchmarked LASSO, Ridge, and SGSR models with five available pathway databases with EXP; (C) Predictability score with WRMSE metric of LASSO, Ridge, SGSR with KEGG, Biocarta, NCI/Nature, GO-BP and GO-MF pathways with COPY across the 24 CCLE drugs, (D) Performance discrepancy between benchmarked LASSO, Ridge and SGSR models with five available pathway databases using COPY data.
Figure 4.

Performance comparison for Sanger pharmacogenomics data. (A) Predictability score with WRMSE metric of LASSO, Ridge, SGSR with KEGG, Biocarta, NCI/Nature, GO-BP and GO-MF pathways using EXP data across the preselected 28 Sanger drugs; (B) Performance discrepancy between benchmarked LASSO, Ridge and SGSR models with five available pathway databases with EXP (C) Predictability score with WRMSE metric of LASSO, Ridge, SGSR with KEGG, Biocarta, NCI/Nature, GO-BP and GO-MF pathways using COPY data across the preselected 28 Sanger drugs, (D) Performance discrepancy between benchmarked LASSO and Ridge and SGSR models with five available pathway databases with COPY.
3.3. Assessment of additional features identified by SGSR model
We next defined 2 tests to assess whether the improved performance by SGSR can be explained by factors other than the information contained in the gene-set priors. First, in order to check whether SGSR improves performance simply by adding additional features, we constructed a null distribution of predictive performance by generating 50 random models that had the same number of features added by SGSR. To do this, we preserved the original model fit by LASSO (M0) and then randomly added genes until we had a model with the same number of features as the SGSR model to which we are comparing. Second, to test whether similar performance could be obtained by incorporation of non-informative gene sets, we trained SGSR models using randomly permuted gene-set priors. Specifically, we preserved the input pathway database structure (i.e. maintained the same number of genes per gene set) but randomly shuffled the genes within each gene set. Figure 5 summarizes the predictive performance of SGSR models compared to the randomized models. In general, the WRMSE of SGSR models is significantly lower than that of both null models.
Figure 5.

Model assessments per drug (WRMSE) including SGSR with BIOCARTA (blue); SGSR using random genes (purple, left distribution using 50 random models); SGSR using random pathways (yellow, right distribution using 50 random models); LASSO (red); RIDGE (green). (A) CCLE with EXP (B) Sanger with EXP (C) CCLE with COPY and (D) Sanger with COPY
3.4. Biological Interpretability from identified priors for anticancer compounds
One attractive characteristic of SGSR is the ability to perform feature selection with increased interpretability compared to state-of-the-art methods. To exemplify this, we analyzed the results of EXP-based SGSR models (with prior using NCI/Nature Cancer pathway database) of sensitivity/resistance to the MEK inhibitors AZD6244 and PD0325901, agents tested in both CCLE and Sanger. We then compared with the matching bootstrapped Elastic Net regression models. It is known that response to these agents correlates with mutation status of KRAS/NRAS/BRAF genes [5, 7]. However, we wanted to assess whether models built on gene expression measurements could give additional biologically meaningful information. Overall, predictive performance of SGSR models for AZD6244 and PD0325901 in both CCLE and Sanger data sets are comparable to the gold-standard method. In addition, top features (genes) identified in SGSR models for each agent significantly overlap both within and across data sets, underscoring the reproducibility and potential biological relevance of the findings. As shown in Table 2, overlapping genes of major interest include: (i) MAP2K1 (also known as MEK) and MAPK1 (also known as ERK), important downstream effectors of the mitogen-activated protein kinase (MAPK) pathway; (ii) RHOA, a small GTPase known to interact with MAPK pathway to promote cell invasion [34]; (iii) AURKB, regulated by MAPK pathway to promote cell division [35]; (iv) Src family kinases SRC and FYN, which have a critical role in cell migration, proliferation and survival via the MAPK pathway [36, 37]; and (v) EDIL3 (EGF-like repeats and discoidin I-like domains 3), a stromal factor that is associated with angiogenic switch and poor prognosis in many cancers [38, 39]. By contrast, the genes described above were not inferred within the top 500 features by the bootstrapped Elastic Net regression models based on gene expression data. Although anecdotal, this analysis suggests that incorporating pathway information during the design of predictive models can identify functionally relevant biomarkers that would not be detected from a purely data-driven approach.
Table 2.
For the SGSR model, the top 7 predictive features are displayed for AZD6244 (AZD) and PD0325901(PD). Cells higlighted in orange correspond to features with evidence of being functionally related to MEK inhibitor compounds, as described in the text. For comparison, the ranks of corresponding predictive features inferred by bootstrapped Elastic Net are displayed (18,897 and 12,024 features are considered in model building with CCLE and Sanger, respectively).
| Biomarker | SGSR | Bootstrapped Elastic Net regression | ||||||
|---|---|---|---|---|---|---|---|---|
| CCLE | Sanger | CCLE | Sanger | |||||
| AZD | PD | AZD | PD | AZD | PD | AZD | PD | |
| EDIL3 | Y | Y | Y | Y | 7260 | 11170 | 5109 | 4361 |
| RHOA | Y | Y | Y | Y | 16833 | 18775 | 1335 | 5022 |
| FYN | NA | NA | Y | Y | 11962 | 9574 | 7932 | 10345 |
| MAPK1 | Y | Y | NA | Y | 618 | 815 | 8914 | 10352 |
| MAP2K1 | Y | Y | NA | Y | 10133 | 11896 | 11924 | 8338 |
| AURKB | Y | Y | Y | Y | 12979 | 16464 | 6772 | 8464 |
| SRC | NA | NA | Y | Y | 10820 | 16675 | 6501 | 8516 |
4. Discussion
The availability of large-scale pharmacogenomic screens on cancer cell line panels has begun to illuminate many of the genomic aberrations underlying compound sensitivity/resistance. The application of machine learning approaches optimized for feature selection on high-dimensional genomic data has been a critical tool in this analysis. Even though the tractability of penalized regression models has been proposed in earlier studies [5 ,7, 13], the resultant models fail to incorporate well-known pathway characteristics that frequently underlie drug efficacy in vitro and in patients. In this study, we propose a novel SGSR algorithm that allows known pathway relationships to influence feature selection during model fitting, thereby enhancing interpretability of the final model without a concomitant decrease in model performance.
Our study benchmarks a statistically principled comparison with state-of-the-art machine learning algorithms - namely LASSO, ElasticNet and Ridge regression – to predict drug sensitivity using input features from gene expression or copy number. In general, we find that the SGSR model has better overall accuracy (smaller MSE) at comparable levels of model sparsity. Of note, we observed the highest gains in predictive performance in the models that originally gave weak predictions, such as those based on COPY data [13]. Moreover, we observe that the specific grouping of the pathways (gene sets) contributes meaningful information, demonstrated in our comparison of SGSR to randomly constructed pathways. This is important, as we might expect that in aggregate the union of all genes from all pathways represent the set of genes/proteins that are more frequently studied, and therefore alone might explain the improved SGSR performance. However, the relevance of the specific gene set composition underscores the complex and pertinent information embedded in these gene sets. Finally, we consider the biological insights derived from our model (at the gene level) and interpretability of results (at the pathway level) as major advantages for cancer researchers.
In summary, SGSR provides a knowledge-incorporated sparse regression framework with significantly increased model interpretability without a trade-off of prediction accuracy. Notably, our modeling approach highlights the value of existing knowledge databases and their relevance in modeling disease phenotypes. Future directions might consider incorporation of even finer-grained relationships (dependence) embedded in these pathway databases, such as the protein interactions encoded in the Reactome pathways. We believe that SGSR advances current state-of-the art approaches for inferring molecular predictors of compound sensitivity, and may be used to identify functionally relevant gene sets used to guide translation of preclinical screens into precision medicine trials.
Footnotes
This work is supported by grant U54CA149237 from the Integrative Cancer Biology Program and by grant U01CA176303 from the Cancer Target Discovery and Development of the National Cancer Institute
Contributor Information
IN SOCK JANG, Sage Bionetworks 1100 Fairview Ave. N Seattle, WA 98109, USA in.sock.jang@sagebase.org.
RODRIGO DIENSTMANN, Sage Bionetworks 1100 Fairview Ave. N Seattle, WA 98109, USA rodrigo.dienstmann@sagebase.org.
ADAM A. MARGOLIN, Oregon Health & Science University 3181 S.W. Sam Jackson Park Rd, Portland, OR 97239, USA.
JUSTIN GUINNEY, Sage Bionetworks 1100 Fairview Ave. N Seattle, WA 98109, USA.
References
- 1.Ferte C, Andre F, Soria JC. Nat Rev Clin Oncol. 2010;7(7) doi: 10.1038/nrclinonc.2010.84. [DOI] [PubMed] [Google Scholar]
- 2.Peggs K, Mackinnon S. N Engl J Med. 2003;348(11) doi: 10.1056/NEJMe030009. [DOI] [PubMed] [Google Scholar]
- 3.Roche-Lestienne C, et al. N Engl J Med. 2003;348(22) doi: 10.1056/NEJMc035089. [DOI] [PubMed] [Google Scholar]
- 4.Savage DG, Antman KH. N Engl J Med, 2002. 2002;346(9) doi: 10.1056/NEJMra013339. [DOI] [PubMed] [Google Scholar]
- 5.Barretina J, et al. Nature. 2012;483(7391) [Google Scholar]
- 6.Marum L. Future Med Chem. 2012;4(8) [PubMed] [Google Scholar]
- 7.Garnett MJ, et al. Nature. 2012;483(7391) [Google Scholar]
- 8.Yang W, et al. Nucleic Acids Res. 2013;41(Database issue) doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Forbes SA, et al. Nucleic Acids Res. 2011;39(Database issue) doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanehisa M, et al. Nucleic Acids Res. 2012;40(Database issue) doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerami EG, et al. Nucleic Acids Res. 2011;39(Database issue):12. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S, et al. Genome Res. 2007;17(10) doi: 10.1101/gr.6202607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang IS, et al. Pac Symp Biocomput. 2014 [Google Scholar]
- 14.Neto EC, et al. Pac Symp Biocomput. 2014 [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, et al. BMC Syst Biol. 2011;5 doi: 10.1186/1752-0509-5-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li C, Li H. Bioinformatics. 2008;24(9) doi: 10.1093/bioinformatics/btn081. [DOI] [PubMed] [Google Scholar]
- 17.Tai F, Pan W. Bioinformatics. 2007;23(23) doi: 10.1093/bioinformatics/btm488. [DOI] [PubMed] [Google Scholar]
- 18.Tai F, Pan W. 2007;23(14) doi: 10.1093/bioinformatics/btm234. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, et al. Bioinformatics. 2013;29(20) doi: 10.1093/bioinformatics/btt443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei P, Pan W. Bioinformatics. 2008;24(3) doi: 10.1093/bioinformatics/btm612. [DOI] [PubMed] [Google Scholar]
- 21.Jang IS, Margolin A, Califano A. Interface Focus. 2013;3(4) doi: 10.1098/rsfs.2013.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, et al. Proteomics. 2011;11(19) doi: 10.1002/pmic.201100180. [DOI] [PubMed] [Google Scholar]
- 23.Wei Z, Li H. Bioinformatics. 2007;23(12) doi: 10.1093/bioinformatics/btm129. [DOI] [PubMed] [Google Scholar]
- 24.Yuan M, et al. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2006;68 doi: 10.1111/j.1467-9868.2006.00539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman J, et al. arXiv:1001.0736. 2010 [Google Scholar]
- 26.Jacob L, et al. Proceedings of the 26th Annual International Conference on Machine Learning. 2009 [Google Scholar]
- 27.Sales G, et al. Bmc Bioinformatics. 2012;13 doi: 10.1186/1471-2105-13-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liberzon A, et al. Bioinformatics. 2011;27(12) doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoerl AE, Kennard RW, Hoerl RW. Applied Statistics-Journal of the Royal Statistical Society Series C. 1985;34(2) [PubMed] [Google Scholar]
- 30.Tibshirani R. Journal of the Royal Statistical Society Series B-Methodological. 1996;58(1) [Google Scholar]
- 31.Tibshirani R. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2011;73 doi: 10.1111/j.1467-9868.2011.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou H, Hastie T. Journal of the Royal Statistical Society Series B-Statistical Methodology. 2005;67 doi: 10.1111/j.1467-9868.2011.01004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu WJJ. Journal of Computational and Graphical Statistics. 1998;7(3) [Google Scholar]
- 34.Vilal E, et al. Cancer Cell. 2003;4(1) [Google Scholar]
- 35.Bonet C, et al. J Biol Chem. 2012;287(35) doi: 10.1074/jbc.M112.371682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim LC, et al. Nat Rev Clin Oncol. 2009;6(10) doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 37.Yadav V, et al. Mol Carcinog. 2011;50(5) doi: 10.1002/mc.20716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun JC, et al. World J Gastroenterol. 2010;16(36) doi: 10.3748/wjg.v16.i36.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Damhofer H, et al. Mol Oncol. 2013;7(6) doi: 10.1016/j.molonc.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]