

Abstract
Prokaryotic subtilisins and eukaryotic proprotein convertases (PCs) are two homologous protease subfamilies that belong to the larger ubiquitous super-family called subtilases. Members of the subtilase super-family are produced as zymogens wherein their propeptide domains function as dedicated intramolecular chaperones (IMCs) that facilitate correct folding and regulate precise activation of their cognate catalytic domains. The molecular and cellular determinants that modulate IMC-dependent folding and activation of PCs are poorly understood. In this chapter we review what we have learned from the folding and activation of prokaryotic subtilisin, discuss how this has molded our understanding of furin maturation, and foray into the concept of pH sensors, which may represent a paradigm that PCs (and possibly other IMC-dependent eukaryotic proteins) follow for regulating their biological functions using the pH gradient in the secretory pathway.
Keywords: Intramolecular chaperones, pH sensors, subtilases, proprotein convertases, protease activation and regulation, secretory pathway, histidine protonation
1. Introduction
The limited proteolysis of an inactive precursor is a regulatory mechanism responsible for the generation of biologically active proteins and peptides (1). Proprotein convertases (PCs), which include seven mammalian Ca2+-dependent endoproteases, furin, PC1/PC3, PC2, PC4, PACE4, PC5/PC6, and PC7/LPC/PC8, represent one such family that has been identified in all eukaryotes (2–6). Although they potentially share overlapping cleavage specificity and function, each PC has its own specific set of protein substrates that are generally cleaved at a pair of basic residues, such as a Lys-Arg (7, 8). More recently, SKI/S1P (9, 10) and NARC-1/PCSK9 (8), which share sequence similarity with PCs, have also been identified as enzymes using this regulatory mechanism (11). While SKI/S1P is a protease, NARC-1/PCSK9 does not appear to display proteolytic activity toward other substrates other that its own IMC domain and functions as a chaperone that binds to LDL receptor and targets it for lysosomal degradation (12). Together, these nine proteases belong to the subtilisin-like super-family called subtilases (13).
Subtilases constitute the largest family of serine proteases after chymotrypsin (14) and contain several divergent proteases found in prokaryotes, archaea, eukaryotes, and viruses. Until the determination of the sequence of bacterial subtilisin (15), it was believed that all serine-type peptidases would be homologous to chymotrypsin. The subsequent X-ray structure established that subtilisin is clearly different and unrelated to chymotrypsin (16) and now corresponds to the family S8 according to the MEROPS database (14). The subtilase family is divided into two subfamilies, with prokaryotic subtilisins the archetype for subfamily S8A and eukaryotic kexin (3–6) the archetype for subfamily S8B. Because of the prokaryotic subtilisins’ broad specificity, their ability to hydrolyze both native and denatured proteins, their catalytic activity under alkaline conditions, and their remarkable stability, they are widely use in detergents, cosmetics, food processing, skin care ointments, and contact lens cleaners and for research purposes in synthetic organic chemistry (17). Such commercial importance provided the momentum to gather extensive biophysical, biochemical, and structural information and has made prokaryotic subtilisins the prototype model for the subtilase super-family. Until 2003, the only structural information on PCs was gleaned through homology models derived using high-resolution crystallographic data of prokaryotic subtilisins as templates (18, 19). The recent high-resolution X-ray structures of furin (20) and kexin (21) have transformed our understanding of the basis of remarkable specificity displayed by eukaryotic PCs (22) when compared with their promiscuous prokaryotic counterparts. Simultaneously, they may potentially provide us with the means to better understand the structural and functional evolution of subtilases within a cellular context.
Furin, which is a constitutively expressed protease and the most intensively studied member of the PC family, can catalyze proteolytic maturation of a diverse repertoire of proprotein substrates within the cellular secretory pathway (2). Since most enzymes are exquisitely pH sensitive, the pH of each secretory and endocytic pathway compartment critically determines and regulates coordinated biochemical reactions (23). These compartments within eukaryotic cells therefore serve to segregate specific biosynthetic and catalytic functions within membrane-limited organelles. Such compartmentalization likely evolved from the necessity to optimize performance of individual metabolic pathways by providing unique environmental conditions and to enable energy storage in the form of electrochemical gradients across the dielectric membrane (24). PCs and their substrates are synthesized in the lumen of endoplasmic reticulum (ER), wherein they undergo correct folding and often have to traverse the changing pH of the secretory pathway compartments together en route to their final destination (1, 11, 25, 26). Since premature protease activity can lead to inappropriate protein activation, sorting, or degradation, PCs and many of their substrates are synthesized as inactive zymogens (27). Upon reaching their correct cellular compartments these zymogens undergo activation usually through proteolysis. The synthesis of proteases as zymogens enables cells with the means to spatially and temporally regulate the catalytic activities of PCs. However, the molecular and cellular determinants that modulate activation of PCs are poorly understood. In this chapter we review what we have learned from the folding and activation of prokaryotic subtilisin, discuss how this has molded our understanding of furin maturation (25, 28), and foray into the concept of pH sensors (26), which may represent a paradigm that PCs (and possibly other propeptide-dependent eukaryotic proteins) follow for regulating their biological functions using the pH gradient in the secretory pathway.
2. Propeptide-Mediated Folding of Bacterial Subtilisin
Bacterial subtilisins constitute a large class of microbial serine proteases, among which subtilisin E (Bacillus subtilis), subtilisin BPN (Bacillus amyloliquefaciens), and subtilisin Carlsberg (Bacillus licheniformis) are the most extensively studied (29). The timely cloning of the genes and their ease of expression, purification, and crystallization and subsequent high-resolution X-ray studies have made subtilisin a model system for protein engineering studies (17). An analysis of the cDNA for the subtilisin E gene suggests that subtilisins are synthesized as zymogens, with an approximately 77-residue propeptide that is located between the signal sequence and the protease domain (30). Propeptide deletion results in robust expression of inactive subtilisin E. Furthermore, if the catalytic domains of any of these bacterial subtilisins are denatured using chaotropes, the unfolded proteins fail to refold into their catalytically active native states even when these chaotropes are removed (31). Further studies established that the addition of the 77-residue propeptide to the folding reaction results in a robust recovery of catalytic activity (32). This establishes that the inability of the catalytic domain to spontaneously refold is because the propeptide is essential for folding the catalytic domain to its native state. Subsequently, the propeptide is removed by two distinct autoproteolytic cleavages, each with a different pH optimum (33, 34), which results in the maturation of the zymogen into enzymatically active subtilisin (35, 36). Additionally, subtilisin propeptides are also effective inhibitors of the cognate catalytic domains and may hence additionally function downstream of the folding process as regulators of enzymatic activity (35).
3. The Concept of Intramolecular Chaperones
Propeptide-mediated folding mechanisms have since been demonstrated to exist in various unrelated proteases, suggesting that such folding pathways may have evolved through convergent evolution (37, 38). Consequently, propeptides are termed as intramolecular chaperones (IMCs) (38) to differentiate them from molecular chaperones (MCs) (39–41). IMCs differ from MCs in a number of ways. Unlike MCs, which fold diverse substrates into thermodynamically stable states in an energy-dependent manner, IMCs are highly substrate specific and can mediate folding in an energy-independent manner. Upon completion of folding, the IMCs are proteolytically degraded, which effectively destroys part of the folding information. This makes the process irreversible by forcing the IMC to function as a as single-turnover catalyst (42). Hence IMCs appear to facilitate folding in an inefficient manner when compared with MCs, which are true multi-turnover catalysts (29).
3.1. Examples of IMC-Mediated Folding
Although initially discovered in bacterial proteases (43), subsequent work establishes the rather ubiquitous existence of IMC-dependent folding in a variety of proteins that include both proteases and non-proteases from prokaryotes, eukaryotes, archaea, and viruses. A few of the classic and emerging examples include the following:
α-Lytic protease is a chymotrypsin-like serine protease that is secreted by the gram-negative soil bacterium Lysobacter enzymogenes and serves to lyse and degrade microorganisms. α-Lytic protease is secreted with a 166-residue propeptide and a 33-residue signal sequence. The 198-residue protease belongs to the same family as the mammalian digestive serine proteases, trypsin and chymotrypsin (44). Several studies clearly establish that the propeptide functions as both a chaperone and an inhibitor of the protease and ensures its folding to an active, secretion-competent, stable conformation. Interestingly, the eukaryotic homologues trypsin and chymotrypsin that display low sequence identity, but adopt similar three-dimensional scaffolds, can fold independent of an IMC domain (45, 46) and have provided valuable insights into understanding the overall mechanism of IMC-dependent folding.
Carboxypeptidase Y (CPY) from Saccharomyces cerevisiae is a serine carboxypeptidase that is used extensively as a marker for protein transport and vacuolar sorting in yeasts. This protease, which is synthesized as a pre-pro-protease with a 91-residue propeptide, folds and cleaves its IMC in the endoplasmic reticulum (ER), resulting in an inhibited complex (47). Upon its translocation to the yeast vacuole the enzyme is activated in trans by another serine protease, Proteinase A (48, 49). Guanidine hydrochloride denatured pro-CPY can be rapidly and efficiently refolded by dilution into a suitable buffer. Under identical conditions, mature CPY fails to refold to an enzymatically active form and suggests that the propeptide is required for correct folding of the mature protein (50). Folding of mature CPY in the absence of the propeptide results in the formation of a molten globule-like intermediate state, similar to that observed in the case of α-lytic protease and SbtE (51).
Proteinase A (PrA) from S. cerevisiae is a vacuolar aspartic endo-proteinase (329 residues) that is vital for sporulation and viability during nitrogen starvation. The protease is secreted with a 54-residue propeptide that is proteolytically removed in the vacuole. Although the pro-PrA has been difficult to purify, studies suggest that the propeptide directly assists protease folding (52).
Procathepsin-L, a member of the large family of cysteine proteinases, was the first example of propeptide-assisted folding in this family of enzymes (53). Studies demonstrate that loss of protease activity is directly proportional to truncations within the propeptide domain, and that the complete cognate propeptide is required for correctly folded cathepsin-L (54). Subsequently, it was established that cathepsin-S and cathepsin-B also required the presence of their cognate propeptides for productive folding (55). Folding of cathepsin-S under varying conditions of pH, time, redox state, and ionic strength did not compensate for the loss of propeptide (54). Similar to the subtilisin family (42), studies on cathepsins demonstrate that mutations in the propeptide directly affect protease function. This is highlighted by the hereditary disease pycnodysostosis, caused by “loss-of-function” mutations in the cathepsin-K gene, one of which is directly localized in the propeptide domain (56).
Endosialidases (endoNs) are tailspike proteins of bacteriophages that bind and specifically degrade the α-2,8-linked polysialic acid (polySia) capsules of their hosts. Recently, an IMC was identified in tailspike proteins of evolutionarily distant viruses, which require a C-terminal chaperone for correct folding. The structure of catalytic domain of coliphage K1F endoN reveals a functional trimer whose folding is mediated by a C-terminal IMC domain. Release of the IMC confers kinetic stability to the folded catalytic domain of endoNF (57). The recent crystal structures of the IMC domain in its pre-cleaved and cleaved isolated forms reveal that tentacle-like protrusions enfold the polypeptide chains of the precursor protein during the folding process. Upon completion of the assembly, correctly folded β-helices trigger a serine–lysine catalytic dyad to autoproteolytically release the mature protein. Interestingly, sequence analysis shows a conservation of the intramolecular chaperones in functionally unrelated proteins sharing β-helices as a common structural motif. Such conserved chaperone domains are interchangeable between pre-proteins and release themselves after protein folding (58).
Elastase, an important virulence factor in the opportunistic pathogen Pseudomonas aeruginosa, is a thermolysin-like neutral zinc metalloprotease (TNP) that is synthesized with an amino-terminal propeptide (174 residues). Elastase was the first TNP family member that was demonstrated to require its propeptide for both folding and secretion (59). Subsequent studies on other TNPs such as thermolysin, the prototype of this family of proteases, and pro-aminopeptidase processing protease (PA protease), demonstrated that their N-terminal propeptides function as IMCs as well (60). Analysis of propeptides of TNPs demonstrates the presence of two conserved regions within the propeptide domains that may be critical for function. Mutations within the two conserved regions directly affect the chaperone function (59). Interestingly, the propeptide of vibriolysin, another TNP, has been shown to chaperone the folding of PA protease even though they share only 36% sequence identity.
-
Several growth factors and neuropeptides such as transforming growth factor-β1, activin A (61), nerve growth factor, and amphiregulin (62, 63); hormones such as insulin (64); certain glycoproteins like von Willebrand factor (65); and bacterial pancreatic trypsin inhibitor (BPTI) (66) also depend on propeptides for folding assistance. The above examples effectively highlight the wide scaffolds that fold using the assistance of IMC domains. Furthermore, apart from IMCs that directly catalyze the folding process, there exist other propeptides that can indirectly assist in folding. For example, the propeptide of barnase interacts with the molecular chaperone GroEL and thus ensures productive folding of barnase (61). Further, the propeptides in matrix metalloproteases (MMPs) contain a conserved cysteine residue where the sulfhydryl group is coordinated by the catalytic Zn2+ ion, thus maintaining these proteases in a catalytically inactive state. Proteolytic cleavage within the propeptide triggers a conformational change and releases shielding of the catalytic cleft in MMPs by interrupting the coordination between Zn2+ ions and cysteine residue (67). Thus, based on their roles in protein folding, propeptides have been grouped into two major classes, Class I and II (29, 43). Class I propeptides directly catalyze the folding, while Class II propeptides function in oligomerization, protein transport, localization, etc., and are indirectly involved in folding. The IMC of SbtE is a stereotypical Class I propeptide as it directly functions to catalyze the folding process (29).
To date, IMCs have been identified in all four major classes of proteases: serine, cysteine, aspartyl, and metalloproteases (37, 43). It is important to note that in the above proteases the IMC domains also function as potent protease inhibitors of their cognate catalytic domains and have to undergo activation to produce a catalytically active enzyme. While the bacterial proteases are mostly secreted extracellularly, the eukaryotic proteases undergo activation mostly in subcellular compartments of extreme pH (38, 68). Why do specific protease sequences require IMCs to fold to their native states? What are the structural and functional determinants that may have driven specific protein families to choose between IMC- and MC-dependent folding pathways? Do IMCs employ a common mechanism to assist the folding of these varied scaffolds? What are the functional implications of the inhibitory functions of IMCs within proteases? Answering these questions will have enormous implications for the fundamental understanding of protein folding in general and regulation of cellular proteases in particular and will also facilitate the rational design of protein-specific chaperones.
3.2. Similarities and Differences in IMCs
Concurrent studies on subtilases, α-lytic protease, and carboxypeptidase established that productive folding mediated by the IMC is a kinetically driven process (69). This conservation of function across unrelated protease families suggests that IMCs have evolved through convergent multiple parallel pathways and may share a common mechanism of action (37). Since IMC mechanisms appear to have evolved through convergent evolution, significant sequence similarity between IMC domains of non-homologous protein families would not be expected. Interestingly, even among homologous families, sequence similarity between IMC domains is significantly lower than that observed between the cognate catalytic domains (38). Nevertheless, sequence analyses of the subtilisin IMCs highlight some unique characteristics that may be critical for function. Alignment of known IMC sequences from subtilases helped identify two small hydrophobic motifs, N1 and N2, that appear to be conserved within such propeptides (70). Interestingly, when one compares the IMC sequence from subtilisin E with aqualysin, POIA1 (71), or with a designed peptide chaperone ProD which was computationally forced to diverge from the IMC of subtilisin, a high degree of sequence conservation appears isolated within motifs N1 and N2 (72). Random mutagenesis using error-prone PCR along with an activity-based genetic screening technique demonstrated that substitutions within motifs N1 and N2 were often deleterious (73, 74). NMR spectroscopy showed that while the subtilisin E IMC domain is largely unstructured, motifs N1 and N2 can display conformational rigidity (75). Together, these experiments suggest that the individual motifs may be critical for nucleating folding, while the non-conserved segments between these motifs may be responsible for functional specificity toward their cognate catalytic domains.
Even though the percentages are restricted to a few homologues from subtilisin, α-lytic protease, and cathepsin family of proteases, the trend of lower sequence conservation among IMC domains compared to their cognate catalytic domains generally holds true for other IMC-dependent systems as well (29). When one compares the percent of charges among the two domains within individual proteins, IMCs also contain more charged amino acids when compared to their respective catalytic domains (38). For example, while 12% of residues in the mature domain of subtilisin E are charged, the IMC sequence has 36% charged residues. Also, the charge on the IMC of SbtE directly complements a pocket around the substrate-binding site (76). Similar trends in asymmetric charge distribution are observed in most IMC-dependent proteins, including α-lytic protease (mature 10%; IMC contains 22%), carboxypeptidase Y (mature 20%; IMC contains 30%), and proteinase A (mature 20%; IMC contains 31%). Establishing why IMCs have evolved to be charged and if the charge on IMCs was selected with their chaperone function would give further insights into the nature of kinetic barriers on folding pathways.
Since structures evolve slower than sequences, proteins with highly divergent sequences may adopt similar structural folds. To explore this possibility, the sequence–structure relation within subtilases was analyzed (72). It is easier to reconcile the differences in the extent of sequence conservation among the IMCs and their cognate protease domains in different protein families. Since protease domains are catalytic units that facilitate similar chemical reactions, they require similar structural organization and precise spatial orientation of key residues. In this regard, the catalytic region of an enzyme represents a small portion of the entire protein. How variable IMC domains can mediate folding of structurally conserved catalytic domains is, however, more difficult to reconcile – especially since the IMCs are folding catalysts that help to attain structurally similar native states. It is possible that certain structural folds are necessary to bestow a specific function to a domain in spite of sequence variation. Since propeptides may impart structural information to their catalytic domains, propeptides within one family could adopt similar structural folds, despite digressions in their primary sequences (42).
3.3. IMCs as Potent Catalytic Inhibitors
Since the catalysis of folding requires the IMC (the catalyst) to interact with its cognate catalytic domain (the reactant), the nature of these interactions was investigated in subtilisin using isolated wild-type and mutant IMC domains (33, 73, 77). Studies indicate that the entire IMC functions as a slow-binding competitive inhibitor of subtilisin (42). In general, slow-binding inhibition is evident when initially weakly associated enzyme–substrate complexes undergo conformational changes that enhance affinity within these complexes. In case of the subtilisin, this involves the transition of the isolated IMC domain from an intrinsically unstructured state to a well-defined α–β conformation upon forming a stoichiometric complex with the catalytic domain (78). Since incorrect spatial and temporal proteolysis within a cell may be lethal, these inhibitory properties were hypothesized to be a mechanism that prevents premature protease activation. In general, cells appear to have evolved two distinct mechanisms to control activity of proteases. The first involves co-evolution of specific endogenous inhibitors, typically within compartments spatially distinct from those containing active enzymes. The second involves proteases being synthesized as inactive or less active precursors, which become activated by limited intra- or intermolecular proteolysis cleaving off a small peptide. Interestingly, these two different protease regulatory mechanisms appear combined in the chaperoning capabilities of IMC-dependent systems. Not surprisingly, the chaperoning ability and inhibitory functions of IMC domains correlate well, and IMC variants with diminished binding affinity are often weaker chaperones (73, 79, 80). However, this correlation is not always true, as the propeptide of aqualysin (a closely related thermostable homologue of subtilisin) (81) and a computationally designed synthetic peptide are potent slow-binding inhibitors (72), yet much weaker chaperones of subtilisin. An examination of other convergently evolved IMC-dependent protease systems establishes that the slow-binding inhibition function of IMC domains appears to be a common theme within proteinase A, α-lytic protease, carboxypeptidase Y, elastase, and the cathepsin family. The important role of this slow-binding inhibition and the convergence of these two distinct protease control mechanisms will both be addressed later.
4. Understanding IMC-Dependent Folding and Maturation of Bacterial Subtilisin
Over the past three decades, numerous genetic, biochemical, and structural analyses of bacterial subtilisins along with complementary data from other proteases have provided insights into the mechanism of IMC-mediated protein folding. Structural data on the bacterial subtilisins offer interesting snapshots into the gradual transition of the polypeptide from an unfolded state, through an inhibited complex, to an active protease. Complementary biophysical and biochemical studies have helped to elaborate reasons for non-productive folding of the isolated protease domains and to elucidate a general mechanism for how IMCs may function in this pathway.
4.1. The Structure of SbtE and Pro-SbtE Complex
Several high-resolution crystal structures of both SbtE and SbtE in complex with its IMC domain have been solved (76, 82, 83). The structure of SbtE (Fig. 4.1a) is comprised of three β-sheets and nine α-helices, with Asp32-His64-Ser221 forming the catalytic triad. The largest β-sheet is comprised of seven parallel β-strands and is flanked on one side by three helices and on the other by two. The substrate-binding site (Fig. 4.1b) is a surface channel that accommodates six residues (P4–P2′). In SbtE, the substrate-binding pocket is large, hydrophobic, and made of main-chain residues from Ser125-Leu126-Gly127 and main- and side-chain residues of Ala152-Ala153-Gly154 and Gly166. The Gly166 is at the bottom of the pocket for P1 and is critical for specificity. The P1–P4 substrate backbone forms the central β-strand in an anti-parallel β-sheet with the protease residues 100–102 and 125–127. Further, SbtE displays two calcium-binding sites (Fig. 4.1a), a high-affinity site that is well conserved (A-site) in most subtilases and a weak affinity site that is less conserved (B-site) (13). Calcium at A-site is coordinated in pentagonal-bipyramidal geometry by the loop comprised of residues 75–83, Gln2 at the N-terminus, and an Asp at position 41. The seven coordination distances range from 2.3 to 2.6 Å with the Asp being the closest (Fig. 4.1c). The second calcium makes contact with the main-chain carbonyl oxygen atoms of residues 169, 171, and 174 in a shallow crevice near the surface of the molecule and is coordinated in a distorted pentagonal bipyramid (84). These two calcium-binding sites together make SbtE an extremely stable protease in the absence of any cysteines or stabilizing disulfides in its structure (85). The lack of cysteine residues in SbtE is advantageous because it allows the probing of specific interactions during the folding process. This approach has been used to identify a precise non-native interaction by engineering two cysteine residues, which are distal in the native protein but are proximal during folding, and form a specific intramolecular molecular disulfide bond under oxidative folding conditions (86).
Fig. 4.1.
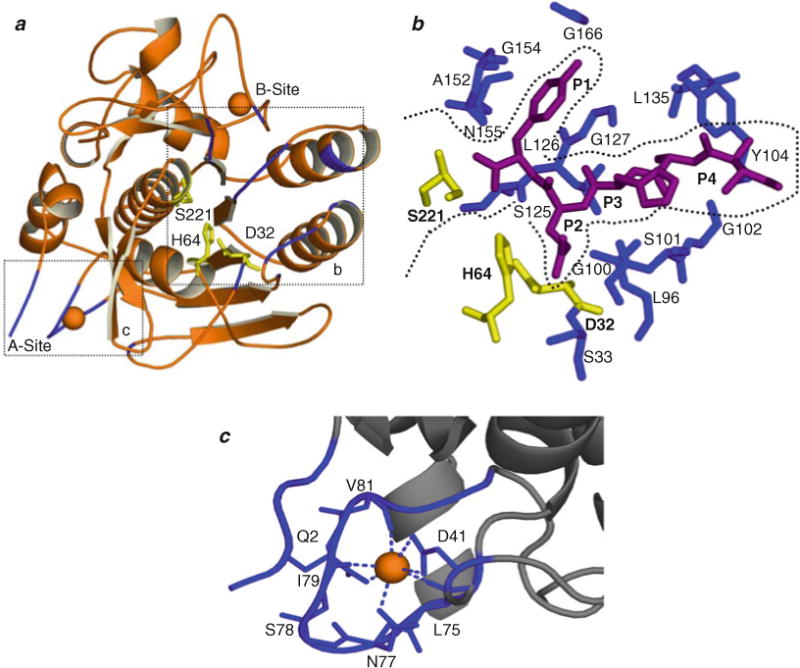
Structural organization of SbtE. (a) Structure of SbtE (ISCJ) is depicted together with the substrate-binding site (b) and the calcium (orange)-binding A-site (c). SbtE also has a second calcium-binding site (B-site) of medium affinity. The catalytic residues are highlighted in yellow. (b) The substrate-binding site is highlighted with an inhibitor (magenta) bound in the S1–S4 pocket. Residues lining the substrate-binding pocket are highlighted in blue. (c) Calcium binding at the A-site is coordinated by residues from a loop comprised of residues 75–83, an N-terminal Gln, and an Asp.
Engineering a S221C substitution at the active site of Pro-SbtE blocks the maturation process subsequent to the first proteolytic cleavage of the IMC. This variant facilitates the isolation of stable, cleaved stoichiometric Pro:S221C–SbtE complex (77, 87), whose X-ray structure (Fig. 4.2a) has been solved (76, 82, 88). The structure of the mature domain in the complex is superimposable with the structure of the isolated protease domain with a root mean square deviation of 0.46 Å, when the Cα atoms were compared. While the isolated IMC is largely unstructured, the IMC in complex with the protease folds into a single domain with a four-stranded anti-parallel β-sheet and two three-turn helices, forming an α+β plait. The structured inhibitory IMC packs against two surface helices (α1 and α2; Fig. 4.2a) of the protease domain formed by residues Tyr104-Asn117 and Ser132-Ser145. Further, residues −1 to −7 from the IMC domain directly interact with the substrate-binding region to complete a three-stranded β-sheet with β-strands from the protease domain. In all, there are 27 hydrogen bonds (Fig. 4.2a) at the IMC–subtilisin interface. Interestingly, 24 hydrogen bonds stabilize the interaction of residues −1 to −9 (Note: the nine C-terminal residues of the subtilisin IMC; the cleavage site locates between residues −1 and 1) with the active site and with the substrate-binding regions. These include the three hydrogen bonds that stabilize the backbone amide groups of Glu(−9) and Asp(−7) that form helix caps for the two SbtE interaction helices (α1 and α2). In contrast, the remaining 68 residues of the propeptide are stabilized by only three hydrogen bonds, between the backbone amides of residues −34 to −36 and the carboxylate group of the Glu112 from the protease domain. The significance of this asymmetric distribution of hydrogen bonds at the IMC–protease interface is unknown. Another noteworthy point is that while the average B-factor of the side-chain and the main-chain atoms is 15.0 and 13.6 Å2 in the protease, it is 35.5 and 33.8 Å2, respectively, in the IMC (76). This approximately twofold increase in the B-factors within the IMC domain may be biologically significant and necessary for function.
Fig. 4.2.
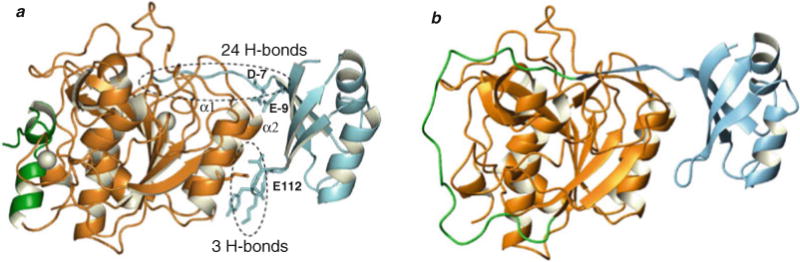
Structural characterization of Pro-SbtE complex. (a) Crystal structure of the autoprocessed, inhibited complex (1SCJ). The IMC (blue) docks against two helices (α1 and α2) in the protease (orange) and occludes the substrate-binding site. The binding interface is stabilized by 27 hydrogen bonds in a asymmetric distribution, of which 3 are contributed by E112 in protease. D–7 and E–9 form helix caps for the two helices (α1 and α2). The calcium ions (white) and the N-terminal helix (green) are also highlighted. (b) Modeled structure of the propeptide–protease complex prior to cleavage. The N-terminus (green) of the protease (orange) is bound to the IMC (blue) at the active site and hence the calcium-binding A-site is not fully formed.
An interesting insight from the structure of the Pro-S221 C-SbtE is that the N-terminus of the protease is more than 20 Å away from the active site. Earlier biochemical studies had clearly established that the processing of IMC was intramolecular (89). Based on this, the structure of Pro-SbtE (Fig. 4.2b) just prior to cleavage was modeled with the N-terminus of the protease bound to the C-terminus of the IMC at the active site. The lack of a preferred conformation in N-terminal residues (residues 1−6) of SbtE and the recovery of activity in a H64A-SbtE active-site mutant with a E(−2)H substitution in IMC, through substrate-assisted catalysis, substantiated the model (87). A recent crystal structure of an active-site mutant of a subtilisin homologue, prokumamolysin, shows the propeptide bound to the protease prior to cleavage and confirms this model proposed for the uncleaved precursor (90).
4.2. Mechanism of IMC-Mediated Structural Acquisition
4.2.1. How Do Polypeptides Enhance the Rates of Folding of Their Cognate Catalytic Domains?
4.2.1.1. Kinetic and Thermodynamic Characterization
To establish how IMCs function in the folding pathway, folding of α-lytic protease and SbtBPN was compared in the presence and absence of their IMC (45, 91). Folding of SbtBPN in the absence of its IMC resulted in formation of a non-functional, structured state that is stable for weeks. This intermediate displayed a hydrodynamic volume intermediate between that of the fully folded and fully unfolded protease. Circular dichroism spectra of the intermediate in the far-UV region (190–250 nm) corresponded to a well-defined secondary structure with a minimum at 208 and 222 nm. However, in the near-UV region (250–320 nm) the intermediate displayed no amplitude, suggesting a lack of well-formed tertiary packing. This was also confirmed through an NMR study that established a strongly reduced dispersion in the amide and methyl regions of the 1H NMR spectrum compared with the fully folded protease. A noteworthy point, however, was that the intermediate appeared to bind calcium with a stoichiometry of 1, but with an affinity intermediate to affinities of A-site and B-site (91). Similar behavior was also observed with the intermediate state of α-lytic protease folded in the absence of its IMC (68). Although the intermediate state was extremely stable for weeks, addition of cognate IMCs yielded active native protease.
Hence, these studies suggested that in the absence of IMC, the protease folds to a kinetically trapped state with properties of a classical “molten globule” intermediate (92). A high-energy barrier between the molten globule intermediate and transition state limits the folding to a native state (Fig. 4.3). Addition of the IMC lowers this barrier to enable productive folding (45, 91, 93, 94). Based on these observations, it was established that the IMC functions to overcome a kinetic barrier on the folding pathway, and that the observed intermediate is either on-pathway or in equilibrium with a conformation on the folding pathway.
Fig. 4.3.
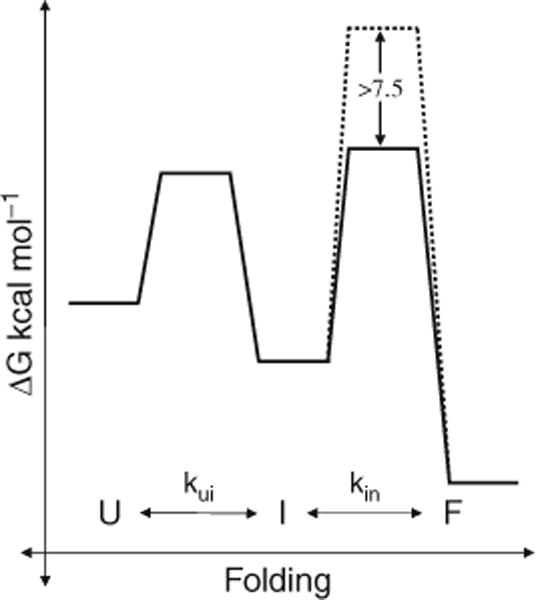
IMCs lower kinetic barriers on the folding pathway. (a) Unfolded Pro-S221 C-SbtBPN (U) folds to the native state (F) through a stable molten globule intermediate (I). Kinetic studies establish that the high activation energy barrier to protease folding (broken line) is lowered by >7.5 kcal/mol in the presence of the IMC (solid black line).
To establish the relevance of the observed intermediate to the biological in-cis folding, refolding of full-length Pro-SbtE and Pro-SbtBPN was analyzed (93, 95). To avoid complications of proteolysis and to inhibit IMC processing, both studies were done using a Ser221Ala active-site variant that has lower proteolytic activity by six orders of magnitude. This variant represents the propeptide:protease complex just prior to cleavage as discussed earlier (Fig. 4.2b). The folded, but uncleaved, Pro-S221A-SbtBPN binds calcium ions and adopts a compact conformation with an apparent molecular weight of 36 kDa. Equilibrium unfolding of fully folded Pro-S221A-SbtBPN monitored through changes in circular dichroism, and fluorescence spectroscopy, followed a three-state unfolding curve. Most of the tertiary and a part of the secondary structure unfolded through an initial cooperative process while separated secondary structures followed a less cooperative second transition. Interestingly, the second unfolding transition was similar to the unfolding transition of the molten globule intermediate formed in the absence of IMC. Additionally, at higher denaturant concentrations Pro-S221A-SbtBPN displayed properties similar to the trapped molten globule intermediate. These results, taken together, established that the equilibrium unfolding of the Pro-S221A-SbtBPN occurs via an intermediate that is similar to the kinetically trapped molten globule intermediate. Further, three-state equilibrium-unfolding transition suggested that the polypeptide folds to a molten globule-like state and then transitions to the native state, through the assistance of the IMC. Thus it was argued that the IMC functions only during the late stages of the folding pathway. Another interesting finding was that the relative thermodynamic stability of the fully folded Pro-SbtBPN complex was only marginally higher than that of the SbtBPN intermediate (93, 95). The thermodynamic stability was also strongly dependent on salt concentrations, which was probably an effect of the highly charged propeptide and its effect on the electrostatic interactions of the solvent environment (37).
Most of the initial folding studies highlighted above employed slow dialysis for refolding and hence were not amenable to kinetic analysis (31). Subsequent optimization of a fast refolding that involves the rapid dilution of unfolded protein into denaturant free buffer paved the way for kinetic studies of IMC-mediated folding. Using the technique of refolding by rapid dilution, Eder and Fersht (93) established the kinetics of refolding of Pro-S221A-SbtBPN by monitoring the increase in intrinsic fluorescence. Pro-S221A-SbtBPN follows two-state kinetics with a rapid phase (65% amplitude as native) and slow phase (35% amplitude as native). While the rapid phase reflects the acquisition of structure in the intermediate, the slow phase established the kinetics of folding of the intermediate in the presence of the IMC. This slow phase followed a rate constant of 0.0047 s−1. Initial studies had established a folding rate of <1.4×10−8 s−1 for SbtBPN refolded in the absence of IMC. Hence, taken together, this demonstrated that the IMC accelerates the kinetics of protease folding by at least five orders of magnitude. Based on equation [1],
| [1] |
the difference in transition state energies for folding with and without the IMC was estimated to be >7.5 kcal/mol (Fig. 4.3) (37). Similar studies with α-lytic protease established that the propeptide lowers the kinetic barrier to folding by 18 kcal/mol. With α-lytic protease, the relative thermodynamic stability of the native state relative to the unfolded and intermediate states was also established. Interestingly, these studies demonstrate that the native state is ~1 kcal/mol less stable than the unfolded state (45, 95). Hydrogen-exchange experiments establish that this state has extremely low conformational dynamics with >50% of the residues having a protection factor (Pf) >104 (46). Thus the protease appears to be in a kinetically trapped, thermodynamically unstable native state.
4.2.1.2. Calcium Deletion Variant
Both SbtE and SbtBPN have two well-defined calcium-binding sites, a high-affinity A-site, and a medium-affinity B-site (Fig. 4.1a). Studies establish that calcium binds to the A-site (Fig. 4.1c) with an affinity of ~107 M−1 and contributes significantly to the thermodynamic stability of the protease (84). What is particularly interesting is that the kinetic barrier to calcium dissociation is extremely high (~23 kcal/mol) and is higher than the binding free energy (96). This suggests that the binding of calcium and the accompanied lowering of conformational entropy might limit IMC-independent folding. To establish this, Bryan and coworkers created a variant (SbtBPNΔcal) that had the calcium-binding loop (A-site: 75–83) removed. Upon refolding of SbtBPNΔcal under low ionic conditions, no activity was observed. However, folding of SbtBPNΔcal under high ionic conditions resulted in independent folding of SbtBPNΔcal to an active, native state (94). Folded SbtBPNΔcal is unstable and further mutations, including a disulfide bond, have to be introduced to enhance stability. However, the new variant has a structure similar to the wild-type protease except in the region of the A-site (97). This suggests that the calcium-binding A-site may be a critical factor that dictates the requirement for an IMC. However, it is interesting to note that productive folding is seen only under conditions of high ionic strength and that the calcium-independent protease variant is extremely unstable.
Thus, a high-energy barrier that separates the unfolded and native states limits the spontaneous folding of specific proteases. IMCs assist in the folding pathway by lowering these barriers to enable productive folding. Selection of such kinetic barriers on the folding pathway may provide a mechanism for evolution of optimal functional properties. Since most IMC-dependent proteases appear to function in harsh protease-rich conditions the presence of the IMC may enhance longevity through high unfolding energy barriers.
4.2.2. How Do IMCs Assist in Folding?
4.2.2.1. Stabilization of Folding Nucleus – “Side-On Model”
The 77-residue IMC domain is an intrinsically unstructured polypeptide that folds to an α–β conformation in the presence of the protease (75). Crystal structure of the inhibited Pro-S221 C-SbtE (Fig. 4.2a) demonstrated that the folded IMC interacts directly with two surface helices of the protease (76). Based on this, it was proposed that the α–β–α binding interface of the IMC may represent the folding nucleation motif of the protease, and that stabilization of this sub-structure upon binding of the IMC may help to induce folding (82). To establish the nature of the interaction between the IMC and protease, Bryan et al. analyzed the bimolecular folding of SbtBPNΔcal as given by equation [2],
| [2] |
where ProU and represent the unfolded propeptide and protease, respectively, represents the partially structured intermediate, represents the folded complex, ProD represents the degraded propeptide, and represents the free active protease. Reaction rates were determined using the differences in tryptophan content between the IMC and protease domains. SbtBPNΔcal, with three tryptophan residues in its primary sequence, shows a 1.7-fold increase in intrinsic tryptophan fluorescence upon refolding. As the IMC has no tryptophan residues, any change in fluorescence reflects structural changes in the protease domain. Further, the binding of IMC to the protease increases the intrinsic fluorescence of the protease due to shielding of one of the tryptophan residues. By monitoring the rate of change in the intrinsic tryptophan fluorescence, the kon and koff for binding of IMC to SbtBPNΔcal and the rates of SbtBPNΔcal folding under increasing concentrations of the IMC were determined. These studies demonstrated that the formation of the initial complex ( ) between IMC and SbtBPNΔcal was the rate-limiting step to folding. However, upon increasing residual structure in the isolated IMC domain the folding of to becomes rate limiting (79, 98). This established that structural content within the IMC might have a direct effect on its chaperoning function. Simultaneous studies that showed a direct correlation between inhibition constants of isolated IMC mutants and their chaperoning efficiency strengthened this conclusion (73, 99). Hence, it was proposed that the binding energy of the IMC contributes to stabilizing the α–β–α structure either by surmounting an entropic barrier through stabilization of native interactions or by overcoming an enthalpic barrier by breaking non-native interactions. While in the presence of an unstructured IMC, the binding energy may be diluted by its folding; the presence of a structured IMC ensures faster binding and folding.
Although this nucleation propagation mechanism of folding through a “side-on interaction” of the IMC with the protease seems possible, other studies, highlighted below, question this hypothesis. Moreover, the naturally occurring biological reaction is clearly unimolecular, and evolution of covalently linked protease domains may be to enhance efficiency and economy of IMC-mediated folding. Hence, while the bimolecular folding studies offer initial insights into propeptide-mediated folding, establishing similar principles in unimolecular folding is fundamental to the above hypothesis.
4.2.2.2. Stabilization of Folding Nucleus – “Top-On Model”
Studies based on unimolecular folding highlight a similar, but slightly varied, mechanism. Random mutagenesis helped identify a number of mutations in the IMC that affected the secretion of active protease. Second-site suppressor analysis for an M(−60)T mutation in the IMC identified a S188L substitution in SbtE that restored activity. Since Ser188 and Met−60 do not interact with each other and are ~47 Å apart in the folded Pro-SbtE complex, it was suggested that these residues may interact during the folding process. One possible way in which these two residues could interact is via a “top-on interaction” (Fig. 4.4). To test this hypothesis, cysteine residues were introduced at positions −60 and 188 and the folding of this double-cysteine variant (M(−60)C-Pro-S188C-SbtE) was analyzed under oxidizing and reducing conditions. Interestingly, folding under oxidizing conditions results in the formation of a cross-linked intermediate with stable secondary structure. A noteworthy point is that upon prolonged incubation with small peptide substrates such as AAPL-pNA and AAPF-pNA, the trapped intermediate displays catalytic activity. Addition of a reducing agent to the cross-linked intermediate triggers proteolysis of the IMC and results in a wild type-like native state. Hence, isolation of the stable cross-linked intermediate suggests that the IMC interacts with the protease in a “top-on orientation” during folding and, further, that this interaction results in productive folding (86).
Fig. 4.4.
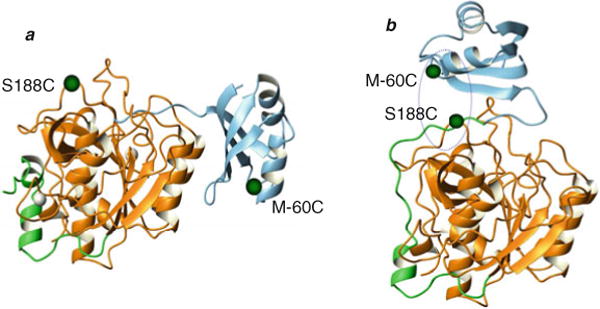
IMC interacts in a “top-on” orientation. (a) M–60 in the IMC (blue) and S188 in the protease (orange), identified through second-site suppressor analysis, are 47 Å apart in the inhibited complex (1SCJ). (b) Cross-linking studies with a double-cysteine mutant helped isolate a stable cross-linked intermediate, based on which the “top-on” interaction between the propeptide (blue) and protease (orange) was modeled.
Furthermore, the “side-on” interaction of the IMC with the protease is stabilized largely by three hydrogen bonds between E112 in the protease and IMC backbone amides (Fig. 4.2a). Disruption of these hydrogen bonds in the E112A-SbtE variant does not affect IMC-mediated folding of the protease. However, the Ki of the IMC to E112A-SbtE is lowered ~35-fold relative to that of SbtE. This suggests that the “side-on” interaction of the IMC is critical for inhibition and, further, that the inhibitory and chaperone functions of the IMC may not be obligatorily linked (100). Additional evidence for the above comes from the fact that IMC of aqualysin is a 10-fold better inhibitor of SbtE, relative to its own IMC, but is unable to efficiently chaperone the folding of SbtE (81).
The above studies demonstrate that in unimolecular folding the “top-on” interaction may initiate folding while the “side-on” interaction is critical for inhibition. Thus the IMC may interact with the protease closer to the active-site region before it transits to the “site-on interaction” seen in the crystal structure. This movement of the IMC may be coincident with its cleavage by the protease.
4.2.2.3. Changes Coincident with IMC Cleavage
Subsequent to folding of the polypeptide to a structured state, the peptide bond between the IMC and protease domains is autoproteolyzed (89). However, the IMC remains bound to the protease as an inhibited complex. The solved crystal structure of the Pro-S221C-SbtE (Fig. 4.2a) inhibited complex offers a structural snapshot of the polypeptide folding at the completion of autoprocessing (76).
To establish changes coincident with autoprocessing of the IMC, structural properties of the complex before and after autoprocessing were characterized (77) using ANS, a fluorescent dye that binds to exposed hydrophobic surfaces on structured proteins (101). ANS displayed a higher intensity and a shift toward a lower wavelength in the presence of the unautoprocessed complex (Pro-S221A-SbtE) relative to processed complex (Pro-S221 C-SbtE). This suggested that there is a large decrease in exposed hydrophobic surface coincident with autoprocessing. Further, in the crystal structure of Pro-S221C-SbtE, the N-terminus of the protease is at least 20 Å away from the active site. Based on this it was proposed that upon autoprocessing, the N-terminus of the protease folds back to form the N-terminal α-helix that contributes Gln2 to calcium binding at the A-site (Fig. 4.2) (87). Recent studies establish that while the rates of folding and autoprocessing are independent of calcium, the stability of the autoprocessed complex and mature subtilisin shows a strong dependence on calcium. Further, the autoprocessed complex has much higher thermal, thermodynamic, and proteolytic stability relative to the unautoprocessed complex (85). This demonstrated that the A-site is indeed formed subsequent to cleavage, and that the IMC regulates its formation. The processing of the IMC triggers calcium binding and induces structural changes in the protease that serve to lock the protease in a more stable conformation. The release and degradation of the IMC from the stable inhibited complex are required for release of active protease (34). While this appears to be mediated through an autocatalytic activation, the precise mechanism of activation is unknown.
4.2.2.4. Alternative Approaches to SbtE Folding
While the above offer direct analyses of IMC-mediated folding, an alternative approach is to identify specific conditions that may allow folding of proteases in the absence of their IMCs. Studies demonstrated that folding low concentrations of the protease in 2M potassium acetate at pH 6.5, or refolding the protease from an acid-denatured state, allowed recovery of IMC-independent activity (102, 103). While the yield of active protease was initially low with potassium acetate, immobilization of the protease on a resin increased the folding efficiency (102). These experiments elucidate an interesting effect of electrostatics on the IMC-mediated folding process. In the case of the acid-denatured proteins, CD studies of the denatured protein established residual structure in the protease, even after denaturation. This further substantiates the hypothesis that stabilization of a folding nucleus may enable productive folding. Hence, the IMC appears to initiate folding by stabilizing a sub-structure within the protease. This may involve the interaction of the IMC with the protease in a “top-on” orientation (Fig. 4.4) (86). Upon acquisition of structure within the protease and formation of the catalytic triad, the IMC is cleaved in an autoprocessing reaction. This is coincident with the movement of the IMC to the “side-on” inhibitory orientation and the formation of a calcium-binding site (Fig. 4.2) (104). Release and degradation of the IMC from this inhibited complex releases an active native protease (Fig. 4.5).
Fig. 4.5.
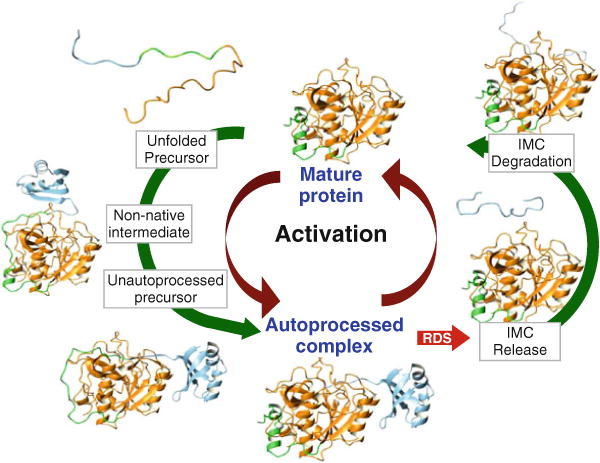
IMC-mediated SbtE maturation – N-terminal helix of protease (green); calcium ion (white). Maturation of Pro-SbtE occurs in three stages: (1) A non-native top-on interaction between the protease (orange) and IMC (blue) punctuates the transition of the precursor from the unfolded state to a structured state (unautoprocessed precursor). (2) Once the active site is formed, the precursor autoproteolyzes to an inhibited, autoprocessed Pro:SbtE complex. (3) The release and degradation of the IMC from the complex release active protease that can subsequently trans-activate other proteases. Activation is the rate-limiting step to maturation.
4.2.3. Why Have Specific Proteins Evolved Dedicated IMCs to Mediate Their Folding?
Since IMCs function as single turnover catalysts, IMC-mediated folding pathways appear to be less efficient that MC-dependent multi-turnover pathways. What is the functional advantage offered by IMC-dependent folding mechanisms? One possibility is that the specific fold of the catalytic domain of subtilases mandates the requirement of IMC-dependent pathways. IMCs may hence serve as “bridges” to reach a specific conformational state. Since the principle of microscopic reversibility requires the mechanisms in the forward direction and reverse direction to be identical, and because protein folding is a reversible reaction, the presence of the folding catalyst – the covalently attached IMC – can also mediate unfolding under specific conditions. However, the proteolytic degradation of the IMC domain prevents microscopic reversibility once the native state is reached because the “bridge” between the unfolded and native state is destroyed, rendering the reverse pathway inaccessible. Such locked-in conformations may represent “high-energy,” kinetically trapped native states that acquire structural information through their cognate IMCs. These are unlike the conventional thermodynamically stable, low-energy conformations found ubiquitously distributed in nature’s conformational space. Since MCs do not impart structural information and only provide an appropriate environment for folding thermodynamically stable proteins, the kinetically trapped conformations may be inaccessible using MCs, hence mandating IMC-dependent folding pathways. Since the subtilase super-family contains several divergent IMC-dependent proteins, a detailed analysis of the sequence, structure, and function has the potential to provide insights into determinants that dictate various functions of the IMC domains.
The Nobel Laureate Christian B. Anfinsen established that all the information ribonuclease requires to fold into its native state resides within its amino acid sequence and that the native state of ribonuclease is independent of its folding pathway (105). Subsequently, experiments from several different laboratories directly supported the thermodynamic hypothesis by demonstrating that the folding/unfolding reactions of various small proteins are generally reversible and hold that the native conformations of proteins are at global free energy minima relative to all other states having identical bond chemistry. However, it is noteworthy that these evidences only argue that the native state is the lowest energy state within a conformational neighborhood which includes all kinetically accessible states (106). Hence the thermodynamic hypothesis is not falsifiable by experiment because conformations outside of this neighborhood cannot be accessed experimentally under normal conditions (69). However, if the final state of a system depends on the initial conditions, then the process is kinetically determined (Fig. 4.6). Evidence for the kinetic hypothesis emerged from conformational studies on plasminogen activator inhibitor 1 (PAI-1), a protease suicide inhibitor that is a member of the serpin family. Upon synthesis in vivo or after refolding following in vitro denaturation, PAI-1 initially adopts a kinetically stable state that functions as a potent protease inhibitor. Remarkably, this active form slowly converts to an inactive but thermodynamically more stable, latent form over a period of several hours (107). The latent form of PAI-1 can, however, be converted back to the active inhibitory form by denaturation and renaturation. The crystal structures of the various conformers explain the structural basis of kinetic stability of the active form and the thermodynamic stability of the latent inactive form (108, 109). Hence the inhibitory form is the lowest energy state accessible during folding, but since it slowly converts to the latent form, it is clearly not the lowest energy state. Another compelling example of kinetic stability is evident from the studies of IMC-mediated and hence pathway-dependent folding of subtilisin (33, 42) and α-lytic protease (46), two evolutionarily unrelated prokaryotic serine proteases.
Fig. 4.6.
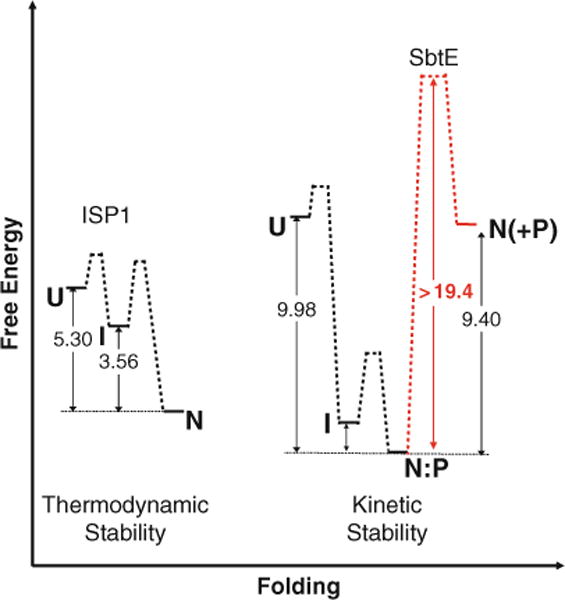
The free energy diagram depicting thermodynamic stability of ISPs and kinetic stability of ESPs – Unfolded ISP1 (U) spontaneously adopts its thermodynamically stable native state (N) through a partially folded intermediate (I). The free energy difference between N and U is approximately 5.3 kcal/mol in the case of ISP1 and is lowest compared to all experimentally observed states (111). In case of ESPs, the unfolded IMC-subtilisin (U) undergoes rapid folding and autoproteolysis to give a thermodynamically stable IMC:SbtE complex (N:P) through an intermediate state (I) (77). The structure of the IMC:SbtE complex has been solved using X-ray crystallography (76). The activation energy for the spontaneous release of the IMC from this complex is energetically unfavorable (approximately 21 kcal/mol and is shown in a broken light line). The release of the first free protease molecule is a stochastic process (110) and the subsequent steps occur by trans-proteolysis. Once folded, the high activation energy barrier kinetically traps folded SbtE in its native state. The free energy difference between the N:P and N is approximately 9.4 kcal/mol of energy. The net free energy difference between the unfolded and folded SbtE is ~0.4 kcal/mol and is likely to preclude spontaneous folding in the absence of the IMC.
4.2.4. Kinetics of IMC-Mediated Maturation of Subtilisin
Based on structural and biochemical studies, the overall IMC-mediated maturation pathway (Fig. 4.5) can be described in terms of three distinct stages:
Folding of the polypeptide to a structured state (77).
Autoprocessing of the peptide bond between the IMC and protease domains resulting in a non-covalently associated IMC protease-inhibited complex (87).
Release and degradation of IMC from the inhibition complex that results in an active protease (34, 100, 110).
As discussed above, most of the early studies focused on the kinetics of folding. Utilizing the optimization of folding by rapid dilution, and the isolation of folding mutants, Yabuta et al. carried out a thorough characterization of the kinetics of all the substages of the maturation pathway (34). These studies established that while in vitro folding and autoprocessing are rapid and reach completion in 30 min, the activation of the protease is not seen until ~240 min. Hence, activation and not protein folding is rate limiting to IMC-mediated maturation (34). As evident from the crystal structure, the protease is fully folded as an inhibited complex (Fig. 4.2a). However it appears that the release of the IMC from this complex is extremely slow. Establishing the reasons for this slow release and the mechanism of release would give further insights into IMC-mediated maturation. Further, during the maturation process, the IMC switches from a chaperone to an inhibitor and eventually to a proteolytic substrate (110). Establishing the energetics of each of these stages will give further insights into the high-energy kinetic barrier and how the IMC functions to modulate this barrier.
4.2.5. IMC-Regulated Activation of Subtilisin
Based on the current literature, a mechanistic model for precursor activation of bacterial subtilisin has been proposed (Fig. 4.5). The precursor of pro-subtilisin, which is secreted extracellularly into the media, produces mature subtilisin through a process that involves folding, followed by autoprocessing, and degradation, which represent two distinct steps of proteolysis (34, 100). As discussed earlier, folding of pro-subtilisin requires the presence of the IMC domain and is a rapid process that occurs through a partially structured non-native folding intermediate (86). The nonnative intermediate then undergoes structural changes to give a native-like folded, but unautoprocessed precursor. The orientation of the IMC with subtilisin is critical for the autoprocessing reaction, which is also a fairly rapid process (100). Once autoprocessed, the IMC–subtilisin complex appears to be in a remarkably stable state due to the inhibitory function of the IMC (110). Release of the IMC is the rate-determining step (RDS) of the maturation reaction. Once a free protease molecule is formed, it can bind to the IMC domain in the autoprocessed complex and facilitate trans degradation. It is important for the IMC–subtilisin complex to interact with mature subtilisin, because this allows a rapid exponential activation. Moreover, the rate constants of the various steps of the maturation appear to be optimized for maximum yield of the protease domain (110). Consistent with this hypothesis, IMC variants that decrease the IMC affinity for their cognate catalytic domains are inadvertently less efficient chaperones. However, high affinity is alone not sufficient for efficient folding because despite displaying tighter affinity for subtilisin, the aqualysin IMC is a weaker chaperone compared to SubtE IMC (81). Nonetheless, the data suggest that folding and autoprocessing should be completed before the release of the first active subtilisin molecule. Hence, the IMC is essential for folding of the protease domain and the inhibitory function is required for the maximum efficiency of this process and serves to regulate protease activation.
The RDS of subtilisin activation was demonstrated to be stochastic in vitro (110). The energetics of activation establishes that the release of the tightly associated IMC domain from the catalytic domain is energetically unfavorable (Fig. 4.6) and is the primary cause of the associated stochastic behavior. Detailed analysis of stochastic activation shows that modulating the structure of the IMC through external solvent conditions can vary both the time and randomness of protease activation. This behavior of the protease correlates with the release-rebinding equilibrium of IMC and suggests that a delicate balance underlies IMC structure, release, and protease activation. Proteases are ubiquitous enzymes crucial for fundamental cellular processes and require deterministic activation mechanisms. The activation of subtilisin establishes that through selection of an intrinsically unstructured IMC domain, nature appears to have selected for a viable deterministic handle that controls a fundamentally random event and outlines an important mechanism for regulation of protease activation.
5. Understanding IMC-Independent Folding and Maturation of Subtilisin
A comprehensive search of the SWISSPROT database has identified two distinct subfamilies, intracellular serine proteases (ISPs) and extracellular serine proteases (ESPs), within the subtilase super-family (111). While ESPs and ISPs display a high level of sequence, structure, and functional conservation, the latter lack the classical subtilisin propeptide signature, which is essential for correct folding of ESPs (Fig. 4.7). Subsequent studies established that although determinants such as topology, contact order, and hydrophobicity that drive protein folding reactions are conserved, ESPs absolutely require the propeptide to fold into a kinetically trapped conformer. However, ISPs fold to a thermodynamically stable state more than 1 million times faster independent of an IMC (110). Moreover, the spectroscopic studies established that ISPs and ESPs fold into their native states through different intermediate states (Fig. 4.6). An evolutionary analysis of folding constraints in subtilases suggests that observed differences in folding pathways appear to be mediated through positive selection of specific residues that map mostly onto the protein surface. Together, these results suggest that closely related subtilases can fold through distinct pathways and mechanisms, and that fine sequence details can dictate the choice between IMC-dependent kinetic stability and IMC-independent thermodynamic stability (111).
Fig. 4.7.
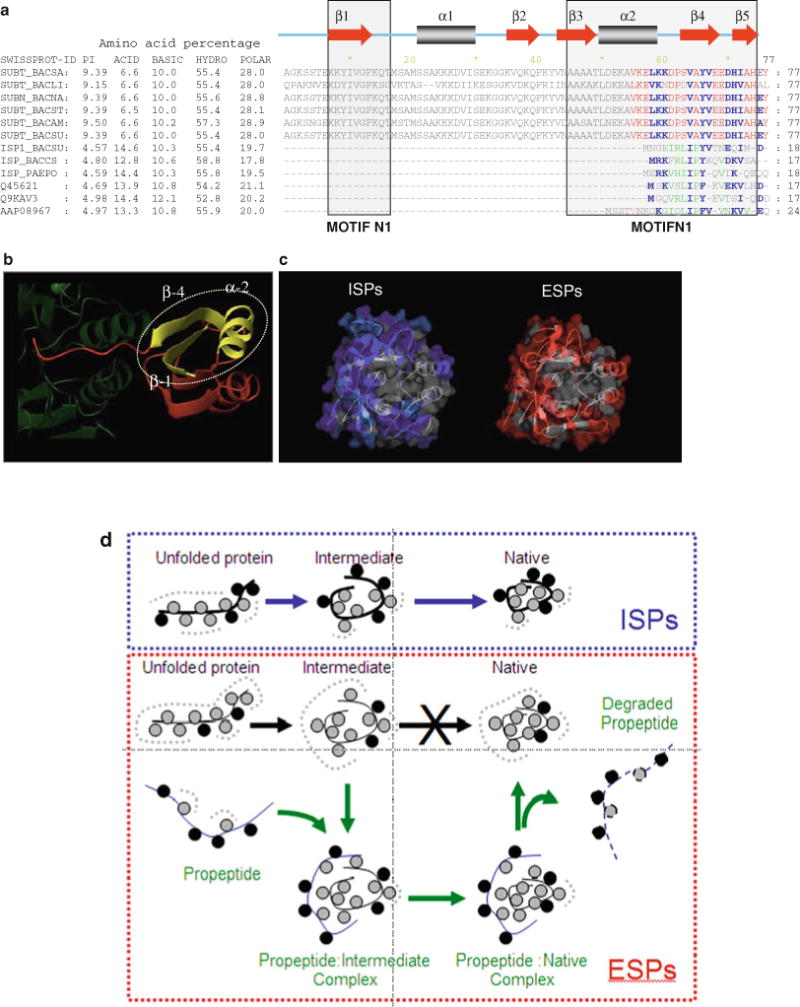
Comparison between ESPs and ISPs. (a) The first six Swiss-Prot accession numbers shown on the left side of the alignment are ESPs while the rest are ISPs. The amino acid composition represents the total percent across the entire protein and suggests that the total number of basic and hydrophobic residues is similar within ESPs and ISPs. However, ISPs have more acid residues when compared with ESPs. The ESPs have most of the basic residues located within the IMC domain while ISPs have the acid residues distributed within the catalytic domain. Motifs N1 and N2 that are conserved within ESPs are boxed and represent putative nucleation sites that initiate folding (38). These regions display conformational rigidity as seen using NMR spectroscopy (75) and constitute a sub-domain within the 3D structure. The secondary structure elements displayed above the alignment are based on the X-ray structure of SubtE (76). Residues that are conserved within ESPs (red typeface) and ISPs (green typeface) are colored separately while those that are conserved in both are indicated in blue typeface. (b) The folding nucleation sites (yellow) constituted by motifs N1 and N2 are superimposed on the structure of the IMC domain of SbtE (red). The C-terminus of the IMC domain interacts with the catalytic domain of SbtE (green) to inhibit catalytic activity. (c) Sequence conservation mapped onto structures. Residues conserved within both ESPs and ISPs are colored gray, while red or blue denotes residues that are conserved within only ESPs or ISPs, respectively. Residues that are conserved within ESPs, but without selection constraints in ISPs, are colored cyan. (d) Model for how changes in surface residues compensate for the loss of IMCs within ISPs. Hydrophobic residues (gray circles) initiate folding through a hydrophobic collapse. When hydrophobic surfaces are solvent exposed, the water tends to form a clathrate cage (broken gray lines). Both ESPs and ISPs are highly charged proteins but with extremely different isoelectric points. This charge (black circles) is localized within IMCs of ESPs, while it is distributed over the surface of the protease domain of ISPs. As a consequence, protease domains of ISPs are more polar than ESPs. This difference may help to enhance the conformational entropy of water around the folding polypeptide and assist the hydrophobic core in driving spontaneous folding of ISPs. When folding of ESPs is carried out in the absence of the propeptide, the protease domain adopts a partially structured molten globule intermediate with solvent-exposed hydrophobic surfaces (77). The partially formed hydrophobic core within the protease domain of ESPs may be insufficient to drive folding without the solvation assistance bestowed by the surface charge. This leads to stabilization of the molten globule intermediate that is prone to aggregation. This concept is consistent with findings that charge per se, and not its polarity, is critical for folding of subtilases (72, 81).
Another important difference between ESPs and ISPs is their distribution of charged amino acids. The IMC domains of ESPs contain several basic amino acid residues that make the net isoelectric point of their precursors extremely alkaline (>9.0 pH). ISPs on the other hand have very small transient N-terminal extensions (Fig. 4.7a) and lack the motifs N1 and N2, which are signatures of IMC-dependent subtilases (Fig. 4.7b). In addition, the net isoelectric point of ISP precursors is highly acidic (<5.0 pH). This is because, despite similar net hydrophobicity, the protease domains of ISPs contain a significant percent of acid residues when compared with the relatively neutral ESPs. These residues appear to have evolved through positive selection and are distributed over the enzymes’ molecular surface (Fig. 4.7c). As a consequence, ESPs and ISPs differ significantly in the properties of their solvent-accessible surfaces (111). This change in surface properties likely alters the folding pathways of the two homologues with ISPs becoming IMC independent while ESPs remain IMC dependent. Figure 4.7d depicts a model for how surface charges may contribute to rapid folding.
It is noteworthy that IMC-independent folding of subtilisin has also been achieved through extensive mutagenesis around the Ca2+-binding site in the catalytic domain or through the use of high concentrations of organic acids. While such studies have provided valuable insights into the folding mechanisms of subtilisin (79, 88, 94, 99, 112), they provide little information regarding the evolutionary pressures that dictate the choice of specific folding pathways. It is striking that although ESPs and ISPs display roughly 50% sequence identity, the catalytic domains of PCs are more related with the corresponding domains of ESPs. Similar to ESPs, members of the PC family are all synthesized as multi-domain proteins and require their propeptides to function as IMCs, though it remains to be seen whether the catalytic domain of PCs actually displays kinetic stability. Why do eukaryotic PCs require their IMCs to fold, rather than an IMC-independent pathway? To better understand the role of IMCs in mediating folding and regulating activation of the catalytic domains of PCs, we will analyze their domain organization and compare that with bacterial subtilisins (Fig. 4.8).
Fig. 4.8.
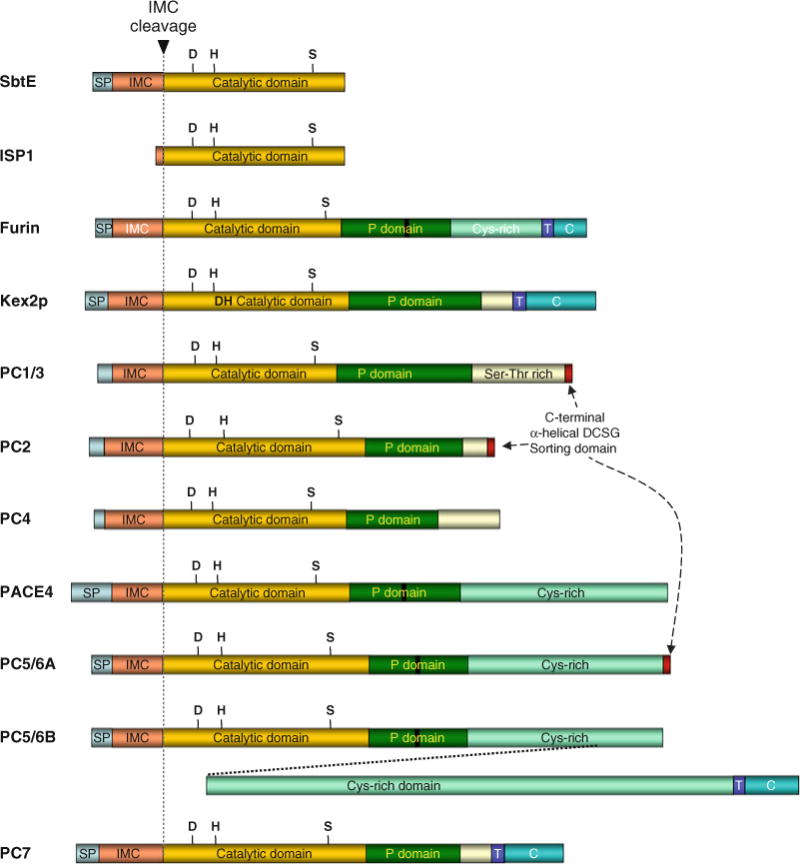
The structural organization of PCs compared with their bacterial counterparts ESPs and ISPs. The letter T represents the trans-membrane region, SP for signal peptide, while C indicates the cytoplasmic domain. The letters D, H, and S represent the catalytic triad and other regions are marked as indicated. Proteolysis at the IMC cleavage site (dotted grey line) initiates protease activation.
6. Proprotein Convertases Are Homologues of IMC-Dependent Subtilases
PCs constitute a family of calcium-dependent serine endoproteases that catalyze cleavages of precursor proteins at sites containing doublets or clusters of basic amino acids to generate active proteins and peptides (11). Analogous to prokaryotic subtilisins, PCs require their propeptides to chaperone folding of their cognate catalytic domains (2, 28). In addition, these IMCs also help to transport the catalytic domains of PCs into specific organelles of the eukaryotic secretory pathway, a complexity that is absent within prokaryotic subtilisins. The eukaryotic secretory compartments maintain a pH gradient inside the cell, wherein the pH drops from ~pH 7.0 in the endoplasmic reticulum (ER) to pH 6.0 in the trans-Golgi network (TGN). The post-TGN compartments are further acidified, with the dense core secretory granules at pH ~5.5 and lysosomes at ~pH 5 (23). Because most enzymes are exquisitely pH sensitive, the pH of each organelle critically determines the coordinated biochemical reactions occurring along the endocytic and secretory pathways. Studies on furin, the most intensively analyzed member of the PC family, demonstrate that the 83-residue propeptide helps to fold the catalytic domain in the ER but requires two pH-dependent and compartment-specific proteolytic cleavages to produce active furin (25, 28). While this two-step activation is similar to bacterial subtilisins (42, 77, 78, 87), the discovery of the differential pH requirements of the two proteolytic cleavages in the IMC domain of furin suggests that the IMC of furin (25, 28) and possibly other PCs may have evolved to sense and respond to a specific local pH.
7. The Domain Organization of PCs
Due to their significant levels of sequence similarity between the catalytic domains of eukaryotic PCs and prokaryotic ESPs and ISPs, they are all members of the subtilase super-family (13, 113). Figure 4.8 compares the domain organization between prokaryotic ESPs and ISPs and eukaryotic PCs. In addition to signal peptides, which direct translocation of pro-enzymes into the ER, PCs also contain IMC domains that are flanked by the signal peptidase cleavage sites at the amino-terminus and a pair of dibasic residues that constitute the autoproteolytic cleavage site at the carboxy-terminus.
7.1. The IMC Domain
The IMC domains in PCs, which are approximately 80–90 amino acid residues in length and are marginally longer than their prokaryotic counterparts, play a vital role in folding, transporting, activating, and regulating catalytic activities of PCs. In addition, the IMC domains of PCs also exhibit potent slow-binding inhibition of their cognate catalytic domains similar to bacterial subtilisins. In vitro studies demonstrate that the IMC:furin affinity is extremely tight (K0.5 = 14 nM) (25). The only residue-specific information on the structure of the IMC domains comes from NMR studies of isolated mouse PC1 IMC domain (114). Although the sequence conservation between IMCs of eukaryotic PCs and prokaryotic subtilases is low, the IMC domains adopt similar folds when they form complexes with their cognate catalytic domains (Fig. 4.9). The relative orientation of the active sites and the IMC domains of bacterial and eukaryotic inhibition complexes are depicted in Fig. 4.9b–d. However, unlike bacterial subtilisins (78), the IMC domains of PCs appear to be partially to fully structured domains in their isolated forms (115). The structure reveals that the IMC adopts an open sandwich anti-parallel α–β, comprised of two α-helices packed against one side of a four-stranded twisted β-sheet (see Fig. 4.9a). The β-sheets of the IMC domain pack tightly against the two parallel surface α-helices. This interface is stabilized by a complementary surface created through a series of conserved hydrophobic residues. Additionally the carboxylates of Glu−7 and Asp−9 in the IMC domain form helix caps for the two parallel helices in the case of subtilisin complex (76, 82, 112). Initial structural models of IMC:PC complexes suggest that such capping may also exist within PCs and may play a role in their autocatalytic activation (22).
Fig. 4.9.
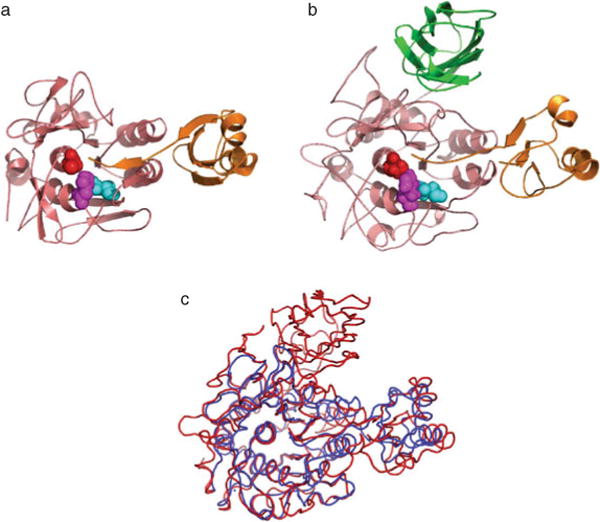
Structural conservation among IMCs of SbtE, furin, and PC1. All subtilases have a well-conserved subtilisin-like catalytic domain and are stabilized by bound metal ions. Yellow spheres depict calcium ion, while the orange sphere represents sodium ion. (a) Crystal structure of subtilisin E (1SCJ), a bacterial alkaline serine protease. (b) Crystal structure of furin (1P8J), a eukaryotic subtilisin-like protease, with the P-domain and an inhibitor (magenta). (c) Structure of a Ak.1 protease (1DBI), a thermostable subtilase from Bacillus showing the location of several calcium ions.
The observed slow binding in PCs may be a consequence of the structuring and binding of its floppy C-terminus of the propeptide to the catalytic domain and the subsequent reorientation of the propeptide:protease interface formed as a consequence of the bimolecular interaction. This was observed for PC5A which displays reduced catalytic activity when complexed with a peptide containing C-terminal residues from its own IMC domain (116). Since incorrect spatial and temporal proteolysis within a cell can be lethal, the inhibitory properties of IMCs were hypothesized to be a mechanism that prevents premature protease activation. In general, cells appear to have evolved two distinct mechanisms to control the activity of proteases. The first involves co-evolution of specific endogenous inhibitors, typically within compartments spatially distinct from those containing active enzymes. The second involves proteases being synthesized as inactive or less active zymogens, which become activated by limited intra- or intermolecular proteolysis cleaving off a small peptide. Interestingly, these two different regulatory mechanisms in proteases appear combined in the chaperoning capabilities of IMC-dependent systems and it is tempting to speculate that this combination enables facile spatiotemporal regulation of PC activation by exploiting the pH gradient of the secretory pathway.
To better understand the sequence–structure relationship in subtilases, a multiple sequence alignment of various bacterial subtilisin and eukaryotic PCs was conducted. The alignment also displays the earlier identified conserved motifs, N1 and N2, that are located at the amino- and carboxy-termini of the IMC domains (Fig. 4.10a). These motifs are more hydrophobic and are generally flanked by charged residues (38). As mentioned earlier, random mutagenesis studies using error-prone PCR demonstrate that substitutions within motifs N1 and N2 in bacterial subtilisin were often deleterious to folding (74). Circular dichroism studies suggest that the isolated subtilisin E IMC domain is largely unstructured (78). While NMR studies confirm this finding, they additionally suggest that motifs N1 and N2 display conformational rigidity (75). It is noteworthy that the unstructured IMC adopts a well-defined conformation when it binds to subtilisin wherein motifs N1 and N2 come together to form a α-β-α sub-domain in this inhibition complex (Fig. 4.10a, b) and may be critical for initiating the folding process (76).
Fig. 4.10.
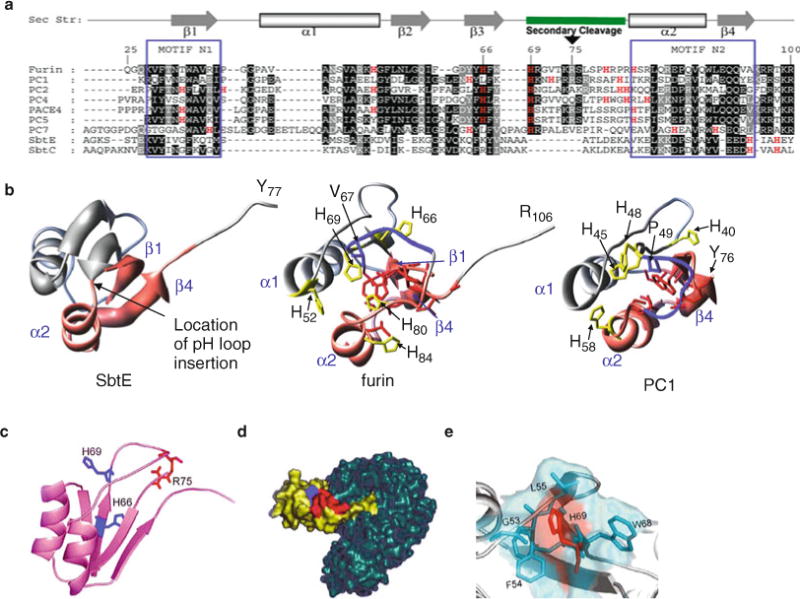
A comparison between the IMC domains of various PCs with their bacterial counterparts. (a) Sequence alignment of the IMC domains of various PCs with bacterial SbtE and SbtBPN. Residues are numbered in reference to the furin IMC, which begins at Gln25. The secondary structure cartoon depicted above the alignment is based on the NMR structure of the IMC domain of PC1/3 (1kn6) (114) and the IMC domain of SbtE (1scj) (76). The pH-sensitive cleavage site loop contains secondary cleavage sites for furin and PC1. Motifs N1 and N2 are well conserved when compared with the rest of the protein (38) and represent the folding nucleation sites in SbtE (75). (b) Pink ribbons in the structures of SbtE, furin, and the IMC domain of PC1/3 depict the relative locations of motifs N1 and N2 mapped on the 3D structure of the IMC domain. The secondary cleavage site is also depicted by the blue tube structure. Histidine residues in the alignment are marked in red typeface and are depicted in the individual structures as well. (c) Ribbon representation of the IMC domain of furin obtained by homology modeling. His66 and His69 (blue) and Arg75 (red) are highlighted. (d) Surface representation of the propeptide–furin complex. The modeled furin IMC (yellow) is docked onto the active site of furin (blue). The internal IMC cleavage site (red) and His69 (blue) are highlighted. (e) Surface representation of the secondary cleavage site illustrates His69 (red) buried in the solvent-accessible pocket formed by Gly53, Phe54, Leu55, Phe67, and Trp68 hydrophobic residues (blue).
While the structures and functions of individual proteins may impose different constraints on their evolution, an analysis of patterns of divergence suggests that individual responses of most proteins are variations on a common set of selective constraints. In proteins with low divergence, mutations within the interior are under strong purifying selection that removes all but a few conservative changes. With increasing divergence, mutations in the interior become more widespread and closer in number to what is found in the intermediate and exposed regions. Since catalytic domains exhibit a higher degree of conservation within subtilases when compared to their IMCs, this suggests that the catalytic and IMC domains may have encountered different mutational frequencies and differential selective constraints. However, why nature may have imposed differential selective constraints that alter both sequence and the asymmetrical distribution of charge in two functional domains within the same protein remains unclear.
7.2. The Catalytic Domain
The greatest sequence similarity resides in the subtilisin-like catalytic domain, with the aspartate (Asp), histidine (His), and serine (Ser) residues which comprise the active site being invariant. Overall, the catalytic domains of the other PCs are 54–70% identical in sequence to furin, which is the most extensively studied PC. Although the sequence identity is lower (20–30%) when PCs and bacterial subtilases are compared, the recently solved X-ray structures establish that the catalytic domains of furin (20, 22) and kexin (21) adopt secondary and tertiary structures that are similar to bacterial ESPs and ISPs (Fig. 4.9b–d).
In addition, these structures also provide insights into their remarkable substrate specificity displayed by PCs when compared with their promiscuous bacterial counterparts. Not surprisingly, homology modeling suggests that the core of the catalytic domains of PCs, which consist of a highly twisted β-sheet comprised of seven parallel and one anti-parallel β-strands (Fig. 4.9), is highly conserved based on the sequence similarity within PCs (22). The β-sheet is surrounded by five adjacent and two peripheral α-helices and by two short β-hairpin loops. These core structural elements are connected through surface loops that are conformationally less well defined. The solvent-exposed surface of the two peripheral helices in subtilisin constitutes the interface that interacts with the IMC domain. The modeling studies further suggest that PCs exhibit a deep active-site cleft with a shape remarkably similar to furin and that the geometry and the charge distribution of their substrate-binding regions appear to be of utmost importance for the distinct specificities of the individual PCs (22).
7.3. The P-Domain
While eukaryotic PCs contain IMCs and catalytic domains that are conserved with corresponding domains within bacterial subtilisins, they also contain an approximately 130 residue P-domain (Fig. 4.9c, d) that is important for catalytic activity of PCs which is missing in their bacterial counterparts (4). The structure of the P-domain of PCs is organized as a separate eight-stranded β-barrel, whose strand connectivity is similar to that for jelly-roll β-barrels (Fig. 4.9b). The eight structurally conserved strands are arranged in two opposing four-stranded β-sheets, which pack in a small hydrophobic core. The catalytic domain and P-domain are folded into two separate but abutting domains that together form the characteristic two-domain structure of furin and kexin (20, 21). Both domains are covalently linked by the “inter-domain linker” and interact with each other through an interface that is approximately 1100 Å2 to form the ectodomain of PCs. The few hydrophobic interface residues contributed by the catalytic domain are of special note, because most have polar/charged counterparts in the topologically related but P-domain-free bacterial subtilisins (22). These hydrophobic residues are mostly conserved in the PCs, obviously constrained by the common function to mediate interaction with the P-domain. Of particular note are a few charged interface residues, which are engaged in buried salt bridges that contribute to a part of the calcium-binding site. As a consequence the P-domain serves to stabilize calcium binding through this domain–domain interface.
7.4. The Variable C-Terminus of PCs
In addition to the signal peptide, propeptide, catalytic, and P-domains, furin, PACE4, PC4, PC5, and PC7 also contain a highly variable Cys-rich C-terminus (Fig. 4.8) (117). Moreover, the full-length furin, the PC5/6B isoform, and PC7 display additional trans-membrane and C-terminal cytoplasmic domains that are absent in PC1/3, PC2, and PC4 which terminate in long Ser/Thr-rich tails instead. PC1/3 and PC2 are predominantly expressed in neuroendocrine cells, PC4 is only found in testicular and ovarian germ cells, while the other mammalian PCs are more broadly distributed. PCs are often simultaneously expressed in various tissues, where they are mainly localized in the TGN, but are also found in other compartments of the secretory pathways and on the cell surface (11). Furin, a constitutively expressed member of the PC family, is localized primarily in the TGN but can traffic between two cycles: one at the TGN and the other at the cell surface where it interacts with its diverse repertoire of proprotein substrates. Moreover, furin is required to process these substrates precisely, even in the presence of other similar cleavage sites. Three of the PCs, PC1/3, PC2, and PC5/6A, are targeted to dense core secretory granules via α-helical domains located in their C-terminal tails (118).
8. Activation of Eukaryotic Subtilases
The activation pathway of PCs shows evolutionary conservation with the activation pathway of bacterial subtilisins, which use IMC cleavage to guide the folding of the catalytic domain and results in a structural reorganization of the folded proprotein. As seen from Fig. 4.8, furin, PC7, and the isoform PC5/6B are type-I membrane-bound proteases; PC1/3, PC2, and PC5/6A are packaged into dense core granules; PACE4 is localized in the extracellular matrix in osteoarthritic cartilages (119), while PC4 is constitutively secreted into the extracellular milieu. Furin can be shed from the membrane and along with PC5/6A can be found in the extracellular milieu. The sequences of IMC domains of PCs are 50–60% identical to furin and are flanked by the signal peptidase cleavage site on the amino-terminal side and by a conserved set of basic amino acids that comprise the autoproteolytic cleavage site on the carboxy-terminal side. Signal peptidase cleavage occurs after the signal peptide directs translocation of the precursor into the ER, where the IMC domain guides the folding of the catalytic triad into its correct conformation. With the exception of PC2, which is processed in the immature secretory granules, all PCs undergo the initial autocatalytic cleavage in the ER to generate a non-covalently associated heterodimeric IMC:convertase complex. This complex exits the ER and sorts to specific secretory pathway compartments where it undergoes a second autocatalytic cleavage within the IMC domain to disinhibit the enzymatic domain. Furin and PC7 undergo this second cleavage in the TGN/endocytic system; PC5/6 and PACE4 appear to get activated in the TGN and/or at the cell surface while PC1/3 and PC2 are maximally active only in secretory granules. PC1/3’s maximal activity in secretory granules is due to a second autoproteolytic cleavage of its C-terminal domain at a pair of arginine residues (R617, R618) (120).
This exquisitely well-orchestrated, multistep, compartment-specific pair of cleavages within the IMC domain is the most intensively studied in furin (2) and is accomplished by exploiting its own rules of substrate specificity to ensure correct spatial and temporal activation in the TGN, where the conditions of pH and calcium are optimal for the in trans cleavage of specific substrates. The first cleavage is rapid (t1/2 =10 min) and takes place in the neutral pH of the ER within furin’s consensus cleavage site –Arg–Thr–Lys–Arg107↓– after residue Arg107 (P1/P4 Arg canonical cleavage site) at the C-terminus of the IMC domain (121). This cleavage of the IMC can be blocked through an active-site substitution and results in the buildup of an intermediate in the ER–Golgi intermediate compartment (ERGIC)/cis-Golgi network. These data suggest that (i) the cleavage occurs through autoproteolysis, (ii) both the ER and ERGIC compartments participate in the initial steps of furin activation, and (iii) specific components of the cellular trafficking machinery detect formation of the furin: propeptide complex before directing their transit to late secretory pathway compartments. Recent studies suggest that PACS-1, a cytosolic sorting protein that connects the CK2-phosphorylated furin cytosolic domain to AP-1, is responsible for localizing the endoprotease to the TGN (122). After IMC cleavage occurs in the ER, the IMC domain remains associated with furin and functions as a potent autoinhibitor (IC50 14 nM) in trans during the transport of this IMC–furin complex = to the late secretory compartments – TGN/endosomes – where the mildly acidic pH promotes a second, slower intramolecular autoproteolytic cleavage (t1/2 ~ 90 min) at His-Arg-Gly-Val-Thr-Lys-Arg75↓, an internal site (P1/P6 Arg cleavage site) within the IMC domain. Surprisingly, mutation of the P1/P6 Arg cleavage site to a P1/P4 Arg canonical furin site fails to yield mature, active furin and instead causes the accumulation of inactive profurin in the ER (25, 28). This second-site cleavage is rate limiting for activation of bacterial and human subtilisin-like proteases (34, 110). Cleavage at Arg75 is followed by the rapid dissociation of the cleaved IMC fragments that releases the inhibition of furin’s catalytic triad allowing it to process its diverse repertoire of endogenous substrates (25). Together, these studies suggest that the ordered, compartment-specific cleavages of the furin propeptide are necessary to guide the folding and activation of the endoprotease, and that this activation process could be controlled by a pH sensor.
As discussed earlier, despite significant sequence and structural conservation with both PCs and ESPs, members of the prokaryotic ISP subfamily lack propeptide domains. How do ISPs prevent premature proteolysis inside the cell? An analysis of the ISPs reveals that they too are produced as zymogens with very small propeptide domains (Fig. 4.7a). Unlike ESPs, the propeptide domains of ISPs are roughly 20–25 residues long and do not function as IMCs. Nonetheless, these propeptides are potent inhibitors of the ISP activity and are removed through autoproteolysis (Subbian et al. unpublished data). Hence the expression of ISPs as zymogens can serve to prevent premature activation. These IMC-independent ISPs are involved in a variety of regulatory cellular functions such as DNA packing, genetic competence, and protein secretion and differ from ESPs which are scavenging proteases (123–126). Although PCs more closely align with ISPs in terms of regulating biological function, the amino acid sequences of PCs more closely resemble ESPs. Clearly both prokaryotic prototype ESPs and ISPs are produced as zymogens. From an evolutionary standpoint it is interesting to note that eukaryotic PCs require their IMC to chaperone correct folding. Why do eukaryotic PCs, like ESPs, require their IMCs to fold, rather than an IMC-independent pathway that exists in ISPs? We hypothesize that the choice of an IMC-dependent pathway was driven by a necessity to modulate folding, to allow interactions with the transport machinery, and to regulate the compartment-specific activation.
9. IMC-Encoded pH Sensors in Eukaryotic Subtilases
The required exposure of the IMC–furin complex to pH 6 in order to complete enzyme activation suggests a role for one or more histidine residues on the IMC domain to serve as a pH sensor controlling furin activation. Histidine is a unique amino acid that contains an imidazole ring for its side chain. The unprotonated imidazole is nucleophilic and can serve as a general base, while the protonated form can serve as a general acid. Hence protonation of the histidine imidazole ring, which has a pKa of 6.0, has profound effects on histidine chemistry under physiological conditions. Such pH-sensing roles for histidines are well established for generating allosteric changes, including control of O2/CO2 exchange by hemoglobin, gating of electrogenic molecules, and the pH-dependent conformational changes within class II MHC molecules that promote ligand exchange.
Analysis of the furin IMC predicts two His residues adjacent to the secondary cleavage site, which are either strictly (His69) or partially (His66) conserved within all PC family members (Fig. 4.10a). This cleavage site maps onto a loop between strands β3 and α2 and is part of a distinct hydrophobic pocket on the surface of the IMC abutting the catalytic domain (Fig. 4.10b, d). Although the cleavage site lies in a surface loop, H69 is buried at the center of a well-formed hydrophobic pocket lined by nonpolar residues, G53 and L55, and aromatic residues, F54, F67, and W68 (Fig. 4.10d). By contrast, the P9 H66 maps to the IMC–protease interface. This interface in bacterial subtilisins displays a higher than average B-factors as seen from the crystal structure of the IMC:SbtE complex (76), and increasing these dynamics can decrease rates of autoprocessing (100). Varying solvent conditions modulate IMC binding to this interface, prolonging its release and hence the activation of the mature protease (110). This suggests that this interface is not solvent accessible. Based on our modeling analysis, we predicted that protonation of the H69 located in a hydrophobic pocket could have dramatic effects on the structure of the IMC, unlike protonation of the solvent-accessible H66. Hence, H69 but not H66 as earlier hypothesized (114) could function as a potential inbuilt primary pH sensor within PCs. Using mutants that mimic the protonation state of histidine the role of His69 in the folding and activation of furin has been elucidated (26). These studies suggest that the protonation state of His69, which is located in a solvent-accessible hydrophobic pocket, plays a critical role in regulating the secondary propeptide cleavage. Mutations that interfere with the His69 block propeptide excision, resulting in accumulation of profurin in the ER by a mechanism that requires the cytosolic sorting protein PACS-2 and COPI. Following propeptide excision, the furin•propeptide complex traffics to the mildly acidic TGN/endosomal system where protonation of His69 disrupts the solvent-accessible hydrophobic pocket to expose the P1/P6 Arg internal cleavage site His-Arg-Gly-Val-Thr-Lys-Arg75↓, leading to release of the inhibitory propeptide and furin activation.
Given that the H69 – the pH sensor in furin – is conserved within other PCs it remains unclear why different PCs display unique pH sensitivity. For example, PC1, which requires a lower pH for its activation, becomes activated in the dense core secretory granules. We hypothesize that additional residues are likely involved in controlling the subtleties of pH-dependent activation of individual PCs. Further whether additional residues are involved for H69 to function as a pH sensor in furin also remains unknown.
9.1. Functional Advantages of Sequence-Encoded pH Sensors in Proteins
While post-translational modifications or cofactors can also regulate biological activity, one of the major advantages of using changes in proton concentrations for regulation and signaling is the potential for exceptionally efficacious spatial and temporal responses. Protons are small single subatomic particles that can diffuse rapidly through water to induce reversible chemical changes which result in considerable electrostatic perturbations, which in turn changes protein structure, dynamics, and interactions. The pH-driven changes in the affinity of hemoglobin for oxygen binding (Bohr’s effect) is a classic example of a single-site, proton-induced allosteric regulation. In this case, the effect of pH is mediated by a His-Asp salt bridge which breaks when His gets deprotonated at neutral pH. The protonation of key histidine residues triggers interaction of the translocation domain of diphtheria toxin with the host cell membrane and causes flaviviruses to fuse into membranes. In addition to modulating activity of enzymes, viral entry, and transformation, exquisite pH-sensitive conformational switches are found to regulate activity of ion and water channels, affinity of proteins for their cognate binding partners, protein stability, solubility, and catalytic activity.
Despite established effects of small changes in proton concentrations on diverse cell functions, our understanding of how these changes affect proteins and macromolecular assemblies driving specific cell processes is limited. Although changes in solvent pH affect the ionization state of all weak acids and bases, and all cellular proteins contain amino acids with titratable groups, only select proteins appear to be bonafide pH sensors. New insights from protein structures and biomolecular simulations are beginning to reveal the structural basis for tight coupling between protonation state and protein conformation. However, our understanding of how physiological changes in pH affect protein conformations and macromolecular assemblies is limited.
9.2. Putative Mechanisms by Which pH Sensors Function
Based on our understanding of the activation of bacterial subtilisins we hypothesize that protonation of the intrinsic pH sensor of furin induces conformational changes that destabilize the IMC domain to initiate rapid activation. This hypothesis is based on the fact that the pH sensor residue in furin – H69 – is solvent accessible and is located within the loop that contains internal cleavage site conserved within all PCs. This cleavage site maps onto a loop between strands β3 and α2 and contributes toward a distinct solvent-exposed hydrophobic pocket on the surface of the IMC and is adjacent to the catalytic domain (Fig. 4.10b, e). The hydrophobic pocket is lined by residues G53, L55, F54, F67, and W68 (Fig. 4.10e). At a near-neutral pH of the ER, the mostly deprotonated H69 displays more hydrophobic properties that allow the imidazole side chain to be stabilized within the hydrophobic pocket. In this conformation the pH-sensitive loop tightly abuts the IMC and reduces the exposure of the internal cleavage site to trans-proteolysis. Moreover, the formation of this pocket appears to be essential for initiating the folding process because an H69K substitution does not result in a folded/processed complex (26). Upon entry into the TGN the imidazole side chain of H69 is exposed to a more acidic pH (~6.0) and gets protonated. The additional proton increases the polarity of the imidazole side chain forcing it out of the hydrophobic pocket (Fig. 4.11). This repulsion may cause a conformational change in the IMC domain, which can potentially destabilize the global conformation of the IMC domain and/or may increase the exposure of the internal cleavage site to trans-proteolysis. Biophysical and computational studies are currently underway to elaborate the mechanism by which the protonation of H69 facilitates activation and preliminary results suggest that (i) His69 protonation may induce local conformational changes and (ii) additional residues may be required to optimize the sensitivity of furin and other PCs to their compartment-specific activation (Shinde et al. unpublished data).
Fig. 4.11.
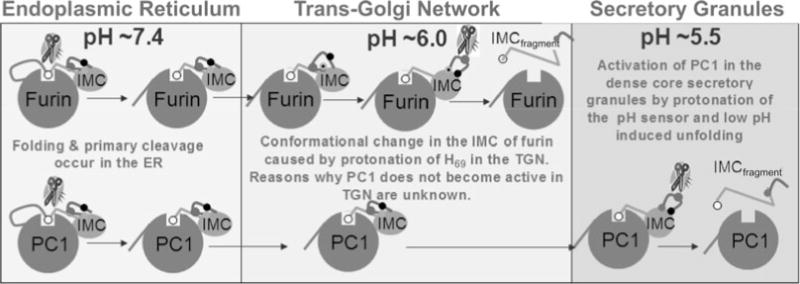
A cartoon model depicting putative mechanism for activation of furin and PC1/PC3 through protonation of H69. Both furin and PC1/PC3 undergo their first cleavage at a pair of conserved dibasic residues. This cleavage allows the non-covalent complexes of furin and PC1/PC3 to exit the ER into the TGN. Here, His69, the pH sensor in furin, gets protonated and induces a conformational change within the pH-sensitive loop. This exposes the internal cleavage site at Arg75, which can undergo proteolysis that releases inhibition. Although the residue corresponding to His69 in furin is conserved, PC1/PC3 does not undergo the second internal cleavage and suggests that additional residues must be involved in fine-tuning the pH sensitivity of individual PCs. Upon internal cleavage the inhibitory C-terminal region of the IMC is released and renders the catalytic site free for proteolysis.
It is interesting to note that residue H69 in furin is absolutely conserved in all PCs. However, unlike furin, other PC family members become activated at different pHs. For example, similar to furin, PC1 is synthesized in the ER and traverses the TGN network but only becomes active in the mature dense secretory granules where the pH is approximately 5.5. If the conserved His residue is alone responsible for pH sensing, why does PC1 not become activated in the TGN? It is tempting to hypothesize that there are hitherto unknown determinants that are additionally required to allow different PCs to have different sensitivities for pH-dependent activation. For example, the determinants that enable the isolated IMC domains of furin and PC1 to display different conformational preferences may play a vital role in activation. Additional determinants could also be located within the P-domains that are found exclusively within eukaryotic PCs and which are completely absent in prokaryotic subtilases. This is consistent with the data that suggest that the P-domains play an important role in regulating stability, calcium dependence, and pH dependence of PCs. Alternately, there may be additional sequence determinants within IMCs that have yet to be identified. For example, IMCs have a higher preponderance of charges than their cognate catalytic domains. Moreover, the type of charges can vary significantly between different IMC domains, which may impart unique sensitivity to specific local environments within the IMC domain alone. Every amino acid has a unique pKa value. When one considers amino acids with charged side chains, aspartic acid and glutamic acid have carboxyl groups on their side chains and are fully ionized at pH 7.4 while arginine and lysine have amino groups as side chains and are fully protonated at pH 7.4. The pKa for histidine, which contains an imidazole ring for its side chain, is the closest to physiological pH and hence profoundly affects its chemical properties. Since every amino acid has a unique pKa value, a combination of specific charged and polar residues and their surface accessibility can dramatically affect the functional properties of the IMC domain through net charge at a particular pH.
The IMC domains of aqualysin and subtilisin, which are prokaryotic enzymes, have pIs that are 4.8 and 9.8, respectively. While these IMCs can be interchanged and the chimeric protease can fold correctly in vitro, they do display dramatically different affinities and therefore different protease activation profiles for their catalytic domains. Surprisingly, the IMC domains have lower affinity for their own catalytic domains, suggesting that the binding affinity of an IMC domain may be optimized for its cognate catalytic domain, so as to enable efficient spatial and temporal activation. The protease aqualysin was isolated from Thermus aquaticus, a strain that tolerates extreme temperatures and was first discovered in the Lower Geyser Basin of Yellowstone National Park. Subtilisin on the other hand was identified in B. subtilis, a bacterium commonly found in soil and thrives in much milder environmental conditions. In B. subtilis the protease is extracellular and is secreted under conditions of starvation to allow the bacterium to scavenge amino acids by breaking down decaying proteins from the soil. To activate the protease, the IMC of subtilisin has to respond to conditions in the soil, which are normally not extreme. On the other hand, aqualysin requires very extreme conditions of temperature and pH in order to become active; and unlike the intrinsically unstructured IMC of subtilisin, the IMC domain from aqualysin is well structured and more stable at room temperature. However, at 55–60°C aqualysin’s IMC domain loses structure and can be released to facilitate rapid activation. Although the IMC domains of these proteases can be swapped and can facilitate correct folding at room temperature in vitro, it is likely that the subtilisin IMC would not function at the temperature where T. aquaticus survives. Similarly, while the aqualysin IMC domain folds subtilisin, the activation of the catalytic domain through IMC release and degradation requires drastically different conditions. This is because the aqualysin IMC is structured and binds subtilisin with an affinity that is more than 10-fold stronger.
Both aqualysin and subtilisin are ESPs. How do they differ from ISPs? This question can be answered by examining ESPs and ISPs from Bacillus sp. As mentioned earlier, ESPs are extracellular, kinetically stable, secreted proteases, while ISPs are thermodynamically stable intracellular proteases (111). It is important to note that the observed differences between folding pathways of ISPs and ESPs are not mere consequences of their cellular location because the intracellular or extracellular expression of ESPs does not yield an active protease in the absence of its cognate propeptide (30). While the energy barriers associated with kinetic stability increase the stability – and hence longevity – of the catalytic domain in harsh extracellular environments, similar barriers leading to high stability in ISPs may impede intracellular protein turnover. This is consistent with our finding that intracellular expression of ESPs (aqualysin 1 and subtilisin E) is detrimental to cell growth due to extensive proteolysis (unpublished data). This suggests that biological requirements, and not just specific conformations, dictate selection of folding pathways. We hypothesize that IMC-mediated folding of PCs has evolved to enable their precise spatiotemporal activation within a complex, compartment-specific environment within a eukaryotic cell (Fig. 4.11).
IMC-dependent folding mechanisms are also found within the cathepsins, which function within the acid environment of the lysosome (pH ~ 5). While the activation of lysosomal cathepsins, like PCs, is also specific and pH dependent, the precise mechanisms by which these proteases sense lysosomal pH are unknown. In some cases propeptides are known to regulate different types of cellular processes such as transport and localization, hierarchical organization, or oligomerization and regulation of protein activity–function by indirectly modulating protein conformations and can be termed as “post-translational modulators” of protein structure and function (29, 43).
10. Implication of pH Sensing for Biological Functions of PCs and Cathepsins
Changes in intracellular pH regulate numerous normal and pathological processes in the cell. Such changes in intracellular pH are permissive for growth factor-induced cell proliferation, cell cycle progression, and differentiation and are necessary for haptokinetic migration and amoeboid chemotaxis. Additionally, increases in cytosolic pH are a hallmark of transformed cells from different tissue origins and genetic backgrounds, making it a common characteristic of distinct cancers and possibly a common critical driving force for tumor progression. Since PCs are involved in the activation of matrix metalloproteases, growth factors, adhesion molecules, and which are crucial for cellular transformation, acquisition of the tumorigenic phenotype, and metastases formation, it is not surprising that they play major roles in tumor progression and malignancy. Experiments suggest that inhibition of PCs can alter the malignant phenotype of various tumor cells and can be used to target tumor angiogenesis. Measurement of pH inside tissue suggests that the microenvironment within tumors is generally more acidic than normal tissues. Major contributors to tumor acidity likely include the excessive production of lactic acid and ATP hydrolysis in hypoxic zones of tumors. Additional reduction in tumor pH may be achieved by administrating glucose (and/or insulin) and hydralazine, a drug which modifies the relative blood flow to tumors and normal tissues. How would the increased acidification of tumors enhance the activation of PCs? A more acidic pH is likely to cause premature activation of furin within the ER. This in turn may alter cellular homeostasis through premature, unregulated proteolysis. It is also known that vesicles shed by cancer cells mediate several tumor–host interactions, and that the tumor microenvironment influences the release and the activity of tumor-shed microvesicles. Such vesicles contain cathepsin-B, the activity of which was measured to be significantly increased at an acidic pH. The enhanced cathepsin-B activity stimulates both the activation of gelatinase and the invasiveness of endothelial cells observed at low pH values. As a consequence, the acidic microenvironment found in most solid tumors may contribute to protease-mediated proinvasive capabilities of tumor-shed vesicles.
In conclusion it is clear that the pH gradient within the secretory and endocytic pathway is essential for normal homeostasis (23). Cells have evolved multiple mechanisms to maintain this pH gradient and there is accumulating evidence that altered pH regulation is a hallmark of several disease states (127). Although there are tantalizing links between altered pH homeostasis and disease there is no definitive example where acidification is a primary cause of disease pathogenesis. However, this is likely to change through an increase in our understanding of how eukaryotic proteins may have evolved mechanisms to exploit such compartment-specific pH for their biological function.
Acknowledgments
Our apologies to colleagues whose work we did not cite because of space limitations. Special thanks go to Jimmy Dikeakos, David Radler, Laura Figoski, and Stephanie Dillon for insightful discussions, for review of the manuscript, and for editing. U.S. is supported by grants from the National Science Foundation (NSF-0746589) and the Oregon Nanoscience and Microtechnologies Institute and G.T. is supported by grant from the National Institutes of Health (DK37274 and CA151564).
References
- 1.Nakayama K. Furin: A mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J. 1997;327(Pt 3):625–35. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas G. Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–66. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuller RS, Brake A, Thorner J. Yeast prohormone processing enzyme (KEX2 gene product) is a Ca2+-dependent serine protease. Proc Natl Acad Sci USA. 1989;86:1434–8. doi: 10.1073/pnas.86.5.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuller RS, Brake AJ, Thorner J. Intracellular targeting and structural conservation of a prohormone-processing endoprotease. Science. 1989;246:482–6. doi: 10.1126/science.2683070. [DOI] [PubMed] [Google Scholar]
- 5.Fuller RS, Sterne RE, Thorner J. Enzymes required for yeast pro-hormone processing. Annu Rev Physiol. 1988;50:345–62. doi: 10.1146/annurev.ph.50.030188.002021. [DOI] [PubMed] [Google Scholar]
- 6.Thomas G, Thorne BA, Thomas L, et al. Yeast KEX2 endopeptidase correctly cleaves a neuroendocrine prohormone in mammalian cells. Science. 1988;241:226–30. doi: 10.1126/science.3291117. [DOI] [PubMed] [Google Scholar]
- 7.Bergeron F, Leduc R, Day R. Subtilase-like pro-protein convertases: From molecular specificity to therapeutic applications. J Mol Endocrinol. 2000;24:1–22. doi: 10.1677/jme.0.0240001. [DOI] [PubMed] [Google Scholar]
- 8.Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc Natl Acad Sci USA. 2003;100:928–33. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seidah NG, Mowla SJ, Hamelin J, et al. Mammalian subtilisin/kexin isozyme SKI-1: A widely expressed proprotein convertase with a unique cleavage specificity and cellular localization. Proc Natl Acad Sci USA. 1999;96:1321–6. doi: 10.1073/pnas.96.4.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toure BB, Munzer JS, Basak A, et al. Biosynthesis and enzymatic characterization of human SKI-1/S1P and the processing of its inhibitory prosegment. J Biol Chem. 2000;275:2349–58. doi: 10.1074/jbc.275.4.2349. [DOI] [PubMed] [Google Scholar]
- 11.Seidah NG, Mayer G, Zaid A, et al. The activation and physiological functions of the proprotein convertases. Int J Biochem Cell Biol. 2008;40:1111–25. doi: 10.1016/j.biocel.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 12.Lambert G, Charlton F, Rye KA, Piper DE. Molecular basis of PCSK9 function. Atherosclerosis. 2009;203:1–7. doi: 10.1016/j.atherosclerosis.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Siezen RJ, Leunissen JA. Subtilases: The superfamily of subtilisin-like serine proteases. Protein Sci. 1997;6:501–23. doi: 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rawlings ND, Barrett AJ, Bateman A. MEROPS: The peptidase database. Nucleic Acids Res. 2009;38:D227–33. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith EL, Markland FS, Kasper CB, DeLange RJ, Landon M, Evans WH. The complete amino acid sequence of two types of subtilisin, BPN’ and Carlsberg. J Biol Chem. 1966;241:5974–6. [PubMed] [Google Scholar]
- 16.Wright CS, Alden RA, Kraut J. Structure of subtilisin BPN’ at 2.5 angstrom resolution. Nature. 1969;221:235–42. doi: 10.1038/221235a0. [DOI] [PubMed] [Google Scholar]
- 17.Bryan PN. Protein engineering of subtilisin. Biochim Biophys Acta. 2000;1543:203–22. doi: 10.1016/s0167-4838(00)00235-1. [DOI] [PubMed] [Google Scholar]
- 18.Lipkind G, Gong Q, Steiner DF. Molecular modeling of the substrate specificity of prohormone convertases SPC2 and SPC3. J Biol Chem. 1995;270:13277–84. doi: 10.1074/jbc.270.22.13277. [DOI] [PubMed] [Google Scholar]
- 19.Oliva AA, Jr, Steiner DF, Chan SJ. Proprotein convertases in amphioxus: Predicted structure and expression of proteases SPC2 and SPC3. Proc Natl Acad Sci USA. 1995;92:3591–5. doi: 10.1073/pnas.92.8.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henrich S, Cameron A, Bourenkov GP, et al. The crystal structure of the pro-protein processing proteinase furin explains its stringent specificity. Nat Struct Biol. 2003;10:520–6. doi: 10.1038/nsb941. [DOI] [PubMed] [Google Scholar]
- 21.Holyoak T, Wilson MA, Fenn TD, et al. 2.4 A resolution crystal structure of the prototypical hormone-processing protease Kex2 in complex with an Ala-Lys-Arg boronic acid inhibitor. Biochemistry. 2003;42:6709–18. doi: 10.1021/bi034434t. [DOI] [PubMed] [Google Scholar]
- 22.Henrich S, Lindberg I, Bode W, Than ME. Proprotein convertase models based on the crystal structures of furin and kexin: Explanation of their specificity. J Mol Biol. 2005;345:211–27. doi: 10.1016/j.jmb.2004.10.050. [DOI] [PubMed] [Google Scholar]
- 23.Demaurex N. pH Homeostasis of cellular organelles. News Physiol Sci. 2002;17:1–5. doi: 10.1152/physiologyonline.2002.17.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 25.Anderson ED, Molloy SS, Jean F, Fei H, Shimamura S, Thomas G. The ordered and compartment-specific autoproteolytic removal of the furin intramolecular chaperone is required for enzyme activation. J Biol Chem. 2002;277:12879–90. doi: 10.1074/jbc.M108740200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feliciangeli SF, Thomas L, Scott GK, et al. Identification of a pH sensor in the furin propeptide that regulates enzyme activation. J Biol Chem. 2006;281:16108–16. doi: 10.1074/jbc.M600760200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ehrmann M, Clausen T. Proteolysis as a regulatory mechanism. Annu Rev Genet. 2004;38:709–24. doi: 10.1146/annurev.genet.38.072902.093416. [DOI] [PubMed] [Google Scholar]
- 28.Anderson ED, VanSlyke JK, Thulin CD, Jean F, Thomas G. Activation of the furin endoprotease is a multiple-step process: Requirements for acidification and internal propeptide cleavage. EMBO J. 1997;16:1508–18. doi: 10.1093/emboj/16.7.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shinde U, Inouye M. Intramolecular chaperones: Polypeptide extensions that modulate protein folding. Semin Cell Dev Biol. 2000;11:35–44. doi: 10.1006/scdb.1999.0349. [DOI] [PubMed] [Google Scholar]
- 30.Ikemura H, Takagi H, Inouye M. Requirement of pro-sequence for the production of active subtilisin E in Escherichia coli. J Biol Chem. 1987;262:7859–64. [PubMed] [Google Scholar]
- 31.Ikemura H, Inouye M. In vitro processing of pro-subtilisin produced in Escherichia coli. J Biol Chem. 1988;263:12959–63. [PubMed] [Google Scholar]
- 32.Zhu XL, Ohta Y, Jordan F, Inouye M. Pro-sequence of subtilisin can guide the refolding of denatured subtilisin in an intermolecular process. Nature. 1989;339:483–4. doi: 10.1038/339483a0. [DOI] [PubMed] [Google Scholar]
- 33.Shinde U, Fu X, Inouye M. A pathway for conformational diversity in proteins mediated by intramolecular chaperones. J Biol Chem. 1999;274:15615–21. doi: 10.1074/jbc.274.22.15615. [DOI] [PubMed] [Google Scholar]
- 34.Yabuta Y, Takagi H, Inouye M, Shinde U. Folding pathway mediated by an intramolecular chaperone: Propeptide release modulates activation precision of pro-subtilisin. J Biol Chem. 2001;276:44427–34. doi: 10.1074/jbc.M107573200. [DOI] [PubMed] [Google Scholar]
- 35.Ohta Y, Hojo H, Aimoto S, et al. Pro-peptide as an intramolecular chaperone: Renaturation of denatured subtilisin E with a synthetic pro-peptide [corrected] Mol Microbiol. 1991;5:1507–10. doi: 10.1111/j.1365-2958.1991.tb00797.x. [DOI] [PubMed] [Google Scholar]
- 36.Ohta Y, Inouye M. Pro-subtilisin E: Purification and characterization of its autoprocessing to active subtilisin E in vitro. Mol Microbiol. 1990;4:295–304. doi: 10.1111/j.1365-2958.1990.tb00596.x. [DOI] [PubMed] [Google Scholar]
- 37.Eder J, Fersht AR. Pro-sequence-assisted protein folding. Mol Microbiol. 1995;16:609–14. doi: 10.1111/j.1365-2958.1995.tb02423.x. [DOI] [PubMed] [Google Scholar]
- 38.Shinde U, Inouye M. Intramolecular chaperones and protein folding. Trends Biochem Sci. 1993;18:442–6. doi: 10.1016/0968-0004(93)90146-e. [DOI] [PubMed] [Google Scholar]
- 39.Ellis RJ. The general concept of molecular chaperones. Philos Trans R Soc Lond B Biol Sci. 1993;339:257–61. doi: 10.1098/rstb.1993.0023. [DOI] [PubMed] [Google Scholar]
- 40.Ellis RJ. Protein misassembly: Macromolecular crowding and molecular chaperones. Adv Exp Med Biol. 2007;594:1–13. doi: 10.1007/978-0-387-39975-1_1. [DOI] [PubMed] [Google Scholar]
- 41.Ellis RJ, van der Vies SM. Molecular chaperones. Annu Rev Biochem. 1991;60:321–47. doi: 10.1146/annurev.bi.60.070191.001541. [DOI] [PubMed] [Google Scholar]
- 42.Shinde UP, Liu JJ, Inouye M. Protein memory through altered folding mediated by intramolecular chaperones. Nature. 1997;389:520–2. doi: 10.1038/39097. [DOI] [PubMed] [Google Scholar]
- 43.Chen YJ, Inouye M. The intramolecular chaperone-mediated protein folding. Curr Opin Struct Biol. 2008;18:765–70. doi: 10.1016/j.sbi.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 44.Silen JL, Agard DA. The alphalytic protease pro-region does not require a physical linkage to activate the protease domain in vivo. Nature. 1989;341:462–4. doi: 10.1038/341462a0. [DOI] [PubMed] [Google Scholar]
- 45.Baker D, Sohl JL, Agard DA. A protein-folding reaction under kinetic control. Nature. 1992;356:263–5. doi: 10.1038/356263a0. [DOI] [PubMed] [Google Scholar]
- 46.Jaswal SS, Sohl JL, Davis JH, Agard DA. Energetic landscape of alpha-lytic protease optimizes longevity through kinetic stability. Nature. 2002;415:343–6. doi: 10.1038/415343a. [DOI] [PubMed] [Google Scholar]
- 47.Valls LA, Hunter CP, Rothman JH, Stevens TH. Protein sorting in yeast: The localization determinant of yeast vacuolar carboxypeptidase Y resides in the propeptide. Cell. 1987;48:887–97. doi: 10.1016/0092-8674(87)90085-7. [DOI] [PubMed] [Google Scholar]
- 48.Ammerer G, Hunter CP, Rothman JH, Saari GC, Valls LA, Stevens TH. PEP4 gene of Saccharomyces cerevisiae encodes proteinase A, a vacuolar enzyme required for processing of vacuolar precursors. Mol Cell Biol. 1986;6:2490–9. doi: 10.1128/mcb.6.7.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramos C, Winther JR, Kielland-Brandt MC. Requirement of the propeptide for in vivo formation of active yeast carboxypeptidase Y. J Biol Chem. 1994;269:7006–12. [PubMed] [Google Scholar]
- 50.Winther JR, Sorensen P. Propeptide of carboxypeptidase Y provides a chaperone-like function as well as inhibition of the enzymatic activity. Proc Natl Acad Sci USA. 1991;88:9330–4. doi: 10.1073/pnas.88.20.9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winther JR, Sorensen P, Kielland-Brandt MC. Refolding of a carboxypeptidase Y folding intermediate in vitro by low-affinity binding of the proregion. J Biol Chem. 1994;269:22007–13. [PubMed] [Google Scholar]
- 52.van den Hazel HB, Kielland-Brandt MC, Winther JR. The propeptide is required for in vivo formation of stable active yeast proteinase A and can function even when not covalently linked to the mature region. J Biol Chem. 1993;268:18002–7. [PubMed] [Google Scholar]
- 53.Smith SM, Gottesman MM. Activity and deletion analysis of recombinant human cathepsin L expressed in Escherichia coli. J Biol Chem. 1989;264:20487–95. [PubMed] [Google Scholar]
- 54.Ogino T, Kaji T, Kawabata M, et al. Function of the propeptide region in recombinant expression of active procathepsin L in Escherichia coli. J Biochem (Tokyo) 1999;126:78–83. doi: 10.1093/oxfordjournals.jbchem.a022439. [DOI] [PubMed] [Google Scholar]
- 55.Wiederanders B, Kaulmann G, Schilling K. Functions of propeptide parts in cysteine proteases. Curr Protein Pept Sci. 2003;4:309–26. doi: 10.2174/1389203033487081. [DOI] [PubMed] [Google Scholar]
- 56.Hou WS, Bromme D, Zhao Y, et al. Characterization of novel cathepsin K mutations in the pro and mature polypeptide regions causing pycnodysostosis. J Clin Invest. 1999;103:731–8. doi: 10.1172/JCI653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schulz EC, Neumann P, Gerardy-Schahn R, Sheldrick GM, Ficner R. Structure analysis of endosialidase NF at 0.98 A resolution. Acta Crystallogr D Biol Crystallogr. 2010;66:176–80. doi: 10.1107/S0907444909048720. [DOI] [PubMed] [Google Scholar]
- 58.Schulz EC, Schwarzer D, Frank M, et al. Structural basis for the recognition and cleavage of polysialic acid by the bacteriophage K1F tailspike protein EndoNF. J Mol Biol. 2010;397:341–51. doi: 10.1016/j.jmb.2010.01.028. [DOI] [PubMed] [Google Scholar]
- 59.McIver KS, Kessler E, Ohman DE. Identification of residues in the Pseudomonas aeruginosa elastase propeptide required for chaperone and secretion activities. Microbiology. 2004;150:3969–77. doi: 10.1099/mic.0.27340-0. [DOI] [PubMed] [Google Scholar]
- 60.Tang B, Nirasawa S, Kitaoka M, Marie-Claire C, Hayashi K. General function of N-terminal propeptide on assisting protein folding and inhibiting catalytic activity based on observations with a chimeric thermolysin-like protease. Biochem Biophys Res Commun. 2003;301:1093–8. doi: 10.1016/s0006-291x(03)00084-6. [DOI] [PubMed] [Google Scholar]
- 61.Gray TE, Eder J, Bycroft M, Day AG, Fersht AR. Refolding of barnase mutants and pro-barnase in the presence and absence of GroEL. EMBO J. 1993;12:4145–50. doi: 10.1002/j.1460-2075.1993.tb06098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suter U, Heymach JV, Jr, Shooter EM. Two conserved domains in the NGF propeptide are necessary and sufficient for the biosynthesis of correctly processed and biologically active NGF. EMBO J. 1991;10:2395–400. doi: 10.1002/j.1460-2075.1991.tb07778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thorne BA, Plowman GD. The heparin-binding domain of amphiregulin necessitates the precursor pro-region for growth factor secretion. Mol Cell Biol. 1994;14:1635–46. doi: 10.1128/mcb.14.3.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steiner DF. The proinsulin C-peptide–a multirole model. Exp Diabesity Res. 2004;5:7–14. doi: 10.1080/15438600490424389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Voorberg J, Fontijn R, Calafat J, Janssen H, van Mourik JA, Pannekoek H. Biogenesis of von Willebrand factor-containing organelles in heterologous transfected CV-1 cells. EMBO J. 1993;12:749–58. doi: 10.1002/j.1460-2075.1993.tb05709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weissman JS, Kim PS. The pro region of BPTI facilitates folding. Cell. 1992;71:841–51. doi: 10.1016/0092-8674(92)90559-u. [DOI] [PubMed] [Google Scholar]
- 67.Morgunova E, Tuuttila A, Bergmann U, et al. Structure of human pro-matrix metalloproteinase-2: Activation mechanism revealed. Science. 1999;284:1667–70. doi: 10.1126/science.284.5420.1667. [DOI] [PubMed] [Google Scholar]
- 68.Baker D, Shiau AK, Agard DA. The role of pro regions in protein folding. Curr Opin Cell Biol. 1993;5:966–70. doi: 10.1016/0955-0674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 69.Baker D, Agard DA. Kinetics versus thermodynamics in protein folding. Biochemistry. 1994;33:7505–9. doi: 10.1021/bi00190a002. [DOI] [PubMed] [Google Scholar]
- 70.Shinde U, Inouye M. The structural and functional organization of intramolecular chaperones: The N-terminal propeptides which mediate protein folding. J Biochem (Tokyo) 1994;115:629–36. doi: 10.1093/oxfordjournals.jbchem.a124386. [DOI] [PubMed] [Google Scholar]
- 71.Kojima S, Iwahara A, Yanai H. Inhibitor-assisted refolding of protease: A protease inhibitor as an intramolecular chaperone. FEBS Lett. 2005;579:4430–6. doi: 10.1016/j.febslet.2005.06.083. [DOI] [PubMed] [Google Scholar]
- 72.Yabuta Y, Subbian E, Oiry C, Shinde U. Folding pathway mediated by an intramolecular chaperone. A functional peptide chaperone designed using sequence databases. J Biol Chem. 2003;278:15246–51. doi: 10.1074/jbc.M212003200. [DOI] [PubMed] [Google Scholar]
- 73.Li Y, Hu Z, Jordan F, Inouye M. Functional analysis of the propeptide of subtilisin E as an intramolecular chaperone for protein folding. Refolding and inhibitory abilities of propeptide mutants. J Biol Chem. 1995;270:25127–32. doi: 10.1074/jbc.270.42.25127. [DOI] [PubMed] [Google Scholar]
- 74.Kobayashi T, Inouye M. Functional analysis of the intramolecular chaperone. Mutational hot spots in the subtilisin pro-peptide and a second-site suppressor mutation within the subtilisin molecule. J Mol Biol. 1992;226:931–3. doi: 10.1016/0022-2836(92)91042-n. [DOI] [PubMed] [Google Scholar]
- 75.Buevich AV, Shinde UP, Inouye M, Baum J. Backbone dynamics of the natively unfolded pro-peptide of subtilisin by heteronuclear NMR relaxation studies. J Biomol NMR. 2001;20:233–49. doi: 10.1023/a:1011243116136. [DOI] [PubMed] [Google Scholar]
- 76.Jain SC, Shinde U, Li Y, Inouye M, Berman HM. The crystal structure of an autoprocessed Ser221Cys-subtilisin E-propeptide complex at 2.0 A resolution. J Mol Biol. 1998;284:137–44. doi: 10.1006/jmbi.1998.2161. [DOI] [PubMed] [Google Scholar]
- 77.Shinde U, Inouye M. Folding pathway mediated by an intramolecular chaperone: Characterization of the structural changes in pro-subtilisin E coincident with autoprocessing. J Mol Biol. 1995b;252:25–30. doi: 10.1006/jmbi.1995.0472. [DOI] [PubMed] [Google Scholar]
- 78.Shinde U, Li Y, Chatterjee S, Inouye M. Folding pathway mediated by an intramolecular chaperone. Proc Natl Acad Sci USA. 1993;90:6924–8. doi: 10.1073/pnas.90.15.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ruan B, Hoskins J, Bryan PN. Rapid folding of calcium-free subtilisin by a stabilized pro-domain mutant. Biochemistry. 1999;38:8562–71. doi: 10.1021/bi990362n. [DOI] [PubMed] [Google Scholar]
- 80.Ruan B, Hoskins J, Wang L, Bryan PN. Stabilizing the subtilisin BPN’ pro-domain by phage display selection: How restrictive is the amino acid code for maximum protein stability? Protein Sci. 1998;7:2345–53. doi: 10.1002/pro.5560071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marie-Claire C, Yabuta Y, Suefuji K, Matsuzawa H, Shinde U. Folding pathway mediated by an intramolecular chaperone: The structural and functional characterization of the aqualysin I propeptide. J Mol Biol. 2001;305:151–65. doi: 10.1006/jmbi.2000.4233. [DOI] [PubMed] [Google Scholar]
- 82.Gallagher T, Gilliland G, Wang L, Bryan P. The prosegment-subtilisin BPN’ complex: Crystal structure of a specific ‘foldase’. Structure. 1995;3:907–14. doi: 10.1016/S0969-2126(01)00225-8. [DOI] [PubMed] [Google Scholar]
- 83.Radisky ES, King DS, Kwan G, Koshland DE., Jr The role of the protein core in the inhibitory power of the classic serine protease inhibitor, chymotrypsin inhibitor 2. Biochemistry. 2003;42:6484–92. doi: 10.1021/bi034275d. [DOI] [PubMed] [Google Scholar]
- 84.Alexander PA, Ruan B, Bryan PN. Cation-dependent stability of subtilisin. Biochemistry. 2001;40:10634–9. doi: 10.1021/bi010797m. [DOI] [PubMed] [Google Scholar]
- 85.Yabuta Y, Subbian E, Takagi H, Shinde U, Inouye M. Folding pathway mediated by an intramolecular chaperone: Dissecting conformational changes coincident with autoprocessing and the role of Ca(2+) in subtilisin maturation. J Biochem (Tokyo) 2002;131:31–7. doi: 10.1093/oxfordjournals.jbchem.a003074. [DOI] [PubMed] [Google Scholar]
- 86.Inouye M, Fu X, Shinde U. Substrate-induced activation of a trapped IMC-mediated protein folding intermediate. Nat Struct Biol. 2001;8:321–5. doi: 10.1038/86194. [DOI] [PubMed] [Google Scholar]
- 87.Shinde U, Inouye M. Folding mediated by an intramolecular chaperone: Autoprocessing pathway of the precursor resolved via a substrate assisted catalysis mechanism. J Mol Biol. 1995a;247:390–5. doi: 10.1006/jmbi.1994.0147. [DOI] [PubMed] [Google Scholar]
- 88.Bryan P, Wang L, Hoskins J, et al. Catalysis of a protein folding reaction: Mechanistic implications of the 2.0 A structure of the subtilisin-prodomain complex. Biochemistry. 1995;34:10310–18. doi: 10.1021/bi00032a026. [DOI] [PubMed] [Google Scholar]
- 89.Li Y, Inouye M. The mechanism of autoprocessing of the propeptide of prosubtilisin E: Intramolecular or intermolecular event? J Mol Biol. 1996;262:591–4. doi: 10.1006/jmbi.1996.0537. [DOI] [PubMed] [Google Scholar]
- 90.Comellas-Bigler M, Maskos K, Huber R, Oyama H, Oda K, Bode W. 1.2 A crystal structure of the serine carboxyl proteinase pro-kumamolisin; structure of an intact pro-subtilase. Structure (Camb) 2004;12:1313–23. doi: 10.1016/j.str.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 91.Eder J, Rheinnecker M, Fersht AR. Folding of subtilisin BPN’: Characterization of a folding intermediate. Biochemistry. 1993;32:18–26. doi: 10.1021/bi00052a004. [DOI] [PubMed] [Google Scholar]
- 92.Kuwajima K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins. 1989;6:87–103. doi: 10.1002/prot.340060202. [DOI] [PubMed] [Google Scholar]
- 93.Eder J, Rheinnecker M, Fersht AR. Folding of subtilisin BPN’: Role of the pro-sequence. J Mol Biol. 1993;233:293–304. doi: 10.1006/jmbi.1993.1507. [DOI] [PubMed] [Google Scholar]
- 94.Bryan P, Alexander P, Strausberg S, et al. Energetics of folding subtilisin BPN’. Biochemistry. 1992;31:4937–45. doi: 10.1021/bi00136a003. [DOI] [PubMed] [Google Scholar]
- 95.Sohl JL, Jaswal SS, Agard DA. Unfolded conformations of alphalytic protease are more stable than its native state. Nature. 1998;395:817–19. doi: 10.1038/27470. [DOI] [PubMed] [Google Scholar]
- 96.Alexander PA, Ruan B, Strausberg SL, Bryan PN. Stabilizing mutations and calcium-dependent stability of subtilisin. Biochemistry. 2001;40:10640–4. doi: 10.1021/bi010798e. [DOI] [PubMed] [Google Scholar]
- 97.Almog O, Gallagher T, Tordova M, Hoskins J, Bryan P, Gilliland GL. Crystal structure of calcium-independent subtilisin BPN’ with restored thermal stability folded without the prodomain. Proteins. 1998;31:21–32. doi: 10.1002/(sici)1097-0134(19980401)31:1<21::aid-prot3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 98.Wang L, Ruan B, Ruvinov S, Bryan PN. Engineering the independent folding of the subtilisin BPN’ pro-domain: Correlation of pro-domain stability with the rate of subtilisin folding. Biochemistry. 1998;37:3165–71. doi: 10.1021/bi972741r. [DOI] [PubMed] [Google Scholar]
- 99.Ruvinov S, Wang L, Ruan B, et al. Engineering the independent folding of the subtilisin BPN’ prodomain: Analysis of two-state folding versus protein stability. Biochemistry. 1997;36:10414–21. doi: 10.1021/bi9703958. [DOI] [PubMed] [Google Scholar]
- 100.Fu X, Inouye M, Shinde U. Folding pathway mediated by an intramolecular chaperone. The inhibitory and chaperone functions of the subtilisin propeptide are not obligatorily linked. J Biol Chem. 2000;275:16871–8. doi: 10.1074/jbc.275.22.16871. [DOI] [PubMed] [Google Scholar]
- 101.Shastry MC, Udgaonkar JB. The folding mechanism of barstar: Evidence for multiple pathways and multiple intermediates. J Mol Biol. 1995;247:1013–27. doi: 10.1006/jmbi.1994.0196. [DOI] [PubMed] [Google Scholar]
- 102.Hayashi T, Matsubara M, Nohara D, Kojima S, Miura K, Sakai T. Renaturation of the mature subtilisin BPN’ immobilized on agarose beads. FEBS Lett. 1994;350:109–12. doi: 10.1016/0014-5793(94)00744-6. [DOI] [PubMed] [Google Scholar]
- 103.Matsubara M, Kurimoto E, Kojima S, Miura K, Sakai T. Achievement of renaturation of subtilisin BPN’ by a novel procedure using organic salts and a digestible mutant of Streptomyces subtilisin inhibitor. FEBS Lett. 1994;342:193–6. doi: 10.1016/0014-5793(94)80499-0. [DOI] [PubMed] [Google Scholar]
- 104.Yabuta Y, Subbian E, Takagi H, Shinde U, Inouye M. Folding pathway mediated by an intramolecular chaperone: Dissecting conformational changes coincident with autoprocessing and the role of Ca(2+) in subtilisin maturation. J Biochem. 2002;131:31–7. doi: 10.1093/oxfordjournals.jbchem.a003074. [DOI] [PubMed] [Google Scholar]
- 105.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–30. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 106.Karplus M. The Levinthal paradox: Yesterday and today. Fold Des. 1997;2:S69–75. doi: 10.1016/s1359-0278(97)00067-9. [DOI] [PubMed] [Google Scholar]
- 107.Franke AE, Danley DE, Kaczmarek FS, et al. Expression of human plasminogen activator inhibitor type-1 (PAI-1) in Escherichia coli as a soluble protein comprised of active and latent forms. Isolation and crystallization of latent PAI-1. Biochim Biophys Acta. 1990;1037:16–23. doi: 10.1016/0167-4838(90)90096-x. [DOI] [PubMed] [Google Scholar]
- 108.Huntington JA, Pannu NS, Hazes B, Read RJ, Lomas DA, Carrell RW. A 2.6 A structure of a serpin polymer and implications for conformational disease. J Mol Biol. 1999;293:449–55. doi: 10.1006/jmbi.1999.3184. [DOI] [PubMed] [Google Scholar]
- 109.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000;407:923–6. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 110.Subbian E, Yabuta Y, Shinde UP. Folding pathway mediated by an intramolecular chaperone: Intrinsically unstructured propeptide modulates stochastic activation of subtilisin. J Mol Biol. 2005;347:367–83. doi: 10.1016/j.jmb.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 111.Subbian E, Yabuta Y, Shinde U. Positive selection dictates the choice between kinetic and thermodynamic protein folding and stability in subtilases. Biochemistry. 2004;43:14348–60. doi: 10.1021/bi048397x. [DOI] [PubMed] [Google Scholar]
- 112.Bryan PN. Prodomains and protein folding catalysis. Chem Rev. 2002;102:4805–16. doi: 10.1021/cr010190b. [DOI] [PubMed] [Google Scholar]
- 113.Siezen RJ. Subtilases: Subtilisin-like serine proteases. Adv Exp Med Biol. 1996;379:75–93. doi: 10.1007/978-1-4613-0319-0_9. [DOI] [PubMed] [Google Scholar]
- 114.Tangrea MA, Bryan PN, Sari N, Orban J. Solution structure of the pro-hormone convertase 1 pro-domain from Mus musculus. J Mol Biol. 2002;320:801–12. doi: 10.1016/s0022-2836(02)00543-0. [DOI] [PubMed] [Google Scholar]
- 115.Tangrea MA, Alexander P, Bryan PN, Eisenstein E, Toedt J, Orban J. Stability and global fold of the mouse prohormone convertase 1 pro-domain. Biochemistry. 2001;40:5488–95. doi: 10.1021/bi0026472. [DOI] [PubMed] [Google Scholar]
- 116.Nour N, Basak A, Chretien M, Seidah NG. Structure-function analysis of the prosegment of the proprotein convertase PC5A. J Biol Chem. 2003;278:2886–95. doi: 10.1074/jbc.M208009200. [DOI] [PubMed] [Google Scholar]
- 117.Nour N, Mayer G, Mort JS, et al. The cysteine-rich domain of the secreted pro-protein convertases PC5A and PACE4 functions as a cell surface anchor and interacts with tissue inhibitors of metalloproteinases. Mol Biol Cell. 2005;16:5215–26. doi: 10.1091/mbc.E05-06-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dikeakos JD, Lacombe MJ, Mercure C, Mireuta M, Reudelhuber TL. A hydrophobic patch in a charged alpha-helix is sufficient to target proteins to dense core secretory granules. J Biol Chem. 2007;282:1136–43. doi: 10.1074/jbc.M605718200. [DOI] [PubMed] [Google Scholar]
- 119.Malfait AM, Arner EC, Song RH, et al. Proprotein convertase activation of aggrecanases in cartilage in situ. Arch Biochem Biophys. 2008;478:43–51. doi: 10.1016/j.abb.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 120.Jutras I, Seidah NG, Reudelhuber TL, Brechler V. Two activation states of the prohormone convertase PC1 in the secretory pathway. J Biol Chem. 1997;272:15184–8. doi: 10.1074/jbc.272.24.15184. [DOI] [PubMed] [Google Scholar]
- 121.Leduc R, Molloy SS, Thorne BA, Thomas G. Activation of human furin precursor processing endoprotease occurs by an intramolecular autoproteolytic cleavage. J Biol Chem. 1992;267:14304–8. [PubMed] [Google Scholar]
- 122.Wan L, Molloy SS, Thomas L, et al. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell. 1998;94:205–16. doi: 10.1016/s0092-8674(00)81420-8. [DOI] [PubMed] [Google Scholar]
- 123.Kucerova H, Chaloupka J. Intracellular serine proteinase behaves as a heat-stress protein in nongrowing but as a cold-stress protein in growing populations of Bacillus megaterium. Curr Microbiol. 1995;31:39–43. doi: 10.1007/BF00294632. [DOI] [PubMed] [Google Scholar]
- 124.Kucerova H, Hlavacek O, Vachova L, Mlichova S, Chaloupka J. Differences in the regulation of the intracellular Ca2+-dependent serine proteinase activity between Bacillus subtilis and B. megaterium. Curr Microbiol. 2001;42:178–83. doi: 10.1007/s002840010200. [DOI] [PubMed] [Google Scholar]
- 125.Vachova L. Activation of the intracellular Ca(2+)-dependent serine protease ISP1 of bacillus megaterium by purification or by high Ca2+ concentrations. Biochem Mol Biol Int. 1996;40:947–54. [PubMed] [Google Scholar]
- 126.Vachova L, Kucerova H, Benesova J, Chaloupka J. Heat and osmotic stress enhance the development of cytoplasmic serine proteinase activity in sporulating Bacillus megaterium. Biochem Mol Biol Int. 1994;32:1049–57. [PubMed] [Google Scholar]
- 127.Weisz OA. Organelle acidification and disease. Traffic. 2003;4:57–64. doi: 10.1034/j.1600-0854.2003.40201.x. [DOI] [PubMed] [Google Scholar]