

1. OVERVIEW
Protein design is a valuable tool for understanding the fundamental factors that dictate protein structure and function. The field of protein design has seen significant progress over the past 30 years, with a growing range of applications in areas of research from fundamental biochemistry to biotechnology and medicine. Protein design is no trivial task. It is an ultimate test of our understanding of structure–function relationships and requires a combined knowledge of physics, chemistry, and biology. Evolution is nature’s method of designing proteins, which has been very effective in producing scaffolds with exquisite structural details, breath-taking efficiency, and high selectivity. Rapid advancement of computer applications in biotechnology now enables one to model the evolution of a particular protein on a human time scale, instead of an evolutionary time scale; however, it is still quite a challenge to select an optimal solution from the enormous amount of mutations and to understand how these selective factors relate to the function of the protein. From the early examples of designing polypeptides or proteins that are stable in aqueous environments to today’s novel functional metalloproteins, protein design has advanced to a stage where it is possible to create structures never before seen in nature, with functions not found in nature and novel sequences not derived from nature while exhibiting desired properties.
The scope of this Review is to discuss the construction of metal sites in designed protein scaffolds. We categorize the effort of designing proteins into redesign, which is to rationally engineer desired functionality into an existing protein scaffold,1–9 and de novo design, which is to build a peptidic or protein system that is not directly related to any sequence found in nature yet folds into a predicted structure and/or carries out desired reactions.10–12 We will analyze and interpret the significance of designed protein systems from a coordination chemistry and biochemistry perspective, with an emphasis on those containing constructed metal sites as mimics for metalloenzymes.
Each functional designed metalloprotein system requires both a stable protein scaffold or “ligand” and an incorporated metal-binding site. These two requirements are the focus of previous protein design reviews, authored by the leaders in the field. Part of this literature addressed how protein scaffolds are designed in terms of fundamental physical properties such as the energy landscape associated with folding and packing of a protein from a biophysical perspective, which is the foundation of designing functional proteins.11–18 Knowledge of the critical factors that govern protein stability and other properties allows researchers to begin incorporating cofactors to pursue better performance or novel functions of designed proteins. Reviews on this topic elaborated the strategies of certain metal cofactor functionality and characterization of designed metalloproteins.1,10,19–26 The important milestones of protein design at its early stage are mostly included in these articles; nonetheless, we feel that it is necessary to present a comprehensive review of the most up-to-date designed protein systems with a particular emphasis on the exciting advancement of functional metalloenzymes. The focus of this Review is to highlight recent examples of different types of metal centers and summarize strategies for incorporating functional metal-binding sites into designed protein/peptide scaffolds.
Before delving into specific cases of designed protein systems, we will delineate some general considerations as a foundation for both protein redesign and de novo design. The fundamental interactions that play important roles in determining protein structures are hydrophobic, electrostatic, hydrogen bond (H-bond), and van der Waals interactions. Conceptually, creating a stable protein scaffold is a trade-off between the unfavorable entropy and favorable enthalpy upon protein folding, in addition to the entropic term of the surrounding water molecules,27 all of which eventually lead to the notion of positive and negative design that distinguishes the unique, native fold versus other folded states.28 Accordingly, in the process of designing a stable protein construct, we need to consider how to utilize hydrophobic interactions as a driving force for folding, electrostatic interactions to build salt bridges, H-bonds to create secondary structures, and van der Waals interactions for packing. Additionally, when designing helix bundles, the helical dipole is an important factor to take into account because it will contribute to the alignment of helices. When it comes to incorporating a metal-binding site, it is critical to consider whether the site is a preorganized ligand environment or if the metal ions would direct the ligand coordination.29 There are reported examples where metal ions are responsible for protein folding, in which case the binding of metal ions is the major force for overcoming the unfavorable entropy upon protein folding.30–32 Moreover, ligand recognition, metal specificity, and substrate specificity (if applicable) are the next level of design goals.
Rational protein design has been aided by the development of automated algorithms, despite the tremendous complexity associated with protein structures and sequences and the degrees of freedom of the calculation. Several strategies have been developed to reduce the degrees of freedom and improve search algorithms for energy minimization. One of the earliest programs enlisted for protein design is InsightII/Discover (MSI), which was applied to the development of helical bundle proteins.33–38 This program uses molecular mechanics to achieve energy minimization. The rational design algorithm Dezymer, for example, searches proteins of known structure for locations where side chain rotamers can be introduced to form a site of known coordination.39,40 This program is particularly useful in the redesign of metalloproteins because it can identify a novel metal-binding site based on user-defined geometry in a protein of known structure. On the other hand, the numerical computer method, Rosetta, developed in the laboratory of David Baker, takes short fragments of proteins and assembles them by a Monte Carlo strategy to form a native-like protein conformation.41 The prerequisite for Rosetta design is a library of fragments representing all of the possible local structures of various native proteins. After the Monte Carlo assembly, the folding and stability of the designed sequences are evaluated based on the comparison between conformational parameters and known protein structures. Metal Search, developed by Clarke and Yuan, is written specifically for designing tetrahedral metal-binding sites in proteins.42
2. PROTEIN REDESIGN
2.1. Making Use of Native Proteins: Protein Redesign
Metalloproteins are essential biomacromolecules that play important roles ranging from structural stabilization to chemical signaling and catalysis in metabolic processes necessary to sustain life on the planet. Over millions of years, nature has evolved a set of metalloenzymes that function as electron transfer or catalytic centers. Biochemists, biophysicists, and chemists are intrigued by the fundamental molecular transformations that occur within various metalloenzymes. One approach to studying these functions is to design and engineer new metal-binding sites into existing native proteins, which is the focus of this section: protein redesign.1,43,44 The ultimate goal of protein redesign is to impart improved or novel properties, for example, new activities, inverted stereoselectivity, or new substrate specificity, into existing proteins, that is, to teach the old enzyme new “tricks”.44,45 What protein design teaches us about the structure–function relationship will eventually allow us to harness the richness of native protein structures and the diverse functions of metalloproteins to construct new proteins for biomedical and pharmaceutical applications.
To create novel artificial metalloenzymes, a fundamental understanding of the structure–function relationship is necessary to predict and control the active site properties of a particular system. More often than previously believed, native proteins exhibit intrinsic catalytic promiscuity, using a single active site to catalyze more than one chemical transformation. 46,47 The microenvironment modification of active sites can allow a wider range of chemical transformations to occur within naturally existing scaffolds. Moreover, metalloenzymes have been evolved to exhibit optimal catalytic efficiencies in the context of their metabolic transformations; however, the activity of a single isolated metalloenzyme is not necessarily the “best”. This leaves chemical space for protein redesign to achieve modified activity and/or specificity.
The advantage of a protein redesign strategy is that it can bypass the problem of developing a stable protein fold because many native proteins have remarkable adaptability for changes. Protein redesign strategy has proven to be one of the most effective approaches in the design and engineering of artificial metalloenzymes. Although biochemical and biophysical studies generally focus on features that result in the modification of functions, design allows us to incorporate only those features that are believed to be necessary for reactivity. By applying a minimal number of changes to convert a known protein to a metalloprotein with a specific cofactor and activity, the basic requirements for a certain activity can be examined in the absence of complications from the limits of stability and symmetry of the scaffold. One can also seek to improve upon the native function of a metalloprotein by making systematic changes, favoring greater stability, altering inhibition profiles, or affecting reactivity as needed for a particular purpose.
2.2. Protein Redesign Based on Functions
2.2.1. Redesign of Zinc Finger Structural Sites
Zinc fingers (ZFs) are ubiquitous, small protein domains that bind to DNA to regulate gene expression. ZFs are now classified into at least 14 different classes, based on the metal-coordinating ligand as well as the protein fold, if known.48 ZFs utilize four Cys and/or His residues for tetrahedral metal binding, forming a Zn(Cys)x(His)4–x coordination site. Three commonly studied ZF proteins are those with Cys4, Cys2HisCys, and Cys2His2 sites.49 Cys4 ZFs constitute one class, which are usually found in nuclear receptors and play important roles in developmental biology.50–68 Another class of ZFs is the nucleic acid binding proteins that have Cys2HisCys binding.69–81 The third, best-known, and most common ZF motif in eukaryotes is the His2Cys2 metal-binding site.48,75,82–87 The secondary structure of ZF proteins consists of an α-helix packed against a two-stranded antiparallel β-sheet. The opposite side of the α-helix with respect to β-sheet packing associates to DNA for gene regulation, whereas the β-hairpin region does not interact with DNA but instead is a structural component of the domain.88–90 For all three types of ZF domains, Zn(II) is almost always necessary for protein folding yet is not directly involved in the DNA-binding activity.91 Therefore, Zn(II) binding does not directly regulate gene expression, but instead it stabilizes the active conformation of the ZF protein.84
Although there have been extensive studies in the field of ZF chemistry, there are still unanswered questions regarding this family of peptides. For example, many ZFs are very similar in sequence, such as NZF-1 and MyT1, when others share only a few similar residues (including metal-binding ligands).92–104 These sites are studied with the goal of understanding how noncoordinating ligands influence the folding and DNA binding properties of ZFs, and how second-sphere residues affect binding, with respect to both the thermodynamic and the kinetic parameters. Additionally, Zn(II) binds to the ZF site with high affinities (Kd ≈ nanomolar to femtomolar), which leads to the question of how the Zn(II)-bound form of ZF proteins is inactivated in the cell.105 Consider two different ZFs, Cys3His and Cys2His2Cys, the metal-binding sites are constructed with the same amino acids, three Cys residues and one His residue (the second His of the Cys2His2Cys does not coordinate to Zn(II)).70 It is not yet known why these proteins fold differently around the same ligand-set, whereas other ZF proteins with different metal-coordinating ligands retain the generic ZF fold as mentioned above. The role of the noncoordinating ligands that are capable of metal binding is yet to be understood completely. In most cases, the H-bonds of noncoordinating residues of the ZFs are key to DNA binding.85,91,106–112 Understanding the interaction between ZFs and DNA binding could lead to the development of designed ZFs to selectively target certain DNA sequences for gene therapy applications.85,86,91,106,110,113,114
Much work has been conducted on structural studies of ZF proteins as well as studies that focus on the alteration of binding and recognition of the ZF to DNA. These types of studies will not be focus of this Review, but will be summarized with references here, and readers are encouraged to read the extensive reviews by Negi et al., Matthews et al., and Kaptein, for further direction to structural studies.48,49,91 Structural and nucleic acid binding properties to elucidate DNA binding details are extensively outlined elsewhere.115–119 Multiple ZF mutations have been made to change how and where the domain will bind to and recognize DNA.112,120–131 Much effort has focused on structural swapping, where the secondary structure is altered to combine either the native α-helix or β-sheet of a ZF and the opposite fold from another protein, but the binding site remains unaltered.132–138 Last, many studies have been conducted on designing multidomain ZF proteins to bind to extended regions of DNA (>16 base pairs).139–141 Linker-domains, where two sections of ZF regions are separated by a linker, were also introduced for ZFs to bind to an extended region of DNA.142–150 Alterations in DNA binding have also been used to result in bending the DNA upon binding,151,152 and even protein stability studies to promote protein folding in the absence of the metal ions.153–155 Further, Seelig and Szostak reported a novel RNA ligase designed by attaching a Zn(Cys)4 ZF protein with randomly mutated loop region onto an mRNA and using mRNA display to select for the optimal mutation.156 Besides DNA binding and transcription regulation, there are other biophysical parameters to be studied by exploiting the stability and the small size of ZF proteins. These include metal ion affinities and selectivity, geometries of native Zn(II) and other metal ions, thermodynamic properties of packing and folding, and, most relevant to this Review, constraints related to specific ligand binding and nonphysiological catalytic activity.84
The first logical step in designing a ZF protein is to understand the residues that influence the structural integrity, folding induction, and DNA binding capability of the ZF. Green et al. substituted a Cys2His2 ZF motif with a Cys4 binding site to examine whether this change would influence the ZF properties and the interchangeability of different ZF domains in one protein.157 Zif268 is a 533-residue, three ZF-containing murine DNA-binding protein, where the three ZF sites all have Cys2His2 motifs.108,158,159 Several mutants corresponding to a combination of mutations with Cys4 (ZF1, ZF2, ZF3) and Cys2HisAla (ZF1, ZF3) were generated, and their DNA binding capability was examined (Figure 1). The results suggest that the three ZF motifs behave differently upon coordinating to Zn(II), leading to dissimilar DNA consensus sequence binding ability. For example, the individual Cys4(1) and Cys4(3) mutations were able to bind to DNA, while the combination of Cys4(1, 3), Cys4(2, 3) could not. Similarly, Hori et al. reported the substitution of a Cys2His2 motif into a His4 binding site, aiming to understand the folding and DNA recognition of this novel ZF site.160 Two proteins, a 32-residue H4Sp1f2 containing one ZF domain and a 94-residue H4Sp1 containing three ZF domains, derived from Sp1 ZF protein, were studied. Sp1 ZF domains have been well characterized,161–167 and the solution structures are available,167 providing a solid starting point for redesigning a His4 site. Based on circular dichroism (CD) spectra, Zn(II) binds to H4Sp1f2, forming a more ordered conformation than other divalent metal ions examined (Co(II), Ni(II), and Cd(II)). Moreover, investigation into the DNA binding properties of H4Sp1 with different divalent metal ions demonstrated that only Zn(II)-reconstituted H4Sp1 bound strongly to the DNA GC-box.160
Figure 1.

Mutations made on zif268 DNA-binding domain to generate ZF sites. Reproduced with permission from ref 157. Copyright 1998 Portland Press.
To study the importance of each of the four coordinating ligands of a classical Cys2His2 ZF protein, Nomura and Sugiura synthesized and investigated a series of peptides corresponding to a Cys2His2 ZF domain of Sp1.168 The derivatives each contain a single mutation to substitute the coordinating ligand individually, with a Gly and/or an Ala residue. For each mutant containing only three metal coordinating ligands (two His and one Cys), 1:1 Zn(II):protein binding was observed, as well as α-helix formation upon the addition of Zn(II). The capability of folding induction for some of the mutants by Zn(II) was demonstrated by the increase of Trp fluorescence due to increased hydrophobicity, and the increase in α-helical features as measured by CD. In particular, they examined the induction of α-helix formation upon Zn(II) binding for the mutant ZF2(CCGH) (with a His23Gly mutation) and ZF2(CCHG) (with a His27Gly mutation) and found that the former ZF protein does not show α-helix signature in the presence of Zn(II), while the latter forms an α-helix even without one of the coordinating His residues. On the basis of this observation, they concluded that His23 is important in α-helix induction.168 Furthermore, all Zn(II)-bound Sp1 mutants exhibit hydrolytic activity toward the substrate p-nitrophenyl acetate (pNPA). As the first reported catalytically active ZF proteins, these will be discussed in more detail in a later section (2.2.2: Redesign of Zinc Hydrolytic Centers).169
One of the most important strategies in protein redesign is to incorporate a novel metal-binding site into a protein that does not have the desired binding site or function in its natural state. This strategy allows one to investigate the influence of protein secondary structure on the metal-binding properties and further functionalities of the metal center; at the same time, such designs test the robustness of a particular metal-binding site in maintaining its properties. Moreover, novel metal-binding sites have the potential of possessing novel functions. To test this, ZF binding sites were engineered into three proteins absent of natural Zn(II)-binding sites: the B1 domain of Streptococcal protein G,170 thioredoxin (Trx),171 and Antennapedia homeodomain (Ant).172,173
Klemba et al. designed a His3Cys site into the B1 domain of IgG-binding protein G, a 56-residue domain consisted of a four-stranded β-sheet crossed by a single α-helix. This is an ideal scaffold for the purpose of incorporating a ZF binding site due to its high stability and lack of pre-existing His and Cys residues.170 The potential metal-binding sites were generated with a computer program, Metal Search,42 using the NMR-derived coordinates of WT-B1 domain as a starting point. Toward selecting the best model, the authors eliminated any potential metal-binding sites with more than one residue inside the hydrophobic core, to maintain the stability of the protein. The final model contains Thr16His, Thr18His (β-strand 2), Phe30His, and Tyr33Cys (α-helix) mutations in addition to a mutation to modulate the sterics around the binding site (Figure 2). Out of the B1 domain variants, named Zβ1L, Zβ1A, and Zβ1M with the final letter designating the additional mutation other than the metal-coordinating ligands, only Zβ1L folds in its apo-form. The addition of Co(II), Zn(II), and Cd(II) led to the folding of the other two proteins, indicating the binding of these divalent metal ions. Moreover, the Co(II) d–d band of the three mutants was indicative of a tetrahedral geometry, and the 113Cd NMR spectrum of Zβ1L showed coordination of the metal ion by one sulfur and three nitrogen donors.
Figure 2.

Ribbon diagram of Zβ1M. Reproduced with permission from ref 170. Copyright 1995 Nature Publishing Group.
Next, Wisz et al. engineered a Cys2His2 ZF site into Trx, a small protein naturally devoid of metal centers.171 Using Dezymer, an automated protein design program that looks for potential metal-binding sites in native proteins by varying side chains while maintaining the protein backbone structure,39,40 seven different Cys2His2-Trx proteins were selected with a putative metal-binding site at different positions of the protein scaffold. Additional mutations were conducted to remove surface residues that could bind metal ions and disulfide-forming residues. Five of these proteins folded upon metal binding, all of which showed tetrahedral coordination geometry based on Co(II) absorption spectra. Detailed analyses of protein stability as well as metal-binding affinities were carried out, providing the following critical insight into the introduction of a metal-binding site into a native protein: (1) steric clashes must be avoided to maintain the stability of the protein fold; (2) the metal-binding site should balance the metal coordination preference with the fold of the surrounding protein environment; and (3) metal ions can influence the protein stability by interacting with the transition state as well as the unfolded state. While at this point this latter interaction is not completely understood, it is still instructive for future endeavors. Overall, the combination of optimization of the metal-bound folded state and negative design (raising the energy for the metal-bound unfolded state) should be adopted to construct a stable metal-binding site.171
Most recently, Hori and Sugiura achieved the conversion of Ant to a ZF protein resulting in a novel “Antennafinger” (Ant-F) protein.172,173 By analyzing the inherent sequence homology of a consensus ZF domain and the known protein scaffolds from Genomenet Motif, a system to look for analogous sequences from the Protein Data Bank (PDB), a small 54-residue sequence, Ant-WT, was chosen. Ant-WT is a stable structure-determined portion (PDB code: 1SAN) of a mutant derived from Ant whose function is well understood.174 Moreover, this domain does not contain potential metal-binding or disulfide-forming residues. Ant-F was designed by substituting Thr, Leu, Arg, and Glu residues to two Cys and two His residues. Apo-Ant-F folds into an α-helical structure, resembling that of apo-Ant-WT. Zn(II) binding resulted in a conformational change, which, in turn, decreased the DNA binding activity of Ant-F by 2.5-fold as compared to Ant-WT.172 Subsequent studies on Ant-F and Ant-F-H1, a second generation of Ant-F ZF protein containing mutations to stabilize the helical conformation, were performed to understand the thermodynamic aspects of folding and metal binding to this designed ZF protein. On the basis of denaturation studies of both apo- and holo- forms of Ant-F and Ant-F-H1, Hori et al. proposed a model to estimate the relationship between the free energies of relevant states (Figure 3), showing that the stability of apo-structures affects the binding of Zn(II) and that metal binding to the unfolded protein also plays an important role. These findings are consistent with what was proposed in Wisz’s report in 1998 described above,171 demonstrating that the intricate interactions between folding and metal binding determine the overall stability and the energetic aspects of the conformational changes in a ZF protein.
Figure 3.

Estimated model of the relationship between the protein stability and Zn(II) binding upon the conformational change in (A) Ant-F, and (B) Ant-F-H1. F and U stand for folded and unfolded states, respectively. Reproduced with permission from ref 173. Copyright 2004 American Chemical Society.
2.2.2. Redesign of Zinc Hydrolytic Centers
The major difference between structural zinc sites, as in ZFs, and catalytic sites, as in CAII, is that structural sites have saturated coordination spheres, with amino acid ligands His and/or Cys bound to the zinc ion, whereas catalytic centers have at least one vacant site allowing for solvent (water) or substrate coordination. Bound water is a feature of catalytic zinc centers, as it can be activated for ionization, polarization, or displaced by another ligand, such as an alcohol substrate.175 We review herein the redesign of carbonic anhydrase (CA) to achieve higher catalytic efficiency, higher Zn(II) affinity, or novel functions, the redesign of a structural ZF site into a hydrolytic site, and the computational redesign of a Zn(II)-containing glyoxalase II enzyme.
CA is a Zn(II)-containing enzyme that is found in almost all plants, algae, and mammals, whose function is fundamental to many eukaryotic biological processes such as respiration, ion transport, and acid–base balance.116,176,177 The physiological reaction catalyzed by CA is the interconversion between CO2 and HCO3−; however, the catalytic promiscuity of CA allows for the rational design of the active site to exhibit increased activity toward ester hydrolysis. High-resolution crystal structures of CA reveal that Zn(II) is coordinated to three His residues (His94, 96, 119) and a water molecule/hydroxide ion, that is H-bonded to the hydroxyl side chain of a nearby residue, Thr199, which, in turn, forms an H-bond with Glu106 (Figure 4).178–181 The active site is contained in a twisted beta-sheet. The redesign of the active site of CA involves modification of the first or outer coordination spheres. Fierke, Christianson, and co-workers pioneered studies mutating both coordinating and noncoordinating residues around the Zn(II) center in CA.182–191 Specifically, these researchers substituted one or more of the Zn(II)-coordinating His residues (His94, 96, 119) with a neutral, nonpolar, noncoordinating amino acid residue (Ala), carboxylate-containing residues (Asp, Glu), a thiolate-containing residue (Cys), and carboxyamide-containing residues (Gln, Asn), and observed varied Zn(II)-binding affinities and catalytic rates and efficiencies.183–186 The substitution of any coordinating His residue decreased the Zn(II)-binding affinity by ~104–105-fold. The pKa of Zn(II)-bound water, representing conversion to the active Zn(II)-hydroxide species, increased by at least one pH unit when His residues were replaced with other residues (pKa(WT-CAII) = 6.8 ± 0.1,192 pKa(mutants) ≥ 8.4). In addition to catalyzing physiological CO2 hydration, CAII can also catalyze ester hydrolysis, which is another important hallmark of its activity. The catalytic efficiency of both pNPA hydrolysis and CO2 hydration decreased significantly with the substitutions described above. Although these studies were reviewed in an earlier contribution, 182 we feel it is important to revisit them briefly here because they have provided important insight into the role of coordinating His residues, outer-sphere residues, and the mechanistic aspects of CO2 hydration. All of these details are critical for the de novo designed CA mimic systems, which will be reviewed in a later section (3.3.1.2: Multifunctional Metal Centers: Structural and Catalytic Sites in One Design). Specifically, the native CA studies showed that the coordinating His residues are essential in maintaining the Zn(II)-binding affinity, the pKa of Zn(II)-bound water, the stability of Zn(II)-bound hydroxide, and the negatively charged transition state for CO2 hydration. Moreover, these His residues might also play a role in forcing the tetrahedral coordination geometry, found in native CAII, over trigonal bipyramidal geometry.186
Figure 4.

CA H-bonding network based on the crystal structure of CA (PDB code: 3KS3).193
The modification of the second coordination sphere of the Zn(II) site in CA focuses on the alteration of hydrogen bonding (H-bonding) interactions, particularly by substituting Thr199, Thr200, and Gln92 residues that were proven to be important in maintaining the H-bonding network around the active site.187–191 In WT-CAII, Thr199 accepts an H-bond from Zn(II)-bound hydroxide and donates an H-bond to Glu106, building a proton transfer chain to facilitate CO2 hydration.181 The crystal structure of Co(II)-substituted Thr199Cys-CAII revealed that the substitution of Thr199 with a Cys residue leads to the displacement of the Zn(II)-bound water by the thiolate ligand.187,194 The additional coordinating ligand, Cys, resulted in a Zn(II) affinity increase in of 4-fold; however, the catalytic efficiency of CO2 hydration was compromised substantially. Similarly, Thr199 was replaced with Glu, Asp, or His residues.191 Both mutants with residues containing carboxylate groups (Thr199Glu and Thr199Asp) showed displacement of Zn(II)-bound water by the carboxylate ligand in their crystal structures, although the O(carboxylate)–Zn(II) distances were different. For the Thr199Asp mutant, the two oxygens from the carboxylate of Asp bound Zn(II) in a bidentate fashion with a Zn(II)–O distance of 2.5 Å, whereas the carboxylate in the Thr199Glu mutant displaced a water molecule and showed a unidentate binding mode with a Zn(II)–O distance of 2.2 Å. Only Thr199Glu exhibited a significant increase in the Zn(II)-binding affinity (200-fold), demonstrating the influence of metal–ligand separation and side chain torsion angles on protein–metal affinity. In contrast to Thr199Asp and Thr199Glu, Thr199His did not coordinate to Zn(II). The Zn(II) affinity of the Thr199His mutant decreased by as much as 20-fold as compared to that of the WTCAII [Kd = 7.7(±1.3) × 10−11 M for Thr199His, Kd = 4(±1) × 10−12 M for WTCAII]. In addition to Thr199, residues at the 200 position are also polar residues (Thr, Asn, or His) that influence the catalytic efficiency of CO2 hydration for different CA isozymes.195 Krebs et al. substituted Thr200 with a Ser residue in CAII, with little to no change in the catalytic efficiency for CO2 hydration, but a 4-fold increase in the catalytic efficiency for pNPA hydrolysis was observed.188 The crystal structure of the Thr200Ser mutant showed that this substitution resulted in relocation of His64, which was thought to change the solvent structure around the active site and to modify the proton transfer chain. Moreover, two additional important second-sphere residues, Gln92 and Glu117, H-bonded to the imidazoles of His94 and His119, respectively, were mutated to examine alteration of the H-bond length (Gln92Asn, Glu117Asp) or charge (Gln92Glu) of the H-bond acceptor.189,190 These studies illustrate the importance of these H-bonding residues for maintaining proper Zn(II) affinity, but also suggest that changing the length or charge of donor residues does not alter the affinity significantly (≤3-fold decrease in Zn(II) affinity). More importantly, these second-sphere residues indirectly influence the stability of the pentacoordinate transition state during CO2 hydration, altering the catalytic efficiency. The efforts reviewed here provide an important foundation for elucidating the structure–function relationship of the active site of CA, shedding light on the de novo design of Zn(II) hydrolytic sites.
The substitution of residues on the secondary coordination sphere, Thr200Cys/His and Asn67His, resulted in increased catalytic efficiency for pNPA hydrolysis.188,196 Additionally, the substitution of Thr200 by His, Leu, Ile, Lys, Arg, Ser, etc., led to a variation of substrate specificity, which might be explained by the dissimilar van der Waals volume of the side chains and their interactions with different substrates. Furthermore, a directed evolution method was used to produce an esterase toward a less active, bulkier substrate 2-naphthyl acetate (2NA).197 The most successful variant with Thr200Ala and Ala65Val mutations, where the Ala to Val substitution was thought to increase the “affinity” of the hydrophobic substrate, raised the catalytic efficiency toward large aromatic substrates. Later, variants of human carbonic anhydrase II (hCAII) were designed by Höst et al. with the goal of modulating the size and increasing the shape of the hydrophobic pocket for ester binding.198,199 The hydrophobic pocket close to the Zn(II) site in hCAII is defined by four amino acids: Val121, Val143, Leu198, and Trp209.192 In this work, increasing the size of the pocket was achieved by Val121Ala/Val143Ala mutations, which showed a significant increase in the catalytic efficiency toward larger substrates with longer acyl chains.198 Moreover, a Thr200Ala mutation was introduced in addition to the two Val mutations, creating a hCAII derivative with a catalytic efficiency 5 orders of magnitude better than that of the WT-hCAII toward pNPA hydrolysis.199
Hydrolytic activity has also been demonstrated in modified ZF sites taking a protein redesign approach. The key component of a catalytic Zn(II) site as compared to a structural site is the presence of at least one vacant site in the coordination sphere. On the basis of a wild-type (WT) ZF protein ZF(CCHH), Nomura et al. incorporated an unsaturated Zn(II) binding site by mutating the coordinating His and Cys residues into an Ala or a Gly.169 CD spectroscopy showed that Zn(II) still induces α-helix formation for some of the mutants. Because of the unsaturation of the coordination at the Zn(II) ion, all of the mutants exhibited pNPA hydrolysis activity. Furthermore, increasing the number of ligating His residues led to higher catalytic activity, due to the increased Lewis acidity of Zn(II). Inhibition studies were carried out to confirm the catalytic role of the Zn(II)-bound protein complex. Interestingly, the Zn(II)-bound complex of ZF(HHHH) exhibited enantioselective esterase activity toward Boc-glutamine 4-nitrophenyl ester (Gln-ONp), indicating that the Zn(II) active site in this protein is situated in an asymmetric environment. Follow-up studies using the ZF protein derivative ZF(HHHH) (Zn•1) and a previously reported three-tandem ZF protein164 derivative Zn•2 (where two Cys residues were mutated to two His) showed that these Zn–protein complexes could hydrolytically cleave DNA.200 A supercoiled plasmid DNA, pUC19GC, was used as a substrate to examine the nuclease activity. Because of preferred binding of Zn•2 to the GC box of the DNA duplex, selective hydrolytic cleavage of DNA could be achieved with moderate activity. To improve the DNA cleavage activity, Negi et al. combined ZF structural sites with a nuclease site into one design, yielding ZWH4.201 ZWH4 contains four Zn(II)-binding sites, three of which are Cys2His2 ZF sites and the other a His4 hydrolytic site. Zinc-bound ZWH4 was shown to catalyze DNA cleavage with a clear production of different forms of products depending on the protein concentration. Specifically, at 25, 50 equiv of ZWH4/DNA, the cleavage reaction produced form II (a nicked circular form), a mixture of form II and form III (a linear form) products, respectively. This report shows how the further control of the specificity of artificial nucleases can be achieved.
Many designed ZF nucleases have been developed to obtain alternative methods to gene therapy. Porteus and Baltimore analyzed gene targeting with homodimers consisted of an artificial ZF binding domain, QQR, to create a DNA double-stranded break in a genomic target.202 Natsukasa et al. also placed a ZF protein on either side of a functional DNA cleavage linker.203 Ideally, these new systems will be capable of unique and specific DNA cleavage and could ultimately be used to manipulate genetic information to precisely alter or replace damaged or disease-correlated DNA sequences. However, off-target site cleavage induced by ZF nucleases could result in cytotoxicity that must be mitigated for these systems to be useful as routine medicinal nucleases.
The Baker group has developed a computational method for redesigning metalloenzymes to catalyze noncognate reactions, and computationally redesigned a Zn(II)-containing mouse adenosine deaminase for organophosphate hydrolysis.204 The approach harnesses de novo enzyme computational design methods.205–207 Scaffolds are selected that contain backbones that can support active sites for the target reaction, and then the binding pocket sequence is optimized for the transition state. Applying this approach to existing metalloenzymes will address only the geometric compatibility of the site with the transition state and not the reactivity of WT functional groups. This is a valuable consideration as most enzymatic redesign involves altering substrate specificity or stereoselectivity, enhancing a preexisting promiscuous activity,198 or relies on having structural homologues that already have the desired activity (in the case of introducing mechanistically related activities) as a starting point.199 One example involved the redesign of the Zn(II)-containing glyoxalase II enzyme into a β-lactamase through extensive modifications made through insertion, deletion, and substitution of several active site loops and subsequent point mutations.208 This redesign alters both the metal-binding geometry and the substrate-binding pocket, yet both glyoxalase II and metallo β-lactamase enzymes contain binuclear metal ions essential to the hydrolysis reaction.
In this study, organophosphate hydrolysis was chosen as a target reaction and Zn(II) metalloenzymes as templates for supporting this activity, given that Zn(II) ions serve as powerful catalysts in many hydrolase enzymes with diverse mechanisms. Several mononuclear Zn(II) enzymes were examined for their geometric compatibility with the transition state of the target reaction (using methyl paraoxon and diethyl 7-hydroxycoumarinyl, or DECP, as substrates). Residues were introduced for both H-bonding interactions and shape complementarity leading to a set of 12 possible designed proteins for experimental characterization. One of these hydrolyzed DECP with a modest efficiency of ~4 M−1 s−1. The template enzyme for this model was adenosine deaminase, which, although also a hydrolytic enzyme, is distinct in terms of its transition state geometry, leaving group character, and inherent reactivity at the substrate electrophilic center. Eight mutations relative to the parent WT complex were made in the computational design model, seven for shape complementary interactions and one for an H-bond interaction to the nucleophile in the model. The Zn(II)-coordinating residues, Asp and three His residues (in a trigonal bipyramidal geometry with one open coordination site), and the catalytic residues, Glu217 and His238, were all retained from the WT enzyme (Figure 5a). The WT enzyme showed no acceleration for DECP hydrolysis over buffer at <20 mM enzyme. Using the initial velocity, the substrate concentration at which it was measured, and 20 μM enzyme concentration, a kcat/KM ≈ 10−3 M−1 s−1 was estimated. Mutation of Glu217, a residue involved in proton shuttling in the WT enzyme, to Gln results in complete loss of activity, indicating that this residue is crucial for organophosphate hydrolysis.
Figure 5.
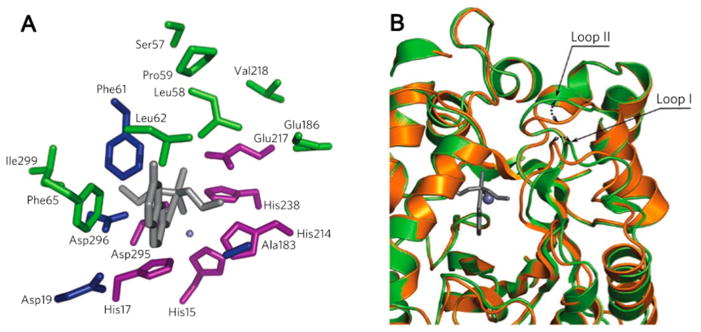
(A) Spatial clustering of WT and activity-enhancing residues in a Zn(II)-containing mouse adenosine deaminase. Residues in which computationally designed simultaneous substitutions were essential for the emergence of organophosphate hydrolysis activity are highlighted in blue. Deaminase residues retained in the most active variant of PT3 (purple), and positions in which activity-enhancing mutations occur during directed evolution (green) form two separate spatial clusters. Residue side chain identities are from the deaminase crystal structure (PDB code: 1A4L), and the transition state model is shown in gray sticks. (B) Superposition of the PT3.1 design model (gold) and the crystal structure (green, PDB code: 3T1G) shows that, although the overall backbone similarity is high (backbone rms deviation = 0.65 Å), there are small shifts in two active site-proximal loops. Reproduced with permission from ref 204. Copyright 2012 Nature Publishing Group.
Directed evolution, a method used in protein engineering for evolving proteins through an iterative process of preparing mutants and selecting those with the desired properties,209 was undertaken to gain a better understanding of the missing elements in this computational design method. Three rounds were performed, the first in which a set of selected residues around the active site was considered and three new mutations were incorporated, leading to a 40-fold increase in efficiency. Two subsequent rounds were carried out, the second by introducing random mutations, and the third considering a single residue identified to be in a suboptimal position by crystallography. These led to four more mutations, and another 10-fold increase in efficiency for each round, resulting in a total 2500-fold improvement over the initial computational design model. The biggest contributor to the increase came from the first round of directed evolution, and involved mutation of Val to the bulkier Phe, resulting in increased hydrophobicity around the catalytic residue Glu217 (Figure 5). The authors propose that, although Zn(II) in the WT enzyme activates the hydroxyl nucleophile, Zn(II) in the design model is more likely to polarize the P–O bond, along with the side chain of His238, and Glu217 is the general base that activates the nucleophile (although a crystal structure with a bound substrate or transition state analogue is required to prove this). These results indicate that the redesigned protein may operate with a mechanism different from that of the WT enzyme.
Analysis of the computational design protocol revealed some of its shortcomings. Most of the mutations that resulted in an improved efficiency were due to an increase in kcat. The biggest increase came from the Val218Phe mutation in the first round of directed evolution. This residue does not directly contact the transition state, but it does increase the bulk around the catalytically critical side chain of Glu217, probably modulating its pKa and reactivity. Analysis of the apoPT3.1 (from the first round of directed evolution) crystal structure reveals that, although much of the backbone and other elements of the structure match the design model, some conformational shifts in the loop backbone structure proximal to the active site were observed (Figure 5). These acted to accommodate the bulkier side chain that came from the Val218Phe mutation. Notably, the design was carried out on a fixed WT backbone so this mutation would have caused clashes in the design model. The authors’ future efforts will be in developing computational enzyme design methodology to allow more backbone flexibility and incorporate pKa effects with more accurate electrostatic interaction models. Although the primary coordination environment around the metal has not been changed in this enzyme redesign, this study demonstrates an effective approach to harnessing the inherent catalytic power of metal ions and introducing new reactivity into an existing metalloenzyme in a way that does not rely on preexisting activity. This study demonstrates the importance that the surrounding protein structure can have on the activity of the “same” metal site, given that both the substrate and the reaction mechanism are different. As a result, this approach can be one way of generating powerful metalloenzymes to catalyze novel reactions for a variety of biocatalyst applications. The catalytic efficiency of this enzyme is compared to other designed systems in Table 1. More recently, the Baker group has expanded the realm of computational protein design by incorporating an unnatural amino acid (2,2′-bipyridine-5yl)alanine (2,2′-bpy-Ala) into native proteins.210 RosettaMatch and RosettaDesign were used to search for the backbone location for the bpy-Ala residue and introduce additional interactions to stabilize the protein, respectively. The resulting protein scaffolds bind to divalent metal ions with a relatively high affinity, laying a solid foundation for the future design of unnatural amino acid-containing hydrolases.
Table 1.
Designed Metalloenzymes Mimicking Native Protein Metal Active Sites
| designed protein | reaction | kcat/s−1 | kcat/Km/M−1 s−1 | k1, k2/(M−1)s−1 | metal coordination and spectroscopic parameters | other notes | ref |
|---|---|---|---|---|---|---|---|
| PT3.3 | DECP hydrolysis | 351 (±26) × 10−3 | 9750 ± 1534 | Zn(His)3Asp(OH2/OH−) | (1) kcat, kcat/Km values at pH 7.5; (2) the crystal structure is for PT3.1; same active site | 204 | |
| PDB code: 3T1G | |||||||
| MID1-zinc | pNPA hydrolysis | 0.30 | 630(max)a | Zn(His)3(OH2/OH−) | (1) pKa = 8.2 ± 0.1; (2) the crysal structure showed tartrate binding, but the activity supports water/hydroxide binding | 211 | |
| PDB code: 3V1C | |||||||
| Hg(II)sZn(II)(H2O/OH−)N(TRIL9CL23H)3 | pNPA hydrolysis | ~0.053 | 31 ± 4(max)a | Zn(His)3(OH2/OH−) | (1) pKa = 9.0 ± 0.1; (2) kcat/Km(max) determined by fitting the kcat/Km(max) values vs pH | 212,213 | |
| PDB code: 3PBJ | |||||||
| Hg(II)sZn(II)(H2O/OH−)N(TRIL9CL23H)3 | CO2 hydration | 1.8 (±0.4) × 103 | 1.8 (±0.5) × 105 | Zn(His)3(OH2/OH−) | kcat, kcat/Km values at pH 9.5 | 213 | |
| PDB code: 3PBJ | |||||||
| Cu(TRI(L2W)L23H)3+/2+ | nitrite reduction | k1Asc = 12 ± 3 × 10−4 | Cu(I)(His)3 trigonal planar (R(Cu–N) = 1.93 Å); Cu(II) λd–dmax = 640 nm (ε = 138 M−1 cm−1); g|| = 2.27 (A|| = 186 G) | (1) pH-dependent rates; (2) k1Asc at pH 5.3; (3) spectroscopic parameters at pH 7.4 | 214 | ||
| di-Fe(III)-DF3 | 3,5-DTBC oxidation | 0.22 ± 0.02 | 105 | di-iron center with an oxo-bridge; ε350 nm = 5270 M−1 cm−1 | pH 7.0, 25 °C | 215 | |
| di-Fe(III)-DF3 | 4-aminophenol oxidation | 0.045 ± 0.003 | 23.0 | pH 7.0, 25 °C | 215 | ||
| G4DFtet | 4-aminophenol oxidation | 0.022 ± 0.002 | 25.7 | pH 7.0, 25 °C | 216 | ||
| Fe-mimochrome VI | ABTS oxidation | 375 | 8.4 × 10−3 for H2O2; 4.42 for ABTS | pH-dependent Soret band transformation | pH 6.5 | 217 | |
| Fe-MP3 | ABTS oxidation | 535 ± 42 | 3.2 × 10−4 for H2O2; 6.4 × 10−8 for ABTS | oxidized form Soret band at 391 nm at pH 4.7 and 6.6 | pH 6.5, 40% TFE | 218 |
Maximal efficiency from the fitting of kcat/KM values versus pH (assuming 100% active enzyme complex is present).
2.2.3. Redesign of Heme Centers
Heme proteins are involved in a remarkably broad spectrum of functions ranging from oxygen binding [hemoglobin, myoglobin (Mb)], oxygen metabolism (catalases, oxidases, oxygenases, peroxidases), electron transfer (cytochromes), signaling or sensing (soluble guanylate cyclase, FixL, CooA), to transport (hemopexin). 219–226 They are essential for fundamental biological processes including steroid biosynthesis, aerobic respiration, and drug metabolism. For these reasons, heme proteins have been the targets of numerous protein redesign efforts. Factors known to affect the reactivity of heme proteins include identity of the proximal axial ligand, structure of the distal site, nearby redox cofactors, and the type of heme used by the protein. Lu et al. extensively reviewed early efforts in this field.1 The relevant properties of designed heme systems are summarized in Table 2. Here, we will review successes from the past two decades.
Table 2.
Redesign and De Novo Designed Heme Proteins
| protein | E°/mV vs NHE | UV–vis absorption (Soret band for heme) | other notes | ref |
|---|---|---|---|---|
| ME1 | −128 ± 2 | oxidized form: 413 nm | Kd, app = (4.7 ± 1.2) × 10−7 M, 2:1 peptide/hemin stoichiometry | 227 |
| reduced form: 427 nm | ||||
| CuBMb | copper-free: 77, Cu(II)-bound: 80 | deoxy-CuBMb: 434 nm | oxygen reduction activity | 229,260 |
| oxy-CuBMb: 418 nm (not 100% conversion) | ||||
| FeBMb | −46 ± 2 | deoxy-FeBMb: 433 nm | nitric oxide reductase activity | 232 |
| deoxy-Fe2+-FeBMb: 434 nm | ||||
| Fe-porphycene-Mb | −190 ± 15 | oxidized form: 387 nm | enhanced O2 binding affinity | 253 |
| reduced form: 375 nm | ||||
| oxy-reduced form: 387 nm | ||||
| 6-propionate Mb | 91.7 ± 1.0 | Met-rMb: 408 nm | (1) Raman shift of deoxy-form: 220 cm−1; (2) pKa(heme-bound water) = 8.67 ± 0.03 | 248 |
| ferrous-rMb: 432 nm | ||||
| oxy-rMb: 417 nm | ||||
| 7-propionate Mb | 84.6 ± 1.0 | Met-rMb: 407 nm | (1) Raman shift of deoxy form: 221 cm−1; (2) pKa(heme-bound water) = 8.73 ± 0.03 | 248 |
| ferrous-rMb: 431 nm | ||||
| oxy-rMb: 417 | ||||
| heme-6-propionate Cyt P450cam | oxidized form: 391 nm (in the presence of d-camphor) | (1) Raman shift of the ferric form: 351 cm−1; (2) NADH oxidation rate 1150 μM/μM min enzyme−1 | 251 | |
| heme-7-propionate Cyt P450cam | oxidized form: 417 nm (in the presence of d-camphor) | (1) Raman shift of the ferric form: 1503, 1489 cm−1; (2) NADH oxidation rate 27 ± 2 μM/μM min enzyme−1 | 250 | |
| PRIME | −97 ± 3, −168 ± 3 | oxidized form: 410 nm | in palmitoyl oleoyl phosphatidylcholine bilayers | 261 |
| [Δ7-H10I14I21]2 | −222 | oxidized form: 412 nm (131 000 M−1 cm−1) | Kd1Fe(II) = 42 nM, Kd2Fe(II) = 15 μM | 262 |
| reduced form: 427 nm (209 000 M−1 cm−1) | ||||
| [Δ7-Pal10I14I21]2 | 58 ± 8 | oxidized form: 410 nm (110 000 M−1 cm−1) | 263 | |
| reduced form: 420 nm (144 000 M−1 cm−1) | ||||
| [Δ7-H1m10I14I21]2 | reduced form: 431 nm (97 000 M−1 cm−1) | vacant coordination site for CO binding | 264 | |
| Fe(PPIX)-[Δ7-H-H3m]2 | −190 ± 10 | oxidized form: 412 nm (123 000 M−1 cm−1) | E° determined at pH 8.0 | 265 |
| reduced form: 427 nm (191 00 M−1 cm−1) | ||||
| Fe(DADPIX)-[Δ7-H-H3m]2 | −30 ± 10 | oxidized form: 426 nm (89 000 M−1 cm−1) | E° determined at pH 8.0 | 265 |
| reduced form: 450 nm (110 000 M−1 cm−1) | ||||
| HP7-H7F | −260 ± 6 | oxidized form: 414 nm (129 000 M−1 cm−1) | pKa (His) = 7.3 ± 0.2 | 266 |
| reduced form: 428 nm (140 000 M−1 cm−1) | ||||
| heme-Ru-MOP2 | −170 ± 6 | oxidized form: 413 nm | electron transfer demonstrated | 267 |
| reduced form: 428 nm | ||||
| Fe-mimochrome IV | −80 (pH 7.0) | oxidized form: 398 nm | 268 | |
| miniperoxidase 3 | oxidized form: 391 nm | HRP activity | 218 | |
| immobilized heme-S824C | −153 | oxidized form: 413 nm | binds to N-donor ligands | 269 |
Although most protein design studies focus on water-soluble proteins, many membrane proteins play important roles in metabolism. Glycophorin A (GpA) is a transmembrane protein that forms noncovalent dimers by association of two single helices into coiled coils. A simple model of a transmembrane heme protein, ME1, was designed on the basis of GpA by mutating five amino acid residues (Glu1Ser, Thr5Ala, Ile22Ala, Ile26His, Ile30Arg), to create a heme-binding site at the C-terminus. The resulting peptide, upon binding to heme in its dimeric form, contains a bis-His heme site capable of H2O2-dependent oxidation of organic substrates.227 The midpoint redox potential of this protein was determined to be −128 ± 2 mV (vs NHE), which is more positive than most of the reported bis-imidazole heme centers in hydrophilic environments. Although the designed construct was optimized for tight heme binding instead of catalysis, it showed unexpected peroxidase activity. For example, ME1 was able to catalyze the peroxide oxidation of 2, 2′, 5, 5′-tetramethylbenzidine (TMB) with a specific velocity of 2300 s−1 M−1 measured by the increase of absorbance at 450 nm (the oxidized form of TMB). Utilizing a protein redesign strategy, researchers were able to incorporate a heme center into an inert membrane protein to confer desired functionalities.
More recently, the metalloprotein design field has expanded to include the engineering of heterometallic sites into native systems. Many of these studies were reviewed by Lu et al.21 or may be found in later sections of this Review. Because the modifications were not directly carried out on the heme center, we will only summarize these efforts here. Cytochrome c peroxidase (CcP) was converted into a functional manganese peroxidase by designing a Mn(II) binding site by a triple mutation (Gly41Glu, Val45Glu, and His181Asp) (section 2.2.4.3: Redesign of Manganese Centers).228 In a more complicated functional conversion, Mb was engineered into a heme copper oxidase (HCO) by introducing His residues near the heme site to create a CuB site for the reduction of O2 (section 2.2.4.2.2: Copper Catalytic Centers).229 This construct can also catalyze the reduction of NO to N2O.230 More recently, important insight into the cross-linked imidazolephenol post-translational modification was gained when an unnatural amino acid that mimics this cross-link was incorporated into the HCO model. The model achieved improved activity, including an 8-fold increased selectivity for O2 and almost a 3-fold increase in catalytic turnover as compared to F33YCuBMb, the mutant without the cross-link (section 2.2.4.2.2: Copper Catalytic Centers).231 Furthermore, Mb was successfully converted to a nitric oxide reductase (NOR) by introducing an FeB site near the heme (section 2.2.4.1.2: Nonheme Iron Catalytic Centers).232 This rationally designed protein was a good structural and functional model of NOR from anaerobic bacteria, and revealed a requirement for Glu residues near the active site for both iron binding and NOR activity. This designed protein is more robust than the native protein, allowing the Lu group to study metal replacement and solve multiple crystal structures (PDB: 3M38, 3M39, 3M3A, 3M3B).233
One strategy for generating supramolecular assemblies of proteins focuses its efforts on cofactor modification. The Hayashi group has generated one- and two-dimensional supramolecular arrays of hemoproteins by linking hemes to the exterior of proteins and relying on natural heme binding to form the polymers. A single point mutation in cytochrome b562 was made to introduce a surface Cys residue (His63Cys), which was then linked to an iodoacetamide-modified heme (Figure 6).234 These successive interprotein heme–heme pocket interactions formed chains of proteins where oligomer size was partially controlled by the length of the link between the iodoacetamide moiety and the heme moiety. These one-dimensional arrays were further expanded by the synthesis and incorporation of a tripodal pivot molecule bearing three heme moieties to create branches in the assembly.235 The iodoacetamide derivative was replaced with a maleimide derivative,235 yielding improved specificity and faster reactions. Moreover, a Mb variant was synthesized, allowing assemblies to be prepared using either Mb or cytochrome b562. The size and degree of branching of the supramolecular assemblies were controlled by the molar ratio of heme–protein and pivot molecule. The success of using cofactor modification to create protein-based polymers encouraged the creation of functional and more complex materials. For instance, the interaction of one-dimensional heme–protein assemblies with nanoparticles was achieved using gold nanoparticles,236 gold electrodes,237 and CdTe quantum dots.238 A Zn-protoporphyrin IX derivative was used to anchor chains of Zn-protoporphyrin IX-bound hemoproteins to a gold electrode, producing a material capable of generating a photocurrent, where the magnitude of the photocurrent is dependent on the density of the layer of redox protein.237
Figure 6.

Schematic representation of the supramolecular hemopro-tein polymerization. The structure of the WT-cyt b562 was obtained from PDB (1QPU). Reproduced with permission from ref 234. Copyright 2007 American Chemical Society.
Mb fibers based on heme–heme pocket interactions display stability that depends on the axial ligation of the heme. For example, cyanide stabilizes the fibers due to its strong interaction with the ferric heme iron.239 Furthermore, these fibers can be cross-linked via a radical mechanism in the presence of H2O2, while retaining their oxygen-binding characteristics. Recently, two orthogonal proteins, Mb and streptavidin (Sav), were combined into a single supramolecular material.240 An asymmetric heme-bis(biotin) dyad was synthesized to enforce 2:1 association of Mb and Sav (Scheme 1, reproduced from ref 240 with permission. Copyright 2012 Wiley.). Modification of Mb for producing Mb dimers resulted in the formation of longer, one-dimensional copolymers of [-(rMbA125C)2-SAv-]n (where rMbA125C is the recombinant Mb mutant).
Scheme 1.240.

A major goal of protein design is to understand how the overall scaffold structure, residue identity and placement, and cofactors combine to create a particular function. Heme protein functions have been studied by replacing heme with related cofactors, including noniron macrocycles and porphyrin isomers (porphycenes). Heme cofactor redesign studies began as early as the 1970s with the goal of modulating the oxygen-binding affinity.241 Later, heme cofactor redesign efforts expanded to include the tuning of protein stability and reactivity. For example, a series of P450NORs with various 2,4-substituted hemes have been prepared, showing a significant decrease in turnover number (TON) as compared to the native protein. This study demonstrates how the electron-withdrawing capacity of the 2,4-substituents influences the reactivity of the heme center.242 A 2,4-diacetyl-[2H]heme-substituted horse-radish peroxidase (HRP) was shown to catalyze NADPH oxidation by H2O2.243 In addition, the solution NMR structure of a dimethyl propionate ester heme containing cytochrome b5 was examined, leading to important insight into the stabilization effects of the H-bonding interactions around the heme center.244
In another series of studies, the Hayashi group sought to understand the role of the two propionate side chains of heme b. By modifying these side chains using hydrophobic clusters,245 glycosylating them,246 placing them on porphycenes,247 and selectively removing each one in turn,248,249 the authors found that the heme cofactor itself has various degrees of influence on the O2 and CO binding ability and the catalytic activity of the proteins. The effect of the propionate side chains of heme b on Mb activity was examined. “One-legged” heme cofactors were synthesized by selectively depropionating heme to yield 6-depropionate or 7-depropionate and were subsequently incorporated into Mb. Measurement of the O2- and CO-binding affinities, X-ray crystallography, and resonance Raman (rR) spectroscopy248 demonstrated that 6-propionate is responsible for a large number of H-bonding interactions at the distal site, resulting in a 3-fold acceleration of oxygen dissociation as compared to the 7-priopionate counterpart and the native protoheme. The 7-propionate moiety did not affect protein structure, but instead slightly increased the Fe–His bond strength, resulting in a 2-fold increase in CO affinity relative to the native protoheme. Similar studies by depropionating heme centers were carried out with cytochrome P450CAM, showing that 7-propionate acts as a gate, regulating water access to the substrate binding site, while the 6-propionate “leg” fixes the putidaredoxin binding site and stabilizes the Fe–S bond required for activity (Figure 7A,B).250,251
Figure 7.

Crystal structures of the cofactor site for (A) reconstituted Cyt P450CAM with 6-methyl-6-depropionatehemin bound (PDB code: 2ZAW);251 (B) reconstituted Cyt P450CAM with 7-methyl-7-depropionatehemin bound (PDB code: 2Z97);250 and (C) iron porphycene bound to apo-swMb (PDB code: 2D6C).254
“Winged” heme cofactors were synthesized by attaching aromatic rings and carboxylate-based clusters to the propionate groups and inserted into a His67Asp mutant of swMb, yielding an active site resembling that of HRP.252 The catalytic efficiency of MbH64D, the MbH64D mutant with a symmetric, double-winged cofactor [MbH64D(double-winged)], and the MbH64D mutant with a single-winged cofactor [MbH64D-(single-winged)] were examined in assays monitoring the oxidation of 2-methoxyphenol.252 As compared to the same reaction catalyzed by HRP, the catalytic efficiency (kcat/Km) of these constructs follows the order: MbH64D(single-winged) (85 000 M−1 s−1) ≈ HRP (72 000 M−1 s−1) > MbH64D-(double-winged) (23 000 M−1 s−1) > MbH64D (5100 M−1 s−1).249 The addition of aromatic and carboxylate clusters on the heme center may form a substrate-binding site for 2-methoxyphenol, thus modulating the reactivity. In Mb, porphycenes with propionates at different positions were reconstituted into the protein, and oxygen binding and selectivity were examined. It was hypothesized that the interaction of propionates with H-bonding residues and the protein matrix could affect ligand binding and cofactor incorporation. Porphycene with propionates at the 13th and 16th positions showed a 2600-fold enhancement for oxygen binding.247,253 The electronics of the porphycene, the lower symmetry that stabilizes the Fe–O2 σ-bonding, combines with the location of the propionate in a synergistic fashion to enhance oxygen binding over heme b.247 Analysis of the crystal structure of Mb reconstituted with Fe(III)-porphycene(13,16) (hereafter referred to as porphycene-Mb) (Figure 7C)254 showed that this construct is capable of H2O2-dependent oxidation of guaiacol, thioanisole, and styrene. Stopped-flow kinetics experiments suggested that high energy intermediates, compound II and compound III, are formed in the absence of substrate, likely due to the enhanced strength of the Fe–His bond.254 To explore further how peripheral groups alter the activity of porphycene, trifluoromethyl groups were added at the third and sixth positions of the macrocycle. This new construct had a redox potential of −41 mV (vs NHE), as measured by spectroelectrochemical titrations, falling between the redox potentials of native Mb and Mb reconstituted with porphycene (+52 and −195 mV, vs NHE, respectively).255 Trifluoromethyl porphycene-containing Mb showed a lower oxygen-binding affinity that may be attributed to an enhancement of the oxygen dissociation rate as a result of the destabilization of the Fe–O2 bond relative to porphycene-Mb. This Mb-based construct also exhibited accelerated CO binding rates. Like porphycene-Mb, trifluoromethyl porphycene-Mb has poor π-back bonding; however, the electron density on iron can be stabilized by electron-withdrawing trifluoromethyl groups.255 Given the enhancement of peroxidase activity of Mb achieved by incorporating porphycene, the effect of porphycene was also investigated in HRP.256 The resulting construct was tested for catalyzing guaiacol and thioanisole oxidation by H2O2. An iron(IV)-oxo porphycene π-cation radical, which is thought to contribute to the catalytic efficiency for thioanisole oxidation, was detected above 0 °C in aqueous conditions. The Hayashi group also incorporated the first formal ferryl cofactor into Mb and HRP by incorporating a meso-unsubstituted iron corrole.257 In this case, Mb formed a stable construct that is EPR-active, observable by UV–vis spectroscopy, and could be assigned as an Fe(III)-neutral ring oxidation state. HRP required sodium dithionite reduction to yield a stable Fe(III) cofactor.
Using a directed evolution strategy toward protein engineering, Arnold and co-workers developed a series of heme-containing proteins, creating functional novel biocatalysts. In particular, the Arnold group reported a series of heme-proteins capable of carrying out carbene transfer.258,259 By introducing a Cys400Ser mutation into WT-P450BM3 and a previously reported enzyme, P450BM3-CIS with 13 mutations on P450, styrene cyclopropanation activity was achieved.259 The goal was to raise the reduction potential of the heme iron center by mutating the axial ligand, Cys, into a weakly donating group, Ser. With a +95 and +155 mV increase in reduction potential as compared to P450BM3-CIS and WT-P450BM3, respectively, the mutant P411BM3-CIS exhibited high TONs for cyclopropanation activity under anaerobic conditions. More importantly, the TON for styrene cyclopropanation was 6 times higher after 6 h in vivo, as compared to the same reaction carried out in vitro with nearly identical stereoselectivity.
2.2.4. Redesign of Nonheme Redox Centers
2.2.4.1. Nonheme Iron Centers
Nonheme iron centers are defined as those in which iron is coordinated by ligands other than polydentate tetrapyrroles (e.g., porphyrins). Figure 8 illustrates the six general types of nonheme iron centers including mononuclear, binuclear, and iron–sulfur centers. Nonheme iron centers exhibit a broad range of functions, including electron transfer, substrate oxidation and reduction, and metal ion transport.270 The redesign of nonheme iron proteins aims to explore the factors that govern the redox properties and reactivity of the iron center, elucidate the influence of interactions between different key amino acid residues, and construct a novel functional nonheme iron site into another nonheme iron protein.
Figure 8.
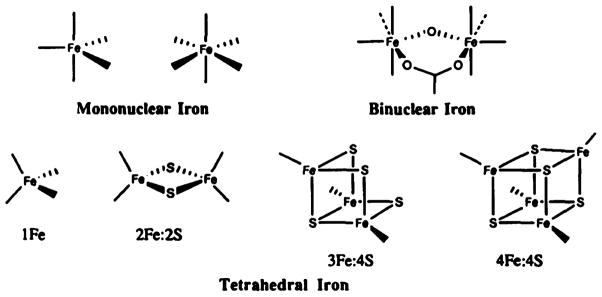
Nonheme iron centers. Reproduced with permission from ref 270. Copyright 1991 Elsevier.
2.2.4.1.1. Nonheme Iron Electron Transfer Centers
Automated rational design algorithms have been used to design several nonheme metalloproteins. Immunoglobulin G binding domain B1 was redesigned using the Metal Search program42 to incorporate a tetrahedrally coordinated iron site for mimicking rubredoxin.271 Although the authors titled this contribution as a de novo designed protein, we feel that because this protein was designed on the basis of a native protein domain, it falls into the protein redesign category. The authors took the backbone movement into account in the solution NMR structure of the native protein, rather than focusing only on the averaged NMR structure. This strategy avoids having restrictive backbone coordinates, which might prohibit the protein from adopting favorable conformations when certain side chains are introduced.272,273 The Fe(S-Cys)4 site was achieved by mutating several residues situated in the flexible loop region of the original protein, Gly9, Gly38, Val39, and Glu56, into Cys residues. Co(II) and Cd(II) were used as spectroscopic probes to confirm the tetrahedral geometry of the (Cys)4 site. The EPR spectrum of the Fe(III)-bound [B1-Cys4] indicated that the protein accommodates Fe(III) in a tetrahedral geometry with g values of 4.3 and 9.5, but this complex is not robust. It auto-oxidizes and decomposes in air.271
A more robust rubredoxin mimic (RM) was obtained by converting a disulfide bond in Trx. The resulting site showed spectroscopic properties that mimic rubredoxin and was able to undergo several cycles of oxidation and reduction.274 Hellinga and co-workers constructed this tetrathiolate site by substituting Trp and Ile residues in native Trx with Cys using a structure-based automated program, Dezymer.39,40 Together with the original disulfide-forming Cys, a metal-based redox site was engineered into an amino-acid-based redox site. Fe(II) bound to the Trx mutant Trx[Rd] with a 1:1 stoichiometry, and the dissociation constant was estimated to be 0.7 μM. Using a minimalist model where only the first coordination sphere of the metal center is taken into account, these contributions demonstrate the strength of protein redesign in achieving a redox-active nonheme iron center in a completely different protein scaffold.
Similarly, Trx was redesigned to incorporate a self-assembled [4Fe–4S] iron–sulfur cluster, resulting in high potential iron protein (HiPIP)-like spectral characteristics, including an EPR silent resting state.275 [4Fe–4S] was incorporated into the Trx variant, Trx-[4Fe–4S], where Leu24, Leu42, Val55, and Leu99 were each mutated to Cys. Holo-Trx-[4Fe–4S] exhibits the spectroscopic signatures of a [4Fe–4(S-Cys)] cluster. In particular, EPR spectra collected for both the reduced and the oxidized forms of holo-Trx-[4Fe–4S] show spectral features analogous to those in synthetic clusters containing [2Fe(II)–2Fe(III)] and the fully oxidized [4Fe–4(S-Cys)]− in HiPIP,276 respectively. The redox potential of Trx-[4Fe–4S] was estimated to be at least +300 mV (vs NHE), which is within the range of native HiPIPs (+50 to +450 mV vs NHE).277
Although [4Fe–4S] clusters are prevalent in both native and model systems, [3Fe–4S] clusters are much less commonly found. To understand the factors that govern the stoichiometry for Fe–S cluster formation, Hoppe et al. designed a cluster-binding peptide (CBP).278 Upon mutating a Cys in the Fe–S cluster binding motif of dimethyl sulfoxide reductase subunit B (DmsB) from Escherichia coli (E. coli), a [4Fe–4S]-binding site was converted to a [3Fe–4S]-binding site. However, the [3Fe–4S] motif is a transient species that only forms under specific conditions and leads to the formation of the thermodynamic product [4Fe–4S]. Although a stable [3Fe–4S] species was not achieved, this work implied that, contrary to the prior consensus and the proposed mechanism in native proteins, iron coordinates in succession to form the stable [4Fe-4S] species.
2.2.4.1.2. Nonheme Iron Catalytic Centers
Besides electron transfer, nonheme iron centers can also serve different reaction purposes including oxygenation (protocatechuate 3,4-dioxygenase,279,280 catechol 2,3-dioxygenase281), peroxidation (rubrerythrin282), oxidation (ribonucleotide reductase (RnR),283 1-aminocyclopropane carboxylic acid oxidase284), superoxide dismutation [iron superoxide dismutase (SOD)285], hydroxylation (phenylalanine hydroxylase,286,287 calavaminate synthase288), and H-abstraction (iron-bound bleomycin289–291). Several comprehensive reviews cover the structures, functions, and mechanistic perspectives of these native nonheme iron centers, so we will omit a detailed description of each enzyme.270,292–294 There are several examples of redesigned nonheme iron proteins that use iron as a catalytic center to control the redox properties and reactivity of the protein.
SOD catalyzes the disproportionation of superoxide into molecular oxygen and hydrogen peroxide, an important gating mechanism for controlling cellular oxidative stress.295 Three types of SODs have been discovered in a variety of organisms: NiSOD in certain fungi and bacteria,296 CuZnSOD in eukaryotic cells,297 and Fe/MnSOD in bacteria, some plant chloroplasts, and mitocondria.285,298,299 FeSOD and MnSOD share a high sequence homology, including the same amino acid ligand set, and, therefore, are considered as one general type. Metal ion substitutions result in a loss of activity,300–302 likely a reflection of the different midpoint potentials of FeSOD and MnSOD.303,304 In FeSOD, iron is bound in a trigonal bipyramidal geometry with two His and One Asp on the trigonal plane and one water/hydroxide and one additional His as axial ligands.285,298,305 This coordination environment was constructed into Trx using the rational protein design program, Dezymer.306 The mutations Leu7His, Phs27Asp, Ile60His, and Asp63His constructed the FeSOD active site, and, together with several other mutations for structural stability, yielded Trx-SOD. Fe-Trx-SOD exhibits spectroscopic features similar to those observed in WT FeSOD and readily catalyzes superoxide dismutation. Although the rate of Fe-Trx-SOD is ~104-fold slower than that of the WT enzyme, it remains an excellent model toward elucidating the chemical details in native proteins. Using Dezymer, Benson et al. introduced a mononuclear (His)3 iron-binding site and an oxygen-binding pocket into Trx,8 with the goal of creating an empty binding site on the iron center for facilitating substrate access. The oxygen-binding center was created by examining the locations that could sterically accommodate a molecular oxygen. Six locations were selected, including three sites in a groove (G1–G3), two sites in a shallow pocket (S1 and S2), and one site in a deep pocket (D1). The redox potential of this Fe(His)3 center was location-dependent: the relatively buried G3 and D1 sites had significantly lower redox potentials (+28 ± 2 and +10 ± 2 mV, respectively), whereas the more exposed S1 site had a higher potential (+440 ± 2 mV). Fenton, SOD, and Udenfriend reactions were tested for these mutants, demonstrating that the stabilization of surface electrostatics, transition states, and the elimination of nonspecific electron transfer processes all contribute to the specificity of the catalytic reaction.
In NOR, a nonheme iron center, FeB, is coupled to a heme center, which promotes the two-electron reduction of NO, producing N2O and water.307,308 On the basis of a previously reported CuBMb construct, where a (His)3 site was introduced close to the heme center in Mb for copper binding,309 Yeung et al. successfully designed a NOR mimic by coupling a nonheme iron center [Fe(His)3] to the heme center in Mb.232 Specifically, the Val68Glu mutation was chosen in addition to the two preexisting mutations in CuBMb (Leu29His and Phe43His). The resulting FeBMb protein contains a nonheme iron coordinated to three His residues (His29, His43, and His64) and Glu68, confirmed by the crystal structure (Figure 9). The presence of the FeB site dramatically increased the redox potential of the heme center, from −158 to −46 mV (vs NHE), which, along with EPR data, supported coupling of the two iron centers. Fe(II)-FeBMb was also a functional model of NOR because it was able to catalyze NO reduction into N2O. To design a closer model of the native NOR, Lin et al. introduced a second Glu residue in close proximity to the FeB-heme site.233 Previous reports indicated that a few Glu residues are critical for retaining NOR activity;310,311 however, because of the complexity of the native protein, the precise functional role of each Glu was unknown. An Ile residue on the second coordination sphere of FeB was mutated to Glu in the simplified model, FeBMb, to elucidate its function. The crystal structure of Fe(II)-I107E-FeBMb was solved, showing that Fe(II) is coordinated to three His residues, Glu68 and a water molecule situated between Fe(II) and Glu107. Glu107 was proposed to facilitate proton uptake. Under single turnover conditions, after ~20 h, the yield of N2O catalyzed by Fe(II)-I107E-FeBMb was 24%, more than twice that catalyzed by Fe(II)-FeBMb (~10%), highlighting the important functional role of Glu107. Moreover, the introduction of Glu107 did not alter the reduction potential of the heme center, showing that the effect of the conserved Glu residues is not additive, but rather depends on the location of the Glu residue. Metal ion substitutions were carried out to confirm the role of the nonheme metal center. Structural differences such as altered H-bonding networks and coordination geometries were observed in the crystal structures when Fe(II) was substituted with Cu(II) and Zn(II), showing that FeB site is involved in the structural fine-tuning.
Figure 9.

The active site of Fe(II)-FeBMb. Fe(II)B is represented by a green sphere. The Fe(II)-bound water is represented by a red sphere. PBD code: 3K9Z. Bond lengths in the figure are in angstroms.232
Several proteins contain di-iron oxo active sites that carry out various reactions. For example, both hemerythrin and methane monooxyganse contain a di-iron oxo site, which functions as an O2 carrier center in hemerythrin312 and a methane hydroxylation site in methane monooxygenase (MMO).313 One of the major goals of protein redesign is to investigate whether it is possible to swap similar active sites from proteins that exhibit dissimilar functions, which may lead to a more in-depth understanding of how structure relates to activities in metalloprotein active sites. DeMaré et al. redesigned the di-iron site in rubrerythrin (Rr) to mimic that of RnR R2.314 Although the physiological function of Rr is yet to be determined, this study contributes to improving the understanding of the functional aspect of the di-iron center. Specifically, the authors created Rr mutants aiming to probe the possibility of forming a tyrosyl radical near the active site (Leu60Tyr) and to determine the role of a coordinating Glu residue (Glu97Ala). The X-ray crystal structures for both Leu60Tyr and Glu97Ala mutants showed minimal structural perturbation as compared to WT-Rr. Leu60Tyr is located in a position similar to the stable tyrosyl radical-forming position in native RnR R2; however, EPR and UV–vis spectroscopy did not show any tyrosyl radical feature in Leu60Tyr-Rr. Ferroxidase reactions were carried out with the mutants. Only the Glu97Ala mutant, in which the directly coordinating Glu ligand was removed, showed decreased activity, while the distal mutation in L60Y-Rr did not result in a change to the activity. These results suggest that the physiological reaction of Rr involves ferroxidase-like activity. In L60Y-Rr, Tyr did not form an H-bond with an iron-coordinating carboxylate, as would have been required to form the tyrosyl radical during the ferroxidase reaction.
Moreover, a di-iron center with activity resembling that of MMO was introduced into RnR R2 through a series of mutations.315 The crystal structures of RnR R2 from different species show similar di-iron sites as well as the conserved residues Phe208, Phe212, and Ile234, which form a hydrophobic patch. Tyr122 is believed to form a radical to initiate the catalytic cycle for ribonucleotide reduction.316,317 To investigate the role of Phe208 and how it may influence Tyr122 radical formation, Örmo et al. mutated Phe208 to Tyr, yielding an RnR R2 mutant that did not have a tyrosyl radical, based on the absence of a characteristic 410 nm feature in the absorption spectrum. The Raman spectrum showed the presence of a ferric bidentate catecholate species, likely derived from the protein rather than an exogenous ligand. The authors postulated that the Tyr108 could be the precursor of the dihydroxyphenyla-lanine ligand, the formation of which is proposed. Scheme 2 (Reproduced from ref 315 with permission. Copyright 2012 American Society for Biochemistry and Molecular Biology.) shows the self-hydroxylation activity of this di-iron center, which involves the formation of a ferric-peroxide that transforms to a high-valent Fe(IV)–oxo species.
Scheme 2.315.

Other proteins that have been redesigned to incorporate non-native iron clusters include metallothionein and cyto-chrome c. The numerous Cys residues in the α domain of metallothionein were used to reconstitute a tetranuclear cluster comprised of Fe(II).318 The resulting complex H-apocyt c was shown to be active in reducing Mb in aqueous solution via a one-electron process, and methyl red via a two-electron process. This construct is also capable of forming a tetranuclear cluster based on Co(II), with a lower reducing ability than the Fe(II) counterpart due to the unmatched redox potential. Native cytochrome c contains a CXXC motif where the heme moiety is covalently linked to the protein. A hydrogenase model complex, Fe2(CO)6, was incorporated into apo-cytochrome c, resulting in an assembly that can catalyze H2 evolution in aqueous media.319 Using a ruthenium sensitizer and ascorbate as a sacrificial donor, a TON of 80 over 2 h at pH 4.7 was achieved with this model complex–protein assembly. A small heptapeptide fragment with a CXXC motif (sequence: YKCAQCH) accommodating a (μ-S-Cys)2Fe2(CO)6 complex was also examined for H2 evolution. The initial turnover frequency of H-apocyt c showed a significant improvement over the heptapeptide version (~2.1/min for H-apocyt c vs ~0.47/min for the peptide fragment), demonstrating the importance of the protein environment surrounding the di-iron center.319
Iron centers in native proteins also play important roles in hydrogen activation. The metal cluster responsible for hydrogen activation is either a Ni–Fe heterodimeric center or an Fe–Fe homodimeric center. The structures of both types of hydrogenases have been reported.320–322 Growing interest in the development of new types of fuel cells underlines the importance of understanding how hydrogenases can activate and incorporate hydrogen into the substrate. In native systems, a di-iron subset is first assembled on maturation enzymes HydE, HydF, and HydG, then inserted into [Fe–Fe]-hydrogenase (HydA).323 A hybrid protein was created by incorporating synthetic di-iron motifs into a HydF protein containing an iron–sulfur cluster.324 Three hybrid proteins were designed, differing by the central bridgehead atom of the dithiolate, and tested for their ability to activate apo-HydA1, which contains a single [4Fe–4S] cluster, but no di-iron subsite. The transfer of the di-iron subsite from the hybrid protein to apo-HydA1 was examined by monitoring hydrogen evolution. The authors examined carbon, nitrogen, and oxygen as the bridgehead atom and found that the nitrogen-bridged HydF hybrid protein exhibits the best efficiency, and is even more efficient than native C. acetobutylicum HydF. More importantly, this work provided insight into the mechanism of hydrogenase maturation by supporting a proposal involving formation of a transient HydF-di-iron-HydA1 species through the stabilization of the [Fe4-S4] cluster and alluded to a possible similarity between the natural precursor and the synthetic model.
2.2.4.2. Redesign of Copper Centers
2.2.4.2.1. Copper Electron Transfer Centers
In addition to heme and nonheme iron electron transfer proteins, cupredoxins or type 1 copper proteins (T1Cu) are another type of biological electron transfer proteins that can be isolated from bacteria, algae, archaea, and plants.325–329 Cupredoxins function in electron transport chains (e.g., photosynthesis) or in chemical reactions by providing an electron to the catalytic site [e.g., copper nitrite reductase (CuNiR)]. The copper site is encompassed in an overall Greek β-barrel fold, with the metal-binding ligands in a pocket between loops, protected from solvent access. T1Cu centers are mononuclear copper centers coordinated to two imidazole N atoms from His residues and the thiolate from a Cys residue in a trigonal plane (CysHis2).329,330 In addition, one or two weak axial ligands can be bound to the copper center, resulting in a distorted tetrahedral geometry that is not preferred by either Cu(II) or Cu(I) metal ions.331 This unique structural feature provides cupredoxins with unusual spectroscopic and redox properties. Blue copper proteins [plastocya-nin332–337 and azurin (Az)336–338] were the first to be classified as T1Cu centers and are among the first structures made available through X-ray crystallography.339–341 The contribution of the copper electronic structure to its function and spectroscopic properties has been thoroughly studied.329–331,336,337,342–353
Classic T1Cu centers display a strong electronic absorption at ~600 nm and a weak band at ~400 nm, as well as a compressed hyperfine coupling constant (AII < 100 × 10−4 cm−1) in the EPR spectrum.332,333,336–338,351 The strong absorption band was later assigned to a Cys Sπ-to-Cu(II) charge transfer transition and the narrow AII was attributed to the highly covalent Cu–SCys bond, reducing the interaction between the unpaired electron with the nuclear spin of Cu(II).351,353 In the following years, green copper proteins (rusticyanin,354,355 stellacyanin,356,357 and pseudoazurin358) were also found to have a core Cu(CysHis2) site, with one or two axial ligands, and were classified as T1.5Cu centers because their spectral and structural properties are between those of T1Cu and T2Cu.327,328,337,352 T1.5Cu centers exhibit an additional absorption band at ~450 nm and display a rhombic EPR signal, instead of the typical axial signal reported for blue T1Cu centers. Both blue and green copper proteins have positive reduction potentials, ranging from +180 to +800 mV (vs NHE). Moreover, nitrosocyanin, a red copper protein, was included in the T1Cu family because it contains the conserved core residues.326,352,359–361 However, nitrosocyanin has the characteristics of a type 2 copper (T2Cu) center in a tetragonal geometry (“normal” copper), with a strong absorption band at 390 nm and a large hyperfine-coupling constant (AII > 100 × 10−4 cm−1). Nitrosocyanins have a reduction potential of ~50 mV (vs NHE) and are suggested to function as catalysts in ammonia-oxidizing autotrophic bacteria.359
Moreover, purple CuA centers are binuclear copper centers involved in electron transfer, relaying electrons between cytochrome c and heme a in cytochrome c oxidase (CcO) and cytochrome c and the CuZ center in nitrous oxide reductase (N2OR).326,350,362–371 The electronic and spectroscopic characteristics of CuA centers have been previously reported and reviewed.326,330,342–344,349,350,368,372–374 Like cupredoxins, the CuA center is found in the loop regions of a Greek β-barrel fold with the copper ions coordinated to an imidazole N atom from a His residue and bridged by μ2-sulfur thiolates from Cys residues, forming a diamond-shaped Cu2S2 core structure.375–378 This core is further supported by weak axial ligands such as a thioether sulfur atom from a Met residue and carbonyl oxygen atoms from Ile, His, and Glu residues. CuA centers possess a Cu–Cu bond, which is unusual for the first row transition metals, and is the first metal–metal bond identified in biological systems.379,380 These centers exhibit strong electronic absorption bands at 480 and 530 nm, assigned to S-to-Cu(II) charge transfer, and a band at 800 nm that originates from a Cu–Cu ψ–ψ* transition.373 The EPR spectrum of CuA displays seven lines, which, although typical for a Class III mixed-valence binuclear metal center, is extraordinary because it is a fully delocalized mixed-valence system [Cu(1.5)---Cu(1.5)].381–383 Furthermore, CuA centers have a reduction potential of about +240 mV (vs NHE), lower than that of most T1Cu centers.326,366,384–387 We review herein the redesign efforts on T1Cu and CuA centers, which are summarized in Table 3.
Table 3.
Redesigned Proteins with Copper Electron Transfer Centers
| protein | LMCT λmax (ε/M−1 cm−1)/nm | Em°/mV vs NHE | bond length/Å | EPR g (A/G) | other notes | ref |
|---|---|---|---|---|---|---|
| Cys112Sec-Az | 677(~4000) | 316 ± 2 | Se–Cu(II) 2.30(2) | gx = 2.047 | 388,389 | |
| 2N(imi)–Cu(II) 1.97(2) | gy = 2.109 | |||||
| Se–Cu(I) 2.33(2) | gz = 2.234(104) | |||||
| 2N(imi)–Cu(I) 2.00(2) | ||||||
| Met121Nle-Az | 630(~5000) | 449 ± 3 | S(cys)–Cu(II) 2.19(1) | gx = 2.0402(13.4) | 390,391 | |
| 2N(imi)–Cu(II) 1.95(1) | gy = 2.0584(19.6) | |||||
| S(Cys)–Cu(I) 2.24(1) | gz = 2.2680(58.8) | |||||
| 2N(imi)–Cu(I) 2.02(1) | ||||||
| Met121Leu-Az | 631(~5000) | 450 | S(Cys)–Cu(I) 2.23(1) | gx = 2.0395(13.2) | 390 | |
| 2N(imi)–Cu(I) 2.01(1) | gy = 2.0578(19.2) | |||||
| gz = 2.2706(57.0) | ||||||
| Met121SeM-Az | 629(~5000) | 348 ± 4 | S(Cys)–Cu(II) 2.18(1) | gx = 2.0383(13.2) | 390,391 | |
| 2N(imi)–Cu(II) 1.95(1) | gy = 2.0565(12.1) | |||||
| gz = 2.2585(60.6) | ||||||
| S(Cys)–Cu(I) 2.26(1) | ||||||
| Met121DFM-Az | 628 | 329 ± 5 | 2N(imi)–Cu(I) 2.02(1) | gx = 2.0274(1.9) | 391 | |
| gy = 2.0583(2.0) | ||||||
| gz = 2.2530(56.8) | ||||||
| Met121TFM-Az | 629 | 379 ± 4 | gx = 2.0295(1.9) | 391 | ||
| gy = 2.0583(1.8) | ||||||
| gz = 2.2530(56.8) | ||||||
| Met121OxM-Az | 624 | 222 ± 5 | gx = 2.0305(0.8) | 391 | ||
| gy = 2.0576(2.5) | ||||||
| gz = 2.2629(55.7) | ||||||
| Met121Cys-Az | 451, 627, pH 4.0; 430, 591, pH 9.0 | 95 ± 3, pH 7.0 | S(Cys1)–Cu(II) 2.17(2) | gz = 2.26(58) pH 4.0; gz = 2.25(93) pH 9.0* | *major species | 392 |
| S(Cys2)–Cu(II) 2.74(2) | ||||||
| 2N–Cu(II) 1.95(2) (pH 7.0) | ||||||
| Met121Hcy-Az | 410, 590 | 113 ± 6, pH 7.0 | S(Cys)–Cu(II) 2.22(2), S(Hcy)–Cu (II) 2.79(2), 2N–Cu(II) 1.97(2) | gz = 2.25(193) | 392 | |
| N47S/F114N/M121L-Az | 630(~2080) | 706 ± 3, pH 4.0; 668 ± 1, pH 6.0; 640 ± 1, pH 7.0 | gx = 2.024(11) | (1) EPR spectrum collected at pH 6.0; (2) kET = 78 ± 12 s−1 at 298 K | 3,393 | |
| gy = 2.062(11) | ||||||
| gz = 2.287(44) | ||||||
| F114P/M121Q-Az | 600(~4030) | 90 ± 8, pH 7.0; −2 ± 13, pH 9.0 | S(Cys)–Cu(II) 1.95 | gx = 2.031(6) | (1) EPR spectrum collected at pH 7.0; (2) kET = 81 ± 11 s−1 at 298 K | 3,393 |
| N(His)–Cu(II) 1.95 | gy = 2.047(6) | |||||
| N(Gln)–Cu(II) 2.15 | gz = 2.258(46) | |||||
| CuA-CyoA | 765(900), 536(1600), 358(2000) | 260 ± 10 | gz = 2.20 | 394 | ||
| CuA-Az | 350(380), 485(155), 530(1380), 765(770) | 270, pH 5.0; 239, pH 5.5 | gx = 2.017(28, 15) | kET = 650 ± 60 s−1, pH 5.1, rt | 395–398 | |
| gy = 2.021(21, 14) | ||||||
| gz = 2.169(63, 57) | ||||||
| M123Q-CuA-Az | 350, 485, 530, 800 | 245 ± 6 | gx = 2.013(17, 22) | 397 | ||
| gy = 2.020(21, 19) | ||||||
| gz = 2.170(65, 56) | ||||||
| M123L-CuA-Az | 485, 800 | 255 ± 10 | gx = 2.021(33, 27), 1.982(20); gy = 2.044(32, 20), 2.055(18); gz = 2.168(65, 51), 2.227(30) | valence-delocalized and localized species | 397 | |
| M123D-CuA-Az | 350, 485, 530, 765 | 234 ± 3 | there are two valence-delocalized states | 397 | ||
| gx = 2.004(23, 19), 1.977 | ||||||
| gy = 2.019(24, 21), 2.034 | ||||||
| gz = 2.156(64, 57), 2.267 | ||||||
| M123E-CuA-Az | 350, 485, 530, 855 | 231 ± 3 | gx = 1.989, 1.987; gy = 2.029, 2.038 | there are two valence-delocalized states | 397 | |
| gz = 2.130, 2.263 | ||||||
| N47S-CuA-Az | 483, 530, 753 | 307 ± 7, pH 7.0; 275 ± 14, pH 5.1 | gx = 2.027(24.3) | 399 | ||
| gy = 2.027(14.2) | ||||||
| gz = 2.176(55.8) | ||||||
| E114P-CuA-Az | 473, 535, 784 | 235 ± 3, pH 7.0; 214 ± 8, pH 5.1 | gx = 2.017(29.5) | 399 | ||
| gy = 2.021(10.9) | ||||||
| gz = 2.169(56.6) | ||||||
| CuA-amicyanin (1) | 360, 483, 532, 790 | gx,y = 1.99–2.02 | 400 | |||
| gz = 2.18 (32.4) | ||||||
| CuA-amicyanin (2) | 360(1020), 485(2300), 530(2380), 780(1055) | 273 ± 2 | gx,y = 2.05 | 401 | ||
| gz = 2.20(36) |
2.2.4.2.1.1. Redesign of a Type 1 Copper Center
T1Cu centers have a unique coordination environment with characteristic spectroscopic and structural properties. Elucidating how the primary and the secondary coordination environments relate to the function of T1Cu as an electron transfer center is a major goal in protein design. The following section covers the transformation of a T2Cu into a T1Cu site and the incorporation of a T1Cu site into a dissimilar native scaffold. In addition, redesign work on native T1Cu sites to modulate their functionalities will be briefly highlighted here.
Sharing a structural similarity with cupredoxins, the active site of copper–zinc superoxide dismutase (CuZnSOD) is also confined in a Greek β-barrel fold.326,402–404 CuZnSOD catalyzes the conversion of superoxide into oxygen and hydrogen peroxide, where Zn(II) plays a structural role in the active site and copper performs the dismutase reaction.404 Although there is no sequence or active site homology between the two systems, researchers intended to introduce a T1Cu site in CuZnSOD by mutating an active site His residue into a Cys.405–409 On the basis of the crystal structure of the active site in CuZnSOD, there are three types of His residues coordinating to the metal ions: His residues that are only coordinated to copper, that is, copper-His (His46, 48, and 120); His that are only coordinated to Zn(II) (zinc-His, His71, and His80); and a bridging His residue that binds to both copper and zinc ions (His63). Besides these three His residue types, an additional water molecule is coordinated to copper and an Asp residue to zinc.410 To obtain a T1Cu center, the logical approach was to mutate the copper-His into Cys, as reported by Lu et al. Two mutants, SOD-H46C and SOD-H120C, were produced by site-directed mutagenesis of CuZnSOD from Saccharomyces cerevisiae.405,408 Interestingly, these mutants, upon binding to copper, exhibited spectroscopic characteristics of a T2Cu center instead of a T1Cu center, although sulfur to copper ligand-to-metal charge transfer (LMCT) was observed. Specifically, the EPR hyperfine coupling constants of the mutants His46Cys and His120Cys were significantly larger than that of a typical T1Cu center. Furthermore, as indicated by ESEEM and 1H NMR, the His120Cys mutant no longer possessed a bridging His, likely due to competition with the strong Cys120–copper bond, leading to breakage of the His63–copper bond.408 A second mutation, substituting a Zn(II)–His with Cys, was also reported by Lu et al.406 In this case, a T1Cu site was created by substituting His80 with Cys at the Zn(II) site of the original CuZnSOD. The mutant, SOD-H80C, preferably binds to Zn(II) at the newly constructed T1Cu site; however, adding Cu(II) before Zn(II) or adding only Cu(II) generates a site that exhibits intense absorption bands at 459 and 595 nm. These transitions were attributed to the sulfur-to-copper LMCT excitations. The hyperfine coupling constant from the T1Cu site was estimated to be very similar to that of stellacyanin. Additional spectroscopic techniques, including using Co(II) as a spectroscopic probe, were employed to confirm the existence of a T1Cu center.406,407 H80C-Cu2Cu2SOD was also redox active: the Cu(II) at the Zn(II) site (T1Cu) lost its visible absorption features when reduced by ascorbate, a process with a higher rate than that for WT-Cu2Cu2SOD.406 Banci et al. reported substitution of the bridging His63 in CuZnSOD with Cys, showing that Cys63 preferably binds to Zn(II) over Cu(II), leaving a five coordinate copper center where copper is coordinated to three His and two water molecules.409 These studies indicate that the simple incorporation of a Cys ligand at the copper center in CuZnSOD is not sufficient to construct a T1Cu site. When designing metal-binding sites, one needs to consider additional factors, including geometric constraints and secondary coordination sphere interactions.
Admittedly, the introduction of a mononuclear center into a native binuclear metal site complicates the interpretation of the experimental results, as one needs to take into consideration the behavior of the second metal-binding site. Using Trx as a parent scaffold that does not possess a native metallo-active site, Hellinga constructed a CysHis2Met center using the automated rational protein design program, Dezymer.411 Trx consists of five β-strands surrounded by four α-helices,412 providing multiple possible sites for the construction of a T1Cu center. At the same time, the β/α fold is distinctly different from the native cupredoxin scaffold, which provides an excellent opportunity to examine the impact of the secondary coordination sphere on the properties of the T1Cu center. After several rounds of design cycles to examine the location of the site, possible competing coordinating ligands, the coordination geometry, solvent access, and equatorial versus axial ligands, a few mutants were prepared that exhibit strong sulfur-to-copper LMCT excitations upon binding to Cu(II); however, these sites more closely resemble T2Cu centers than type 1, due to the strong equatorial ligand and weak axial ligand. Although these efforts failed to introduce a conventional T1Cu center with a tetrahedral coordination, this work indeed provided some very important and interesting insight in the design of a T1Cu center. The authors emphasized the importance of negative design approach, destabilizing the competing structures to maintain the rigidity of a native T1Cu site, and also pointed out the necessity of constructing an anhydrous, rigid coordination environment.
In addition to incorporating a T1Cu site into unrelated native scaffolds, Az, a native T1Cu protein, has been widely used in studies intended for modulating the redox and electron transfer properties of the copper center. Azurin is a small protein with a T1Cu center that shuttles electrons in certain types of denitrifying bacteria.413–415 At its T1Cu site, copper is coordinated to two His (His47, 117) and a Cys (Cys112) in a trigonal plane, while weakly interacting with two axial ligands, Met121 and a carbonyl oxygen from Gly45.416 The electron transfer function of Az was proposed to be related to cellular redox stress.417 Extensive studies have been carried out to probe the roles of each ligand to the spectroscopic features, redox properties, and the impact of certain substitutions on the driving force and the reorganization energy of the electron transfer process.365,418–432 We focus here on reports that demonstrate tuning of the redox potentials and the electron transfer rates of Az.
The thiolate–copper bond has been proposed to be essential to the spectroscopic and electron transfer features of a T1Cu center. To test this hypothesis, several groups of scientists carried out mutation studies to probe the role of copper-bound Cys. Cys112 in Pseudomonas aeruginosa Az was substituted with Asp, resulting in significant changes to its spectroscopic parameters, with loss of the T1Cu-featured strong absorption at 600 nm and the small EPR hyperfine coupling constant.432 The crystal structure of the oxidized form revealed that the overall structure of the protein had undergone minimal changes with this mutation; however, the active site adopted a square pyramidal geometry, with an asymmetric and out-of-plane carboxylate from Asp112 coordinated to the copper center.419 The X-ray absorption spectrum (XAS) of the oxidized form of C112D-Az is consistent with the crystal structure.420 Interestingly, XAS of the reduced form indicates an approximately three-coordinate Cu(I), forecasting a slow electron transfer rate due to the structural perturbation upon oxidation/reduction of the copper center.420 C112D-Az showed reversible interprotein electron transfer activity, as demonstrated by reacting the oxidized form of C112D-Az with reduced P. aeruginosa cytochrome c551.419 At the same time, its redox potential was lower than that of the WT-Az (+180 mV for C112D-Az and +310 mV for WT-Az, pH 7.0, vs NHE).365,419 On the basis of the results from C112D-Az, several mutants were prepared with the goal of expanding the range of accessible redox potentials.421–424 Lancaster et al. obtained high-resolution crystal structures of these mutants, which show significant reorganization around the copper site, including reorientation of the monodendate ligation of the carboxylate on Asp112.421 Specifically, the nonligating carboxyl oxygen from Asp112 forms H-bonds with Asn47 and Phe114 in C112D/M121L-Az, whereas in C112D-Az, C112D/M121I-Az, and C112D/M121F-Az, this oxygen only interacts with Phe114. The oxidized forms of the mutants C112D/M121X (X = Leu, Phe, Ile) exhibited very similar EPR features as compared to a typical T1Cu center, but did not have an intense absorption band due to a lack of sulfur-to-copper LMCT excitations, leading to a novel copper center called “type zero copper”. The authors examined the electron transfer rates of these type zero copper centers and found that they were much higher than the C112D-Az mutant with a T2Cu.421 Furthermore, mutants C112D/M121X-Az (X = Glu, His) have higher redox potentials than C112D-Az,422 a property later attributed to structural frustration around the copper site.423 These studies inspired researchers to re-examine rack-induced metal binding in proteins, which refers to the preorganization of the metal-binding site in electron transfer proteins to facilitate rapid electron transfer.425,431,433 On one hand, electron transfer proteins require rigidity at the metal site to minimize reorganization energy upon oxidation/reduction of the metal center; on the other, after certain residues or interactions are eliminated, the metal ion will adopt its favored coordination while the protein environment responds to this adjustment. The formation of different types of copper centers in Az mutants shows how these effects influence the coordination environment of the electron transfer center.
Lu and co-workers carried out a series of studies aiming to address the roles of the Cys ligand,388,389 the axial Met ligand,390–392 and secondary coordination sphere interactions.3 The impact of these residues on the spectroscopic and redox properties of T1Cu in Az highlights the power in using protein redesign to deepen our understanding of the factors that influence redox properties. Furthermore, this work showed varied intraprotein electron transfer capability of certain mutants, which is indicative of variation of the reorganization energy with rational design.393
The Cu–SCys bond in T1Cu centers was shown to be highly covalent, providing a path for efficient electron transfer. 432,434,435 In contrast, the role of axial ligands, especially SMet, remained unclear but was proposed to serve in controlling and tuning the redox properties of T1Cu sites. To study the role of the SCys core ligand further, Lu and co-workers used a semisynthetic method that allows the fusion of a recombinantly expressed protein with a synthetic peptide containing the desired selenocysteine (Sec) substitution.388,389 This technique was developed to incorporate unnatural amino acids into native protein scaffolds.436 The substitution of Cys112 with Sec, an isostructural analogue of Cys, was achieved by attaching a 17-residue Sec-containing peptide corresponding to the Nterminus of Az onto a N-terminus truncated Az. Copper-bound C112Sec-Az was further characterized using various spectroscopic techniques (UV–vis, EPR, XAS). The selenium-to-Cu(II) charge transfer shifted to lower energy (677 vs ~628 nm for WT-Az), and the EPR spectrum of the oxidized form of C112Sec-Az yielded a larger hyperfine coupling constant (104 vs 56 G for WT Az) with more rhombic character.389 Moreover, Ralle et al. reported, for the first time, the Se–Cu bond distance situated in a protein environment to be 2.32 Å. Interestingly, the reduction potential of C112Sec-Az is similar to that of the WT-Az (+316 vs +328 mV for WT, vs NHE). This work represents the first stable native scaffold that contains an unnatural Sec residue at the active site of the metalloprotein.388,389
The role of axial Met121 was studied using a similar strategy. Met121 of Az was substituted with a series of unnatural amino acids: norleucine (Nle), selenomethionine (SeM), difluoromethionine (DFM), trifluoromethionine (TFM), and oxomethionine (OxM).390,391 The Nle residue provides a hydrophobic moiety above the copper site, whereas SeM is an isostructural analogue with electronic and structural properties equivalent to those of Met. Methyl-fluorinated thioethers, both DFM and TFM, have sulfur ligands that are less electronegative than that on Met, while OxM contains an oxygen ligand that is more electronegative. Comparison of the absorption and EPR spectra of the variants to WT-Az reveals minimal shifts in the S(Cys)–Cu charge transfer band. The largest shift is 6 nm for the OxM mutant, and a difference of only 8 G in the copper hyperfine coupling constants (A//) is observed among all variants. The trend in shifting from blue to red, as observed in the absorption spectra, correlates well with an increase in ligand electronegativity. As compared to WT-Az (627 nm), the absorption maximum of the OxM variant is slightly higher in energy, and the Nle variant has the lowest energy absorption maximum. Changes in A// did not follow this electronegativity trend: the OxM variant (55.7 G) had the smallest A//, and WT Az (63.5 G) had the highest. Overall, these findings demonstrate that unnatural residues in the axial position have a minimal effect on the structure of the active site and further support the role of the axial Met as a weakly bound ligand. Additionally, the trend in reduction potentials (Nle > TFM > SeM > WT > DFM > OxM) is governed by the hydrophobic nature of the axial ligand, irrespective of the protein scaffold, experimental conditions, or steric differences. This trend was also witnessed in other T1Cu centers, including those in rusticyanin,437 fungal laccase,438,439 and cucumber stellacyanin.440 Furthermore, axial Met121 was substituted with much stronger donors, Cys and homocysteine (Hcy). The spectroscopic parameters and greatly enhanced interaction at the axial position indicate that these substitutions perturbed the active site drastically.392 Specifically, M121C-Az exhibited UV–vis absorption features similar to those of a typical blue copper center at pH below 5, while resembling that of nitrosocyanin at pH 9.0; at the same time, the EPR hyperfine coupling constant of M121C-Az fell into the range for a type 1.5 or a green copper center. To achieve the conversion of a blue copper center to a red copper center, Hcy was incorporated to take the place of axial Met121, resulting in a “coupled distorted” cupredoxin site that resembles the red copper in nitrosocyanin. The redox potentials of the mutants are lower than that of WT-Az (+95 mV for M121C-Az and +113 mV for M121Hcy-Az at pH 7.0, vs NHE), yet similar to that of nitrosocyanin at the same pH (+85 mV).359 A red copper center was also introduced into Az, without using unnatural amino acids, by substituting the blue copper-binding loop with a red copper-binding loop from nitrosocyanin using loop-directed mutagenesis.441
Redox processes, including electron transfer and catalytic oxidation–reduction reactions, are highly sensitive, not only to the first coordination sphere of the metal center, but also to the noncovalent, secondary interactions surrounding the redox-active site. Lu and co-workers demonstrated that noncovalent hydrophobic and H-bonding interactions assist in tuning the reduction potential of T1Cu proteins.3 The copper coordination environment varies slightly between T1Cu centers in the different mutants, yet all display similar spectroscopic properties, including strong absorptions in the visible region and small EPR hyperfine coupling constants. Most importantly, the redox potentials of this rationally designed series of T1Cu centers in Az span a wider range than even native cupredoxin centers. Lu and co-workers hypothesized that subtle differences outside the metal center can allow native T1Cu proteins to achieve their required reduction potentials.
Marshall et al. incorporated residues in a stepwise manner that served to disrupt or introduce H-bonding interactions at the outer coordination sphere of the T1Cu center in Az.3 An Asn47Ser mutation was first introduced to provide an H-bonding interaction between two ligand-containing loops in a motif found in rusticyanin (a T1Cu with one of the highest reported reduction potentials).326,353,442,443 At pH 7.0, the Asn47Ser variant has a redox potential ~130 mV higher than that of the WT (+265 mV vs NHE). Aiming to achieve an additive effect, the authors introduced a Met121Leu mutation to the Asn47Ser variant. As a hydrophobic, nonbonding residue, Leu was previously shown to increase the potential of the copper center by ~70 mV.425 As expected, Asn47Ser/ Met121Leu (Figure 10A) had a redox potential ~200 mV higher than that of WT rusticyanin, demonstrating an additive effect. The crystal structure of the Asn47Ser variant showed that the Ser mutation introduced an H-bonding group in close proximity to the S-Cys(112) coordinating ligand and the backbone amide nitrogen of a Thr residue, forming a Cys112-Ser47-Thr113 H-bonding network (Figure 10A), similar to the pattern found in rusticyanin.444,445 Nonetheless, the Asn47Ser/ Met121Leu variant did not achieve a redox potential surpassing that of rusticyanin. The copper in rusticyanin is missing an interaction with a backbone carbonyl of Gly45 that provides an ionic interaction with copper in native Az, lowering the potential. Therefore, the authors replaced Phe114 with an Asn residue (Asn47Ser/Phe114Asn, Figure 10B) to perturb the H-bonding interactions and disrupt the ionic interactions at the copper center. At pH 6.2, the triple mutant Asn47Ser/Phe114Asn/Met121Leu was determined to have a E° of +668 mV, a value exceeding that of any known rusticyanin variant.446 The crystal structure of Asn47Ser/Phe114Asn showed that an H-bond donor close to the Gly45 backbone carbonyl was introduced, disrupting the H-bonding network around the copper ligands and at the secondary coordination sphere (Figure 10B). Subsequently, Lu and co-workers aimed to tune E° to less positive potentials. This was achieved using a Met121Gln variant, which was previously shown to lower the redox potential of the native cupredoxin center in stellacyanin. 447 Additionally, substitution of Phe114 with a Pro served to reorganize the H-bonding network at the copper site. The Phe114Pro/Met121Gln mutant (Figure 10C) has a redox potential of +90 ± 8 mV at pH 7.0. When the pH was raised to 9.0, the redox potential of this mutant decreased to −2 ± 13 mV, which is the lowest potential ever reported for any cupredoxin or cupredoxin variant. Overall, by modifying noncovalent interactions in an additive, stepwise fashion, Lu and co-workers achieved exquisite control over the reduction potential of Az. This work resulted in the highest and lowest redox potentials ever achieved with a cupredoxin center, illustrating how subtle changes may influence noncovalent interactions around a redox center, and impact the functional properties of the site. These findings were further corroborated with subsequent XAS and DFT studies.448
Figure 10.
X-ray crystal structures of selected variants of Az. (A) N47S/M121L-Az (PDB code: 3IN2); (B) N47S/F114N-Az (PDB code: 3JTB); and (C) F114P/M121Q-Az (PDB code: 3IN0).3
Some of these mutants with outer-sphere modifications also showed intraprotein electron transfer activities. Farver et al.393 measured the electron transfer rates to address the question of whether or not a large range of redox potentials can also lead to controlled variation of the intramolecular electron transfer rates. Pulse radiolytically induced CO2− radical anions were introduced to these anaerobic Az derivatives to reduce the disulfide bond, located at the opposite side of Az relative to the copper center (Figure 11A), into a disulfide radical anion. Electron transfer between Cu(II) and the disulfide radical anion was tracked by measuring the absorption changes of both the Cu(II) band and that for the disulfide radical species. Six mutants with redox potentials ranging from +114 to +641 mV (vs NHE) were examined, and their electron transfer rates (kET) were measured. kET correlates with the driving force for electron transfer, falling along the bell-shaped curve that arises from theoretical calculations based on the Marcus theory449,450 (Figure 11B). This correlation indicates that these mutants have a low activation barrier for electron transfer that may be attributed to increased flexibility of the mutants as compared to the WT-Az. This effect leads to an electron transfer rate that can be up to 1 order of magnitude higher than that observed for WT-Az. The measurements of electron transfer rates, together with the modulation of the redox properties of Az, provide important insight into designing electron transfer centers. The cupredoxin site needs enough rigidity to maintain its copper coordination. For example, the coordination number of the Cu(I) and Cu(II) states would ideally remain unchanged during the redox process to minimize the reorganization energy. At the same time, incorporating enough flexibility at the copper site to leave some “wiggle” room for electron transfer can also contribute to lowering the reorganization energy. Moreover, both primary and secondary coordination sphere interactions contribute to the redox properties of a cupredoxin site.
Figure 11.
(A) Calculated electron transfer pathway from the disulfide bond to the copper site in Az mutants. Residues involved in electron transfer are in red. (B) Logarithm of the electron transfer rate constants as a function of the driving force of the reaction (dots) and the theoretically calculated bell-shaped curve. Mutants: 1, Phe114Pro/Met121Gln; 2, Phe114Pro; 3, Asn47Ser/Phe114Asn; 4, Asn47Ser/Met121Leu; 5, Phe114Asn/ Met121Leu; 6, Asn47Ser/Phe114Asn/Met121Leu. Reproduced with permission from ref 393. Copyright 2013 National Academy of Sciences.
In addition, an Az-polymer conjugate has been reported that exhibits thermosensitive electron transfer behavior. An imidazole terminated poly(N-isopropylacrylamide) (PNIPAM) polymer was introduced at the T1Cu site by replacing His117 in Pseudomonas aeruginosa Az (Figure 12).451 The “smart” polymer PNIPAM switches between hydrophobic and hydrophilic states based on temperature changes;452–454 hence the incorporation of this polymer directly at the electron transfer center in a native protein should modulate its redox properties as a function of temperature. The temperature at which the polymer turns to its hydrophobic state is called the low critical solution temperature (LCST).452–454 His117 was first mutated to Gly, then apo-H117G-Az was reconstituted with the imidazole-terminated PNIPAM complex in the presence of Cu(NO3)2. The absorption spectra of Az-PNIPAM (3800 g mol−1) and Az-PNIPAM (9800 g mol−1) were collected at 25 °C (below LCST) and 35 °C (above LCST), which showed characteristic T1Cu features. No spectral variation was observed at different temperatures, indicating that imidazole remained bound to copper. The existence of a T1Cu center in the conjugate was also supported by similar EPR parameters (A|| = 58 × 10−4 cm−1, g|| = 2.248, g⊥ = 2.055) as compared to WT-Az. Electron transfer processes were demonstrated between Az-PNIPAM and cytochrome c at 25 and 35 °C. Watanabe and co-workers showed that the inclusion of the PNIPAM tail slowed the electron transfer rate by about 1 order of magnitude as compared to WT-Az. Additionally, the longer the polymer tail, the more pronounced its thermo-sensitivity. For the smaller polymer PNIPAM(3800), the electron transfer rates were on the same order of magnitude above and below the LCST; however, the conjugate with a long tail, PNIPAM(9800), showed a 4-fold decrease in its electron transfer rate when the temperature was above LCST. These observations were attributed to the collapse of the polymer tail upon hydrophilic to hydrophobic switching when the temperature was raised.
Figure 12.
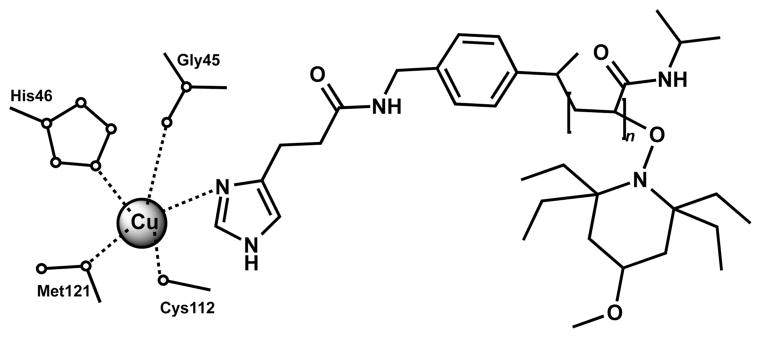
The active site of a reconstituted Az-PNIPAM conjugate. Reproduced with permission from ref 451. Copyright 2009 Wiley.
2.2.4.2.1.2. Redesign of Native Scaffolds To Incorporate a CuA Center
As introduced earlier, a CuA center is a copper electron transfer cofactor that is found in the terminal enzymes of respirational chains, for example, in CcO, N2OR, and NOR.375–378 CuA is a binuclear copper center with a diamond shaped [2Cu-2S(Cys)] core structure and NHis, SMet, and Obackbone ligands bound to the copper ions (Figure 13). CuA centers have a unique structural feature, the Cu–Cu bond, which is not commonly observed in biological systems for first row transition metals.379,380 The richness of its spectroscopic characteristics, as well as its electron transfer capability, make it a good candidate for comparison to a cupredoxin center. For example, both types of copper centers function to relay electrons, and both are situated in a Greek β-barrel fold. It is interesting to ask why nature has chosen to place the same metal ion in two different coordination environments while possessing the same biological function. We will summarize spectroscopic characterizations,395,455 kinetic studies,456,457 and investigations into the role of selected coordinating ligands and secondary coordination sphere interactions.396,397,399,458–460 A CuA center was first introduced into the CyoA subunit of E. coli cytochrome o quinol oxidase complex, a scaffold that is devoid of a copper binding site,394,461 was then built into an Az.395–399,455–457,459,460 Electron transfer activities were also demonstrated for the CuA–Az hybrid protein.398 Moreover, several groups have incorporated a CuA center into another T1Cu protein, amicyanin, a cupredoxin found in methylotrophic bacteria,400,401 and showed the electron transfer capability in these designed systems.400,401
Figure 13.

CuA center in N2OR from Pseudomonas nautica. The purple spheres are copper ions. PDB code: 1QNI.462
The sequence homology of the CyoA subunit of cytochrome o quinol oxidase to a cupredoxin was investigated, and the CuA binding motif was incorporated by substituting a SASYSGPGF sequence with a CAEICGPGH sequence. The Cys and His residues were built properly, together with the other important conserved residues, to create a CuA center.394 The absorption spectrum of CuA-CyoA resembled that of native N2OR.384,463,464 The redox potential of the CuA mutant (+260 mV) matches that of the purple copper center in N2OR.384 Subsequently, several derivatives were made from the purple copper CyoA, which demonstrated the importance of the His and Cys residues in keeping the integrity of a CuA center.461 Three categories of mutants were studied, out of which only one type of mutant with a nearby Glu mutated into an Ala maintained the CuA characteristics; the other two types with mutations on the coordinating His, Cys, or Met yielded either a blue or a brown copper center or the loss of copper binding capability. The authors demonstrated that at least five residues, two His, two Cys, and one Met, were essential to construct a CuA center.461
A CuA center was then incorporated into an Az, taking the place of the T1Cu center, by Hay et al.395,455 A short CuA binding sequence was attached to a recombinant Az, replacing the original loop region that accommodates the T1Cu center.395 One of the variants with Trp120 mutated to Leu to avoid steric hindrance exhibited an intense purple color upon binding to Cu(II). The absorption features at 350, 485, 530, and 765 nm and the EPR parameters were very similar to those of the CuA center in CcO.465 Metal substitution studies with Hg(II) and Ag(I) further indicated that the two copper binding sites might have particular preferences or selectivity, suggesting that the two binding sites might not be equivalent.455 The reduction potential of CuA-Az was measured by cyclic voltammetry (CV) and was shown to be pH-dependent.396 At pH 5.0, the reduction potential of CuA-Az was +270 mV, similar to that of the native CcO.387,465 However, as the pH was lowered from 7.0 to 4.0, the reduction potential increased by about 180 mV (160 mV at pH 7.0 to 340 mV at pH 4.0, vs NHE), a change attributed to the different protonation states of His120.396
Although at this point researchers have already obtained a significant amount of knowledge on the spectroscopic and structural features of a CuA center, the mechanism of copper incorporation into a binuclear binding site is still not very well understood. The kinetics of copper binding to CuA-Az were examined by Wang et al. using stopped-flow UV–vis spectroscopy, aiming to unveil the important intermediates formed during the uptake process of copper ions by apo-CuA-Az. 456 On the basis of the appearance of the initial absorption bands at 386 and 765 nm, and the transformation of these bands into characteristic CuA features at 485, 530, and 770 nm, they proposed that the copper incorporation process goes through a tetragonal Cu(II)–S(Cys) intermediate, corresponding to an initial 386 nm LMCT band. Moreover, an in situ reduced Cu(I) was also thought to be important in building the binuclear center in CuA-Az.456 Further studies suggested that not only is a blue copper involved in the formation of this binuclear center, but that a red copper may also be an intermediate depending on the pH and copper equivalents.457 For example, at pH 5.0, in the presence of 0.4 equiv of CuSO4, Cu(II) was initially captured as a T2Cu, then it transformed to an unidentified intermediate Ix, followed by the transition into a T1Cu and then CuA. On the basis of the spectral evolution at different pH conditions, Wilson et al. proposed a more detailed mechanism for copper uptake by CuA-Az (Figure 14). The addition of Cu(II) to a solution of apo-CuA-Az rapidly leads to a red T2Cu intermediate, and then three pathways might occur. The middle pathway directly leads to the formation of the CuA center due to the reduction of a Cu(II) into a Cu(I) in situ by the thiolates in Az. At lower copper concentrations, the reaction goes through the top pathway, where the intermediate Ix forms possibly through a structural rearrangement, and then it converts to a CuA center upon binding to an in situ reduced Cu(I). At higher pH values, the reaction goes through the bottom route, where a T1Cu center appears after the red copper formation involving a possible Cu(His46)(His120)-(Cys112) coordination. This T1Cu center then combines with a reduced Cu(I) to form a CuA center.457 The intermediates reported in this work were very similar to those reported for the native CuA metalation process in cytochrome oxidase, although one should recognize that copper is inserted in vivo from the Cu(I) oxidation level using a metallochaperone.466
Figure 14.

Proposed mechanism for copper binding to apo-CuA-Az. Reproduced with permission from ref 457. Copyright 2011 American Chemical Society.
Similar to T1Cu centers, the coordinating ligands in CuA centers are categorized into three groups, and the roles of each of them were examined in several contributions.396,397,458–460 There are two His residues at the equatorial position, four axial residues with sulfur or oxygen as coordinating atoms, and two bridging Cys residues (Figure 13).462,467
The coordinating His120 was substituted to Asn, Asp, Ala, and Gly to study the influence of the equatorial ligand on the properties of the CuA center in the Az scaffold.458 Prior to this work, the mutation of the equatorial His of a CuA site in CcO to Asn was reported by Farrar et al., resulting in no significant changes in the CuA UV–vis and EPR spectra.468 Consistent with the CuA site mutagenesis in its native scaffold, the CuA-Az derivatives His120Asn, His120Asp, and His120Ala showed very similar UV–vis absorption features and EPR parameters as compared to those of the CuA site in N2OR.458,469 This strongly suggests that the equatorial His120 is not essential in maintaining the binuclear core of the CuA center. Moreover, the double mutants His120Gly/Asn119X (X = Asp, Ser, or Ala) were created to test the hypothesis of whether Asn119 can replace His120 to coordinate copper.459 The mutation His120Gly was introduced to create an empty coordination site on the copper ion to allow the binding of potential exogeneous ligands such as imidazole, chloride, and azide. Berry et al. observed no significant changes in the UV–vis and EPR spectra of the double mutants, suggesting that Asn119 is not the replacement ligand for His120. This study showed that the His120X mutants are relatively stable with regards to the identity of the replacing residues.459 Furthermore, Hwang et al. determined the redox potential for the H120A-CuA-Az mutant and demonstrated the pH-dependent behavior of the original CuA-Az.396 His120 was also identified as a trigger for the pH-dependent redox potential change, providing important insight into the functional role of each of the two equivalents of copper ions in native CcO. In bovine heart CcO, His204, which corresponds to the His120 in CuA-Az, plays an important role in the proton-coupled electron transfer chain. This pH-dependent behavior might be reflective of the mechanism to regulate the proton-coupled electron transfer process.
The axial Met is a conserved residue in both CuA and T1Cu centers; however, its influence on the properties of these two types of copper centers is dissimilar, as reported by Hwang et al.397 As compared to the substitution of the axial Met in native Az, which resulted in a redox potential change of as much as 170 mV, the mutation of the axial Met in CuA-Az only led to a variation in the redox potential by up to 24 mV. In addition, the role of the bridging Cys was examined in CuA-Az by replacing the Cys112 and Cys116 with Ser.460 C112S-CuA-Az and C116S-CuA-Az exhibit spectroscopic features different not only from the parent construct CuA-Az, but also from each other, which is very interesting considering that these two Cys residues are almost geometrically equivalent. The Cys112Ser mutant was most likely to contain two distinct T2Cu centers, while the Cys116Ser mutant resulted in a mononuclear T1Cu center. Detailed analyses on the crystal structure of CuA-Az467 revealed that Cys112 and Cys116 experienced different H-bonding interactions with neighboring residues, leading to the differences upon mutations. This study, combined with the previous work on the role of the axial Met ligand and the equatorial His ligand, indicates that the bridging Cys residues are more important for keeping the structural integrity of the CuA center in CuA-Az.
As discussed earlier, changing the secondary sphere interactions can alter the redox potential of the T1Cu center in Az by up to a few hundred mV.3 New et al. examined whether similar changes to the noncovalent interactions would result in the same extent of redox potential variation in CuA-Az. 399 In the blue Az, two mutations, Asn47Ser and Phe114Pro, altered the H-bonding interactions around the coordinating Cys112, but Asn47Ser increased the redox potential while Phe114Pro decreased the potential.3 In CuA-Az, the residue at the 114 position is Glu. Similar effects were observed in CuA-Az, with Asn47Ser elevating the redox potential by 30 mV (from 277 mV in CuA-Az to 307 mV in N47S-CuA-Az, vs NHE) and Glu114Pro decreasing the redox potential by 42 mV (from 277 to 235 mV in E114P-CuA-Az) at pH 7.0. The change in the redox potentials upon these mutations is less than that in the blue copper Az (+99 and −67 mV for Asn47Ser and Phe114Pro, respectively), likely due to the fact that purple Az has two copper centers and two thiolate ligands that can share the variation in the electronegativity upon changing the H-bonding interactions involving the bridging Cys residues.
One of the major goals of incorporating a purple copper center into a native blue copper protein is to test whether this site maintains its native function: electron transfer. Farver et al. examined the electron transfer rate of CuA-Az.398 The benefit of having a CuA center in an Az scaffold is that it allows for a direct comparison of the electron transfer rates between the two core structures, a diamond-shaped binuclear Cu2S2 structure and a T1Cu [Cu(His)2(Cys)] structure, without the perturbation from the differences in the secondary structure. Using the same pulse radiolysis method as introduced earlier, intramolecular electron transfer rates were measured between the CuA center and the disulfide-bond containing RSSR− radical anion. The intramolecular electron transfer rate of a CuA center is almost 3 times faster than that of a T1Cu center at low pH. Even though the driving force calculated based on differences between the redox potentials of the electron donor and acceptor for CuA-Az is smaller than that of the WT-Az, the reorganization energy of the mixed valent Cu(1.5)---Cu(1.5) purple copper site is only one-half of that of a blue copper center, leading to a more efficient electron transfer center.398
Similar work substituting a T1Cu center with a CuA center has also been carried out by a few other groups.400,401 An amicyanin is a periplasmic blue copper protein that transfers electrons from metholamine dehydrogenase (MADH) to cytochrome in the respiratory chain.470–472 Dennison et al. introduced a CuA center in amicyanin by substituting the sequence from Thr94 to Phe98 in native amicyanin into a CuA binding sequence previously used to introduce a CuA motif into a copperless protein scaffold CyoA.394,400 Specifically, a Bacillus subtilis CcO CuA binding loop was incorporated into a Thiobacillus versutus amicyanin. Both the absorption and the EPR spectra of CuA-amicyanin were very similar to those of the CuA domains of CcO and N2OR.400,465,473 The intermolecular electron transfer activity of CuA-amicyanin was later demonstrated by Jones et al.401 As compared to the previous work, a slightly different CuA binding sequence from P. denitrificans CcO loop was used and incorporated into a P. denitrificans amicyanin. The characteristics of a mixed-valent binuclear copper center were observed in the absorption spectrum, and the redox potential of this site is +273 mV, similar to that of native CuA centers.387,465 Jones et al. showed complete oxidation of cytochrome c-55li, one of the native redox partners of amicyanin, when mixed with 1 equiv of CuA-amicyanin. The engineered CuA-amicyanin was not, however, able react with MADH, likely due to its inability to bind to MADH as a consequence of the mutated loop.401
2.2.4.2.2. Copper Catalytic Centers
Heme-copper oxidases (HCOs) are a class of important terminal oxidases that catalyze the reduction of molecular oxygen to water, coupled to proton translocations, which drive the synthesis of ATP in eukaryotic mitocondria and bacteria.474–479 HCOs contain a bimetallic iron–copper center with a CuB site located above the heme porphyrin plane, and coordinated by three His residues and a hydroxide ion in a tetrahedral geometry.480 Because native HCOs are large membrane proteins that contain other metal-binding sites, it is challenging to isolate the CuB-heme site for biochemical studies. Lu and co-workers introduced a CuB center into Mb, a small globular oxygen-binding protein that does not contain a copper site in its native state.229,260,309 With the guidance of molecular modeling of sperm whale myoglobin (swMb), two mutations were introduced, Leu29His and Phe43His, which, combined with the distal His64 residue, formed a (His)3 copper binding site.309 The double mutation did not perturb the structure of the protein or the integrity of the heme center. The difference spectrum before and after Cu(II) addition indicates a single Cu(II)-binding site. An important method to characterize the CuB-heme site in HCOs is to examine the binding of a small bridging ligand. Cyanide was titrated into a solution of CuBMb, while observing changes in the UV–vis spectra from a high- to low-spin heme. In the presence of copper, the change required less cyanide. These results indicate that cyanide interacts with both CuB and heme iron, which is further supported by EPR studies. Moreover, the disappearance of the Fe(III)-heme EPR signal upon addition of copper to CuBMb indicates a spin coupling between the iron and the copper center, which is another important feature of HCOs. The kinetics of oxygen binding and its reduction at the CuBMb site was studied by Sigman et al.229 In the absence of copper, the oxygen-binding ability of CuBMb decreased in comparison to the WT protein. However, once a redox-inactive metal was added (e.g., Ag(I)), the oxygen affinity was enhanced. Kinetic studies of oxygen reduction suggest that the copper ion facilitates the reduction of oxygen, which then converts heme to verdoheme. The role of protons in this reaction was also investigated, showing that protons are necessary for maintaining the integrity of the heme center by inhibiting the conversion from heme to verdoheme.229 The redox properties of CuBMb were examined by Zhao et al. using UV–vis spectroelectrochemistry, showing that the redox potential of the heme iron center is strongly dependent on the surrounding environment and electrostatics at the distal site.260 Specifically, the redox potentials were determined for CuBMb in the presence and the absence of the bridging ligand cyanide, Cu(II), and a redox-inactive Zn(II). The cyanide free complexes generally have higher redox potentials than the cyanide-bound complexes. Most importantly, in the absence of cyanide, the redox potential of the heme center remained the same (+77 to +80 mV, vs NHE). This is true whether or not a divalent cation was present. However, in the presence of CN−, the redox potential of Cu(II)–CuBMb was 16 mV higher than CuBMb; and that of the Zn(II)–CuBMb was even higher, an effect attributed to the addition of positive charge close to the heme center. These studies provide important insight into the role of CuB and proton coupled redox processes in native HCOs. Furthermore, they established a foundation for future work to increase the oxidase activities.
X-ray crystallography and relevant biophysical studies revealed that an important feature of native HCOs is a cross-link between the copper-coordinating His24 and a Tyr244 residue that form a covalent bond between the carbon C6 of the Tyr residue and nitrogen Nε2 of the His residue. This covalent link is responsible for the decreased pKa values of both the phenol and the imidazole groups.481–484 The importance of the Tyr residue at the active site was also demonstrated for the engineered HCO system CuBMb. Miner et al. introduced a Tyr residue at two different positions close to the copper-coordinating His of the CuB site, aiming to examine whether this functionality would lead to higher efficiency in the reduction of oxygen to water.485 IA Phe four residues away from the copper-coordinating His29 was substituted with a Tyr to mimic the native HCO, leading to F33Y-CuBMb. The second design was based on the crystal structure of a cbb3 HCO,486 and resulted in the G65Y-CuBMb mutant. This second mutant exhibited higher product selectivity than F33Y-CuBMb and could cleanly reduce oxygen to produce water with a higher TON that was attributed to the better positioning of the Tyr residue. Having demonstrated the importance of the Tyr residue in close proximity to the copper-coordinating His, to further investigate the role of this cross-linked Tyr-His functionality, Liu et al. introduced an unnatural amino acid imiTyr (Figure 15) into the engineered CuBMb protein, yielding imiTyrCuBMb.231 Cu(II) binds to imiTyrCuBMb with a Kd of 1.6 μM, with a perturbation of the Soret band of heme. The rate of oxygen reduction was measured in the presence of a reductant and a redox mediator; and catalase and SOD were introduced to distinguish between the formation of water and reactive oxygen species. The oxygen reduction assay showed that imiTyrCuBMb carries out an efficient and selective oxygen reduction to produce water only when Cu(II) is bound to the protein. More importantly, the TON of Cu(II)-imiTyrCuBMb was almost 3 times that of Cu(II)-F33YCuBMb (a mutant lacking the Tyr-His cross-link). These studies demonstrated the functional significance of the Tyr-His cross-link in maintaining sufficient catalytic activity.
Figure 15.

imiTyr residue. Reproduced with permission from ref 231. Copyright 2012 Wiley.
A site similar to a CuB-heme is a nonheme iron center (FeB) that is coupled to a heme center, an active site present in native NOR. The presence of a CuB or a FeB center coupled to the heme cofactor is a determinant for the function of the site.307,308 Zhao et al. examined the influence of the nonheme metal ion on the reaction of NO using an engineered protein CuBMb.230 The absorption spectrum of ferrous-CuBMb-NO was similar to that of the WTswMb-NO.487,488 In the presence of Zn(II), the absorption spectrum possesses features associated with a five-coordinate ferrous heme-NO species.489 Moreover, the binding of Cu(I) at the CuB site weakens the Fe–His bond, as suggested by both UV–vis and EPR spectroscopies. The difference between the effect of Zn(II) and Cu(I) binding was attributed to the difference in the oxidation states of the two metal ions. Catalytic NO reduction was observed for Cu(I)-bound CuBMb. The authors suggested that native NOR may have an nonheme iron center instead of a copper center because Fe(II) binding leads to a further weakening of the heme Fe–His bond and facilitates the heme Fe–NO interaction. Further studies on the derivatives of CuBMb to generate a mimic of NOR are reviewed in a previous section (2.2.4.1.2: Nonheme Iron Catalytic Centers).
2.2.4.3. Redesign of Manganese Centers
Manganese is another important redox-active metal with several biological roles. Manganese has a wide variety of formal oxidation states, providing richness in its redox chemistry. Manganese-containing proteins also play important roles in microorganisms, plants, and animals. Manganese catalase and manganese superoxide dismutase (MnSOD) are important antioxidant defense enzymes in certain types of oxygen-consuming organisms.298,490–496 A manganese cluster functions as the oxygen-evolving center (OEC) in Photosystem II of plants and photosynthetic bacteria.497–501 Moreover, manganese-containing enzymes are involved in diverse metabolic pathways including DNA synthesis502 and protein modification by glycosylation.503–505 The effort of redesigning manganese centers into proteins has focused on incorporating a manganese center into a foreign protein scaffold to carry out desired functions, improving native catalytic activity, and creating a novel magnanese binding site with functional importance. These studies have addressed different fundamental aspects of structure–function relationships in metalloenzymes and propelled the design of novel functional metalloenzymes toward potential applications in therapeutics.
Using a metal-substitution strategy, manganese was bound to apo-CA to obtain artificial enzymes capable of catalyzing epoxidation and peroxidation reactions.506,507 A few Thr199X mutants of Mn-CA were tested for epoxidation activities to investigate how the Thr199 side chain influences the epoxidation yields and enantioselectivity.506 Thr199Asp and Thr199Glu abolished the epoxidation activity, whereas Thr199Ser and Thr199Ala showed almost the same activity as compared to the WT-Mn-CA, but the enantioselectivity decreased drastically, indicating that the Thr199 side chain was important for inducing asymmetric catalysis. Moreover, Mn-CA was also examined for its peroxidase activity.507 Okrasa and Kazlauskas showed that Mn-CA catalyzes the oxidation of o-dianisidine with a catalytic efficiency comparable to native HRP (for HRP, kcat/Km = 57 × 106 M−1 s−1;508 Mn-CAII, kcat/Km = 1.4 × 106 M−1 s−1).
Researchers have always been fascinated by the similarities and differences between manganese and iron centers in metalloenzymes. For example, manganese peroxidase (MnP) is a heme peroxidase that shares structural and functional similarities with a CcP509 and a lignin peroxidase (LiP):510 all three enzymes contain a heme center, and they all catalyze the oxidation of organic molecules by peroxide.509–514 However, different types of peroxidases oxidize different categories of substrates and undergo slightly different reaction pathways. In MnP, Mn(II) binds to two Glu, one Asp, one heme propionate, and two water molecules in an octahedral geometry.511
Yeung et al. designed a manganese center into CcP to examine the structural and functional role of manganese in a similar local environment but in a foreign protein scaffold.228 After comparing the crystal structures of CcP and MnP, they chose to carry out triple mutations on CcP (Gly41Glu, Val45Glu, and His181Asp), which together with the propionate from heme available for coordination, mimicked the binding site of manganese in the MnP. The absorption spectrum of MnCcP was similar to those with Mn(II) bound to the periphery of the heme center. The oxidation of Mn(II) by H2O2 was examined for Mn(II)-MnCcP, which showed higher rates than those of WTCcP.228 Using proton NMR techniques, and on the basis of the broadening of specific signals due to Mn(II) binding, Wang et al. demonstrated that the desired manganese binding site was obtained.515 The binding of Mn(II) did not perturb the structure of the heme distal site but altered the heme proximal site. A more detailed comparison of the active site of CcP and MnP revealed that there are two Trp residues in close proximity to the heme center in CcP, while in MnP, two Phe are at the corresponding positions. Gengenbach et al. carried out mutations of Trp into Phe in MnCcP to incorporate more structural details of the Mn-heme center, further illustrating the role of these noncoordinating, radical-forming residues in the redox process.516 In native CcP, Trp191 and Trp51 were shown to be involved in the modulation of the reactivity of the high-valent heme center.517–519 Three mutants were designed [MnCcP(W191F), MnCcP(W51F), and MnCcP(W191F, W51F)] to study the kinetics of Compound I formation, decay, and manganese oxidation. Specifically, by substituting a Trp with a Phe, which is more difficult to oxidize, the decay time of Compound I increased significantly, indicating that this mutation disturbs the relay of radicals. Compound II formed as the active intermediate during the Mn(II) oxidation process instead of Compound III (formed by the reaction of Compound II with excess peroxide). Compound II was shown to be important in maintaining the catalytic activity. Moreover, MnCcP and its derivatives exhibited pH-dependent activity, similar to its native counter-part. 520 High-resolution crystal structures of the metal-free and Co(II)-bound forms of MnCcP.1, a slightly different design with Asp37Glu, Val45Glu, and His181Glu mutations, were reported, showing that the metal-free MnCcP.1 matches the structures of the active site of the native MnP.521 Moreover, the MnCcP.1 had a 2.5-fold increase in its catalytic efficiency from the previously reported construct. Inhibition assays were carried out for several divalent metal ions: Ca(II), Mg(II), Co(II), Ni(II), and Zn(II), showing that Ca(II) did not inhibit the reaction while Zn(II) had the greatest inhibition (maintained 37 ± 2% of the activity without the inhibitor).521 Wilcox et al. reported a similar design that incorporates a Mn binding site into CcP to mimic MnP.522 Initial screening of the possible appropriate manganese binding sites brought up two candidates, one of which, MP6.1, was almost the same design as Yeung’s construct,228 while the other one maintained the His at 181 position. Importantly, Wilcox et al. have observed intramolecular electron transfer activity between magnanese and heme iron.522
Another important manganese center in biology is the manganese cluster in the OEC of Photosystem II (PSII). The crystal structure of PSII unveiled a Mn4CaO5 cluster, where three manganese, one calcium, and four oxygen atoms form a cubane-like structure and the fourth manganese connects to the cubane through a μ-oxo bridge.501 Numerous synthetic complexes have been reported to mimic the structure and function of this cluster, which have provided a significant amount of mechanistic insight.523–525 The incorporation of such a site in a native protein can be challenging. Nevertheless, installing a redox-active manganese binding site situated in a protein environment is a good starting point for the purpose of understanding the role of each manganese ion and how its ligation contributes to its properties. Thielges et al. reported the design of a manganese binding site in a bacteriochlorophyll a dimer (P), which showed light-induced redox activity similar to that of OEC.526 The pigment–protein complex P can be oxidized upon excitation through an electron transfer process to a quinone via intermediate cofactors.527,528 The engineered manganese center in such a protein would serve as a secondary electron donor to P+. Three substitutions Met168Glu, Val192Glu, and Gly288Glu were designed in the M subunit, together with the native Glu173 and water molecules, to achieve the appropriate octahedral binding site. Four mutants were generated on the basis of the combination of these substitutions. In the absence of Mn(II), the light-induced reaction of the mutants exhibited spectroscopic properties similar to those of WT-P. When Mn(II) was present, P+ oxidized Mn(II) after illumination, producing a decrease of P+ absorption feature dependent on the Mn(II) concentration. X-ray crystallography was used to determine the structural changes associated with the incorporation of the manganese center, showing structural similarities between the mutant and PSII in terms of the relative locations of the Mn ion/cluster, the quinone, and the chlorophylls (Figure 16). This study provided a foundation for designing a light-induced metal catalytic center for potential biomedical applications.
Figure 16.

Comparison of (A) M2 mutant (the reaction center of the protein L and M subunit from Rhodobacter sphaeroides, PDB code: 1Z9J526) and (B) PSII (a and b chains of PSII from Thermosynechococcus elongates, PDB code: 1S5L529). Mn(II) (purple sphere), Fe(II) (red sphere), (bacterio)chlorophylls, and quinone acceptors are shown. The manganese ions are located at similar positions in both proteins.
2.2.4.4. Redesign of Rhodium Centers
One of the simplest strategies for protein redesign is metal substitution. We already introduced one example in section 2.2.4.3: Redesign of Manganese Centers (Mn-substituted CA506,507). Using the same strategy, Kazlauskas and co-workers have carried out the design of a rhodium-substituted CA (CA-[Rh]) that catalyzes the stereoselective hydrogenation of olefin and the regioselective hydroformylation of styrene.530,531 To minimize the nonspecific binding of rhodium to the surface residues of the protein, particularly His and Lys, they carried out either site-directed mutagenesis or a combination of site-directed mutagenesis and modification of His residues using the His-selective reagent diethylpyrocarbonate (DEPC) to prevent rhodium binding. Rhodium-substituted WT-CA and several mutants with low surface-bound rhodium ions were selected to test for cis-stilbene hydrogenation.530 While the hydrogenation was observed under 5 atm of H2, the isomerization product was also generated during the reaction. The isomerization was thought to be catalyzed by the surface-bound rhodium because the reaction catalyzed by mutants with low surface-bound rhodium produced a significantly lower level of the trans isomer. In addition, the authors demonstrated that the rhodium bound to the active site of CA selectively catalyze the hydrogenation of cis-stilbene over trans-stilbene. This selectivity was attributed to the structure of the hydrophobic pocket defined by a few active-site Val, Leu, and Trp residues. Furthermore, Kazlauska and co-workers investigated the hydroformylation activity of CA-[Rh].531 They showed that both rhodium-substituted WT-CA and several mutants with lower surface-bound rhodium were able to catalyze the hydroformylation of styrene into 3-phenylpropanal (linear) and 2-phenylpropanal (branched). It was also shown that the production of the linear product was due to the CA-active-site rhodium instead of the surface rhodium. The highest selectivity reported was approximately 8.4-fold linear aldehyde over branched aldehyde. Computer modeling was employed to explain the regioselectivity, which showed that the active site accommodates the linear aldehyde with the carbonyl group closer to the rhodium center, a feature that is important for the reductive elimination reaction.
2.2.5. Artificial Metalloenzymes for Regio- and Enantioselective Catalysis
The synthetic chemists’ pursuit of regioselectivity and stereoselectivity in organic chemical transformations is a nontrivial task. Thus, it comes as no surprise that the field of protein engineering provides both great opportunities (e.g., exploiting the inherent chirality of synthetic catalysts) and great obstacles (e.g., defining proper substrate binding and orientation). Protein design applies the general principles of organic, inorganic, and supramolecular chemistry to yield hybrid catalysts with the molecular recognition ability of proteins and the broad reactivity scope of small molecule catalysts. The combination of transition metals and organometallic catalysts with proteins has yielded a library of artificial enzymes for regio- and enantioselective catalysis.532 There are a few main approaches to attaching synthetic catalysts to protein scaffolds: supramolecular anchoring (with biotin–avidin (Avi) being the most commonly employed form), dative anchoring (direct coordination to the metal center), and covalent attaching. Examples of each strategy are included below.
2.2.5.1. Supramolecular Anchoring Approach
The pioneering work carried out by Wilson and Whitesides set the first example of utilizing biotin–Avi noncovalent interactions.533 They attached a diphosphinerhodium(I) moiety onto biotin, which was then incorporated into Avi to carry out enantioselective homogeneous hydrogenation. Ward and coworkers have developed a series of artificial enzymes that rely on strong noncovalent interactions to anchor organometallic catalysts within natural proteins.534 Biotin has an extremely high affinity for the proteins Avi and Sav (Ka ≈ 1013 and 1015 M−1, respectively).535–538 Furthermore, the derivatization of the valeric acid side chain on biotin caused no significant decrease in this affinity, making it a promising attachment point for an organometallic compound. A short spacer between the biotin and organometallic catalyst precursor ensured that, once bound, the active site resided within the well-defined chiral environment of the protein (Figure 17). There are several advantages to this hybrid supramolecular approach to catalyst design. No chemical step is required for the coupling of the biotinylated catalyst precursor to the protein, so the integrity of the organometallic species is not compromised. Moreover, because the catalyst and the host protein are developed separately of one another, they can be optimized independently through synthetic and molecular biology approaches. Diversity in the catalyst precursor was achieved by varying the length and chirality of the spacers between the biotin and the chelating group, the nature of the chelating group, and the metal itself. On the other hand, the host protein could be diversified using random or site-directed mutagenesis, which do not influence the overall protein structure.538 This chemogenetic approach yielded a diverse library of compounds to be screened for activity and selectivity in abiotic reactions. The resulting hybrid catalysts have features of both enzymes (activity in water, selectivity relies on second coordination sphere interactions, optimization by mutagenesis) and homogeneous catalysts (broad substrate tolerance, chemical optimization).539 In general, the activity of the enzyme is dictated by the organometallic catalyst precursor, while the selectivity is reliant upon mutations to the host protein.
Figure 17.

The assembly of an active achiral catalyst into a host protein. Reproduced with permission from ref 539. Copyright 2003 Swiss Chemical Society.
2.2.5.1.1. Enantioselective Hydrogenation of Alkenes
As a proof-of-concept, an artificial enzyme was developed for the enantioselective hydrogenation of acetamidoacrylic acid to acetamidoalanine. The organometallic catalyst [Rh(COD)2]-BF4 was biotinylated with ligands Biot-3 through Biot-6 and then incorporated into Avi, neutravidin, and Sav (Scheme 3, reproduced from ref 540 with permission. Copyright 2003 American Chemical Society.).540 Because of its relatively large binding pocket and tolerance to mutation, Sav was found to be the best host protein. Generally, Biot-3 with a more flexible linker showed higher enantioselectivity than Biot-4. The most selective artificial catalyst, [Rh(COD)(Biot-3)]⊂Sav S112G, gave a quantitative conversion with 96% ee (R) after 15 h.540 Further screening of the above biotin-spacer-ligand scaffolds with seven host proteins (2 WT, 5 mutants) led to [Rh(COD)(Biot-51-4)]⊂Avi, an artificial enzyme that selectively produces (S)-acetamidoalanine (80% ee).541 These results established the ability of this approach to yield enantioselective catalysts with features of both enzymatic and homogeneous catalysts by exploiting the secondary coordination sphere of the host proteins.542 To diversify the host protein scaffold further, saturation mutagenesis was carried out on position 112 of Sav.543 The combination of these 20 proteins with the 18 biotinylated catalyst precursors (Biot-3–Biot-6, Scheme 3) yielded 360 artificial metalloenzymes, which were screened as catalysts for the hydrogenation of α-acetamidoacrylic acid and α-acetamidocinnamic acid. Fingerprint displays of the enantioselectivities (both (R) and (S)) revealed the importance of the linker and side chains for activity and selectivity. To further establish the location of the catalyst within the biotin-binding pocket of Sav, association constants were measured for prototypical cationic biotinylated rhodium-diphosphine catalyst precursors and host proteins at pH 7 (Ka = 107.7 M−1 for Avi and Ka = 107.1 M−1 for Sav).541 CD revealed cooperative binding of the biotinylated complexes.544 Next, the stereochemistry of the acetamidoalanine formed from the hydrogenation of acetamidoacrylic acid was monitored when the ratio of [Rh(COD)(Biot-3)]+ to Avi and Sav was varied.545 The ee decreased slowly with the addition of excess [Rh(COD)(Biot-3)]+, suggesting that when bound the catalyst is more active and selective than when unbound. Low substrate solubilities precluded the direct measure of rate constants (kcat), but the relative rates of the free and protein bound [Rh(COD)(Biot-3)]+ could be determined. The catalyst bound within Avi and Sav displayed 12.0- and 3.0-fold acceleration over the unbound catalyst, respectively. This “protein-accelerated catalysis” is similar to ligand-accelerated catalysis, in which reaction rates for heterogeneous and homogeneous catalysis are accelerated upon the addition of a ligand to a catalyst that performs even in the absence of that ligand. These studies were reviewed previously by Thomas et al.542 and Steinreiber et al.7
Scheme 3.540.

Having established the effects of short achiral spacers on activity and stability, enantiopure amino acid spacers were introduced between the Biot-3 and Biot-4 (Scheme 3) anchors and the rhodium-diphosphine moiety.546,547 (R)- and (S)-Phe, which may form π–π bonds with the four Trp residues that line the biotin-binding pocket of Sav, and (R)- and (S)-Pro, which have restricted degrees of freedom, were chosen. Again, the hydrogenation of α-acetamidoacrylic acid and α-acetamidocinnamic acid was studied. For both substrates with the Biot-3 anchor, the (R)- and (S)-Phe spacers gave moderate selectivity (64% and 73% ee for (R) and (S), respectively), the (S)-Pro spacer gave low selectivity (23% ee (R)), and the (R)-Pro spacer yielded ~90% ee (S).546 Only modest activities and enantioselectivities were observed with the Biot-4 anchor.547 These differences suggest that the chiral environment and position of the catalyst within the protein change upon inverting the spacer configuration, showing the importance of secondary interactions to achieve selectivity. Next, these four biotinylated catalyst precursors were combined with the 20 proteins generated by saturation mutagenesis at position 112 on Sav, screened for selectivity, and analyzed by fingerprint display.548 Here, genetic modification of Ser112X Sav yielded more diverse outcomes than it did for the achiral spacers. Additionally, Avi was shown to yield 86% ee (S) using [Rh(COD)Biot-(R)-Pro-3]+⊂Avi. Moreover, up to 45% DMSO was found to be tolerable allowing one to expand the number of suitable substrates, and the catalyst was immobilized on Biotin-Sepharose while maintaining selectivity.7,549 Besides Avi and Sav, burkavidin, secreted from the human pathogen Burkholderia pseudomallei, was isolated, purified, characterized, and used as a scaffold for an artificial metalloenzyme.550 [Rh(COD)(Biot-3)]+ and [Rh(COD)(Biot-(R)-Pro-3)]+ were incorporated into burkavidin and screened for catalytic activity in the hydrogenation of N-acetamidoacrylic acid and N-acetamidocinnamic acid. N-Acetamidoacrylic acid showed 39% ee (R) and quantitative conversion, while N-acetamidocinnamic acid gave 65% ee (S) and only 21% conversion. This modest enantiomeric enrichment suggests that catalysis occurs in the biotin-binding pocket and that this catalyst has potential for further optimization use in biotin-Avi technology.
2.2.5.1.2. Reduction of Ketones
Artificial enzymes based on biotin-Avi technology that incorporate d6-piano stool complexes are capable of catalyzing the asymmetric reduction of ketones by transfer hydrogenation using a formate·boric acid mixture as the hydrogen source.551 Modifications of the spacer and introduction of point mutations to Sav and Avi led to the chemogenetically optimized catalyst [η6-(p-cymene)-Ru(Biot-p-L)Cl]⊂P64G Sav, which is efficient for the reduction of p-methylacetophenone (94% ee (R), 92% yield) (Schemes 4 and 5, reproduced from ref 551 with permission. Copyright 2005 National Academy of Sciences.). Sav was found to be the best host protein, para-ligands were the best anchors, and η6-p-cymene resulted better than η6-p-benzene as the catalytic center. Point mutations farthest from catalytic site had the biggest effect on enantioselectivity. Like natural enzymes, these versatile transfer hydrogenation catalysts have enantioselectivities determined by second coordination sphere interactions. Docking studies were carried out to identify the localization of the piano stool complex within Sav.552 This approach allowed the development of a three-step search strategy to identify good catalyst/host combinations without examination of each. First, screens were carried out on 21 catalyst precursors with two Sav forms, then the most promising ones were tested with 20 Sav isoforms, and, finally, the best combinations were screened with eight different ketones (Figure 18). This procedure led to good enantioselectivities for the reduction of aromatic ketones to enantioenriched alcohols (up to 97% ee (R) and 70% ee (S)), but only poor to modest enantioselectivities for nonaromatic ketones. This behavior suggests that CH–π interactions between the η6-arene and the substrate are important for dictating enantioselection.
Scheme 4.551.

Scheme 5.551.
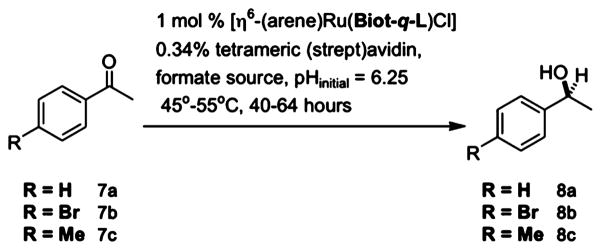
Figure 18.

Search strategy to identify good catalyst⊂protein combinations. Reproduced with permission from ref 552. Copyright 2006 American Chemical Society.
Enantioselectivities for the reduction of aromatic ketones were improved using a designed evolution approach starting from the crystal structure of [η6-(C6H6)RuCl(Biot-p-L)⊂S112K Sav.553 The structure (1.58 Å resolution, R = 0.168, Rfree = 0.187, PDB code: 2QCB) confirmed that the protein does not undergo structural reorganization upon incorporation of the bulky biotinylated complex and allowed for the identification of several amino acids that interact with the Ru-complex. While the biotin portion of the cofactor is well-localized in the host binding pocket, the ethylenediamine (en) and phenylsulfonyl moieties are disordered. Interestingly, only the S configuration of the ruthenium piano stool complex was crystallized, despite the use of a racemic mixture of the cofactor used for crystallization. This preference for only one of the two chiral forms of the cofactor may be the cause of the observed enantioselectivity of the metalloenzyme. The crystal structure also allowed for the selection of positions 112 and 124 for genetic optimization by saturation mutagenesis. The resultant proteins were combined with [η6-(arene)RuCl(Biot-p-L)] (arene = benzene or p-cymene), immobilized on biotin-sepharose, screened for transfer hydrogenation of seven ketones, and analyzed by fingerprint display. This resulted in high levels of conversion and selectivity for the reduction of methyl aryl (up to 96% ee (R)) and methyl alkyl (up to 90% ee (R)) ketones to alcohols. Classical force field and DFT Born–Oppenheimer molecular dynamics methods, used to probe host–guest interactions in [η6-(p-cymene)RuCl(Biot-p-L)]⊂Sav, highlighted the conformational flexibility of the cofactor and suggested residue 64 as a promising location for mutations that could affect activity.538
2.2.5.1.3. Reduction of Imines
The asymmetric transfer hydrogenation of imines and ketones follows different enantioselective mechanisms when catalyzed by homogeneous catalysts. Artificial metalloenzymes obtained by the combination of Sav and four biotinylated ruthenium and iridium-containing d5 and d6 piano-stool complexes were screened for the production of salsolidine (Scheme 6, reproduced from ref 554 with permission. Copyright 2011 Wiley.).554 The most promising catalyst [Cp*Ir(Biot-p-L)Cl]⊂ Sav was screened with the saturation mutagenesis library Sav S112X revealing that both enantiomers of salsolidine can be produced. For most mutations, (R) is dominant and the smallest amino acids at position 112 give the best (R)-selectivities, while (S)-selectivity results from cationic residues at position 112. The best results were obtained at 5 °C for [Cp*Ir(Biot-p-L)Cl]⊂ S112A (91% (R)) and [Cp*Ir(Biot-p-L)Cl]⊂ S112K (78% (S)). Catalysis appears to occur within the biotin-binding pocket and by the same mechanism as ketone reduction.
Scheme 6.554.
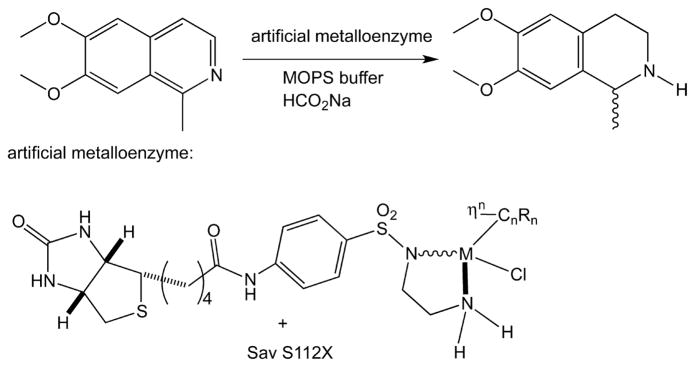
[Cp*Ir(Biot-p-L)Cl]⊂ S112X catalysts were applied to enzymatic cascade reactions.555 When [Cp*Ir(Biot-p-L)Cl]⊂ S112T (enantioselective for (R) product formation) was combined with an (S)-reducing monoamine oxidase (and a catalase to consume H2O2), 9-ox and rac-9-red were converted to (R)-9-red quantitatively with 99% ee (Scheme 7A, Scheme 8, reproduced from ref 555 with permission. Copyright 2013 Nature Publishing Group.). Similarly, this amine→imine→amine cascade occurred with high conversion and stereoselectivity for amines with purely aliphatic substituents (rac-10-red, Scheme 7A) and a tertiary amine ((S)-11-red, Scheme 7B). Together, the transfer hydrogenase and the monoamine oxidase work in concert to produce the enantiopure amine. Cascade reactions (Scheme 9, reproduced from ref 555 with permission. Copyright 2013 Nature Publishing Group.) were also developed for the production of pepecolic acid from L-Lys (12→13, Scheme 7C) and a monooxygenase-catalyzed oxy-functionalization reaction involving the regeneration of NADH (Scheme 7D).
Scheme 7.

(A) Reduction of Prochiral Imines 9-ox, 10-ox with Subsequent Deracemization of Cyclic Amines; (B) Stereoinversion of Natural Nicotine (S)-11-red to (R)-11-red, Leading to the Formation of the Chiral Alcohol 11-redol; (C) Formation of L-Pipecolic Acid (13) from L-Lys (12); and (D) Hydroxylation of 2-Hydroxybiphenyl (14) Coupled to an ATHase-Catalyzed NADH Regeneration Processa
aMAO-N, a monoamine oxidase; ATHase, an artificial transfer hydrogenase varian; LAAO, L-amino acid oxidase; DAAO, D-amino acid oxidase; HbpA, 2-hydroxybiphenyl monooxygenase. Reproduced with permission from ref 555. Copyright 2013 Nature Publishing Group.
Scheme 8.555.
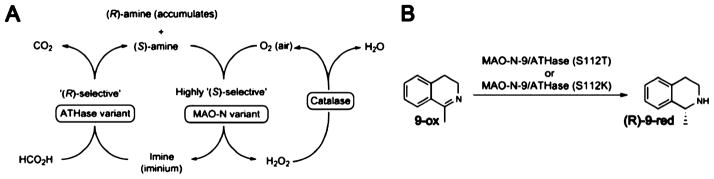
Scheme 9.555.
2.2.5.1.4. Oxidation of Alcohols
d6-Piano stool complexes hosted within Avi/Sav were also shown to catalyze the oxidation of alcohols in water.556 Four biotinylated organometallic catalyst precursors containing ruthenium, rhodium, and iridium ions (Scheme 10, reproduced from ref 556 with permission. Copyright 2005 Elsevier.) were incorporated in Sav and used to catalyze the oxidation of sec-phenethyl alcohol using tert-butylhydroperoxide and a terminal oxidant. The ruthenium-containing catalysts were found to be the most active (up to 81% conversion). The use of benzyl alcohol or cyclohexanol and point mutations to Sav did not improve the activity; however, when precursor 16 was hosted within Avi, the reaction proceeded nearly to completion (92% conversion).
Scheme 10.556.
2.2.5.1.5. Enantioselective Sulfoxidation
Artificial metal-loenzymes were built by incorporating four chiral biotinylated manganese–salen complexes (Sal-1 to Sal-4, Scheme 11) into WT Sav and five other mutants.557 The resulting enzymes showed moderate conversions (up to 56%) and low enantioselectivities (up to 13%) for the sulfoxidation of thioanisole using hydrogen peroxide in water. Although the enantioselectivities were low, they suggested that the protein participated in the mechanism of sulfoxidation.
Scheme 11.557.
2.2.5.1.6. Asymmetric Allylic Alkylation
An artificial palladium enzyme for asymmetric allylic alkylation was designed on the basis of biotin-Avi technology and optimized using chemical and genetic optimization.558 In asymmetric allylic alkylation, a soft nucleophile attacks a palladium-bound η3-allyl moiety. Screens were carried out to determine the best biotinylated scaffolds, spacer, protein host (Avi, Sav, or Sav mutants), and surfactants in conjunction with [PdCl(η3-allyl)] and [PdCl(η3-Ph2allyl)]. [PdCl(η3-Ph2allyl)(Biot6o-3)]+⊂S112X stood out as the best catalyst with a 90% ee (R) and 95% conversion when X = A, while a 31% ee and 96% conversion was obtained when X = Q.
2.2.5.1.7. Asymmetric C–H Activation
Three sites on [Cp*RhCl2]2 are required for the catalysis of electrophilic aromatic C–H activation, which precludes the addition of an asymmetric ligand to the primary coordination sphere and the development of an enantioselective catalyst. The integration of the catalyst into a protein scaffold allowed for enantioenrichment, resulting from the secondary coordination sphere of rhodium.559 The biotinylated catalyst precursor [RhCp*biotinCl2] was incorporated into Sav, and this artificial metalloenzyme was genetically optimized. The introduction of a basic residue in close proximity to rhodium improved the reaction yield, whereas incorporating bulky aromatic residues led to improved enantioselectivity for the reaction of pivaloyl-protected benzohydroxamic acid and methyl acrylate to dihydroisoquinolone (Figure 19). Yields up to 99%, regioisomeric ratios of up to 20:1, and enantiomeric ratios up to 91:9 were observed for different Sav mutants. While the limited solubility of the substrate precluded the use of Michaelis–Menten kinetics, rate accelerations of up to 92-fold were observed for the protein-bound catalyst as compared to the free catalyst.
Figure 19.
(A) The conceptual approach to combine a basic residue and the catalytic biotinylated RhCp*biotinCl2 to achieve a synergistic catalyst. (B) The structure of RhCp*biotinCl2 and the reaction it catalyzes. (C) Proposed transition state for the C–H bond activation. (D) Auto-Dock model showing the spatial relationship of the biotinylated RhCpbiotin(OAc)2 catalyst in the Sav tetramer active site. Reproduced with permission from ref 559. Copyright 2012 AAAS.
Catalysts were prepared by binding metal complexes directly within the biotin-binding pocket of Sav without first covalently linking them to biotin. According to docking studies, the precatalyst [VO(H2O)5]2+ interacts with the biotin-binding site of Sav via only second-sphere contacts.560 This complex yields an artificial metalloenzyme capable of catalyzing the enantioselective oxidation of prochiral sulfides with tert-butylhydrogen peroxide as an oxidant. Aromatic substrates saw improved enantioselectivity with increased steric bulk of the aromatic moieties (up to 93% ee for compound 23), and dialkyl sulfides gave reasonable enantioselectivities as well (up to 86% ee for compound 26). This work differs from all of the previous work from Ward and co-workers in that the catalytic metal ion is not biotinylated, rather the vanadium ion sits in the biotin-binding pocket (Scheme 12, Reproduced from ref 560 with permission. Copyright 2008 American Chemical Society).
Scheme 12.560.
The homogeneous catalyst OsO4− is used for the dihydroxylation of most olefins, but cis-substituted olefins are shown to be challenging substrates for this reaction. By anchoring an Os(VIII) center directly within Sav, Ward and coworkers were able to develop a catalyst specific for this substrate type (Figure 20).561 While crystal structures of the metalloprotein did not clearly reveal the specific binding site for Os(VIII), mutations to the biotin-binding site resulted in diminished selectivity. Further mutations were used to fine-tune the protein for dihydroxylation of seven different olefins, resulting in the highest ee values ever reported for allyl phenyl sulfide and cis-β-methylstyrene. Overall, OsO4·Sav can be optimized for the enantioselective dihydroxylation of challenging olefins, but suffers from modest TONs (27 turnovers per metal center).
Figure 20.
Schematic representation of a transition-state structure for an artificial cis-dihydroxylase resulting from anchoring OsO4− onto a host protein. Reproduced with permission from ref 561. Copyright 2011 Wiley.
2.2.5.1.8. Artificial Hemoproteins
Mahy and co-workers contributed significantly in the field of designing artificial hemoproteins that exhibit a variety of catalytic functions.562 They designed a series of catalytic antibodies associated with a metalloporphyrin cofactor via noncovalent interactions, which they named “hemoabzymes”. These antibody complexes exhibited peroxidase-like activity and were capable of catalyzing the regioselective nitration of phenols and the stereoselective oxidation of sulfides.563–565 A hapten iron(III)-meso-tetrakis-(ortho-carboxylphenyl)porphyrin was recognized by three antibodies, two of which exhibited high binding affinity with Kd values of 2.9 × 10−9 and 5.5 × 10−9 M,563 the best values reported for iron-porphyrin binding to antibodies. The peroxidase activity of these two antibody complexes was examined for the oxidation of 2,2′-azino-di(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) by H2O2.566 The catalytic efficiency of these constructs was not as high as their native counterparts, which was attributed to the lack of the axial His. To solve this issue, Mahy and co-workers decided to use microperoxidase 8 (MP8). This scaffold contains a heme group and residues 14–21 of horse cytochrome c, where residue His18 was thought to bind to the heme center to form a five-coordinate iron center. MP8 binds antibody 3A3 with a relatively high affinity (Kd,app = 1 × 10−7 M), yielding a heme–antibody complex via the recognition of the carboxylate groups on the porphyrin. The 3A3–MP8 complex catalyzed the oxidation of o-dianisidine by H2O2 with a catalytic efficiency of 2 × 106 M−1 min−1, slightly higher than the unbound MP8 catalyzing the same reaction.563 It was also demonstrated that the 3A3–MP8 complex could catalyze the regioselective nitration of phenol in the presence of H2O2 with 2-nitrophenol as a preferred product over 4-nitrophenol.565 Moreover, the 3A3–MP8 complex was shown to catalyze the oxidation of thioanisole by H2O2 and tert-butyl-hydroperoxide with 23% enantiomeric excess in 100% aqueous solution and 46% enantiomeric excess in 5% tBuOH.564
The next generation of the artificial hemoproteins is the “hemozymes”, which is constructed with two strategies. On the basis of a “Trojan horse” strategy, Mahy and co-workers designed an estradiol–iron metalloporphyrin conjugate. This structure was then associated with an antiestradiol antibody because it has a high affinity to the steroid estradiol based on supramolecular interactions.567,568 Specifically, 5,10,15-tris(4-pyridyl)-20-(4-aminophenyl)porphyrin was synthesized and attached to 3-O-carboxymethylestrone. The estradiol–iron–porphyrin conjugate was then associated with antibody 7A3. The antibody-bound estradiol–iron–porphyrin complex was shown to catalyze the enantioselective sulfoxidation of thioanisole using H2O2 as an oxidant.567
Recently, a new generation of artificial peroxidases was constructed using a host–guest strategy by associating a synthetic iron–porphyrin complex with xylanase A.569,570 Xylanase A (Xln10A) possesses a globally positive charge, which can properly accommodate the negative charge from the iron(III)–porphyrin complexes. Ricoux et al. examined a few such complexes, and they showed that Fe(III)-tetra-para-carboxyphenylporphyrin [Fe(TpCPP)] had the highest binding affinity to Xln10A with a Kd value of 5 × 10−7 M. When catalyzing the oxidation of o-dianisidine by H2O2, the peroxidase activity of Xln10A-Fe(TpCPP) had a catalytic efficiency of 6.5 × 104 M−1 min−1.569 In addition, Ricoux et al. showed that the same strategy can be used to create an enzyme that catalyzes the selective oxidation of aromatic sulfides.570 They studied two iron–porphyrin complexes [Fe(TpCPP) and meso-tetra(4-sulfonatophenyl)-porphyrinatoiron(III) (FeTpSPP)] and their binding behavior and catalytic activity toward the selective oxidation of thioanisole into sulfoxide by H2O2. It was shown that the Fe(TpCPP)-Xln10A hybrid protein with an additional imidazole was the best catalyst for the S-oxidation of sulfides showing the second highest yield (85 ± 4%), the highest TON (145 ± 3), and the highest enantioselectivity (ee = 40 ± 3%) at pH 7.4.
2.2.5.1.9. Other Hybrid Proteins
In addition to introducing iron–porphyrin complexes into Xln10A protein, Mahy and coworkers also reported a case where they incorporated Mn(III)-meso-tetrakis(p-carboxyphenyl)porpyrin (MnTpCPP) or N,N′-ethylene bis(2-hydroxybenzylimine)-5,5′-dicarboxylic acid-Mn-(III) (Mn–salen) complex into Xln10A.571 Docking studies showed different orientation of the salen and porphyrin complexes when bound to Xln10A. The Mn(III)(TpCPP) complex was shown to enter deeply into the complementary binding site that Xln10A provided, forming relatively strong H-bonds with the polar residues that were also observed in the Fe(III)(TpCPP) complex.569 Conversely, the Mn(III)–salen complex did not have a high affinity to Xln10A, the reason for which was that the Schiff base ligand was more solvent-exposed and was weakly interacting with the polar ligands. The Mn(III)(TpCPP) complex was then shown to catalyze the epoxidation of styrene and a series of styrene derivatives with the best epoxidation yield for styrene (17%) and the best ee for 4-methoxystyrene [80(R)%] with KHSO5 as the oxidant. This Mn(III)-(TpCPP)-Xln10A hybrid enzyme represents a new “haemozyme”.
Ménage and co-workers designed a novel Mn(III)-salen-human serum albumin (HSA) hybrid protein that can catalyze the selective sulfide oxidation by NaClO.572 The Mn(III)–salen complexes used are shown in Scheme 13. Binding studies showed that the presence of charged R-groups could improve the binding affinity at pH 7.0, leading to more stable Mn(III)–salen⊂HSA hybrid proteins. HSA was reported to bind a variety of drugs at different locations of the protein.573 They used several drugs with common HSA binding sites to study the approximate binding location of the Mn(III)–salen complexes. On the basis of the binding studies monitored by CD spectroscopy or Trp214 fluorescence quenching, they identified that Mn(III)–salen complexes 28 and 29 bind to the cleft site of the protein. With NaClO as an oxidant, thioanisole was oxidized to methylphenylsulfoxide (SO) and/or methyl-phenylsulfone (SO2) catalyzed by Mn(III)–salen⊂HSA and Mn(III)–salen complexes. In particular, the 28⊂HSA complex exhibited a turnover frequency of 20 min−1, and the reaction catalyzed by this hybrid protein produced only the sulfoxide. A high selectivity for sulfoxide production was also observed for the reaction catalyzed by 29⊂HSA and 30⊂HSA. The authors attributed this selectivity to the polarity of the protein environment.
Scheme 13. Mn-Salen Complexes Used To Bind to HSA572a.
aScheme used with permission from ref 572. Copyright 2009 Wiley.
NikA is a member of the ABC family of transporters that is responsible for nickel homeostasis as the initial nickel receptor and mediator.574 NikA is a periplasmic nickel-binding protein with a known structure, and it was found that the Ni(II) binding pocket is rich in aromatic and Arg residues.574–577 In addition to Ni(II), this active site is capable of binding FeEDTA complexes and a Ni-butane-1,2,3-tricarboxylate (BTC) complex.578,579 Recently, Ménage and co-workers reported crystal structures that captured the intermediates during an oxygen-mediated hydroxylation reaction of an aromatic C–H bond, providing important mechanistic details.580 They created a hybrid protein by incorporating an iron complex FeL (L = N-benzyl-N′-(2-hydroxybenzyl)-N,N′-ethyl-enediaminediacetic acid) into NikA. The ferrous state of the hybrid protein was able to activate molecular oxygen, forming a ferric-hydroperoxo species. This high-valent oxo species initiates the evolution of hydroxyl radicals, which then attack the C–H bond at the meta position of the phenolate ligand. The formation of this nondiffusible hydroxyl radical close to an iron atom was thought to be critical in selecting the aromatic ring without interacting with neighboring amino acid side chains. UV–vis and rR spectroscopies were also used to confirm the proposed mechanism. Furthermore, a series of iron complexes with either N,N′-dimethyl-N,N′-bis(2-pyridylmethyl)ethane-1,2-diamine (BPMEN) ligand or N,N′-bis(2-pyridylmethyl)-N,N′-dimethyl-trans-1,2-diaminocyclohexane (BPMCN) ligand derivatives were incorporated into apo-NikA.581 These complexes had Kd values in the micromolar range, with complexes A and B shown in Scheme 14 having the highest and the lowest affinities, respectively. They obtained crystal structures of some of hybrid proteins, which revealed H-bonding interactions, as well as CH/π H-bonds that are essential in forming these NikA complexes.582 Ménage and co-workers carried out docking studies to examine the possible positions and orientations for a variety of potential substrates containing a C6H5-S-CH2-X motif. They examined the distance of the iron center of the complexes and the sulfur atom of the substrates to screen for possible substrates. They found that the hybrid protein containing complex A (Scheme 14) and NikA exhibited a high yield of 79(±5)% and a TON of 173 in 4 h (turnover frequency of 43 h−1) when 2-((4-acetamidophenyl)-thio)-N-phenylacetamide was used as a substrate. Importantly, this catalytic reaction was selective for the sulfoxide product (69 ± 5%).
Scheme 14. Complexes A and B, the Iron Complexes with the Highest and the Lowest Affinity for NikA, Respectivelya.
aScheme adjusted with permission from ref 581. Copyright 2012 Springer.
2.2.5.2. Dative Anchoring Approach
Taking a different approach, Watanabe and co-workers incorporated synthetic metal complexes into apo-Mb mutants. Rhodium 2,6-bis(2-oxazolinyl)phenyl (Rh·Phebox) complexes were bound within the heme-binding pocket of apo-Mb.583 The X-ray crystal structure (1.8 Å resolution, Figure 21) displayed an overall protein structure that is similar to the native Mb; however, they observed that the Rh·Phebox moiety has a unique orientation within the cavity, almost perpendicular to the position of heme at the same site. They found that His93, which is the axial ligand of the heme cofactor in Mb, enforces this orientation by coordinating to the equatorial position of on the Rh ion, allowing for this unique orientation. H-bonding and hydrophobic interactions between the protein side chains (especially His93) and the Phebox ligands greatly stabilize the Rh·Phebox-Mb complex. The stability also has an enantioselective preference for the (S,S)-Rh·Phebox complex over the (R,R)-isomer, which is predicted to have less favorable π–π interactions with His93. This study showed that the heme-binding pocket in Mb is capable of accommodating moieties that are chemically and structurally different from its native cofactor, adopting a dissimilar arrangement of the prosthetic group.
Figure 21.
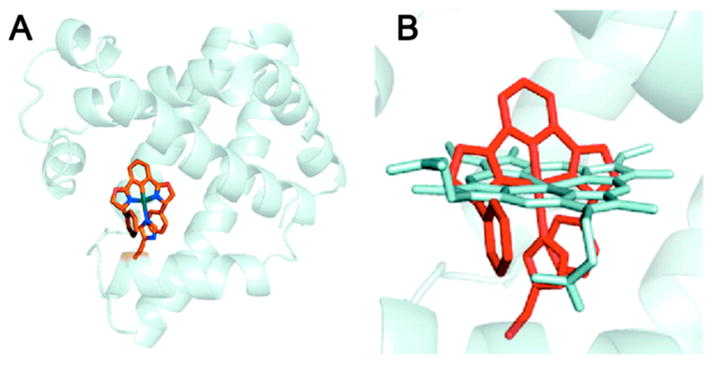
(A) The crystal structure of Rh·Phebox·A71GMb (PDB code: 2EF2); and (B) comparison of arrangement between Rh·Phebox (red) and heme (white). Reproduced with permission from ref 583. Copyright 2007 American Chemical Society.
Similarly, a Cr(III)(salophen) moiety was incorporated into an apo-Mb mutant through a metal-binding residue His (Figure 22). The resulting complex Cr(III)·[(5,5′-tBu2-)salophen]·apo-H64D/A71GMb catalyzes an enantioselective sulfoxidation reaction.584 The Schiff base complexes have molecular sizes and coordination geometries similar to those of heme centers, allowing for relatively facile modulation and comparison of the two cofactors. The authors found that a His64Asp mutation created a vacant distal site above the Cr(III)(salophen) complex, which was shown to improve substrate and oxidant access. Ala71 was mutated to a Gly, leading to an increased binding affinity for the Cr(III)(salophen) complex, an observation previously reported for iron-Schiff-base complexes.585 The sulfoxidation of thioanisole was then carried out at 35 °C in aqueous conditions at pH 5.0 with H2O2 as the oxidant. While Cr(III)·(salophen)·apo-H64D/A71GMb and Cr(III)·(5,5′-tBu2-Salophen)·apo-H64D/A71GMb exhibited the highest activities, Cr·(5,5′-tBu2-Salophen)·apo-H64D/ A71GMb had the highest enantioselectivity (13% ee (S)). The crystal structures of the complexes with both manganese and chromium as the central metal were obtained, which revealed other secondary interactions that might participate in modulating the reactivity of the enzyme.586 Specifically, the methyl groups on the 3 and 3′ positions of the salophen ligand (Figure 23) interact with the hydrophobic Ile107, modulating the penetration distance of the Schiff base complex. Salen ligands with two ethyl or n-propyl groups on the 3 and 3′ positions were designed and incorporated to adjust the location of the complex in the protein (Figure 23). As compared to the salophen ligand, the phenylenediamine unit was substituted for an en unit for better substrate access. Furthermore, the bulkiness of the residues above the metal complex was shown to influence the enantioselectivity. Finally, on the basis of the interactions revealed by the crystal structures and molecular modeling studies, a mechanism for the enantioselectivity in the sulfoxidation reaction was proposed (Figure 24).586,587 Molecular models suggested that ethyl or n-propyl substitutions at the 3 and 3′ positions would cause the position of the complex within the protein to be offset by 0.83 and 2.17 Å, respectively, from the location of the methyl-substituted complex. Because the position of the substrate thioanisole would not change, the interaction of the Mn=O active species with the lone pair on the sulfur atom would be different depending on the relative locations of these two components, dictating the enantioselective product.
Figure 22.
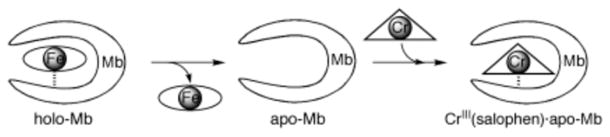
Schematic representation of insertion of a Cr(III)-(salophen) complex into apo-Mb. Reproduced with permission from ref 584. Copyright 2003 Wiley.
Figure 23.
M–Salophen and M–Salen complexes. Reproduced with permission from ref 586. Copyright 2005 American Chemical Society.
Figure 24.

Proposed interactions between the Mn Schiff base complex and thioanisole. Reproduced with permission from ref 586. Copyright 2005 American Chemical Society.
He and co-workers incorporated a uranyl cation binding site into a Ni(II)-dependent transcriptional repressor NikR.588 In the crystal structure of holo-NikR (PDB code: 1Q5Y), Ni(II) is coordinated to three His and one Cys in a square planar geometry.589 The authors created a mutant NikR′ by substituting two of the His residues to Asp to bind uranyl cation and a Val residue below the binding pocket into a Ser to accommodate the oxo group and form an H-bond, leading to a triple mutant NikR′. XAS studies showed that the uranyl cation was bound to two His and two Asp residues. The DNA-binding activity was then examined for both apo- and holo-NikR′. The NikR′ only interacted with DNA when a uranyl cation was bound. Furthermore, when divalent metal ions (Ni(II), Fe(II), Cu(II), Zn(II), etc.) were present, no protein–DNA complex was observed, which demonstrated that the selectivity of the mutant was altered as compared to the native NikR.
2.2.5.3. Covalent Attaching Approach
Another effective strategy for incorporating metal complexes into protein scaffolds is a covalent linking approach. Carey et al. employed this strategy to introduce a manganese salen complex, N,N′-bis(4-(2-methanesulfonylthioethoxyl)salicylidene)-1,2-ethane-diamino-manganese(III) bromide, into apo-swMb.590 Specifically, they used the high selectivity and reactivity of methane thiosulfonate toward Cys residues to covalently attach this molecule to apo-Mb. After carrying out molecular modeling on InsightII (Accelrys) program, they identified two possible sites for Cys mutation: Tyr103 and Leu72. The dual anchor conjugate Mn(salen)·apo-Mb(Y103C/L72C) was generated and was shown to catalyze the enantioselective sulfoxidation of thioanisole with much higher rates and ee than the single-point attached conjugate [Mn(salen)·apo-Mb(Y103C)]. To investigate how the protein scaffold influences the chemoselectivity of this reaction, they chose apo-Mb(T39C/L72C) for Mn(salen) attachment to select the product sulfoxide over sulfone.591 No sulfone production was observed when Mn(salen)·apo-Mb(T39C/L72C) was used to catalyze the reaction. They also found that the more polar the environment of the Mn(salen) complex is, the more sulfoxide product the reaction generates. For example, the Mn(salen)·apo-Mb-(T39C/L72C) conjugate produced a significant amount of sulfoxide over sulfone because the protein scaffold acted like a “solvent-cage”, mimicking a polar proton-donating organic solvent. Two additional mutants, Mb(Y103C/S108C) and Mb(T39C/S108C), were examined for the enantioselective sulfoxidation of thioanisole, which showed that the location where the anchor arms were attached had a significant influence on the rates and selectivity of the reaction.592 In addition, it was proposed that the covalent anchor can participate in directing the substrate binding during the reaction.
A series of Cu(II) centers in protein settings that are capable of catalyzing enantioselective Diels–Alder reactions have been reported. Aqueous solutions of Cu(II) complexes with amino acids,593,594 Cu(II) DNA intercalating agents,595–597 and Cu(II)–phthalocyanine anchored noncovalently to serum albumins598 were shown to carry out stereoselective Diels–Alder reactions using (aza)chalchones and cyclopentadiene as the reactants (Scheme 15). Here, we discuss examples of redesigned proteins as catalysts for the benchmark reaction described in Scheme 15.
Scheme 15.
Lactococcal multidrug resistance Regulator (LmrR) protein, a dimeric protein of ca. 13.5 kDa per subunit, was engineered to catalyze the Diels–Alder reaction shown in Scheme 15.599 LmrR consists of a typical β-winged helix-turn-helix domain with an additional C-terminal helix.600,601 The large flat hydrophobic core in this dimer was functionalized with two phenanthroline- or bpy-based chelating moieties per dimer (one per monomer) (Figure 25).599 Cys residues, which were not present in the WT protein, were incorporated into the protein monomers as anchor points for these copper-binding moieties. By reacting bromoacetamide-substituted 1,10-phenan-throline (1,10-Phen)or 2,2′-bpy with the Cys residues, mutants were generated with the copper-chelating moieties in the hydrophobic core.
Figure 25.

(A) Schematic representation of the proposed artificial metalloenzyme in which a Cu(II) complex is grafted on the dimer interface of a protein scaffold. (B) Ligands used for grafting on the dimer interface. (C) Pymol representations of dimeric LmrR in a ribbon and a space-filling model (PDB code: 3F8B). Either position 89 (red) or position 19 (yellow) was used for the covalent attachment of the copper-chelating ligands. Reproduced with permission from ref 599. Copyright 2012 Wiley.
The final constructs can bind two Cu(II) ions and still retain their dimeric form. The catalytic activity of the Cu(II)(1,10-Phen)-LmrR-M89C for the reaction described in Scheme 15 (monitored after 3 days of reaction) was remarkable: the reaction yielded 98% conversion of the substrate and 95% ee of the (+)-endo isomer at room temperature. Interestingly, an even higher enantiomeric excess (97%) was achieved when the reaction was carried out at 4 °C, albeit with a lower conversion (93%). Conversely, the Cu(II)(2,2′-bpy)-LmrR-M89C mutant yielded 66% ee of the opposite (−)-endo isomer of the Diels–Alder product. This demonstrated that these constructs can be tuned for a desired stereoselectivity through a proper choice of the position and the nature of the Cu(II) sites. The design of these constructs was prompted by the observation that in DNAzymes, copper(II) sites close to a chiral environment have the ability to catalyze Diels–Alder or Michael reactions with a high degree of selectivity.595–597 The chiral scaffold, even quite distant from the copper site, could therefore provide efficient secondary sphere interactions to induce significant stereoselectivity.
In addition to covalently attaching a copper complex to a protein scaffold, direct mutations on the native protein can also yield a stereoselective Diels–Alder catalyst. Although these studies do not involve covalently attaching a copper complex into a protein scaffold, we feel that it is appropriate to include them here because they are also Diels–Alder catalysts. The imidazole glycerol phosphate synthase (tHisF) from Thermotoga maritime is a thermostable enzyme essential in the biosynthesis of His residue,602 which has a typical TIM barrel structure with a narrow bottom and a wide top. This scaffold has been used to engineer a His2Asp Cu(II) binding site in proximity to the wide top rim.603 The selective mutation of two noncoordinating residues close to Asp at position 11 yielded the mutant tHisFL50H/I52H, where the copper binding site is accessible by relatively large substrates. Furthermore, to avoid nonspecific Cu(II) binding, surface Cys and His residues were mutated to Ala. An extensive EPR characterization revealed that Cu(II) is coordinated to Asp11/His50/His52 residues, matching the proposed model. When the reaction reported in Scheme 15 was carried out, the conversion was 73%, with 46% ee and an endo/exo ratio of 13:1. The mutation of either the Asp or the His into an Ala of the His2Asp site resulted in lower conversion values and almost negligible enantiomeric excess values, demonstrating the necessity of a stable Cu(II)N2O center to catalyze this enantioselective reaction. As reported in the literature, to obtain a significant stereoselectivity in Diels–Alder reactions, the substrate must bind to Cu(II) through a carbonyl oxygen and a pyridine nitrogen to activate this reaction.598,604 This type of activation is required also for the Cu(II)-tHisF protein. The chalcones (an aromatic ketone and an enone that form the central core for a variety of important biological compounds) lacking the pyridine nitrogen in the ortho position with respect to the carbonyl did not react with significant conversion values.603
The bovine pancreatic polypeptide (bPP) is a 36 amino acid peptide that contains a poly-Pro type II helix (residues 1–8), a turn (9–12), and a helix (13–31).605–607 The poly-Pro helix is backfolded on the α-helix, and the construct dimerizes in solution. A 31-amino acid truncated sequence (1–31) of bPP was designed, where Tyr7 was mutated into a His, a L-3-pyridylalanine, or a L-4-pyridylalanine residue, while an Asp or Glu residue in the loop (position 10) provided additional donor atoms.608 Upon dimerization, a His2 or pyridyl2 copper-binding site was obtained within the intrahelical space of the dimer. Further mutations were introduced at the interface between the dimers with an attempt to make the interior less sterically crowded. Unfortunately, these mutants were found to exist predominantly in their monomeric Cu(II)-(peptide) form. The best results for the reaction shown in Scheme 15 (complete conversion of the substrate and up to 83% ee) were obtained with the L-3-pyridylalanine mutant (bPPc). As observed for tHisF, the coordination of the substrate to the Cu(II)-bound protein is a prerequisite for having the highest activity.608 Moreover, Cu(II) or WT bPP 1–31 alone did not show significant catalytic activities, neither did the His-containing mutant. Overall, these results demonstrated that the nature of the coordinating groups (pyridyl vs imidazolyl, and their diastereoisomers) has a profound impact on regulating the stereoselectivity and the activity of these reengineered protein catalysts. Moreover, it has been previously demonstrated that the presence of a cavity in the construct is not a strict requirement to achieve control of the stereoselectivity. These bPP mutants were also examined as catalysts for the Michael addition reaction, exhibiting significant stereoselectivity.608
In summary, for metalloprotein conjugated systems, the strategies developed to control the stereo- and regioselectivity include the manipulation of the local catalytic center and the construction of proper secondary sphere interactions. Specifically, enantioselectivity is directly associated with the chirality of the substrate binding site, the local charge in the protein environment, and the specific interactions of substrate–enzyme and product–enzyme complexes with the amino acid residues around the active site. Moreover, the protein scaffold, if stable enough to withstand the incorporation of a metal complex in the hydrophobic center or mutation of certain amino acid residues, can also influence the specificity and enantioselectivity by providing a substrate/product channel, size and shape control, and proper dielectrics to minimize side reactions.
2.2.6. Redesigned Protein Assemblies as Nanoreactors
2.2.6.1. Catalysis within Protein Cages
Ferritin (Fr) is a spherical protein that can store up to 4500 iron atoms as ferric oxyhydroxides. Access to the 8 nm diameter interior cavity of Fr can be obtained through the 3-fold axis channels that exist between the 24 subunits of Fr. The robust and well-defined nanocapsule of ferritin has served as a template for the synthesis of nanoparticles609,610 from CdS,611 Fe,612 CdSe,613 Au,614 and Ag,615 as well as metal-complexes610 including gadolinium MRI contrast agents,616 hexacyanoferrate(III),612 desferrioxamine B,617 cis-platin and carboplatin,618 redox-active ferrocene derivatives,619 and Ru(II)(η6-p-cymene) half-sandwich complexes.620
Watanabe and co-workers have made significant contributions to the field of protein design through their work incorporating metal catalysts within the cavities of apo-ferritin (apo-Fr).621 After entering apo-Fr, Pd(II) ions were reduced to form encapsulated, monodisperse, roughly spherical Pd clusters (Figure 26), which were active as catalysts for the hydrogenation of acrylamide derivatives.622 Apo-Fr discriminates substrates by size because they must enter the cavity through the 3-fold hydrophobic channels. In this example, the smallest substrate, acrylamide, was the fastest with a turnover frequency of around 33 000 mol product per Pd·apo-Fr complex per hour.
Figure 26.
Preparation of Pd·apo-ferritin. Reproduced with permission from ref 622. Copyright 2004 Wiley.
Starting from crystal structures of Pd(II)621 and Au(III)623 ions within apo-rHLFr (recombinant L-chain apo-Fr from horse liver), a nontraditional synthetic route for making bimetallic Au–Pd nanoparticles (NPs) emerged.623 Au–Pd alloy NPs (average size 2.2 nm) were prepared upon pretreating apo-rHLFr with Au(III), loading with Pd(II), and then reducing the mixture to give (AuPd-NP)·apo-rHLFr. Alternatively, core–shell NPs (average size 2.4 nm) were prepared by a sequential synthesis. In one case, a Au core was reduced within apo-rHLFr, then Pd(II) ions were introduced and reduced to form the Pd-shell of ([Au](Pd)-NP)·apo-rHLFr. In another, the reduction of Au(III) onto a Pd-core yielded ([Pd](Au)-NP)·apo-rHLFr. Most of these bimetallic composites catalyze the hydrogenation of acrylamide to different degrees. While ([Pd](Au)-NP)·apo-rHLFr showed no activity, (AuPd-NP)·apo-rHLFr, (Pd-NP)·apo-rHLFr, and ([Au](Pd)-NP)·apo-rHLFr had turnover frequencies of 310, 510, and 1300 mol of product per mol of metal atom per hour, respectively.
In addition to metal ions and metal nanoparticles, organometallic complexes were incorporated into apo-Fr. The reaction of apo-Fr with [Rh(nbd)Cl]2 (nbd = norbornadiene), complexes of which are known catalyts, resulted in the integration of 57.5 ± 3.5 Rh atoms per apo-Fr.624 The 1.8 Å resolution crystal structure of Rh(nbd)·apo-Fr shows that the overall protein structure is not disturbed (Figure 27A–C). Rh(nbd)·apo-Fr catalyzed the polymerization of phenyl-acetylene, affording polyphenylacetylene with a cis-transoidal main chain. Polymerization occurred within the cage, without disrupting its structure, yielding polymers with average number molecular weights (Mn) ranging from 8.3 × 103 to 13.1 × 103 and a polydispersity index (Mw/Mn) of 2.1–2.6 under the range of conditions studied (Figure 27D). Overall, polymerization was regulated by the size and environment of the apo-Fr interior, and the size distribution of polymers prepared within the cage was narrower than for those prepared by Rh(nbd) outside the cage.
Figure 27.
(A–C) Ribbon diagrams of L-ferritin taken from PDB entry 1DAT: (A) the 24-subunit assembled cage; (B) the inner cavity; and (C) the 3-fold axis channel. (D) Schematic representation of insertion of [Rh(nbd)Cl]2 into the apo-Fr cage and polymerization catalyzed by the Rh(nbd)·apo-Fr composite. Reproduced with permission from ref 624. Copyright 2009 American Chemical Society.
In addition to these Rh-complexes, multinuclear organometallic Pd-complexes were hosted within apo-Fr. The reaction of [Pd(II)(allyl)Cl]2 (allyl = η3-C3H5) complexes with apo-rHLFr (Figure 28A) resulted in di- and trinuclear Pd(allyl) complexes capable of promoting Suzuki coupling reactions.625 The crystal structure of Pd(allyl)·apo-rHLFr reveals that, without disrupting the protein fold, 48 dinuclear Pd complexes bound the 3-fold axis channel and the accumulation center of the interior surface (Figure 28B). In addition to allyl, amino acid, and water ligands, every two square planar Pd(II) ions share a Cys residue, which is essential for protein binding. The Suzuki coupling of p-I-PhNH2 and PhB(OH)2, known to be catalyzed by Pd(allyl) complexes, was carried out using these assemblies. Pd(allyl)·apo-rHLFr performed the fastest with a turnover frequency of 3500 mol product per mol apo-rHLFr per hour. While the His49Ala (3400) mutation had little effect on the activity, His114Ala (900) and Cys126Ala (830) mutations had a 4-fold decrease in activity, and the combination of Cys48Ala/His49Ala (1900) resulted in a 1.8-fold decrease in activity.
Figure 28.
(A) Preparation of Pd(allyl)·apo-rHLFr. (B) The crystal structure of 100-Pd(allyl)·apo-rHLFr (PDB code: 2ZG7). Pd(allyl) complexes are located at the binding sites. The Pb atoms are shown in green spheres. Reproduced with permission from ref 626. Copyright 2010 American Chemical Society.
Further, mutations to apo-rHLFr were used to clarify the mechanism of the incorporation and accumulation of organometallic Pd complexes in ferritin. Crystal structures of apo-rHLFr, apo-C48A-rHLFr, apo-C126A-rHLFr, apo-H49A-rHLFr, and apo-H114A-rHLFr containing Pd(allyl) complexes provided insight.626 While apo-rHLFr incorporated a maximum of 106 Pd atoms, apo-C126A-rHLFr and apo-C48A-rHLFr mutants accumulated only 37 and 63 Pd atoms, respectively. The crystal structure of apo-rHLFr exposed Cys126 and Cys48 as bridging ligands for dinuclear square planar Pd(allyl) (allyl = η3-C3H5) complexes at the 3-fold channel and accumulation center, respectively. Crystal structures of the mutants revealed no Pd(allyl) complexes within the 3-fold axis channel of 100-Pd(allyl)·apo-C126A-rHLFr or within the accumulation center of 100-Pd(allyl)·apo-C48A-rHLFr. The structures allowed for the proposal of a mechanism for incorporation and accumulation of Pd(allyl) into apo-rHLFr. His114, Cys126, Asp127, and Glu130 capture and shuttle Pd(allyl) through the 3-fold channel into the center of the cage, with Cys126 accelerating the incorporation by inducing a conformational change of the 3-fold channel. Pd(allyl) accumulates in the center of the cage as a thiolate-bridged dinuclear complex coordinated by Cys48, His49, and Glu45.
Crystal structures and ICP showed that three rationally designed mutants controlled the coordination arrangement and quantity of Pd(allyl) complexes on the interior surface of the apo-Fr cage. The Pd(allyl) binding site at the accumulation center was rearranged in Pd(allyl)·apo-E45C/C48A-rHLFr. An additional site was added in Pd(allyl)·apo-E45C/R52H-rHLFr. In the mutant Pd(allyl)·apo-E45C/H49A/R52H-Fr, the original site was rearranged, and an additional site was added. These assemblies reveal the importance of Cys residues for capturing and His residues for controlling the direction of ligation of Pd(allyl) complexes. Each mutant was tested as a catalyst in the aqueous Suzuki coupling of p-I-PhNH2 and PhB(OH)2. Comparison of turnover frequencies (in mol product per mol apo-rHLFr per hour) reveals that each of the mutants shows improved catalytic activity over Pd(allyl)· apo-rHLFr (3500). However, despite having 1.5 times more Pd(allyl) complexes than Pd(allyl)·apo-E45C/C48A-rHLFr (4200), Pd(allyl)·apo-E45C/R52H-rHLFr (4300) and Pd-(allyl)·apo-E45C/H49A/R52H–Fr (4200) show no improvement, likely because penetration of reactants into the cavity is the rate-limiting step.
The Tezcan group has pioneered the design of metal-templated protein–protein interfaces.627–634 Their work represents the frontier of investigating the dynamic process of protein self-assembling and of engineering metal ion selective responses. However, because the focus of this Review is on designed metalloenzymes, we will not discuss these studies in detail here. Of greater relevance to this Review is the Tezcan group’s recent design of the protein assembly Zn30:CFMC-112 as a host for enzyme binding.635 Zn30:CFMC-112 was assembled in the crystal lattice through metal-mediated interactions between the protein–protein interface. This protein architecture was used to encapsulate a heme protein fragment, a 9-amino-acid-residue-long microperoxidase MP9cb562.635 The building block of the protein assembly was a D2-symmetric tetramer of a cytochrome cb562 unit, which was held together by four interfacial Zn(II) ions via His and Asp coordination.627 Six mutations were introduced that switched hydrophilic residues into hydrophobic ones,636 one zinc-binding residue (Asp74) was deleted, and a few bulky residues were incorporated onto the protein surfaces. The assembly that was used as a reactor was a protein dodecamer Zn30:CFMC-112, which was tetrahedron-shaped with an inner cavity diameter of 35 Å. A 9-amino-acid microperoxidase MP9cb562 (sequence: KTTCNACHQ) with a heme cofactor was then introduced as a guest to bind to the protein host, and the complex was crystallized with equimolar Zn(II) and substoichiometric MP9cb562. MP9cb562 was shown to interact with the cage surface residues through hydrophobic and H-bonding interactions in addition to the anchoring Fe-His ligation, leading to a unique orientation of the heme group. No peroxidase activity of this complex was reported, but this assembly may be a good foundation for future functional studies.
2.2.6.2. Bionanocup Reaction Centers
The virus bacteriophage T4 infects E. coli by first penetrating the outer cell membrane using a heteroprotein “needle”. This assembly, containing a trimer of gene products 27 and 5, (gp27-gp5)3, consists of a bionanotube topped with a bionanocup with an internal height of 6 nm and internal diameter of 3 nm. Mutants of (gp27-gp5)3 containing Cys residues at the third and seventh positions of the (gp5)3 nanotube subunit were covalently linked to Fe(III)-protoporphyrin-maleimide (FePP-MI) derivatives, yielding FePP·39 and FePP·40, respectively (Scheme 16).637 Although the exact location of the moieties within the heteroprotein assemblies was unresolved, FePP·39 and FePP· 40 were found to contain 2.7 and 1.9 Fe atoms, respectively, and to catalyze the H2O2-dependent sulfoxidation of thioanisole, but without enantiomeric selectivity. As compared to the unbound complexes, 10- and 6-fold rate enhancements were observed for FePP·39 and FePP·40, respectively, presumably due to the protection from aggregation provided by the hydrophobic nanocup.
Scheme 16. A Schematic Drawing of the Conjugation Reactions of FePP-Maleimides and (gp5)3 or (gp27-gp5)3 Cys Mutantsa.
aReproduced with permission from ref 637. Copyright 2008 Wiley.
The triple-stranded β-helical (gp5)3 subunit is an attractive architecture, but stabilization of the isolated structure is challenging. Ueno and co-workers identified the most stable β-helical fragment (gp5βf) and used it to prepare a head-to-head dimer of nanotubes, genetically fused and protected at the C-terminus with a foldon fragment.638 The 2.0 Å resolution crystal structure of [(gp5βF)3]2 reveals a homodimeric tube structure where the dimers are “locked” together with antiparallel β-helices at the interface. Lysine residues, spaced 10–15 Å apart along the ridge lines of the triple-stranded β-helices, were appended with flavin-(1)-N-hydroxy-succinimidyl ester derivatives. These thermally stable flavin composites of [(gp5βF)3]2 were utilized in Cu(I)-catalyzed azide–alkyne [3 + 2] cycloaddition reactions. The high activities achieved suggest that Cu(I) is monoligated to the flavin moiety.
Next, [(gp5βF)3]2 mutants were prepared in which one Lys residue along each ridge line was replaced with Cys.639 Upon folding, these Cys residues are in close proximity to Lys residues on neighboring strands. The Cys residues were appended with Re(I)(4-ethylmaleimide-bpy)(CO)3Cl and the Lys with Ru(II)(bpy)2[3-(4′-methyl-bpy-4-yl)-propionic acid N-succinimidyl ester](PF6)2. The resulting ReRu mutants accelerated the reduction of CO2 to CO upon electron transfer from the photoreduced ruthenium moiety to the rhenium moiety. The top performing composite, K41C_ReCysRuNH, showed a 3.3-fold improvement in rate over the mixture of K41C_ReCys and Ru(bpy)2-[4-(2-carboxylethyl)-4′-methyl-2,2′-bipyridyl](PF6)2, demonstrating the proximity effect of the metal complexes.
Further Lys-to-Cys mutations yielded [(gp5βF)3]2 constructs uniquely suited for Sc(III) coordination.640 Thiol-maleimide coupling introduced a 2,2′-bipyridine (bpy) moiety adjacent to two Thr alcohols. Tetradentate coordination by these three ligands yielded a Sc[bpy-(ROH)2] complex capable of catalyzing an epoxide ring-opening reaction. The top performer, G18C_bpy, has significantly higher conversion than a mixture of ligands for the ring-opening of cis-stilbene oxide with aniline. Cooperative coordination of Sc(III) by bpy and the Thr residues tunes its Lewis acidity to activate epoxides for nucleophilic attack. Each of these examples showcases the use of [(gp5βF)3]2 constructs for the rational coordination of metal complexes for use as artificial metalloenzymes.
2.3. Summary
We have highlighted in this section the protein redesign approach to achieving novel functional metalloenzymes. Since this approach starts from an existing protein scaffold, the advantages of structural stability and functional diversity are evident. A fundamental understanding of the structure–function relationship of the initial system lays the groundwork for protein redesign, allowing the implementation of a rational design strategy. For this reason, a high-resolution crystal structure or solution NMR structure is an important starting point. Generally, novel functionality is achieved upon redesign of the protein ligands using traditional site-directed mutagenesis, or, in the case of the copper electron transfer centers, loop-directed mutagenesis. Rational design strategies were used to achieve the incorporation of a structural/catalytic ZF site, the modification of heme-containing proteins, the design of electron transfer or catalytic iron/copper centers, and the control of reaction stereoselectivity. Simultaneously, automated algorithms were used successfully to yield several nonheme proteins. In addition, many examples involved the incorporation of unnatural amino acids to modulate the hydrophobicity or metal-binding properties of the active site. Both the inner and the outer coordination spheres of metal ions can be modified through protein redesign, conveying various metal-binding properties, redox properties (if applicable), activities, or substrate/product specificities. With these design considerations, the protein redesign approach has proved successful in yielding stable metalloprotein constructs with improved or novel functions. Additionally, researchers have gained tremendous insight into the structure–function relationship during the design process, which in turn serves the design purpose.
3. DE NOVO DESIGN
3.1. A Minimalist Approach: Designing Proteins from Scratch
Protein design is progressing rapidly beyond the improvement of existing proteins. Arguably more challenging than protein redesign, de novo or “from scratch” design has helped to disentangle structure–function relationships on a fundamental level and to create novel properties and functions in peptidic/ protein systems that are not necessarily related to native protein sequences. Designing proteins from scratch was considered a bold endeavor in its infancy; however, the challenges did not prevent researchers from trying. As Richard Feynman expressed, “What I cannot create I do not understand.”641 Indeed, de novo design is the ultimate test of our understanding of the fundamental factors that dictate protein folding, structure, and function using a minimalist approach.12 It involves constructing a peptide sequence that spontaneously folds into a unique, predictable three-dimensional structure while retaining sufficient complexity and functionality to provide an adequate model for a protein.10,12,35,642,643 What distinguishes de novo design from other types of protein design or engineering strategies is that it is based on first principles,644 which refer to the postulation that the primary amino acid sequence determines the three-dimensional structure of a protein. Richardson presented a vivid analogy of the protein folding problem at the Biophysical Society National Lecture in 1992: the folded protein can be thought of as an origami canary with the amino acid sequence being the creased piece of paper from which it is folded.15 Thus, protein redesign amounts to modifying the wings or the tail of the folded origami canary, while de novo design starts with a flat piece of paper to figure out the crease patterns needed to fold it into a three-dimensional origami canary.
The goal of de novo design is to create stable peptidic/ protein constructs with desirable functions and properties comparable to or better than those of native proteins. Combined with the knowledge learned from synthetic model complex studies and protein redesign work, de novo design should eventually lead to novel applications in biocatalysis and pharmaceuticals. Ever since the first reported de novo designed peptides,642,645 this approach has been utilized to understand the interplay between protein structure and function, heavy metal toxicity, electron transfer, and structural and catalytic sites in metalloproteins. Our review on this topic is arranged accordingly.
Rather than designing each new protein from scratch, previous de novo scaffolds can serve as a starting point for the design of metalloproteins (much as natural proteins are the starting points in protein redesign). Although there are fewer de novo designed scaffolds to choose from as compared to the number of natural proteins, the existing ones cover the most important and common structural motifs found in nature. Choosing a starting scaffold is a critical first step of de novo metalloprotein design because the scaffold provides the protein environment and orients the residue side chains, which have a profound impact on metal coordination. There are a few excellent and exhaustive reviews discussing the de novo design of turns, β-sheets, and other motifs.646–651 Since most of the functional metalloenzyme mimics are associated with α-helical coiled coils, the designs based on this scaffold are the focus of this Review. Finally, it is worth mentioning that computational protein design has contributed greatly to the establishment and expansion of the field. The major goal of computational design is to identify amino acid sequences compatible with a stable, folded, three-dimensional structure or to find the lowest free-energy structure for a specific amino acid sequence. We will omit a detailed discussion on computational design because the effort of this approach in the past few decades has already been reviewed extensively.17,24,652–664
3.2. Interactions between De Novo Designed Peptides and Metal Ions
The de novo design approach provides a useful tool for understanding protein–metal interaction on a fundamental level by allowing for the incorporation of a controlled coordination site in a protein matrix. From the perspective of coordination chemistry, one can think of the protein scaffold as a multidentate ligand that accommodates metal binding; however, unlike simple bioinspired ligands such as EDTA,665,666 (2-OH)SALPN,667 or NTA,668 this ligand is a true supramolecular structure that has its own properties that originate from its proteinaceous nature. From a bioinorganic point of view, the construction of a metal center in a protein scaffold not only illustrates the interplay between protein and metal ions, but also assesses important issues such as heavy metal uptake, regulation and toxicity.669 Moreover, the successful installation of a controlled metal center in a de novo designed protein is a necessary step toward creating functional metalloenzymes mimics.
3.2.1. Heavy Metal Toxicity
Although the focus of this Review is the design of functional mimics of metalloenzymes, we feel that it is appropriate to introduce some work based on heavy metal binding to de novo designed scaffolds because of the following reasons: (1) heavy metal ions are introduced as spectroscopic tags to understand metal–protein interactions, especially the control of metal coordination in a protein scaffold, serving as a foundation for the design of functional sites; (2) heavy metal ions are proven to form structurally stabilizing sites for certain designs; and (3) we still lack a thorough understanding of the mechanisms involved in heavy metal toxicity. Commonly encountered toxic heavy metals, Cd(II), Hg(II), As(III), and Pb(II), are considered as some of the most dangerous substances in the environment. They are toxic to many organisms by interfering with metabolic pathways, which display a wide range of nonspecific syndromes.670,671 Therefore, it is critical to understand the mechanism of heavy metal toxicity as well as detoxification pathways.
Prokaryotic heavy metal stress response is largely mediated by metalloregulatory proteins, which generally fall into two categories: the MerR family and the SmtB/ArsR family. The MerR family proteins are transcriptional activators/repressors, whereas the SmtB/ArsR family are solely repressor proteins.672,673 MerR, found as a homodimer, binds to the DNA of the mer operon both in the absence and in the presence of Hg(II), regulating proteins for Hg(II) uptake, metabolism, and reduction, which all contribute to Hg(II) detoxification.673–676 Mercury binds to three Cys residues at the dimer interface of MerR: Cys82 of one monomer and Cys117 and Cys126 from the second monomer.674 When mercury is bound to MerR in nanomolar concentrations, the MerR/DNA ternary complex changes conformation, subsequently leading to the unwinding of DNA,677,678 promoting transcription of Mer genes in the system. MerR also has high affinity for Pb(II) and Cd(II) resulting in trigonal pyramidal and tetrahedral binding geometries, respectively.679,680 The SmtB/ArsR family proteins function as repressors that bind to DNA in the absence of metal ions.681 CadC is a member of the SmtB/ArsR metalloregulatory family, which is expressed in Staphylococcus aureus. This metal sensing protein is responsible for Cd(II), Pb(II), and Zn(II) detoxification.682–685 Metal binding up-regulates both CadC expression and the expression of an integral membrane metal efflux protein, CadA. CadC is a homodimer that contains two binding sites. The site in charge of heavy metal sequestration contains Cys58 and Cys60 from one monomer and Cys7 and Cys11 from the other. This active site sequesters Cd(II) into a [Cd(SR)4]− coordination and Pb(II) in a trigonal pyramidal coordination. The arsRDABC operon is employed by E. coli and other bacteria as a way of detoxifying As(III).686 The pathway of arsenic detoxification has been proposed as follows: As(III) binds to ArsR, which leads to the dissociation of the repressor protein from DNA. ArsA and ArsB are responsible for the transportation of As(III) out of the cell. In addition, ArsC reduces arsenate (AsO43−) to arsenite (AsO2−), which contributes to arsenic detoxification.687–689 The metallochaperone ArsD then transports As(III) to ArsA/B for extrusion. 690,691 Although multiple studies support this pathway, there is little information available for As(III) coordination in biological systems. Similarly, a bacterial metalloregulatory protein for Pb(II) detoxification, PbrR691, binds Pb(II) in a tris-Cys environment.680,692,693
Moreover, one of the most well-studied proteins that is inhibited by Pb(II) binding is δ-aminolevulinate dehydratase (ALAD), a zinc-dependent enzyme with a Cys-rich active site. Crystallographic studies of the lead-inhibited form of ALAD, where Zn(II) in the Cys3H2O site is replaced by Pb(II), demonstrated that a Pb(Cys)3 structure is present. The stereochemically active lone pair of Pb(II) resides where the solvent molecule or substrate would be expected to bind to Zn(II).694 Binding of Pb(II) to ALAD prevents heme synthesis results in an accumulation of the precursor 5-amino levulinate, leading to anemia and neurological symptoms in humans. 695–698 All of these relevant metalloproteins have thiolrich binding environments, which are favorable for heavy metal binding. Additionally, the preorganized protein environment plays an important role in determining heavy metal coordination.
Using a de novo design strategy to enforce a tris-thiolate environment around ions such as Hg(II), As(III), and Pb(II), Pecoraro, DeGrado, and co-workers prepared a series of coiled coil peptides based on the heptad repeat approach to study heavy metal toxicity. They demonstrated the design of thiolate environments amenable for heavy metal binding, addressing heavy metal selectivity, specificity, and coordination geometry in a protein environment at a fundamental level. Their work has contributed significantly to the understanding of the interaction between heavy metals and proteins. Herein, we review the achievement of controlling heavy metal coordination number and geometry in a peptidic scaffold and its implications in explaining the relationship between heavy metal geometric preferences and inherent protein structures.
A well-defined coiled coil scaffold based on the heptad repeat approach was used to study heavy metal-binding behaviors. Using the program InsightII/Discover,699 DeGrado applied negative design principles10,16,28 to prepare the 29 amino acid peptide CoilSer (CS)33,34 (Table 4), which was acetylated at the N-terminus and amidated at the C-terminus, with the intention of generating an antiparallel four-stranded coiled coil (4SCC). However, X-ray analysis demonstrated that the resulting aggregate was an antiparallel (two “up” C-termini and one “up” N-terminus) three-stranded coiled coil (3SCC).34 The basis of this approach was to exploit the heptad repeat, known to induce α-helix formation. Simply, the concept is that a peptide assembly will form through “hydrophobic collapse”700–703 based on the exclusion of hydrophobic residues from the aqueous phase. Control of strand number is achieved by matching the packing of the hydrophobes into the desired aggregates (2SCC, 3SCC, or 4SCC). Orientational control (parallel vs antiparallel) and solubility are conferred by hydrophilic, charged residues placed at the interhelical interface. The heptad is composed of seven amino acid residues, denoted as abcdefg, where a and d positions are usually hydrophobic residues, and e and g positions are salt-bridging residues.704,705 The connectivities between helices and overlapping hydrophobes are often represented as helical wheel diagrams (Figure 29). For heptads containing leucine, the addition of each 7 amino acid repeat confers ~9 kcal/mol of stability to the system.706,707 It should be noted that the intrinsic nature of the heptad places the a residue of the first heptad slightly (~20°) out of phase with the a residue of the following heptad. To optimize packing of the hydrophobes, the resultant α-helices twist, or supercoil, around one another. This is where the name “coiled coil” comes from as α-helices (coils) supercoil around each other to form the stable aggregate.
Table 4.
CoilSer and TRI Peptide Sequences
| peptidea | N-terminus | a b c d e f g | a b c d e f g | a b c d e f g | a b c d e f g | a b c d e f g | C-terminus |
|---|---|---|---|---|---|---|---|
| 1 | 2 8 | 9 12 15 | 16 19 22 | 23 26 29 | (30 36) | 30(37) | |
| Baby | Ac-G | LKALEEK | LKALEEK | LKALEEK | G-NH2 | ||
| BabyL9C | Ac-G | LKALEEK | CKALEEK | LKALEEK | G-NH2 | ||
| BabyL12C | Ac-G | LKALEEK | LKACEEK | LKALEEK | G-NH2 | ||
| TRI | Ac-G | LKALEEK | LKALEEK | LKALEEK | LKALEEK | G-NH2 | |
| TRIL9C | Ac-G | LKALEEK | CKALEEK | LKALEEK | LKALEEK | G-NH2 | |
| TRIL12C | Ac-G | LKALEEK | LKACEEK | LKALEEK | LKALEEK | G-NH2 | |
| TRIL9CL12C | Ac-G | LKALEEK | CKACEEK | LKALEEK | LKALEEK | G-NH2 | |
| TRIL16C | Ac-G | LKALEEK | LKALEEK | CKALEEK | LKALEEK | G-NH2 | |
| TRIL16Pnb | Ac-G | LKALEEK | LKALEEK | PnKALEEK | LKALEEK | G-NH2 | |
| TRIL19C | Ac-G | LKALEEK | LKALEEK | LKACEEK | LKALEEK | G-NH2 | |
| TRIL12AL16C | Ac-G | LKALEEK | LKAAEEK | CKALEEK | LKALEEK | G-NH2 | |
| TRIL12DLL16C | Ac-G | LKALEEK | LKADLEEK | CKALEEK | LKALEEK | G-NH2 | |
| TRIL12CL16C | Ac-G | LKALEEK | LKACEEK | CKALEEK | LKALEEK | G-NH2 | |
| TRIL23H | Ac-G | LKALEEK | LKALEEK | LKALEEK | HKALEEK | G-NH2 | |
| TRIL9CL23H | Ac-G | LKALEEK | CKALEEK | LKALEEK | HKALEEK | G-NH2 | |
| TRIL9CL19H | Ac-G | LKALEEK | CKALEEK | LKAHEEK | LKALEEK | G-NH2 | |
| TRIL9HL23C | Ac-G | LKALEEK | HKALEEK | LKALEEK | CKALEEK | G-NH2 | |
| TRI-EH | Ac-G | WKALEEK | LKALEEK | LKALEEE | HKALEEK | G-NH2 | |
| TRI-EHE27K | Ac-G | WKALEEK | LKALEEK | LKALEEE | HKALKEK | G-NH2 | |
| TRI-EHE27Q | Ac-G | WKALEEK | LKALEEK | LKALEEE | HKALQEK | G-NH2 | |
| TRI-EHK24Q | Ac-G | WKALEEK | LKALEEK | LKALEEE | HQALEEK | G-NH2 | |
| TRI-EHK24E | Ac-G | WKALEEK | LKALEEK | LKALEEE | HEALEEK | G-NH2 | |
| TRI-HK22Q | Ac-G | WKALEEK | LKALEEK | LKALEEQ | HKALEEK | G-NH2 | |
| CoilSer (CS) | Ac-E | WEALEKK | LAALESK | LQALEKK | LEALEHG | -NH2 | |
| CSL9C | Ac-E | WEALEKK | CAALESK | LQALEKK | LEALEHG | -NH2 | |
| CSL16C | Ac-E | WEALEKK | LAALESK | CQALEKK | LEALEHG | -NH2 | |
| CSL12AL16C | Ac-E | WEALEKK | LAAAESK | CQALEKK | LEALEHG | -NH2 | |
| CSL16Pn | Ac-E | WEALEKK | LAAAESK | PnQALEKK | LEALEHG | -NH2 | |
| CSL19C | Ac-E | WEALEKK | LAALESK | LQACEKK | LEALEHG | -NH2 | |
| Grand (GR) | Ac-G | LKALEEK | LKALEEK | LKALEEK | LKALEEK | LKALEEK | G-NH2 |
| GRL16PnL26AL30C | Ac-G | LKALEEK | LKALEEK | PnALEEK | LKAAEEK | CKALEEK | G-NH2 |
| GRL12DLL16CL26AL30C | Ac-G | LKALEEK | LKADLEEK | CKALEEK | LKAAEEK | CKALEEK | G-NH2 |
| GRL16PnL19IL23PnL26I | Ac-G | LKALEEK | LKALEEK | PnKAIEEK | PnKAIEEK | LKALEEK | G-NH2 |
| GRL12AL16CL26AL30C | Ac-G | LKALEEK | LKAAEEK | CKALEEK | LKAAEEK | CKALEEK | G-NH2 |
| GRL12AL16CL26C | Ac-G | LKALEEK | LKAAEEK | CKALEEK | LKACEEK | LKALEEK | G-NH2 |
| a3DIV | MGS WAEF K QR LAAIKTR CQAL GG | ||||||
| SEAECAAF E KE IAAFESE LQAY KGKGNPE | |||||||
| VEAL R KE AAAIRDE CQAY RHN | |||||||
Metal-binding residues are in red.
Pn = Pen.
Figure 29.

Helical wheel diagram of (A) 2SCC, (B) 3SCC, and (C) 4SCC. Reproduced with permission from ref 704. Copyright 2004 American Chemical Society.
Because of the high symmetry of these systems, one must determine whether a single protein fold exists in a “native-like fold”, or has multiple similar energy conformations, which is then termed a “molten globule”.708–710 Usually, increasing the number of heptads or increasing the asymmetry in the sequence will lead to native-like folds. DeGrado demonstrated that a parallel 3SCC can be achieved by preparing Coil-VaLd.36 This sequence alternates Val and Leu residues at the a and d positions, respectively, assuring that all helices are aligned by commensurate matching of the amino acid side chains in each layer.
On the basis of these early studies, DeGrado and Pecoraro designed the amphipathic peptide scaffold known as the TRI family of peptides, so named because a parallel 3SCC scaffold was desired. This scaffold utilizes the generic heptad repeat, LKALEEK, and is capped by Gly residues at either end.35,37,38 The peptides in this group are BABY, TRI, and GRAND, which have three, four, and five heptad repeats, respectively (Table 4). The N-termini are acetylated and C-termini amidated to align the helical dipole, facilitating the assembly of the coiled coils.35,37,705
The TRI-family peptides self-assemble upon dissolving in water into 2SCCs at low pH (< 5) and 3SCCs at higher pH (> 5.5).35,37 The Leu residues in the interior of the helix bundle can be substituted for heavy or transition metal-binding residues such as Cys or His. The locations of the mutations are described in Table 4. At physiological pH, these peptides adopt a 3-fold symmetry, which is an excellent framework to generate trigonal, tetrahedral, or trigonal pyramidal metal coordination environments.23
A major advance in the understanding of the metalation of Cys-substituted TRI peptides occurred when the crystal structure of As(III)(CSL9C)3 was obtained.711 This structure demonstrated that metals bind to these systems with one Cys thiolate sulfur from each helix (Figure 30). The structure also revealed the influence of the stereochemically active lone pair of As(III) ion on the As(III) coordination711 and demonstrated that the As(III) binds to the Cys in an endo conformation. When comparison to the apo-CSL9C structure, it became clear that the As(III) was accommodated in a preformed site, designed perfectly for this metalloid. DFT calculations on simplified As(III)Cys3 models were subsequently carried out to determine the most stable endo/exo conformation. The model corresponding to the lowest computed energy was described with two endo Cys residues and one exo Cys residue, whereas the most energetically unfavorable conformer had two exo Cys and one endo.712 Similar DFT results were also reported for Pb(II) complexation by these systems. EXAFS studies showed that both TRIL16C and TRIL12C bind As(III) with an As–S bond length of 2.25 Å, which is consistent with the reported values for ArsR (2.25 Å) and small molecule As(III)S3 ligation site distances (2.20–2.33 Å).686,713–719
Figure 30.

As(III) tris-thiolate site in As(III)(CSL9C)3. (A) A topdown view from the N-terminus. (B) A side view illustrating the pyramidal coordination of As(III). Reproduced with permission from ref 711. Copyright 2007 National Academy of Sciences.
Varying the location of the Cys substitution generates an a site versus d site for metal binding within the 3SCCs. CS, was utilized to understand the inherent differences between a sites and d sites.33,34,720 As mentioned above, unmodified CS crystallizes as an antiparallel 3SCC; however, reorientation of the peptide takes place once a single modification of the sequence at an a or d site occurs. Therefore, we can conclude that As(III) in As(CSL9C)3 did not force the peptide to be parallel; rather the Cys substitution at position 9 seems sufficient to shift the 3SCC into the parallel orientation. The crystal structures of apo-CSL9C and apo-CSL19C corroborate this scenario with a site and a d site, respectively.721 Before discussing the experimental observations, it is worth considering why a and d sites might be different. If one considers the sequence GLKALEEKLKALEEKG, one realizes that going from an a position to a d position requires a spacer of two amino acids LKAL, whereas proceeding from d to a requires three amino acids, LEEKL. This sequence difference requires that layers be separated asymmetrically within the helical core. Second, because of the rotation of the peptide backbone around the helix, it is expected that the side chain of an a position hydrophobe or metal ligand will be oriented slightly differently from that of a d position side chain. Thus, either steric effects based on the layer above a metal or the positioning of a metal ligand will be different depending on the choice between an a or a d position for the metal-binding site. The major rotamer of Cys in an a site is suitable for metal binding as it is predominantly directed toward the center of the structure. The Cys side chains placed at a d site are slightly rotated downward toward the C-terminus of the 3SCCs. One of the residues is pointing away from the hydrophobic interior toward the helical interface, whereas the other two Cys residues are directed toward the hydrophobic interior, offering a larger cavity, which might be preferable for larger metal ions like Pb(II).721 The difference between the orientations of a and d site Cys side chains is expected to lead to different affinities for metal ions due to the inherent van der Waals radii of the metal ions. For example, Cd(II) binds to the a site peptide TRIL16C with a stability constant of 1.6 × 108 M−1, while Cd(II) binds to the d site in TRIL12C with a stability constant of 2.6 × 107 M−1.722 Hg(II) binds to an a site in BABYL9C with a binding constant of 1.9 × 105 M−1, whereas it binds to the d sitecontaining peptide, BABYL12C, with an affinity of 5 × 104 M−1.704
The difference between an a site and a d site also leads to different protonation equilibra of the metal-bound peptides. When these peptides bind Hg(II), one sees a linear or Tshaped Hg(II) at low pH, which converts to a trigonal planar Hg(II) in more basic conditions.35,37,707,723–725 A model in which Hg(SR)2(HSR) forms Hg(SR)3 while liberating a proton accounts for this chemistry and corresponds to a one-proton loss. The metal-binding associated pKa is 7.6 for the a site peptide TRIL9C and 8.4 for the d site peptide TRIL12C.704 A similar trend is observed for Cd(II); however, this system is complicated by the fact that the first step of Cd(II) binding is believed to form a complex with a single coordinated thiolate at low pH. Subsequently, two protons are released simultaneously to generate either trigonal planar CdS3-type structures or pseudo-tetrahedral CdS3O structures.705,726–728 Thus, the protonation equilibrium constant is reported as pKa2. The equilibrium is described722,728 as
and Ka2 is described as
An a site peptide always has a lower pKa2 than a d site peptide, leading to a preference of Cd(II) for the a site. Specifically, both TRIL9C (a site) and TRIL16C (a site) have an apparent pKa2 of 13.4, while the d site peptides TRIL12C and TRIL19C have pKa2 values of 14.6 and 14.3, respectively.705,728 Interestingly, the pKa2 exhibits a reverse trend for Pb(II) binding to an a site versus a d site; that is, the pKa2 for an a site peptide is higher than that of a d site peptide for Pb(II) binding.712,722 This, together with the analysis based on the crystal structure, suggests that an a site prefers binding to Hg(II) and Cd(II), while a d site prefers binding to a larger cation. Both TRIL12C and TRIL16C bind Pb(II) with a 1:1 metal:trimer ratio. A similar pH study of Pb(II) yielded pKa2 values of 12.6 and 12.0 for TRIL16C and TRIL12C, respectively, indicating that Pb(II) binds to a d site at a lower pH than an a site.722
On the basis of the inherent differences between an a site peptide and a d site peptide, it is expected that they would enforce distinct heavy metal ion coordination environments. This hypothesis was examined by binding Hg(II), Cd(II), and Pb(II) to various a site and d site-containing peptides and investigating the coordination by means of 199Hg, 113Cd, and 207Pb NMR and 199mHg and 111mCd perturbed angular correlation spectroscopy (PAC). NMR of the heavy nuclei is a sensitive technique to probe the nature of ligands, coordination number, and geometry of the heavy metal-binding site. PAC spectroscopy has a faster time scale than NMR, and can complement NMR studies by providing details about local electronics, geometries, and dynamics of the site.729
The dynamics of an a and a d site upon Hg(II)23,704 and Cd(II)705 binding was reviewed previously. Here, we will focus on the difference between an a site versus a d site based on the Pb(II) binding environment. 207Pb NMR is an extremely sensitive technique for studying Pb(II) binding to proteins due to the wide chemical shift range (~16 000 ppm); however, this sensitivity also makes the detection of resonances nontrivial. The first detection of a 207Pb NMR spectrum for Pb(II) bound to a homoleptic thiolate protein environment was only recently reported.730,731 To distinguish the difference between an a site and a d site, several peptides were analyzed in the presence of Pb(II). CSL16C and CSL12AL16C, both containing an a site binding pocket, exhibit 207Pb NMR shifts of 5612 and 5555 ppm, respectively, while d site peptides BABYL12C and CSL12C have NMR resonances that shift downfield to 5786 and 5814 ppm, respectively.730 This shows an approximate 200 ppm shift downfield for binding to an a site peptide versus a d site peptide. Additionally, the slightly shifted Pb-(CSL12AL16C)32− peak at 5555 ppm, as compared to Pb(CSL16C)32− at 5612 ppm, indicates that introducing space above the lead binding site results in a small perturbation of the Pb(II) coordination environment.
All of the previous examples illustrate how a versus d sites engendered metal-binding specificity for a variety of metalloids. One can modify affinity and structure type by varying the properties of the first coordination sphere ligands or by modifying the sterics of the second coordination sphere. These considerations require a more advanced level of design to achieve coordination environments influencing a specific metal ion. First coordination sphere modifications were achieved by substituting each of the heavy metal-binding Cys residues with the noncoded amino acid residue, penicillamine (Pen). This donor, which modifies the electronics of the first coordination sphere and the sterics of the second coordination sphere, was used to examine Cd(II) binding to the TRI peptides. Specific secondary coordination sphere modification was achieved by substituting Leu residues with either a D-Leu or an Ala. These altered amino acids influence the binding of solvent molecules and have an impact on the metal coordination geometry.
The a site peptide, TRIL16C, binds to Cd(II), forming two species with different Cd(II) coordination numbers. The primary goal of incorporating a Pen residue was to obtain exclusively a three-coordinate Cd(II). While retaining a basicity similar to that of Cys (pKa(Pen) = 7.9, pKa(Cys) = 8.3), Pen provides greater steric bulk around the metal center, with two additional methyl groups in place of the β-methylene hydrogens. Interestingly, Peacock et al.732 obtained the crystal structure of CSL16Pen, showing that all of the thiol groups from Pen point into the interior of the helix bundle, as expected on the basis of apo-CSL9C. However, the placement of the methyl groups at the helical interface causes a change in the packing of the Leu layers above and below the metal-binding layers.732 Thus, the additional methyl groups perturbed the second coordination sphere of the metal, not through direct steric hindrance as one might expect for a small molecule model complex, but rather by changing the packing of hydrophobic layers as might be seen in a protein. Most important, TRIL16Pen was able to bind to Cd(II), forming a homogeneous CdS3 structure, confirmed by 113Cd NMR and 111mCd PAC spectroscopy, which was the first time such a coordination environment was observed for Cd(II) in an aqueous solution.727 A similar three-coordinate structure could be obtained by directly modifying the Leu packing layer using D-Leu. By switching L-Leu into D-Leu one layer above the coordination site, the orientation of the side chain inverts, now the C-terminus, and blocking the access of a water molecule to the Cd(II) ion to yield Cd(II) with 100% trigonal planar geometry.705,733 To obtain a pure four-coordinate Cd(II) site, residues at the secondary coordination sphere were mutated to facilitate the access of water molecules. Substituting an Ala residue for Leu one layer above the binding site opened a “hole” for solvent access above the thiol pocket. 100% fourcoordinate Cd(II) was obtained upon binding Cd(II) to TRIL12AL16C.728 These studies illustrate how protein design can generate rare metal coordination environments by exploiting the packing and the second coordination sphere encapsulating an ion within the hydrophobic environment of a coiled coil. Furthermore, these studies demonstrate for the first time that D-amino acids can be stably incorporated into helical assemblies and used to alter the properties of coordinated metal ions. A number of broad applications of noncoded amino acids have been recognized in designed proteins with improved biophysical properties or novel functions.734–737 The incorporation of unnatural amino acids in these de novo designed constructs is straightforward due to the application of solidphase peptide synthesis techniques.
In the previous paragraphs, we have described how designed proteins can bind a variety of metal ions and metalloids, how site selectivity can be controlled by sequence (a vs d sites), and how modifications of first and second coordination sphere ligands can be used to control metal coordination number. All of these issues address metal ion recognition by proteins for single metal sites; however, numerous metalloproteins contain multiple metal sites that are often in coordination environments that are very similar. Examples include ALAD, which contains both a structural five-coordinate Zn(II) active site and a catalytically active four-coordinate Zn(II) site,738,739 CuNiR, which contains both a T1- and a T2Cu center,740–742 and Cu,ZnSOD, which has very similar ligand environments surrounding the copper and zinc ions.297,410 ALAD is particularly interesting because heavy metal ions, especially Hg(II), Cd(II), Pb(II), usually substitute the Zn(II) ion solely at the catalytic site. To expand the de novo design of heavy metal-binding proteins, it is necessary to confer recognition at two independent metal-binding sites within a single protein construct. This goal was achieved using two separate strategies that exploited the concepts learned from designing mononuclear binding sites. One approach was to exploit a versus d preferences of metal ions for metal-binding sites, and the second was to exploit noncoded amino acids such as Pen or D-Leu to achieve metal selectivity at different protein sites. These heterochromic peptides paved the way for the first step of building analogues of metalloenzymes that contain distinct structural and functional sites. Because two different layers were to be modified to obtain dimetalated systems, GRAND peptide derivatives (Table 4) were used in these studies, providing a more stable initial scaffold (~9 kcal/mol more stable than TRI peptides), which could then be modified to achieve selective ion recognition.706,707 The first objective was to design a dual site peptide that contained the same metal in two different coordination environments. Two constructs were successfully designed to bind to Cd(II) [three-, four-coordinate] with 100% specificity according to 113Cd NMR and 111mCd PAC spectroscopy. The design of GRL16PenL26AL30C combined the strategy of enforcing three-coordinate Cd(II) used in the design of TRIL16Pen and that of forming four-coordinate Cd(II) used in TRIL12AL16C.743 Importantly, the two binding sites, one pseudotetrahedral and one trigonal planar, exhibited different physical properties, indicating that not only coordination number, but also spectroscopic features can be controlled in the different regions of the same peptide. This achievement further suggests that redox, structural, or catalytic sites can be engineered into designed proteins with confidence. Furthermore, alternate chirality peptides were shown to result in peptides containing distinct [three-, four-coordinate] centers as was demonstrated with GRL12D-LL16CL26AL30C. This design utilized solely the secondary sphere control to obtain two exclusive coordination environments.733 Pecoraro and co-workers also demonstrated other GRAND peptide derivatives that were able to bind to Cd(II) with two different coordination environments.705,733,743,744 A detailed discussion of this work is provided by Peacock et al.705
Heterometallic structures with selective incorporation of two different metals into this class of 3SCCs were also prepared using Pb(II) and Cd(II).745 To understand the specificity achieved in this system, the influence of the second-sphere coordination environment on Pb(II) when bound to 3SCCs was evaluated via DFT calculations and 207Pb NMR.712 In the presence of a mixture of CSL9C and TRIL12AL16C and 1 equiv of Pb(II), a mixture of the metalated species was observed; however, a marked preference was noted for the peptide with the Ala substitution, which introduced more space around the Cys binding site. Overall, the preferential binding of Pb(II) as determined via 207Pb NMR is a site with an Ala “hole” > d site > a site. Presumably, this is due to the better accommodation of the larger Pb(II) ion and bulky lone pair on Pb(II).712
In addition to the coiled coil systems, smaller peptides containing Cys residues were also designed to investigate the Hg(II) binding properties. An 18-residue peptide with a sequence identical to the metal-binding loop (sequence: TLAVPGMTCAACPITVKK) in MerP was synthesized and characterized.746–748 The linear sequence of Cys and Ala (bold in the sequence) was shown to determine the Hg(II) binding affinity. The dissociation constant Kd for Hg(II)(CAAC) (90 μM) lies between those of Hg(II)(CCAA) (78 μM) and Hg(II)(CACA) (207 μM).746 More recently, a tetrapeptide Ac-CD-PPC-NH2 (CdPPC) was designed by Iranzo and co-workers. 749 The D-Pro-Pro motif forms a preorganized β-turn that directs the orientation of the two Cys, shown to bind to Hg(II) with a high affinity (the stability constant log β = 40.0), forming a linear mononuclear dithiolate–Hg(II) complex. However, the peptide with stereochemically reversed configuration CPPC (Ac-CPPC-NH2) showed a much lower stability constant, demonstrating the efficacy of the D-Pro-Pro template in stabilizing the linear Hg(II) coordination. Computational modeling using quantum mechanical methods was carried out on Hg(II)(CdPPC), showing that there were two energy-minimized conformers for this complex, both of which contained a slightly distorted linear HgS2 coordination, consistent with the evidence obtained from 199mHg PAC.
Furthermore, the binding of heavy metal ions to a thiol-rich environment was demonstrated in a more native-like α-helix bundle α3DIV.750 This design was based on DeGrado’s α3D, which is a globular, antiparallel three-helix bundle.751 Bryson et al. examined the crystal structure of CS34 and successfully imparted more native-like properties into de novo designed proteins in a hierarchic approach, where they introduced specific interactions to achieve a uniquely folded scaffold.752 Three rounds of design were carried out initially to construct α3C: first, they shortened the helix region by one heptad to 21 residues, a length more commonly seen in native helix bundles; then, helix start/stop residues were introduced to avoid the formation of long helices and provide an initial trajectory of the loop; and finally, rearrangement of the residues in the hydrophobic core and on the helical interface was accomplished to direct the counter-clockwise folding topology. On the basis of α3C, α3D was designed to introduce more sequence diversity.751 The proton exchange and thermodynamic properties of α3D closely resembled those of native proteins, providing an excellent foundation for incorporating a metal-binding site.
The design of α3DIV represents the first example of engineering a metal-binding site within an antiparallel single-chain three-helix bundle by rational design.750 The tris-Cys site was incorporated near the C-terminal end of the helix bundle in a “hydrophobic box”, which is an ideal location for exploring the sequestration of heavy metal ions in a hydrophobic environment (Figure 31). On the basis of the spectroscopic parameters revealed earlier, Chakraborty et al. demonstrated that α3DIV binds Hg(II), Cd(II), and Pb(II) at the tris-Cys center as expected with relatively high affinities (Ka > 107 M−1). Specifically, at pH 8.0, Hg(II) binds to α3DIV, forming a HgS3 site; similar to the TRI family peptides, at a lower pH, Hg(II) forms a linear HgS2 coordination when bound to α3DIV as revealed by 199Hg NMR and 199mHg PAC. α3DIV accommodates Cd(II), forming a pseudotetrahedral CdS3O coordination with the lower limit at an apparent binding constant of 2.0 × 107 M−1. Pb(II) binds to the tris-Cys site of α3DIV with an affinity of 3.1 × 107 M−1 (lower limit), forming a trigonal pyramidal PbS3 site. These thiol-rich heavy metal-binding sites behave very similarly to those in the TRI family peptides; however, the sequence of α3DIV contains all 20 coded amino acids, representing a more native-like de novo designed scaffold. Moreover, the single stranded three-helix bundle allows one to incorporate asymmetry at the first coordination sphere of the metal-binding site, which is more challenging to achieve in a self-assembled coiled coil system.
Figure 31.
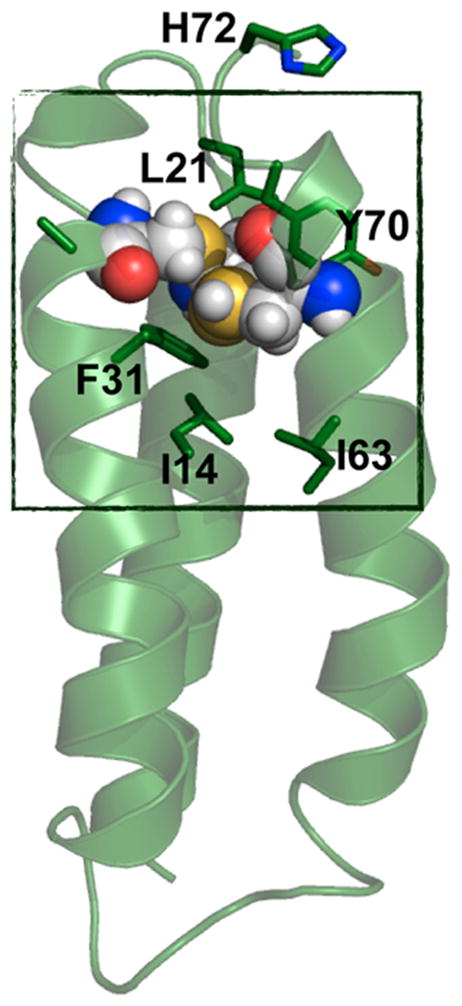
PyMOL model of α3DIV. Cys mutations are shown in spheres. The box represents the “hydrophobic box”, which includes residues Leu21, Tyr70, Phe31, Ile14, and Ile63. This model is made based on the solution NMR structure of α3D (PDB code: 2A3D751).
3.2.2. De Novo Designed Metal Centers Based on β-Structures
β-sheet structures, one of the most important secondary structure types in protein assembly, are composed of almost fully extended polypeptides that pack through hydrophobic and H-bonding interactions involving the backbone carbonyl group and the neighboring amide proton functionalities. 646,648,753–755 β-Sheet structures are prevalent in proteins. Commonly seen topologies include β-sandwich, β-barrel, α/β arrangement, β-propeller, β-helix, β-clam, and β-roll.648 β-structures have also been demonstrated to play important roles in protein–DNA,756 protein–RNA,757 and protein–protein758 recognition. The de novo design of β-peptides, despite their structural and functional significance in native proteins, is not as well-advanced as that of α-helical structures, partially because it is more challenging than the design of α-helices. More often than not, an isolated β-strand is not stable, making it less modular and more difficult to design.647 Specifically, the design of β-sheets requires the correct pairing of β-strands that not only contribute to its stability but also to its secondary structure elements. Previous reviews on the design of β-hairpins, β-bulges, β-sheets, β-turns, etc., are focused on the earlier achievements of constructing a stable β-structure.10,647,649–651 We review herein the incorporation of a metal site in a β-structure in the context of creating functional metalloproteins.
3.2.2.1. The Minibody
The first reported case of a designed β-protein with a novel fold that binds to metals is the minibody.759 It is a de novo designed monomeric protein with 61 residues whose design was based on a fragment (residues 19–83) of an immunoglobulin McPC603, a phosphocholine-binding mouse myeloma protein.760 The minibody is composed of six β-strands, forming two β-sheets. With preserved structurally important residues from the fragment of immunoglobulin (Gly26, Phe27, Phe29, Glu35, Arg69 were preserved in the minibody) and appropriately modified residues from the hydrophobic core in the original immunoglobulin [Leu20(McPC603)→Asn2(minibody), Cys22(McPC603)→Gln4(minibody), Trp41(McPC603)→Tyr23(minibody), Phe65(McPC603)→Tyr47(minibody)], the minibody is moderately soluble in water up to a concentration of 10 μM. It demonstrated the tolerance of sequence variability and the predictability of the main chain conformation of the hypervariable region of immunoglobulin. Moreover, the highlight of this contribution is that this β-protein can accommodate the metal-binding His residues located in the loop region (Figure 32). Upon binding to divalent metal ions, for example, Cu(II), Zn(II), or Cd(II), the mini-body forms a M(His)3 center, which is of important biological relevance. Additionally, the metal binding was demonstrated to display various affinities in the order of Cu(II) > Zn(II) ≫ Cd(II) > Co(II). Although the metal ion affinity was limited (with a dissociation constant in the micromolar range) and no activity studies were carried out when it was first reported, the minibody represents a significant advancement toward creating a metalloenzyme based on a β-sheet scaffold.
Figure 32.

Modeled structure of the minibody with predicted metal-binding site. Reproduced with permission from ref 759. Copyright 1993 Nature Publishing Group.
The follow-up work from the same group focused on increasing the solubility of the minibody, which was realized through rational design and mutagenesis.761 The rationale behind increasing the solubility is to substitute residues that are predicted to be on the surface of the protein with residues that are considered to have favorable interactions with water, while maintaining the structurally important residues to retain stability. This goal was achieved by Arg20Lys, Val19Glu, and a few other mutations. With this strategy, the solubility of the minibody derivatives was enhanced 100-fold as compared to the original minibody (from 10 μM to 1 mM). At the same time, most of the rationally designed variants retained the β-sheet secondary structure, sufficient stability, and the ability to bind to metal ions. One variant did not exhibit the β-protein structural motif nor bind metal ions, due to a slight distortion of the backbone upon mutagenesis. This work illustrates the fundamental importance of hydrophobic–hydrophilic interactions in protein assembly, which can be utilized to achieve improved properties.
Although the increased solubility was still did not allow for solution NMR or crystallographic studies, Sollazzo and co-workers have employed other strategies to understand the structure of the minibody. For example, the surface topology of the minibody can be examined through rational chemical modifications, such as the acetylation of certain Lys residues, O-acetylation of the phenolic hydroxyl group on Tyr, nitration of Tyr, or modification of the protein guanine group on Arg.762 The conditions for these modifications were carefully selected to ensure that the protein was in its native state; thus the surface residues were more susceptible to modifications. Using mass spectrometry, they were able to pinpoint the residues that exhibited higher, moderate, and lower reactivities toward these chemical modifications. Moreover, proteolysis combined with mass spectrometry was used to probe the tertiary structure of the minibody.763
3.2.2.2. The Betabellin
The betabellin is a de novo designed bell-shaped β-sandwich protein that consisted of two 32-residue β-sheets, each of which is designed to have 12 polar residues on one side and 12 nonpolar residues on the other so that the β-sheets can assemble through hydrophobic interactions. Betabellin-15D, in particular, has six His residues (His1, D-His7 and His32 from each sheet) at the rr edge of the β-sandwich (Figure 33).764 The two sheets are assembled upon air oxidation, such that two clusters of (His)3 sites are formed, each of which can bind a divalent metal ion. A computer model was built, indicating that there were two slightly different mononuclear tris-His sites in betabellin-15D, one of which was expected to bind divalent metal ions slightly tighter than the other. Betabellin-15D binds divalent metal ions with various affinities in the order of Cu(II) > Zn(II) > Co(II) > Mn(II) (the Kd ranges from micromolar to millimolar).764 For Cu(II), in particular, the binding is pH-dependent.765 Limited proteolysis by immobilized pepsin, which differentially cleaves the peptide bond with moderate specificity depending on how accessible it is in the protein structure, both in the presence and in the absence of Cu(II), indicates that Cu(II) might induce the folding of betabellin-15D, and lead to a slower cleavage of the Phe12–Ser13 peptide bond.764,765 Specifically, at pH 5.8, 6.4, and 6.7, betabellin-15D only folds into β-sheets in the presence of Cu(II), signified by a substantial increase in the absolute value of molar ellipticity at 218 nm in the CD spectrum, which was not observed in its apo-protein form.765
Figure 33.

Predicted β-sheet structure of each betabellin-15 chain viewed from the nonpolar face. t = turn, r = reverse turn. Reproduced with permission from ref 764. Copyright 1999 Springer.
It is worth noticing that the design of betabellin-15D incorporated D-amino acids (D-His, D-Lys, D-Pro), which serve the purpose of creating an inverse-common (Type I′) β-turn754,766 to favor the formation of the antiparallel β-sheet.767,768 Solid-phase peptide synthesis provides a facile route to achieve the incorporation of unnatural amino acids.765 It is further demonstrated that betabellin-15D can serve as a molecular model for protofilaments of amyloid fibers.769,770
3.2.2.3. Other β-Structures
Imperiali and co-workers incorporated unnatural amino acids with heteroaromatic ligands such as bpy and neocuproine as side chains to bind metal ions in a β-hairpin motif.771,772 The idea is to introduce bidentate heteroaromatic ligands, which are excellent metal chelators, into a protein system to rationally modulate the metal-binding affinity. A series of amino acids were synthesized (Figure 34), which have the α-amino acid functionality appended to the 6, 5, and 4 positions of bpy (compounds 41, 42, and 43), respectively, to the 1 position of 10-phenanthrol-2-yl (compound 44), and to the 1 position of a novel neocuproine derivative (compound 45).155,771,772 These unnatural amino acids were introduced to the N-termini of the six-residue peptide chains; at the same time, some of the designed peptides also contained metal-binding natural amino acid residues such as His or Asp at the C-termini to assist the formation of a preorganized binding site. Upon the formation of a β-turn, the two ends come close together to form the metal-binding site. These reports demonstrated that by introducing unnatural amino acid residues, the metal affinities could be modulated. The binding of Zn(II) to these peptides, however, is relatively weak with a dissociation constant in the millimolar to micromolar range.771,772 Using a similar strategy, with different unnatural amino acid residues, the same group has investigated dynamic protein interactions,773 cell migrations, 774 and lanthanide sensing.775
Figure 34.

Unnatural amino acids synthesized by Imperiali and co-workers. Reproduced with permission from ref 772. Copyright 1996 American Chemical Society.
3.2.3. Metal-Induced Protein Folding
3.2.3.1. De Novo Designed Zinc Fingers
ZF binding motifs have also been engineered into de novo designed proteins. The first reported designed ZF protein introduced a His2Cys2 metal-binding site into a four helix bundle protein, Zα4.776 The protein scaffold is a simplified version of a naturally occurring motif. Initial studies showed that Zn(II) binds with a relatively high affinity to a monomeric protein and that the Cys residues are necessary for binding. The desired tetrahedral coordination environment was obtained on the basis of the absorption spectrum of the Co(II)-substituted protein, which showed a d–d band at 615 nm with shoulders at 557 and 664 nm, fully consistent with a tetrahedral binding environment. Both the apo- and the metalated protein adopted an α-helical structure, revealed by CD spectra; however, the Zn(II)-bound form was more resistant toward guanidine hydrochloride denaturation. Interestingly, as early as 1990 when de novo protein design was in its infancy, the authors foresaw that this strategy could potentially be used to construct a catalytically active metalloprotein (vide supra). Further analysis of Zα4 was carried out by Klemba and Regan to study the changes in the system after removal of metal-binding ligands one by one (similar to the work of Nomura and Suguira described previously168).777 His25, Cys47, and His51 each separately mutated to Ala residues, creating a vacant site at the metal-binding center. The absorption spectra for Co(II)-bound proteins were collected, showing that Co(II)-Zα4H25A and Co(II)-Zα4H51A had absorption features very similar to the parent construct Co(II)-Zα4, while Co(II)-Zα4C47A did not exhibit such features. The d–d band of Co(II)-Zα4C47A revealed perturbation of the tetrahedral geometry, resulting in either a distorted tetrahedral or a five-coordinate Co(II) site. Dimerization was observed in the presence of Cd(II), a thiophilic metal, in the Zα4C47A mutant, presumably due to the decreased number of Cys residues available to coordinate Cd(II). All other mutations were shown to bind Zn(II) in a 1:1 ratio. Moreover, the authors investigated the pKa of a putative Co(II)-bound water by collecting absorption spectra at different pH conditions. No absorption was observed below pH 9.5, and was attributed to the low electrophilicity of the metal site. The authors suggested incorporating more His ligands to convert this site into a catalytic Zn(II) site.777
A minimalist 26-residue peptide with a Cys2His2 binding site, containing more than 60% Ala residues, was reported by Michael et al. to analyze the importance of the seven most conserved residues characteristic of the TFIIIA-type ZFs.778 TFIIIA is a transcription factor that regulates the transcription of 5S RNA in vitro. Previous studies on TFIIIA unveiled remarkable repeating structures in the protein ZF domains.779 The minimal ZF protein was denoted as MZF. To understand the role of hydrophobic residues in maintaining the structural integrity of the MZF, three other mutants were designed: MZF-Coreless, where the relatively hydrophobic residues Tyr and Phe were substituted with Ala residues, MZF-Δ8, where one of the Ala residues between the Cys and Phe was deleted, and MZF-Coreless-Δ8, where both types of mutations were combined into one protein. Co(II) binding studies showed that MZF binds Co(II), forming 2:1 and 1:1 complexes, out of which the 1:1 complex was similar to that of Co(II)-bound native ZF proteins and the 2:1 complex was reflective of a tetrathiolate environment. The NOESY spectrum of the MZF–Zn(II) complex showed well-distributed peaks, indicating a stable folded structure. In particular, the patterns of the NOEs were consistent with those observed for native ZFs. Only one of the mutants, MZF-Coreless-Δ8, with both the deletion of the hydrophobic residues and the spacing residue Ala, showed metal-binding properties similar to those of native ZF proteins, while the other two mutants induced higher formation of the 2:1 complex than the 1:1 complex. These results showed that the seven conserved residues were necessary and sufficient to maintain the ZF site.778
Latour and co-workers studied the nonclassical Cys4 system using a de novo design strategy. The Latour lab initially reported a series of branched cyclic proteins containing a Cys4 motif to model the heat shock protein Hsp33, in which they replaced the native two strand β-sheet with a de novo designed branched cyclic polypeptide.780 Additional substitutions to the polypeptide and the addition of Zn(II) resulted in good reproductions of both the first coordination sphere and the extended H-bonding system of the native Hsp33 system. A similar approach was used to model a treble-clef ZF type-site, similar to the bacterial peroxide sensor PerR.781 Utilizing eight-residue cyclic proteins containing 9–14 residue “tails” and four Cys residues, the simplified polypeptides bind Zn(II) with high affinity at biologically relevant pH resulting in treble-clef structures. The tail of the protein folds into a helix to accommodate the four-coordinate Zn(II) binding site. The shorter, nine-residue tail lacks the full helix but still folds to allow a four-coordinate Zn(II) binding site. The longer tail, which can fold into a helix upon metal binding, was found to stabilize the structure of the de novo designed treble-clef ZF (Figure 35).781
Figure 35.

Zn(Cys)4 site in Bacillus subtilis PerR (left), its schematic representation (middle), and the model peptides LTC and LTTC. Reproduced with permission from ref 781. Copyright 2009 Wiley.
To address the influence of amino acid sequence and the secondary coordination sphere interactions, LaTour and co-workers examined a series of de novo designed ZF proteins with different Zn(II) coordination environments and dissimilar secondary structures (Figure 36).782 Specifically, treble-clef Zn-LTC and cyclic β-hairpin Zn-LHSP constructs containing Cys4 sites and ββα ZF consensus peptide (CP) with Cys2His2, Cys3His, and Cys4 sites were examined. The difference between the three constructs lies in the location of the hydrophobic residues: all hydrophobic residues of Zn-LTC are solvent-exposed, yielding no hydrophobic core in the protein construct; Zn-LHSP has a relatively small hydrophobic core, while the traditional ββα ZF peptide CP contains more hydrophobic residues, forming a larger core. The CP series of peptides exhibited pH-dependent metal-binding affinities, attributed to the deprotonation equilibra of the His and Cys residues. The Zn(II) binding studies in the CP1 variants yielded higher-than-previously reported783 binding constants that reached 1014–1015 M−1 at a physiological pH. Additional metal exchange experiments with EDTA and peptides with altered hydrophobic cores also revealed that hydrophobic residues might play a role in controlling the kinetics of metal binding. These findings cast light on Zn(II) homeostasis and the role of transcription regulation by ZFs. Tight metal-binding ensures that these ZF proteins are in the proper conformation to bind DNA and carry out transcription. At the same time, lower hydrophobicity close to the Zn(II) binding site contributes to a fast exchange, convenient for Zn(II) uptake and release to maintain zinc homeostasis.
Figure 36.
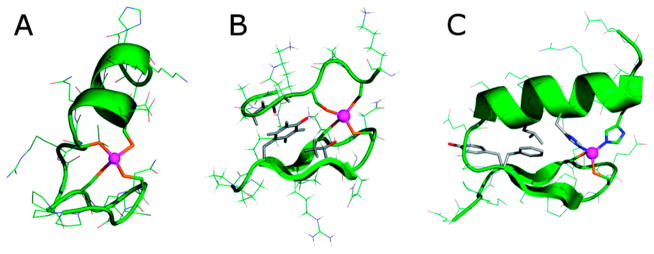
(A) Zn·LTC complex; (B) Zn·LHSP; and (C) a classic ZF site. The zinc ion is shown in purple. Reprinted with permission from ref 782. Copyright 2010 American Chemical Society.
3.2.3.2. Other Types of Metal-Induced Protein Folding
There are a number of studies examining metal-induced folding of α-helical coiled coils. Since this work has been previously reviewed, we will summarize it briefly here.23,29,784–789 Some of the earliest studies involving the use of metal-binding sites to promote α-helix and coiled coil formation were reported by Ghadiri and co-workers. They reported a 17-mer peptide, containing two His or one His and one Cys site(s), which displays a ~50–90% increase in α-helicity upon binding Cd(II), Cu(II), Ni(II), and Zn(II).790 The His/His version of this peptide, when bound to the exchange inert metal complex [Ru(NH3)5(OH2)]2+, forms a macrocyclic complex (cis-[Ru-(NH3)4(His)2peptide]3+) with 80% α-helical content (only 45% in the absence of metal).791 Additionally, the group reported a 15-mer amphiphilic peptide, which, upon binding Ni(II), Co(II), or Ru(II) at the bpy moiety at its N-terminus, self-assembles to form a 70% α-helical 3SCC.786 This construct was also used to incorporate a Cu(II)(His)3 site for the development of a heterobinuclear 3-helix complex.792 The group also reported the formation of a parallel 4-helix bundle metalloprotein made up of a 15-residue amphiphilic peptide with a pyridyl group at the N-terminus and bound Ru(II).793
Similarly, the Ogawa group has designed several related peptides, which use metal-binding sites to form α-helical bundles. Kharenko et al. reported the 32-mer peptide Cys16Cys19-GGY, which has a Cys-X-X-Cys motif formed by the placement of Cys residues in adjacent a and d positions of one of the heptads. When this peptide binds a single Cd(II) ion, the random coil is converted to a 2SCC.794,795 This same peptide, in the presence of Cu(I), forms a 4-helix bundle containing a tetranuclear [4Cu–4S] cofactor.796 Additionally, the group reported that upon binding of the metal complex Pt(en)(NO3)2 to AQ-Pal14 Pal21, which contains two 4-pyridylalanine residues on its surface and forms a 2SCC in the absence of metal, a significant conformational change to a metal-bridged four-helix bundle occurs.787
Tanaka et al. reported the design of a parallel 3SCC (in the presence of metal ions) called IZ (sequence YGG-(IEKKIEA)4).797 Substitution of the Ile residue on the third heptad with a His generated IZ-3aH and with two His substitutions, IZ-3adH. In the presence of Cu(II), Zn(II), Co(II), or Ni(II), the peptides were converted from random coils to 3SCCs.31,788 When Cys residues were substituted into the IZ peptides, Hg(II) and Cd(II) binding occurred.789 The group has now utilized the metal-induced folding nature of their IZ peptides by fusing IZ-3adH to the DNA-binding domain of a heat shock element to generate a metal ion-controlled DNA binding protein.798 In the presence of metal ions, the fusion protein can inhibit RNA transcription by T7 RNA polymerase. This is an important achievement demonstrating the use of de novo design and metals as external stimuli to regulate protein function.
Moreover, the monovalent ion Ag(I) was shown to influence the assembly of the de novo designed peptide TZ1H.799 TZ1H is a 41-residue peptide consisting of six heptad repeats, with a design based on the isoleucine zipper peptide GCN4-pH.800 Conticello’s group previously demonstrated the pH-dependent assembly of this peptide before exploiting the influence of Ag(I).801 At pH 5.6, the apo-peptide exists in a random coil conformation, indicated by a minimum at 201 nm in the CD spectrum. Silver(I) triflate was added into the solution, and the spectrum transformed to a double-well manifold, indicative of the formation of coiled coils. Moreover, Ag(I) can direct TZ1H to assemble into a long-aspect-ratio helical fiber, which can be directly observed by TEM. Energy dispersed X-ray analysis (EDX) further confirmed the presence of Ag(I) in the helical fibers. Interestingly, the assembly of TZ1H is only triggered by Ag(I), not by Ni(II), Cu(II), or Zn(II). Conticello and co-workers attributed this type of selectivity to the geometric constraints of the trigonal site of TZ1H.
Recently, a metallacrown-templated peptide assembly was reported by Cal et al.802 This approach took advantage of the stability and the possible chirality of the metallacrown system by attaching amphiphilic polypeptide chains to the amino-hydroxamate moiety, which then self-assembled to a metallacrown when Cu(II) was present.803–805 The CD spectrum of the peptide [AD]-NHOH (Ac-KALEKALKEALAKL-NH-(CH2)2-NHCO-hydroxamic acid) showed some fraction of α-helix and β-turn conformation. Upon the addition of Cu(II), the spectrum changed drastically into a typical α-helical coiled coil spectrum. Mass spectrometry and proton NMR experiments also confirmed that the introduction of Cu(II) drove the hydroxamic moiety to assemble into a metallacrown, which acted as a template for the formation of a coiled coil peptide.
Protein conformational switches are involved in important biological processes that modulate the function of proteins in various ways.806–811 It is still unknown how whether certain protein sequences can inherently adopt more than one stable conformation. Some conformational changes are initiated by the introduction of metal ions for transport or regulatory purposes.812–814 In addition, research has shown that along with pH, specific mutations, and oxidative stress, the metal ions directly impact neurodegenerative diseases, such as Alzheimer’s and Parkinson’s disease, by inducing conformational changes on the relevant oligomeric peptides.815,816 Herein, we review the examples of de novo designed protein systems that undergo conformational changes between two stable structures upon association/dissociation of metal ions in the context of understanding the fundamental interactions between metal ions and proteins that direct conformational switches. It is undoubtedly a challenging task, as the design requires inherent structural ambiguity and stability, as well as metal-binding functionality.
The Woolfson group reported the design of ZiCo, a polypeptide that forms 3SCCs in the absence of Zn(II) and a monomeric structure upon binding Zn(II).817 The reverse switch from the Zn(II)-bound structure back to the α-helical coiled coil was achieved by adding EDTA to extract Zn(II) from the peptide. The recovery of the apo-peptide features was observed using CD spectroscopy. Furthermore, Kuhlman and co-workers used computational design to create a similar switch Sw2.818 Using biophysical characterization, they demonstrated that Sw2 undergoes a reversible conformational change from α-helical oligomers to a ZF monomeric structure upon binding Zn(II).
An important type of conformational switch design originated from the motivation to understand the amyloid formation process and its role in neurodegenerative diseases. Pagel et al. used de novo principles to design a peptide that changes its secondary structure upon binding Cu(II) or Zn(II).819 The peptides i+2 and i+4 were designed on the basis of a heptad repeat approach, where the numbers represent the positions of the His binding residues in the sequence. His at i and i+2 positions bind to Cu(II) or Zn(II); however, the formation of the complex distorts the peptide backbone, thus disrupting the helix. In the absence of the metal ions, peptide i+2 slowly transitions from a coiled coil structure to amyloids. The addition of 2 equiv of Cu(II) disrupts the amyloid formation, while Zn(II) accelerates its formation. On the contrary, peptide i+4 rapidly converts to a β-sheet structure in the absence of the metal ions, and switches to an α-helical coiled coil structure upon addition of either Cu(II) or Zn(II). This work demonstrates that the position of the metal-binding residues influence the structural switch, while the nature of the metal ions plays a key role in determining the conformation.
3.3. De Novo Designed Functional Metalloproteins: The Grail Quest of Protein Design
3.3.1. De Novo Designed Hydrolytic Centers
3.3.1.1. Mononuclear Zinc Center at Dimeric Interfaces
Although most designed enzymes rely on monomeric structures with metal sites inserted into their cores, Der and co-workers serendipitously discovered that a designed Zn(II) site at the interface of a homodimer (MID1-Zn) could effectively catalyze pNPA and p-nitrophenyl phosphate (pNPP) hydrolysis (Table 1).211,820 The motivation behind the design was to use metal-binding sites at a designed protein interface to improve computational design protocols for protein–protein interactions. Given that metals form stronger interactions with the side chains of residues such as His, Cys, Asp, and Glu than do protein–protein hydrogen bonds or van der Waals contacts, it is reasonable to postulate that metal-binding interactions should overcome suboptimal hydrogen-bonding patterns or packing at the interface in a design.
The design was based on a two-residue Zn(II) binding site on a monomeric protein (derived from the Rab4-binding domain of rabenosyn), which was then dimerized and computationally optimized to form what was intended to be two ZnHis4 binding sites at the interface of a homodimer.820 The design model, which also included interactions between Met38, Tyr41, and Phe42 on both chains for forming a hydrophobic core at the interface, was named MID1 (metal interface design 1). As intended, MID1 forms a dimer at high micromolar concentrations both with and without Zn(II) present, but the dissociation constant in the presence of Zn(II) is more than 100-fold stronger than it is in its absence (Kd < 30 nM). X-ray crystallography demonstrates that MID1-Zn closely resembles the model, although with some key differences (Figure 37). One difference is that only three His residues coordinate each Zn(II) ion at the interfaces. The coordination sphere is completed either by carboxylate groups from the C-terminal Asp or by a symmetry-related molecule or tartrate from the crystallization buffer. A small deviation in the binding orientation and in the protein–protein interface contacts was also observed. Despite these differences, a high affinity metal-mediated homodimer site was prepared using only computational design without any optimization by directed evolution.
Figure 37.

X-ray crystal structure of MID1-zinc, a designed protein with a metal-mediated protein interface. The red mesh represents the active site cleft above the open coordination site of the ZnHis3 metal site. Reproduced with permission from ref 211. Copyright 2012 American Chemical Society.
Although the original intention was to obtain a ZnHis4 site, Der et al. observed that the presence of tartrate in the ZnHis3 represented a small binding pocket (~6 Å wide and 4 Å deep), suggesting that the interface metal site may display esterase activity toward substrates like pNPA and pNPP. Indeed, MID1-Zn effectively catalyzes pNPA hydrolysis with more than 50 turnovers and a rate of 0.22 s−1 and KM = 0.47 mM at pH 8.5.211 The reaction is pH-dependent (increasing efficiency with increasing pH) and has a kinetic pKa of 8.2 ± 0.1 and a maximal efficiency of 630 ± 90 M−1 s−1. Control experiments using the apo version of the homodimer, a mutated version with no His binding residues, the WT monomer on which the design was based mutated with His residues, and free zinc all demonstrated no significant activity above the background, confirming that the interface pocket and Zn(II)-binding site are both required for catalysis. Further, mutation of some of the surrounding His residues to Glu demonstrated a loss of activity because the open coordination site was closed (His12Glu and His35Glu). Despite the original design intention of two Zn(II) sites, only one active site is observed in the crystal structure (only one tartrate is bound to one Zn(II) site), and the dimer is fully formed with 1 equiv of Zn(II) for 2 equiv of monomer (no additional activity is observed with more Zn(II) present). Although pNPP is intrinsically less reactive, MID1-Zn still effectively catalyzes its hydrolysis with an efficiency of 14 M−1 s−1 at pH 8.5. The KM for this reaction, 12 μM, is 40 times lower than that for pNPA because of favorable electrostatic interactions between the negative phosphate group and positive Zn(II) active site. The authors discuss the importance of the binding cleft in achieving these efficiencies. However, while enhancing efficiencies, it is expected that such an open catalytic site is likely to exhibit significant product inhibition, which has been a bane to small molecule model compounds. Synthetic models are relatively ineffective given that they have no binding cleft, although this can be improved somewhat in apolar solvents, which can simulate the apolarity of an active site cleft.821 Further discussions and comparisons will be made in a later section where the de novo design of a ZnHis3O site in a 3SCC is described. Regardless, this study of a minimalist engineered enzyme demonstrates that cleft formation at protein interfaces provides another effective strategy for the rational engineering of new catalytic activities.
3.3.1.2. Multifunctional Metal Centers: Structural and Catalytic Sites in One Design
Despite the prevalence of multisite metalloproteins in nature, there are few reports of designed two-site metalloproteins in which two different metals exist in separate binding sites.32,212,213,792 Of these, only one has reported catalytic activity (Table 1).212,213 This de novo designed 3SCC contains two metal sites, one for structural stabilization (HgS3) and the second for hydrolytic activity (ZnN3O) within just a few hundred-fold of the native CA. Although the design of this metalloprotein has been previously reviewed,23,822 it is appropriate to summarize the initial work before reviewing more recent developments.
To design a 3SCC, which discriminates two different metals in two different sites, the peptide TRIL9CL23H was used together with a metal ion with high affinity for thiolate ligands but low affinity for His (Hg(II)). Previous work demonstrated that Hg(II) can be used to induce folding in unfolded peptides and increase thermodynamic stability in prefolded peptides (as demonstrated by CD denaturation studies).38,212,213 Additionally, Hg(II) provides a well-studied spectroscopic tag for the formation of a 3SCC (UV–vis and 199Hg NMR differentiate between linear HgS2 found in a 2SCC and trigonal HgS3).725 In the presence of this highly stable Hg(II)pep3 complex, Zn(II) then binds solely to the His3 site. One motivation behind the development of a Zn(II)N3 site was to model the active site of CA, which catalyzes the reversible hydration of carbon dioxide and is one of the fastest known and most well-studied zinc metalloenzymes, making it an excellent system for de novo design mimicry studies. X-ray crystallographic studies on the Hg(II)Zn(II) complex of the related peptide sequence CSL9PenL23H (the CS sequence has properties similar to those of TRI in solution) confirmed the presence of Hg(II) in the sulfur site (fully trigonal in one of the 3SCCs in the asymmetric unit and T-shaped in the second) and a Zn(II)N3X site in both 3SCCs in the asymmetric unit, analogous to that found in CA (Figure 38).213 This X-ray structure was significant for two reasons. First, it demonstrated that the proposed trigonal Hg(II) center for Hg(II)(TRIL16C)3− existed as was predicted (vide supra) and provided the first structural model for the Hg(II) binding site in the metalloregulatory protein MerR.673–676 Second, this work demonstrated that the native protein fold of CA was not necessary to achieve a closely similar structural model for the first coordination sphere of the Zn(II)(His)3(H2O) center. One can conclude that for the first coordination sphere, this type of center can be accommodated in protein structures as varied as a twisted β-sheet with a long water channel as found in the native enzyme or in the hydrophobic interior of an α-helical coiled coil as shown for [Hg(II)]S[Zn(II)(OH2/OH−)]N(TRIL9CL23H)3n+. Finding that the structural similarity of the Zn(II) site to that in CA is limited to the first coordination sphere allowed (Figure 39), for the first time, to begin to address what is the minimal unit required for catalytic activity in this type of system. To do this, Pecoraro and co-workers initially looked at whether the construct could catalyze the hydrolysis of pNPA, the non-physiological substrate of CA often used for analysis of its reactivity as compared to mutants and small molecule model complexes. Analysis of these reactivity studies demonstrates that hydrolysis of pNPA by [Hg(II)]S[Zn(II)(OH2/OH−)]N(TRIL9CL23H)3n+ follows enzyme-like saturation kinetics with a maximal kcat/KM of 31 ± 4 M−1 s−1, about 550-fold more efficient than comparable small molecule model complexes.212,213,823–825 Further, it is within ~100-fold of the maximum efficiency of CAII and has a similar pH-dependent profile (increasing with increasing pH), yet the pKa is shifted ~2 units higher (9.0 ± 0.1 versus 6.8 for CAII).826,827 Importantly, the model compares even more favorably with CA mutants such as Thr199Ala in which the Thr residue that forms a hydrogen bond to the Zn(II)-bound solvent is replaced by Ala and as a result loses ~100-fold of its catalytic efficiency along with an increase in pKa of about 2 units.182,828,829 Therefore, it is reasonable to assume that the design of further secondary interactions similar to those found in CA is needed to bridge the differences between this design and the native enzyme (assuming it is possible in this alternate scaffold). One other designed Zn(II)N3 site (discussed in the previous section) catalyzes pNPA with a kcat/KM ~20-fold faster than [Hg(II)]S[Zn(II)(OH2/OH−)]N(TRIL9CL23H)3n+ and has a pKa of 8.2 ± 0.1.211 CA’s physiologically relevant reaction, CO2 hydration, was also examined, which yields an efficiency of ~1.8 × 105 M−1 s−1 (pH 9.5) that is within ~500-fold of that of CA and faster than any reported small molecule complex.213,830–837 There are no reports of CO2 hydration by other designed protein systems.
Figure 38.

Ribbon diagrams of the [Hg(II)]S[Zn(II)(H2O/OH−)]N(CSL9PenL23H)3n+ parallel 3SCC (one of two different 3-helix bundles present in the asymmetric unit) at pH 8.5. Shown are the main chain atoms represented as helical ribbons (cyan) and the Pen and His side chains in stick form (sulfur = yellow, nitrogen = blue, oxygen = red). (A) One of two trimers found in the asymmetric unit of the crystal structure; (B) a top down view of the structural trigonal thiolate site, Hg(II)S3, confirming the proposed structure of Hg(II) in Cys-containing TRI peptides.35 This metal site should mimic well the structural site in the metalloregulatory protein MerR.676 (C) A side view of the tetrahedral catalytic site, Zn(II)N3O, which closely mimics CA and matrix metalloproteinase active sites.838 All figures are shown with 2Fo – Fc electron density contoured at 1.5σ overlaid. Modified with permission from ref 213. Copyright 2012 Nature Publishing Group.
Figure 39.
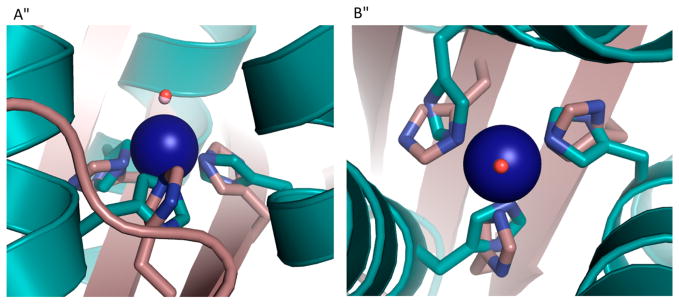
Overlay of the Zn(II)N3O site in [Hg(II)]S[Zn(II)(H2O/OH−)]N(CSL9PenL23H)3n+ with the active site of human CAII. [Hg(II)]S[Zn(II)(H2O/OH−)]N(CSL9PenL23H)3n+ is shown in cyan (PDB code: 3PBJ) and CAII in tan (PDB code: 2CBA). (A) Top-down view of the overlay with CAII. The solvent molecule associated with [Hg(II)]S[Zn(II)(H2O/OH−)]N(CSL9PenL23H)3n+ is shown in red, and that associated with CAII lies underneath. (B) Side-on view of the overlay with CAII. The model displays an excellent structural overlay for the first coordination sphere atoms with CAII; however, the orientation of the imidazoles differs between the two proteins. Another subtle difference is that the present structure has three ε amino nitrogens bound to the Zn(II) ion, whereas CAII has a mixed two ε and one δ coordination sphere. Overlay was performed manually in Pymol. Adapted with permission from ref 213. Copyright 2012 Nature Publishing Group.
Having demonstrated that a significant amount of hydrolytic activity can be achieved simply by removing an active site from its native scaffold and placing it into a 3SCC, the next major aim was to incorporate secondary interactions to modulate the properties of this site (activity, pKa, binding afinity). As mentioned above, the design of H-bonding channels so often found in native proteins is likely necessary. However, this advanced design will likely require changes to the location of the active site within the 3SCC to retain stability and potentially allow for improved substrate access. To this end, Pecoraro and co-workers recently reported the systematic variation of the Zn(II) active site along the TRI sequence.212 It is important to note that, although the properties of the Zn(II) site remain roughly the same in the absence of the Hg(II)S3 site, they chose to retain the structural site when moving the active site along the linear sequence to take advantage of the Hg(II)S3 chromophore’s utility as a spectroscopic tag to ensure that the oligomeric state (trimer) remained. The first variation in sequence involved switching the locations of the two different metal sites to generate [Zn(II)(OH2/OH−)]N[Hg-(II)]S(TRIL9HL23C)3n+. Assuming that the general orientation of the Zn(II) site remains constant (with the Zn(II)-bound solvent molecule oriented toward the N-terminus), it was expected and found that the substrate access was improved. This was further confirmed through inhibition studies using one of the products, acetate, and fitting the pNPA kinetic data for competitive inhibition. The KI’s for the Zn(II) sites in (TRIL2WL23H)3 and [Hg(II)]S(TRIL9CL23H)3− are similar (0.34 ± 0.01 and 0.32 ± 0.01 M, respectively), while that for [Hg(II)]S(TRIL9HL23C)3− is 0.20 ± 0.01 M.212 Despite the improved substrate access, however, the catalytic rate is decreased relative to the original model. Overall, the maximal catalytic efficiency is about the same as the original model with and without the structural site (24 ± 3 M−1 s−1). The second variation in sequence involves moving the His3 site from the 23rd position closer to the N-terminus of the coiled coil and into a d site to yield TRIL9CL19H. Analysis of the hydrolysis of pNPA by the metal-bound complex of this peptide showed that while maximal catalytic efficiency is approximately the same as for those previously described, the substrate access is slightly worse, while the maximal catalytic rate is improved. The most drastic difference with this sequence, however, is that the kinetic pKa shifted up to 9.6 (all of the others are approximately the same within error, ~9–9.2). It is unclear whether this is due to the altered orientation of the His ligands (from a site to d site) or simply the change in location. The KI of acetate inhibition for this model is approximately the same as for the L23H peptides, 0.36 ± 0.01 M. The apparent dissociation constants of Zn(II) to the His3 site for all of the above-described complexes vary from ~0.2 μM (L23H sites) to 0.4 μM (L19H) to 0.8 μM (L9H) at pH 9, demonstrating some location-dependence. Generally, these binding affinities indicate that the designs bind Zn(II) well and are in the same range as for similar designs with three binding ligands; however, they remain weak as compared to native Zn(II) enzymes (Kd ~ nanomolar to picomolar).19,31,182,759,839–846
In summary, despite drastic variation in the location of the active site along this 3SCC, significant catalytic efficiency for hydrolysis is retained, suggesting that this may represent the “baseline” activity in this type of model. Further, Pecoraro and co-workers have shown that location can be used to control binding affinity, pKa, catalytic rate, and substrate and solvent access (and selectivity). Although there are very few studies examining the effects of the protein matrix on the active site in this way,8 such a systematic approach is very important to future design endeavors, especially when deciding on the placement of the active site. As a whole, these studies demonstrated that replicating the exact protein fold of the native system was unnecessary to achieve a structural and functional model; however, maximal catalysis may still be critically dependent on the native protein structure as the incorporation of water channels, H-bonds for activation or stabilization of the substrate or intermediates, and H-bonds to metal coordinated amino acids may be best facilitated by β-sheet structure rather than α-helices.
3.3.1.3. Hydrolytic Phosphate Cleavage Catalyzed by De Novo Designed Metallonucleases
Many native nucleases contain metal ions such as Mg(II), Ca(II), Cu(II), and Zn(II),847–852 which enhance catalytic activity by acting as Lewis acid catalysts or activators for either the nucleophilic attack or the leaving group departure.847–850,853–862 Early attempts to incorporate a Zn(II) binding moiety in a peptidic scaffold to carry out hydrolytic phosphate cleavage reactions were performed by the Scrimin group.863,864 They installed 2 equiv of an azacrown functionalized amino acid, (S)-2-amino-3-[1-(1,4,7-triazacyclononane)]propanoic acid (ATANP), into a water-soluble heptapeptide chain that adopts a 310-helix structure. This peptide binds up to 2 equiv of Zn(II) at the azacrown sites, which cooperatively catalyze the cleavage of an RNA model substrate 2-hydroxypropyl-p-nitrophenol phosphate (HPNP) (Scheme 17, Figure 40). This represents the first example of a short, water-soluble 310-helix peptide with catalytic activity. Further studies involved binding other divalent metal ions such as Cu(II) and Ni(II) to the azacrown site of the same scaffold with minor adjustments to the amino acid side chains and examination of their reactivity toward HPNP cleavage. Although all three metal-bound peptides exhibit catalytic activity, the Zn(II)-peptide has the highest activity, more than 3-fold faster than the Ni(II)- and Cu(II)-peptides. It was shown that the dinuclear metallo-peptide complex with a 310-helix structure has higher activity than the smaller dipeptide without any secondary structure due to the proper positioning of the substrate. It was thought that only upon binding two metal ions 6.3 Å apart, as is the case occurring in a 310-helix peptide, could an HPNP molecule be properly activated and subsequently cleaved.864 With the incorporation of an interchelating moiety (an acridine ring) at the N-terminus for DNA binding, the dinuclear Zn(II)-peptide was able to perform hydrolytic cleavage of plasmid DNA pBR322.865
Scheme 17.866a.
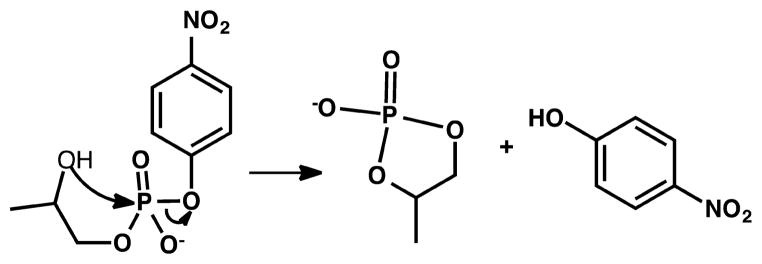
aReproduced with permission from ref 866. Copyright 2002 National Academy of Sciences.
Figure 40.
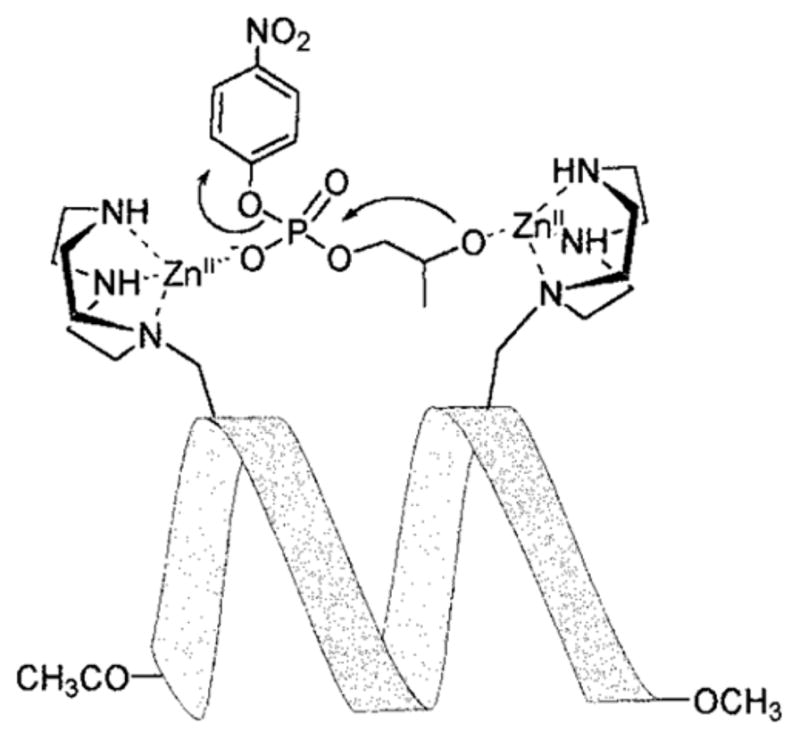
Proposed cleavage mechanism of HPNP. Reproduced with permission from ref 863. Copyright 1999 American Chemical Society.
On the basis of the same principle, Scarso et al. designed a peptide template T(P1)3 that is capable of binding 4 equiv of Zn(II).866 This peptide template consists of three copies of P1a, which is a designed 310-helix heptapeptide chain that has one azacrown Zn(II) binding moiety (Figure 41A), and a tris(2-aminoethyl)amine (Tren) moiety as a platform onto which three peptide strands were attached. Because Tren can bind 1 equiv of Zn(II) itself, the fully Zn(II)-bound complex contains 3 equiv of Zn(II) bound to the peptide and 1 equiv of Zn(II) at the Tren center (Figure 41B). This complex was able to carry out the transphosphorylation of HPNP (Scheme 17) with the highest rates when 4 equiv of Zn(II) were bound. In addition, allosteric control of Zn(II) binding to Tren was observed for T(P1)3, which led to the cooperative interactions between the Zn(II)-azacrown moieties, previously demonstrated to be critical to the rates of the cleavage reaction.863,864 Oligomeric RNA cleavage was also attempted with Zn(II)4(T-(P1)3), but proved to be less efficient than the more flexible apo-(T(P1)3).
Figure 41.

Molecular models of (A) P1a-Zn(II) complex top view and side view; and (B) Zn(II)4(T(P1)3) complex top view and side view. Reproduced with permission from ref 866. Copyright 2002 National Academy of Sciences.
The same Zn(II)(1,4,7-triazacyclononane) moiety was also integrated into a de novo designed helix–loop–helix (HLH) motif.867 The 42-residue peptides PR I–III were designed on the basis of the sequence of SA-42 and KO-42 that Baltzer and co-workers previously reported.868–870 PRI has two ATANP on each helical strand at the i/(i+4) positions, assuming the peptide adopts an α-helical conformation, yielding four Zn(II) binding sites. PRII has two ATANP on one helical strand at the i/(i+4) positions, and PRIIII has one ATANP on each strand. All three peptides adopt predicted HLH conformations at a neutral pH range. The apparent second-order rate constant of Zn(II)4-PRI toward HPNP cleavage was improved by 3-fold as compared to the first Zn(II)-peptide with the aridine ring (k2 = 0.16 M−1 s−1 for Zn(II)4-PRI and 0.05 M−1 s−1 for Zn(II)2-310).863,864 Zn(II)2-PRII and Zn(II)2-PRIII showed comparable second-order rate constants (k2 = 0.094 and 0.084 M−1 s−1, respectively), both smaller than that of Zn(II)4-PRI. Rossi et al. also showed cooperativity between at least two Zn(II) ions in the metal-loaded peptides. They proposed two possible pathways for the hydrolysis reaction to proceed coorperatively, both of which involve two Zn(II) ions acting together to interact with the substrate hydroxyl or Zn(II)-bound water molecules.867
3.3.2. De Novo Designed Electron Transfer Centers
3.3.2.1. Heme Electron Transfer Centers
The most progress in designing de novo proteins has come from working in α-helical scaffolds, which are known to form the secondary structures of numerous heme proteins in nature. This convenience, coupled with the wide array of accessible reactions catalyzed by heme, has made them the subject of study for those particularly interested in electron transfer, oxygenases, and peroxidases. As far back as 1989, amphipathic α-helices were used to construct a heme peptide capable of hydrolytic activity.871 Efforts quickly began to focus on imparting native-like folding and native-like activity to designed proteins. The collaborative effort between Dutton and DeGrado is noteworthy for its success in developing heme-binding “maquettes” with cooperative binding, physiologically relevant redox potentials, and conformational specificity. These efforts were focused initially on hemes involved in electron transfer, which are symmetric and have no open coordination sites, as de novo design worked to overcome the challenges posed by self-assembly and the rigorous symmetry this imposes. The progress in heme systems followed in lockstep with developments in de novo design as greater control was achieved in these systems. One of the first redox-active de novo heme constructs was published in 1994872 and progressed quickly with structural characterization and a demonstration of proton-coupled electron transfer properties.873 Investigations in this field mirrored those on native proteins as these designed constructs were examined on surfaces and electrodes, and in monolayers to explore their potential applications.874–880 In particular, a series of four helix bundles were designed with the goal of having them orient vectorally in a bilayer. These peptides were developed to study electron transfer between hemes in bilayers, and thus were designed to have hydrophobic and hydrophilic domains to orient and stabilize construct–bilayer interactions, termed amphiphilic membrane-associating redox maquettes (AP) redox maquettes.881 Several mutants of AP peptides were designed.880–883 AP0 and AP1 can bind porphryins in their hydrophilic portions,882,883 while AP2 and AP3 bind porphryins in their hydrophobic regions.880 AP0 and AP2 are capable of binding nonbiological cofactors containing extended π systems, and AP2 shows a preference for bacteriochlorophyll. Clearly, these investigations were driven by a desire to understand both soluble electron transfer proteins as well as membrane-bound electron transfer centers. The design of transmembrane proteins toward functional systems has been reviewed by Ghirlanda.884 A model of CcO, with both heme a and heme b cofactors, was reported by Gibney et al. and is capable of enzymatic reduction by NAD(P)H and subsequent oxidation by O2.885 Cytochrome bc1 was also successfully modeled, with an unusual perpendicular arrangement of the ligating His residues with respect to the hemes. This water-soluble model recapitulated the b subunit of cytochrome bc1 by mimicking the residues around the heme-binding sites and incorporating a Gly residue and a Thr residue to allow close contact of the heme with the backbone and H-bonding to fix the position of the His residue.886
More recently, these designs were combined to synthesize a transmembrane protein PRIME (porphyrins in membrane), which consists of four de novo designed helices arranged into a D2-symmetric bundle that binds to 2 equiv of non-natural iron diphenylporphyrin (Fe(III)DPP), whose planes are aligned almost parallel to the helical axis.261,887 PRIME binds to the cofactor Fe(III)DPP in phospholipid bilayer in the predicted stoichiometry as confirmed by the position of the Soret band monitored by UV–vis spectroscopy. EPR spectroscopy was used to investigate the structural details around the heme site and suggests that the angle between the His imidazole planes is greater than 60°, resulting in a “highly anisotropic low spin” iron EPR signal. A potentiometric titration showed two redox signals with apparent E1/2[Fe(III)Fe(III)/Fe(III)Fe(II)] and E1/2[Fe(III)Fe(II)/Fe(II)Fe(II)] of −97 (±3) mV and −168 (±3) mV vs NHE, respectively.261 This design brings us closer to the ultimate goal of creating artificial electron transfer centers that can shuttle electrons across a membrane. Photosynthesis and respiratory chains are important examples of the transmembrane electron transfer process. In these cases, directional electron flow is facilitated by changes in dielectric based on sequence differences going across the membrane. For this reason, a single chain version that can easily accommodate asymmetry was developed.888 The peptide PASC was prepared by taking a previous design (PATET), which was the same base as for PRIME (vide supra), and computationally linking it with loop regions and numerous designed H-bonds to orient the coordinating His residues. In addition to incorporating heme into this construct, a Zn(II)DPP version was also prepared889 because Zn(II)-porphyrins do not favor bis-His coordination. This resulted in a less symmetric construct [AHis:BThr: (DPP)Zn]. Surprisingly, while binding Zn(II)DPP, it did not bind FeDPP or Zn(II)PPIX. Because Zn(II)-porphyrins are emissive, one can use optical methods to probe the chromophore’s environment using steady-state emission, transient absorption, and fluoresence spectroscopies. The AHis:BThr:(DPP)Zn(II) has a normal excited-state singlet and triplet, but the latter has a 5 ms half-life. This triplet is significantly longer lived than that of Zn(II)-porphyrins exposed to water, proving that the porphyrin is embedded in the designed protein.
The de novo design and synthesis of multiheme proteins began with incorporating heme cofactors into four helix bundles. DeGrado’s group has described a few such heme-proteins that place heme iron ligands and hydrophobic contacts in the proper three-dimensional arrangement for binding.872,890,891 These studies laid a solid foundation for the future design of heme proteins, yet they have been extensively reviewed10 so we will not go into details here. Gibney and coworkers successfully demonstrated the incorporation of a bis-pyridine coordination to heme iron in a de novo designed protein environment. Upon introducing an unnatural amino acid 4-β-(pyridyl)-L-alanine (Pal) to a previously reported four-α-helical bundle [Δ7-H10I14I21]2 maquette and incorporating heme iron cofactor, they were able to evaluate the difference between bis-pyridine and bis-His coordinated heme iron.263 This particular unnatural amino acid increased the redox potential of the heme center by 287 mV as compared to the bis-His ligated counterpart.
With all of the success in designing electron transfer hemes, engineering asymmetry and open coordination spheres with either a displaceable water molecule or an open coordination site became a key challenge in de novo design. Early efforts succeeded in achieving a five-coordinate heme iron by introducing steric bulk on the His ligand in the form of 1-methylhistidine (H1m), yielding a ferrous heme center that coordinates to Nδ of His. The dimeric peptide [Δ7-H1m10I14I21]2 binds Fe(II)(protoporphyrin IX), generating a five-coordinate high-spin ferrous heme center, which is an excellent spectroscopic model for the deoxy state of Mb. This particular modification decreased the ferric heme affinity dramatically, resulting in a well-defined ferrous heme center that binds CO but not O2.264 The construct containing two 3-methyl-histidines, [ΔH3m]2, was used to examine the roles of the side chains on heme a, which differs from heme b by the presence of a C-2 hydroxyethylfarnesyl group and a C-8 formyl group.265 The affinity, spectroscopy, and electrochemistry were compared between heme b and a heme a mimic, diacetyldeuterioporphyrin IX (DADPIX). While the binding of the ferrous forms of the two porphryins is the same at both sites in the construct, there is a marked difference in binding affinity in the ferric state for the construct containing DADPIX, which is attributed to the +160 mV (vs NHE) shift in redox potential. Thus, the function of the electron-withdrawing groups on the porphyrin ring, such as the formyl group, is to raise the reduction potential by destabilizing the ferric form.265 This work was extended by incorporating heme a itself.892 The presence of the farnesyl tail on hemes a and o introduced hydrophobic interactions, which increased the binding affinity by 6.3 kcal mol−1 in both the ferrous and the ferric forms. The net effect of both the formyl group and the hydroxyethylfarne-syl is to raise the affinity of heme a relative to heme b in both the ferrous and the ferric forms by 2.1 and 6.3 kcal mol−1, respectively.892
A template-assembled synthetic protein (TASP) approach was employed to design a bis-heme binding helix bundle.893,894 TASP is a de novo design approach that entails a combinatorial assembly of peptides that are linked to a preorganized template.893–897 As a result, this strategy bypassed the entropically unfavorable nucleation step in the assembly of secondary structures, producing the desired peptidic scaffold for further functionalization.895–897 Bromoacetylated helical peptide chains Br–H1 and Br–H2 were synthesized and coupled onto a cyclic decapeptide template, forming a protein assembly Modular Protein 1 (Mop1). Four helices were implanted in an antiparallel manner onto the template, forming an ABAB arrangement. CD spectra of both the apo- and the holo-proteins showed characteristics of a helix bundle. Mop1 binds heme with a 1:2 stoichiometry, and the complex has a Soret band at 412.5 nm for the oxidized form and 426.5 nm for the reduced form. Two distinct midpoint reduction potentials were reported (−106 (±8) and −170 (±8) mV vs NHE), the difference between which was attributed to the poor equilibration by the redox mediators.893 This potential was significantly higher than the bis-His ligated single heme center in a helix bundle (−220 mV vs NHE).872 The two heme centers in Mop1, however, cannot be distinguished either from UV–vis absorption or from redox titrations.
Light-induced electron transfer was demonstrated in the Mop derivatives.267 A Ru(bpy)2Cl2 complex was assembled by coupling to a deprotected Cys residue on the helix surface. Mop2 and Mop3 were designed, with a Ru(bpy)2 complex attached to the Cys residue at the 16th and 13th positions, respectively (Figure 42). Rau et al. showed that the introduction of the ruthenium complex did not alter the absorption features of the heme-Mop assembly. The redox potentials of heme-Ru-Mop2 and heme-Ru-Mop3 were determined to be −170 and −164 mV (vs NHE), respectively. Electron transfer capability between the ruthenium complex and the heme center was demonstrated for both assemblies upon laser excitation. Specifically, the rate constants determined from a two-exponential decay for heme-Ru-Mop2 were 4.8 and 2.2 μs−1, comparable to those for Ru-Cyt c complex (6.2, 2.7 μs−1). Besides the two-exponential decay with rate constants of 9.5 and 1.9 μs−1, heme-Ru-Mop3 also had a fast component with a rate constant of 3.6 μs−1. The authors attributed the complex kinetics of heme-Ru-Mop3 to the different conformations of the ruthenium complex, one of which led to a shorter pathway corresponding to the faster portion of the electron transfer process.267 Taking a step further, Rau et al. attached the heme-protein assembly to a cellulose membrane.894 By modifying the membrane with β-Ala, to which a linker 3-maleimidopropionic acid (Mp) was attached, they introduced the Mops through the selective coupling of Mp and unprotected Cys residues on the cyclic template. A library of helices Ai and Bj was examined to ensure the antiparallel orientation of the helices. The midpoint redox potentials were estimated by the reduced fraction at −95 mV (vs NHE) by comparing the absorbance of the partially reduced form, fully oxidized form, and fully reduced form using the Nernst equation. No apparent trend between redox potentials and the amino acid residues was observed; however, it seemed that the more packed were the residues, the higher was the redox potential due to the exclusion of water molecules at the heme site.
Figure 42.
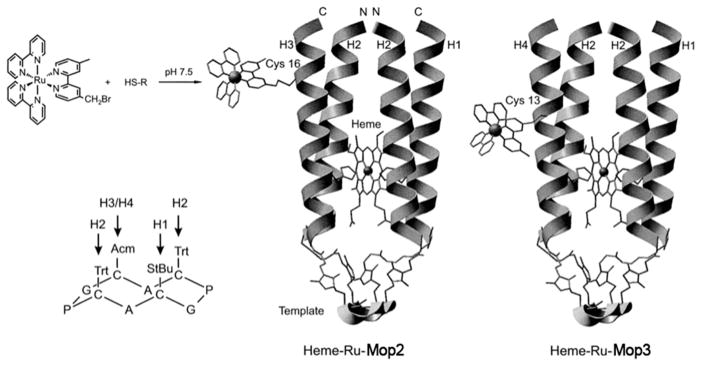
The cyclic template (left) and structural models of heme-Ru-Mop2 (middle) and heme-Ru-Mop3 (right). Reproduced with permission from ref 267. Copyright 1998 National Academy of Sciences.
3.3.2.2. Nonheme Electron Transfer Centers
3.3.2.2.1. Non-heme Iron Electron Transfer Centers
The ubiquity of iron–sulfur [Fe–S] clusters in proteins from all branches of life and their use in energetically interesting systems such at Photo-system I and hydrogenases have promoted the study of their chemical and biochemical properties.838,898–902 Biological centers comprised of iron and sulfur span from mononuclear Fe(S–Cys)4 (rubredoxin) sites, to [2Fe–2S], Rieske Fe–S clusters, [3Fe–4S], and [4Fe–4S] sites.903 While rubredoxin sites represent structurally the simplest type of site, they are not necessarily the easiest to study. [4Fe–4S] clusters can self-assemble in solution from inorganic sulfide and iron, and for this reason are thought to have comprised some of the earliest catalysts that were incorporated into primordial proteins in an anaerobic environment.838
An early attempt to use de novo principles to design a rubredoxin site was only marginally successful, as the Fe(III) form was not stable in aqueous solution and had to be measured in methanol instead.904 More recently, two designs that largely mimic the β-hairpin secondary structure around native rubredoxin’s metal site have been reported. One of the major challenges in designing a rubredoxin center is to create a construct that has sufficient stability in both oxidation states. Nanda et al. designed a C2-symmetric metal site inspired by the three strand sections of P. furiosis rubredoxin linked by a highly stable hairpin motif.905 This construct RM1 is capable of binding Zn(II), Co(II), and Fe(III) with 1:1 stoichiometry, and the UV–visible features of Zn(II)- and Fe(III)-RM1 are consistent with the mimetic and native rubredoxin systems. The midpoint redox potential is +55 mV (vs NHE), a little higher than the range observed for native rubredoxins (−90 to +50 mV vs NHE).906,907 The holo-RM1 is stable for 16 rounds of oxidation and reduction, representing the longest redox cycling of de novo designed rubredoxin mimic systems. Jacques et al. 908 recently reported a cyclic peptide model LZR for rubredoxin. The fold and spectroscopic features of rubredoxin are almost perfectly reproduced with the exception of the reduction potential, which is +140 mV (vs NHE), rather than the −90 to +50 mV906,907 generally found in native rubredoxins. This is likely due to the model’s small size and greater relative solvent accessibility.
There are two categories of [4Fe–4S] clusters: ferredoxin-type and HiPIP. Both are similar, but the protein fold and environment changes the redox potential drastically, with ferredoxin centers accessing the [4Fe–4S]2+/[4Fe–4S]+ couple at around −250 to −400 mV (vs NHE) and the HiPIPs accessing the [4Fe–4S]3+/[4Fe–4S]2+ couple at +150 to +350 mV (vs NHE).909 The first instance of a [4Fe–4S] cluster incorporated into a de novo designed heme protein was reported by Gibney et al.910 They designed and synthesized both a protein with a ferredoxin site (FdM, ferridoxin maquette) and one with a heme and a ferredoxin cluster (HLH-FdM, standing for HLH ferridoxin-heme maquette). UV–vis, CD, and EPR spectroscopy techniques were used to characterize the apo- and holo-peptides. It is worth mentioning that the HLH-FdM peptide, which has FdM incorporated into the tetra-α-helix bundle, binds hemin readily without perturbing the properties of the [4Fe–4S] cluster, indicating that these two redox centers behave independently. The reduction potential of the [4Fe–4S] cluster (−350 mV vs NHE) correlates with that of a typical ferredoxin cluster (−250 to −400 mV). This work, the HLH-FdM in particular, represents the first example of incorporating different functional redox cofactors in a single de novo designed protein, which heralds the further advancement in the protein design field toward multifunctional metalloenzymes.
Recently, small 16-amino acid peptide models were designed that mimic the [4Fe–4S] binding site of FA and FB of Photosystem I (PSI) by incorporating the cluster-binding residues and nearby active-site residues and were successful in recapitulating the more negative reduction potentials found in PSI.911 Antonkine and co-workers reported a redox potential for the iron-bound form of FA mimic (sequence: TEDCVGCKRCKPECPW) to be −440 (±30) mV and FB (sequence: YDTCIGCTQCKPECPW) to be −470(±30) mV (vs NHE), which correlated very well with the their native counterparts. A [4Fe–4S] center incorporated in a de novo designed α-helix bundle was previously shown to have a midpoint redox potential of −442 mV (at pH 8.2, vs NHE).912 They attributed the decreased redox potential as compared to the other mimetic system to the H-bond interactions of sulfur atoms in the cluster with the backbone amide protons, and the easy solvent accessibility of their models revealed by ESEEM and 2H ENDOR. Furthermore, they showed that these models can bind to the native P700-Fx core and participate in the light-induced electron transfer.
Further attempts have been made to incorporate [4Fe–4S] clusters into α-helical bundles,913 a non-natural fold for the site. Gryzb et al. reported the design of a [4Fe–4S] binding peptide CCIS (coiled coil iron sulfur protein) based on a computational approach. Several iterations were performed to increase the stability, helix propensity, and proper charge pairing after the first generation of the protein. UV–vis and EPR spectra for all of the variants of the holo-CCISs were indicative of the presence of a [4Fe–4S] cluster.
Most recently, Ghirlanda and co-workers reported a de novo design bis[4Fe–4S]-binding protein.914 The peptide DSD is a homodimeric three-helix bundle, with each monomer being a helical hairpin, in which one of the helices is about twice the length of the other. They incorporated a CXXCXXC…C motif in the monomer by replacing Leu with Cys so that three Cys residues from one helix and one Cys from the other helix can form a Cys4-binding pocket. Because the peptide has a pseudo 2-fold symmetry, 2 equiv of [4Fe–4S] were bound. The holo-protein showed typical iron–sulfur cluster spectroscopic features. The UV–vis absorption spectrum had absorption bands at 410, 324, and 280 nm for the oxidized state, while the reduced state lost the band at 415 nm. The CW EPR spectrum of the reduced form of the holo-protein showed g values of 1.879(gx), 1.943(gy), and 2.058( gz), similar to what was reported for native [4Fe–4S]+ clusters.915 The design of iron–sulfur clusters toward artificial hydrogenase for sustainable energy sources has been reviewed by Ghirlanda and coworkers.916
3.3.2.2.2. Copper Electron Transfer Centers
Copper electron transfer centers play important roles in enzymatic reactions. In native proteins, copper electron transfer centers are, in most cases, embedded in a Greek β-barrel fold (vide supra). By creating a copper electron transfer center in α-helical bundles, people can start to address the question of whether the β-sheets are necessary to facilitate electron transfer. In addition, the de novo design of copper electron transfer centers provides critical insight into how the geometric constraints influence the spectroscopy.
Schnepf et al. were the first to report a T1Cu center embedded in an antiparallel four-helix bundle using TASP.917,918 Topological templates on the surface were used to covalently attach peptide strands to a predetermined packing arrangement. One can incorporate an asymmetric metal-binding site, such as a T1Cu center, within the helix bundle that is rationally designed to accommodate such functionalities. The design of the helices was based on the backbone structure of the natural supercoiled four helix bundle Repressor of Primer (Rop) protein.919 The helices were attached to the template using similar chemistry as introduced in the previous section, forming an antiparallel helix bundle (Figure 43). A few modular proteins (Mop) variants were synthesized to explore the possibility of forming a T1Cu binding site. One of these variants, in which the copper binding site was buried in the hydrophobic core, exhibited stability toward Cys oxidation. UV–vis absorption spectra were collected for the Cu(II) complexes of Mop5, Mop6, and Mop7, revealing strong absorption bands at 410, 401, and 379 nm, respectively, indicating a tetragonal coordination geometry. The strong absorption features were assigned as a sulfur to copper charge transfer, which was further confirmed by rR spectroscopy. Meanwhile, the EPR hyperfine coupling constants of these Cu(II)-proteins were similar to what was reported for a type 1.5 copper center.337,428 On the basis of the first generation of de novo designed four-helix bundle copper proteins, Schnepf et al. further investigated the modification on the secondary coordination sphere and its influence on the overall stability, copper coordination, and spectroscopic properties.918 They synthesized a library of proteins mutated from Mop5, the most stable copper-binding protein in the first design cycle,917 and examined the copper-binding capabilities. The design of the second generation of proteins focused on modifying the secondary coordination sphere by introducing residues of differing polarity, bulkiness, and flexibility to achieve two goals: to stabilize the copper center further and to enforce the variation of copper coordination by changing the sterics around the copper binding site. Three types of copper centers were found in the initial screening step. Specifically, nonpolar residues such as Leu or a combination of Leu/Ala were placed above the His2Cys site to yield Mop23 and Mop21, respectively. In Mop22, a Met residue was introduced above both of the His ligands, providing a putative weak axial ligand as found in T1Cu centers. Further analyses of the absorption, EPR, rR, and MCD spectra of Cu(II)-Mop21, Cu(II)-Mop22, and Cu(II)-Mop23 suggested that Cu(II)-Mop21 contains a tetragonal Cu(II) center, whereas Cu(II)-Mop23 has a T1Cu center with a distorted tetrahedral geometry, which was attributed to the difference in the steric hindrance above the copper site. Interestingly, Mop22, with an additional Met substitution at Z13 position, was able to accommodate a purple CuA center, as revealed by various spectroscopic techniques. In particular, the complex displayed an absorption band at 774 nm, indicative of a Cu–Cu transition. The rR marker band at 345.5 cm−1 was in good agreement with a native CuA center,920–922 and a relatively small hyperfine coupling constant supported a delocalized unpaired electron at a binuclear site. The MCD spectrum of Cu(II)-Mop22 was also remarkably similar to that of the native CuA site469 (Table 5).
Figure 43.

Stepwise conversion of a linear peptide covalently attached to a cellulose membrane into a cyclic decapeptide that incorporates four antiparallel helical appendages. The fragment T4-Ah was synthesized on a resin loaded with a peptide amide linker. Synthesis steps: (i) Pd(0) in DCM, TBTU/DIEA in DMF; (ii) hydrazine in DMF; (iii) solid-phase synthesis; (iv) 19/1 TFA/DTT, HPLC; (v) phosphate buffer/acetonitrile (2/1), pH 8; (vi) DTT, pH 7.8, 10 equiv. of helix Bv, pH ≈ 8; (vii) Hg(OAc)2, pH 4.0, 10 equiv of helix Ck, pH ≈ 8, P(Ph)3 in n-propanol/buffer, pH 7.8. Reproduced with permission from ref 917. Copyright 2001 American Chemical Society.
Table 5.
De Novo Designed Proteins with Copper Electron Transfer Centers
| protein | LMCT λmax(ε/M−1 cm−1)/nm | Em°/mV vs NHE | bond length/Å | EPR g (A/G) | other notes | ref |
|---|---|---|---|---|---|---|
| Cu-Mop23 | 429, 571(1000) | 2.205* | gx = 2.040(16) | *the bond length is predicted from rR band, rR: 340, 355 cm−1 (λexcitation = 568 nm) | 918 | |
| gy = 2.052(14) | ||||||
| gz = 2.210(132) | ||||||
| Cu2-Mop22 | 347, 477(4040), 525, 637, 774 | gx = 2.010(19) | rR: 240, 306, 345.5, 395.5 cm−1 (λexcitation = 482 nm) | 918 | ||
| gy = 2.035(34) | ||||||
| gz = 2.155(32) | ||||||
| AM2C-Cu | 616 (~3600), 474; 616 (4100), 476 (Cl− bound) | 328 | N(His1)–Cu(II) 1.93 | gx,y = 2.065 | 923 | |
| N(His2)–Cu(II) 1.98 | gz = 2.250(≤ca. 16) | |||||
| S(Cys)–Cu(II) 2.30 | ||||||
| Cl–Cu(II) 2.66 | ||||||
| Bi-AM2C-Cu2 | 350(420), 488(1350), 530(1085), 849(695) | 2S–Cu(1.5) 2.21 | gx = 1.98 | 927 | ||
| N–Cu(1.5) 1.90 | gy = 2.04 | |||||
| Cu(1.5)–Cu(1.5) 2.51 | gz = 2.19 |
Also taking a de novo design approach, Tanaka and co-workers incorporated a His2Cys core motif in a four-stranded α-helical coiled coil scaffold to adopt a T1Cu site.923,924 Likewise, the amino acid sequence was based on a heptad approach, containing Lys and Glu residues to form electrostatic interactions and nonpolar residues such as Ala and Leu to establish a hydrophobic interior. The His2Cys residues replaced Leu residues in three of the four helices, forming a preorganized trigonal planar binding site.923 The apo-AM2C folds into an α-helical secondary structure in an aqueous environment, and the incorporation of copper ions does not perturb the secondary structure. The AM2C-copper complex exhibited a strong absorption feature at 616 nm, assigned to a sulfur(π)-to-copper LMCT, and a weak band at 474 nm, corresponding to a sulfur(σ)-to-copper LMCT.351,923 The hyperfine coupling constant of AM2C-copper complex was unusually small and was not resolved in the X-band EPR. Using CV, the reduction potential was determines to be +328 mV (vs NHE), a value that falls in the 180–800 mV range of T1Cu proteins.326,351 Moreover, XAS techniques were employed to gain further information of the copper coordination. The edge structures of Cu-AM2C were similar to those of the T1Cu center in Az, and the EXAFS fittings resulted in a 2.3 Å copper sulfur bond and a 2.66 Å copper chloride bond when chloride was added as an exogenous ligand. This coordination environment mimics the unusually short Cu–SCys and long axial Cu–SMet bond observed in native T1Cu centers.925,926 Further tuning of the copper coordination geometries by variation of the axial interactions was demonstrated by the same group.924 A gradual change of complex color from blue to green was achieved by choosing various exogenous ligands to bind to the axial position to the copper center. At the same time, the Ala residue right above the His2Cys site was substituted to a Glu or a Phe, resulting in two mutants AM2C-E5 and AM2C–F1, to modulate the interaction of the axial ligand with the copper center. The intention of introducing a negatively charged Glu resiude was to exclude anionic ligand binding to the copper center (Figure 44). As the strength of the axial ligand increased, the color of the solution changed from blue to green, indicative of the change of copper coordination from a trigonal planar to a tetrahedral-like geometry. This work beautifully demonstrates how to rationally tune the spectroscopic features of a T1Cu center by modulating the coordination geometry, particularly the strength of the axial ligand.
Figure 44.

Copper-AM2C variants. (A) Phosphate bound; (B) chloride bound; (C) azide bound; (D) imidazole bound; (E) AM2C-E1. Reproduced with permission from ref 924. Copyright 2012 Springer.
Using the same protein scaffold, Tanaka and co-workers modified certain residues to accommodate a CuA center.927 Similar to their T1Cu construct, the coordinating ligands in the new protein, bi-AM2C, included four His residues and two Cys residues as equatorial and bridging ligands, respectively, and were devoid of the axial (SMet and Oxx) ligands as found in native CuA proteins. The addition of excess Cu(II) into apo-bi-AM2C resulted in an intense purple color, with an absorption spectrum similar to that of the engineered purple copper center in Az.395 The EPR spectrum showed unresolved hyperfine splitting in the gz region. In addition, the EPR signal transformed into a mononuclear-like signal when the temperature was raised, indicating that the copper bound bi-AM2C has a binuclear copper center due to the short relaxation time of the binuclear center. Furthermore, XAS techniques, including both EXAFS and XANES, confirmed the CuA coordination environment of Cu2-bi-AMC2 possessed ligand–metal and metal–metal distances similar to those of the native CuA centers467 (Table 5).
3.3.3. Other Catalytic Centers
3.3.3.1. Heme Catalytic Centers
Catalytic heme proteins require a five-coordinate heme center. Pavone’s group has pioneered these types of hemes, particularly with their microperoxidase series. They designed a series of helix–heme–helix complexes named mimochromes, which feature the covalent attachment of a heme and the helical nature of the polypeptides and were based on the F helix of hemoglobin (Figure 45).268,928–930 Early studies focused on the design, synthesis, and spectroscopic characterization of the complex, in particular the stabilization of the helices and the control of the Δ ↔ Λ interconversion (Scheme 18, reproduced from ref 930 with permission) of the heme iron/cobalt center.928 It was demonstrated that Fe-mimochrome IV has a redox potential of −80 mV (vs NHE) at pH 7, which was also modulated by pH conditions.268 This pH-dependence of redox potentials was attributed to the association of the His residues with the heme iron center, replacing an iron-bound axial water, and the deprotonation equilibrium of the other axial iron-bound water to an iron-bound hydroxide. Fe-mimochrome VI contains a 14-residue peptide with a His coordinating residue at the sixth position and a 10-residue peptide without an Fe-coordinating ligand, creating an asymmetric iron heme center. Fe(III)-mimochrome VI showed catalytic activities toward the oxidation of several substrates with H2O2 as an oxidant.217 In particular, the catalytic efficiency of the oxidation of ABTS and guaiacol is comparable to that of native HRP. Additionally, it also catalyzes the conversion of phenol to nitrophenol in the presence of NO2− and H2O2. Mimochrome VI was then immobilized on a self-assembled monolayer (SAM)-coated gold electrode, and its electronic properties were studied with CV.931 Mimochrome VI showed a quasi-reversible one-electron process. In particular, varying scan rates revealed that there are two conformers of mimochrome VI that possessed a high potential (−106 mV vs NHE) and a low potential (−131 mV vs NHE), respectively, which were hypothesized to correspond to the Δ and Λ conformations of the iron heme cofactor based on an analysis of the Cotton effects of the heme Soret band by CD spectroscopy. To introduce more functionalities around the heme center, particularly H-bonding residues to the proximal His ligand and a distal Arg site, they moved on to use a four-helix bundle to accommodate the desired heme site. Most recently, Faiella et al. reported Fe-MP3 (MiniPeroxidase 3), which has an α2-heme-α2 motif, providing a functional catalyst for HRP activity.218 On the basis of the four-helix bundle, structural complexity was imparted by incorporating an Asp near the proximal His for H-bonding and a distal Arg for transition state stabilization. The Fe-MP3 exhibits a high turnover (kcat = 535 s−1), only 8-fold less than that of the native HRP, representing the first de novo designed four-helix bundle peptide with a covalently attached heme center that has HRP activity comparable to that of its native counterpart. Now, this peptide is being studied for applications in nanostructured detection devices.
Figure 45.

Schematic representation of the mimochrome chemical structures. Reproduced with permission from ref 268. Copyright 2003 Wiley.
Scheme 18. 930.
Taking a different approach, Hecht and co-workers designed a series of heme-binding four-helix bundles that exhibited peroxidase activities. On the basis of a binary patterning strategy, which refers to designing a stable polypeptide sequence by assigning the polar and nonpolar residues into binary codes while not specifying the identity of the side chains to create combinatorial diversity,932 they created a library of sequences and demonstrated that some of the polypeptides were capable of binding heme due to the presence of His and/ or Met residues.933 Detailed selection of the protein sequence and characterizations were reviewed previously.14,659,661 This strategy provides interesting insight into the design of novel sequences in general because the stable proteins generated from the binary codes are not subject to evolution or rational design, thus representing the most natural or “default” state. Peroxidase activities of a small library of heme proteins were screened by monitoring the oxidation of TMB by H2O2.934 Several proteins exhibited peroxidase activity, out of which protein n86 had the highest activity with a turnover frequency of 17 000 min−1, which was still much lower than that of the native HRP (~60 000 min−1).935 These heme proteins were also able to bind small gas molecules such as CO, exhibiting spectroscopic features similar to those from the native heme-containing proteins with a narrower range of the spectroscopic parameters, which was thought to be the “default” starting point of a heme center.936 At the same time, the midpoint redox potentials were measured for these heme proteins by redox titrations using various redox mediators.937 These potentials range from −112 to −174 mV, which again was an unbiased assessment of the default reduction potential of a heme protein.
One protein, S824, a 102-residue protein designed to fold into a four-helix bundle, was engineered with a Gly-Gly-Cys linker at the C-terminal end, leading to S824C. S824C was immobilized onto a maleimide-functionalized gold electrode for electrochemical studies.269 The redox potential was found to be pH- and ionic strength-dependent. Because the heme iron has a displaceable axial ligand,937 they investigated the influence of binding an exogenous ligand on the redox potential of the heme/S824C. The binding of imidazole and its derivatives decreased the reduction potential, while the addition of pyridine and its derivatives increased the potential for both heme/S824C and an immobilized isolated heme. These two types of heme centers, however, responded to the ligand binding to a different extent, which was attributed to the partial burial of the heme center in the heme/S824C assembly. Moreover, the sterics of the exogenous ligand also influenced the binding affinities of the heme iron, thus modulating the redox potential of the system.269 Das et al. also showed that a series of these immobilized de novo designed heme proteins exhibit peroxidase activity, with protein n86 possessing the highest activity, one-half that of the native HRP.938
Various enzymatic activity screenings were performed on a family of unevolved binary patterning generated proteins. While some of the apo-proteins had esterase and lipase activities in solution,939,940 80% of the heme-bound protein exhibited HRP activity in solution, several of which showed 105–106-fold enhancement in rates as compared to the background reaction.940 Taking evolutionary principles from nature, directed molecular evolution was performed on two previously characterized proteins S824 and S836 to achieve a higher peroxidase activity.941 Random mutations were carried out, and the resulting mutants were screened for peroxidase activity. While a large percentage of the mutants were deleterious in either the folding or the enzymatic activity, there were a few mutants that had higher activities than the parent proteins. In particular, two variants 1-2B11 and 2-2H12 had nearly 3-fold higher catalytic efficiency than the parent protein S836. What distinguishes this from other directed evolutional designs is that the starting point of the evolution was a novel sequence that was generated simply on the basis of polar–nonpolar interactions of the side chains, instead of a native protein. One can think of this approach as an evolutionary paradigm that starts from a randomly assembled stable sequence and evolves gradually to achieve desired functional constructs.
In 2010, Koder et al. incorporated many lessons learned over the previous 20 years to modify bis-His ligated heme bundles to promote oxygen binding.942 The authors describe a four-step plan for the design and optimization of their constructs beginning with the assembly of a simple, robust, and generic construct. This construct was then modified to introduce cofactor-binding amino acids, improve the structural resolution, and then fine-tuned by iterative testing and redesign. They introduced strain at the distal His by introducing three Glu residues to exclude binding to the heme center and create an entatic state, which favors exogenous ligand binding. Further designs to exclude water from the active site enabled the stabilization of the oxy-ferrous form of heme. The resulting highly successful HP-7 construct is capable of binding oxygen with affinities on time scales that mirror native globins, except that for HP-7, the oxygen binding is tighter than carbon monoxide binding. The mechanism of heme binding to HP-7 has also been reported.266 It was shown that a three-state model that involves both a pentacoordinate heme and a hexacoordi-nate heme accurately describes how the entatic state of HP-7 contributes to its function (Figure 46). When the Glu residues, which were introduced to preclude distal His binding, were mutated to Ala residues, the distal His showed a higher affinity for the heme iron. The free heme is first bound as pentacoordinate and then as hexacoordinate; however, the overall heme binding affinity is lower in the mutants, which is ascribed to a lower level of pentacoordinate heme interaction.
Figure 46.

Proposed mechanism of heme binding to HP-7. Reproduced with permission from ref 266. Copyright 2011 American Chemical Society.
In the context of heme protein engineering, these works beautifully manifest the strength of de novo design in modulating the redox potentials and reactivities of a particular system. Especially for designs with unnatural amino acids, it is relatively easy to incorporate such functionalities in solid-phase peptide synthesis for relatively short sequences, while it might be challenging to engineer them into an existing protein system. The difference between the heme-coordinating ligands is exhibited noticeably from the variation of redox potentials and catalytic activities (Table 2). These efforts provide interesting insight into the design of heme systems as well as the general strategies of tuning the properties of an engineered redox-active metalloprotein.
3.3.3.2. Type 2 Copper Centers
(A review on de novo designed copper-peptides was recently accepted, see ref 1004.) Copper enzymes are involved in many important metabolic pathways and are among the most efficient biological redox catalysts.740,943–945 In particular, redox-active copper centers that are involved in catalysis are ideal targets for de novo metalloprotein design. The study of native copper redox proteins does not necessarily reveal the subtle interplay between the protein dielectrics, H-bonding interactions, and van der Waals contacts in the protein environment due to their structural complexity. Synthetic models, on the other hand, lack proper dielectrics to associate with biologically relevant redox potentials. De novo designed systems with catalytic copper centers play a unique role in understanding the structure–function relationship in their native counterparts. Not only have researchers obtained stable functional catalytic copper centers using de novo design strategies, they have also investigated the modulation of redox properties and activities. One of the challenges to obtain a stable catalytic copper center is to design a peptide with a coordination environment suitable to stabilize copper on both oxidation levels. The coordination geometry of Cu(I) is usually linear, trigonal (T-shaped), or tetrahedral, while for Cu(II), distorted square planar, tetrahedral, pyramidal, and octahedral geometries are commonly observed.946 However, in copper proteins, coordination numbers of four or higher (involving weak interactions between the copper ion and distal ligands) are prevalent.947 At the same time, the two principal oxidation states (+1 and +2) of copper have different hard–soft behaviors: while Cu(I) is a soft metal, Cu(II) has a more intermediate-to-hard character. The general strategy to design a defined copper binding site is to mutate the hydrophobic residues into coordinating residues such as His, Met, Cys, Asp, or Glu in the interior of the two- to four-stranded coiled coils. T2Cu centers, in particular Cu(His)3 centers, play multiple roles in native proteins. Their functions span from electron transfer to cofactor-assisted oxygen activation to nitrite reduction.742,948–955 For example, in peptidylglycine α-hydroxylating monooxygenase (PHM), the CuH center [Cu(His)3] is involved in shuttling electrons to a CuM copper center 12.5 Å away where oxygen activation occurs.950,956,957
The first case of de novo designed 3SCCs with a T2Cu center CuHis3 was reported in 1993.792 The construct consists of a 19-amino acid peptide based on the heptad repeat approach. It contains a 4,4′-bpy moiety at the N-terminus, a His residue for Cu(II) binding at the C-terminus, and a Tyr residue adjacent to His as a spectroscopic probe (sequence: 4,4′-bpy-GELAQKLEQALQKLAAAHYNH2).792 When reacted with Ru5Cl122−, three helices came together, forming a homotrimeric α-helix bundle from the template created by the fac-2− unit at the N-terminus.791,793 The presence of [Ru(bpyPep)3] this stable Ru(II)(bpyPep)3 unit assisted the formation of an α-helix apo-(peptide)3 construct, which contains a preorganized (His)3 site at the C-terminus available for copper binding. Indeed, the addition of Cu(II) to the Ru(II)(peptide)3 led to Cu(II)Ru(II)(peptide)3 with a dissociation constant of ca. 3 × 10−7 M determined by the change of Tyr emission. Ru(II)-(peptide)3 possessed a high degree of helical content; the addition of Cu(II) to the Ru(II)(peptide)3 did not change the CD spectrum even when Cu(II) was 20-fold in excess compared to Ru(II)(peptide)3. Spectroscopic studies revealed that the Cu(II)Ru(II)(peptide)3 complex has a ligand-field band centered at 495 nm (ε = 300 M−1 cm−1) monitored by UV–vis spectroscopy and an A// value of 173 G in the EPR spectrum, fully consistent with the presence of a tetrahedral or square planar T2Cu site in a (His)3 environment.792 This work represents the first example of a stable T2Cu center in a de novo designed α-helical coiled coil scaffold, laying a solid foundation for the development of catalytic T2Cu centers.
CuNiR, expressed in algae and fungi, promotes a one-electron reduction of nitrite (NO2−) to nitric oxide (NO) at its T2Cu center, where copper is coordinated to three His and a water molecule.958 A de novo designed structural and functional model for CuNiR was recently reported by Pecoraro and co-workers, representing the first stable Cu(His)3 center that exhibits NiR activity in a peptidic environment in aqueous solutions.214 This copper peptide was designed on the basis of the TRI family peptides previously introduced (section 3.2.1: Heavy Metal Toxicity). The Leu23His mutation leads to TRIL23H (Table 4) with His as the copper-binding residue. The Zn(II)-bound form of (TRIL23H)3 was demonstrated to be a very efficient structural and functional model of CA in aqueous solutions.212,213 Because Zn(II) is isoelectronic to Cu(I), the same peptide construct was used to obtain a stable Cu(I)(TRIL23H)3+ peptide with a Cu(I)(His)3 coordination. A comparison between the model of Cu(I)(TRIL23H)3+ peptide based on the Zn(II)(His)3(H2O/OH−) site and the T2Cu center of NiR reveals remarkable structural similarities (Figure 47).213,214,958 A Trp residue was introduced at the second position as a spectroscopic tag in addition to the His mutation, leading to TRIL2WL23H. Apo-(TRIL23H)3 (and apo-(TRIL2WL23H)3) binds both Cu(II) (above pH 5.8) and Cu(I) (above pH 4.5), forming Cu(II)(TRI(L2W)L23H)32+ and Cu(I)(TRI(L2W)L23H)3+, respectively. In both oxidation states, copper is bound to three imidazoles.214 Characterizations of both oxidation states were carried out, delineating the structural details of the copper center. The Cu(II)-bound peptide showed typical spectroscopic features of a T2Cu center, with a weak visible ligand field absorption at ca. 640 nm, and an A// value of ca. 186 G in the EPR spectrum, consistent with a Cu(II)(His)3(OH2)1–2 coordination with three imidazoles on the quasi-equatorial plane. XAS data indicates that Cu(I) exists in a trigonal planar environment, coordinated by three imidazoles from His23. This stoichiometry was confirmed by a titration of Cu(I) into apo-3SCCs monitored by 1H NMR. Copper ions of both oxidation states bind to the apo-3SCCs with relatively high affinities, with dissociation constants in the picomolar range for Cu(I) and micromolar to nanomolar range for Cu(II) at different pH conditions. The redox potentials of the copper center, calculated based on the Kd values, are in 400–500 mV (vs NHE) range at pH 5.9 (dependent on the counteranion used) and ca. 430 mV (vs NHE) at pH 7.4, higher than those reported for the T2Cu centers in NiR (218 mV at pH 6.0 and 137 mV at pH 8.4 for R. sphaeroides,958 220–310 mV at pH 7.0 for A. cycloclastes and A. xylosoxidans,959 vs NHE). It was suggested that the high redox potentials for the Cu(TRI(L2W)L23H)3n+ peptides originate from the strong stabilizing effect of the trigonal planar structure of the Cu(I)(His)3 site within the coiled coils and the high energy barrier associated with the redox process due to the change of the coordination number of the copper center.214
Figure 47.

(A) A model of Cu(TRIL23H)32+/+ based on the crystal structure of Hg(II)SZn(II)N(CSL19PenL23H)3+ (PDB code: 3PBJ213). Overlay of the Zn(II)(His)3(OH2/OH−) site in Hg(II)SZn-(II)N(CSL19PenL23H)3+ (protein ligands: dark blue; zinc: dark blue sphere; zinc-bound water: red sphere) and the T1Cu center in CuNiR from R. sphaeroides (PDB code: 2DY2.958 Protein ligands: light green and teal; copper: cyan sphere; copper-bound water: light pink sphere). (B) Top view. (C) Side view. Adapted with permission from 214. Copyright 2012 National Academy of Sciences.
The reduction of Cu(II)(TRIL23H)32+ into Cu(I)-(TRIL23H)3+ occurred when sodium ascorbate (E° ca. 100 mV, two-electron donor) was added as a reductant in the pH range 5.8–7.4.214 The reduction monitored by the disappearance of the ligand field band of the copper center was quantitative with 0.5 mol of ascorbate per mol of Cu(II)-(TRIL23H)32+, and it occurred within mixing time. Cu(I)-(TRIL23H)3+ could be reoxidized using nitrite, as expected on the basis of the formal E° of the latter (ca. 1.3 V at pH 6.0).955 The reoxidation of the Cu(I)(His)3 site was relatively slow (45–60 min), which could be tracked by monitoring the reappearance of the ligand field band of Cu(II)(TRI L23H)32+ upon the addition of 1 equiv of nitrite to a Cu(I)(TRIL23H)3+ solution. More importantly, the reduction of nitrite is a one-electron process, which only produces NO, with a negligible amount of N2O.214
The reaction Cu(I)(TRIL23H)3+ + NO2− + 2H+ ⇋ Cu(II)(TRIL23H)32+ + NO + H2O was studied as a function of the pH under catalytic (i.e., substoichiometric) conditions.214 The rates of nitrite reduction under catalytic conditions increased as pH decreased. Most importantly, the reduction of nitrite was observed for substoichiometric quantities of Cu(II)(TRIL23H)32+ with respect to both the oxidant (ca. 160-fold excess relative to Cu(II)) and the sacrificial reductant (ascorbate, ca. 13-fold equiv per Cu(II)). Under these conditions, Cu(II)(TRIL23H)32+ exhibits NiR activity corresponding to up to 5 turnovers in 3.7 h at pH 5.8. Although the NiR activity of Cu(II)(TRIL23H)32+ is significantly lower than that of the native NiR (ca. 1500 s−1 at pH 5.8 for Alcaligenes faecalis NiR with Pseudoazurin as the electron donor960), the Cu(II)(TRIL23H)32+ construct is an effective functional NiR model in aqueous conditions.
One should be cautious that the mechanism of this model may differ from that of the native CuNiR because the oxidation states for copper during turnover conditions are not the same. In CuNiR, nitrite binds to Cu(II) followed by electron transfer, which initiates the reduction of the substrate.961–963 Under turnover conditions in this designed system, however, the rapid reduction of Cu(II) by ascorbate ensures that the resting form of the enzyme is Cu(I). Thus, the interaction of nitrite with the reduced Cu(I) likely initiates catalysis. On the other hand, in the synthetic small molecule studies, it is usually observed that nitrite binds Cu(II) via the oxygen atoms in a bidentate mode, while it prefers binding Cu(I) via the nitrogen atom.964–967 If this is true for this de novo designed system, the actual chemical conversion may not follow the same pathway as that in the native NiR. Nonetheless, these alternate reaction schemes suggest that the de novo designed Cu(II)/(I)(TRIL23H)32+/+ may provide a unique approach to evaluate both types of chemistry within the same framework. Thus, one may evaluate nitrite reduction from the cuprous form using existing protocols, but probe the chemistry of nitrite reduction from the cupric enzyme by photoinducing electron transfer after the nitrite has bound to Cu(II).
An extensive body of work for the synthesis of model complexes of NiR (e.g., using tetrahedral-enforcing ligands such as scorpionates) has been reported in the literature in the past two decades.964,966,968–971 Cu(II)(TRIL23H)32+ is, however, the first de novo designed metallopeptide for which the NiR reactivity is observed (Table 1). It represents an advanced model of NiR with a stable and functional Cu(His)3 site in an aqueous solution. The robustness and product specificity of this model solidifies its status as an excellent starting point for NiR model chemistry in de novo designed systems.
Despite its importance in redox-active processes such as electron transfer and catalyzing oxidation/reduction of small molecules, systematic studies of the factors that govern the properties of a Cu(His)3 site are scarce. Yu et al. reported the first systematic modulation of the redox properties and activities by changing the charged residues on the second coordination sphere of a Cu(His)3 center in a stepwise manner.972 In the native redox-active proteins, it is often the case that one change around the active site would lead to a cascade of events that results in the modulation of the redox properties; however, it is challenging to track the specific role of each event. A de novo design strategy provides a simplified functional model, an ideal system to study the influence of different factors that govern the redox-properties and related activities. Based on the same scaffold TRI-H (TRI-H = TRIL2WL23H), the positively charged residue Lys at the 22nd position was substituted to a Glu, resulting in a difference of −6 in charge as compared to the parent peptide (assuming that the Lys is fully protonated and the Glu is fully deprotonated). Taking TRI-EH (TRI-EH = TRIL2WK22EL23H) as the basic scaffold, modifications were made to the charged residues at the 24th and 27th positions to yield a series of peptides with charge 0, −3, −6, −9, and −12 different from TRI-H (Table 4, Figure 48). This series of designed peptides aims to understand how the local electrostatics influences the redox properties and reactivities of the T2Cu center Cu(His)3.
Figure 48.

PyMOL models of copper-peptides based on the crystal structure of Hg(II)SZn(II)N(CSL19PenL23H)3+ (PDB code: 3PBJ).213 (A) TRI-H; (B) TRI-HK22Q; (C) TRI-EH; (D) TRI-EHE27K; (E) TRI-EHE27Q; (F) TRI-EHK24Q; (G) TRI-EHK24E. Reproduced with permission from ref 972. Copyright 2013 American Chemical Society.
Similar to the parent peptide TRI-H, all peptides in the TRI-EH series bind both Cu(I) and Cu(II) with relatively high affinities, mimicking well the coordination of the T2Cu center in the native CuNiR. The modifications changed the peptide matrix around the copper center from a relatively positive environment to a relatively negative one (in the order of peptides D, E, C, F, G in Figure 48), leading to a direct impact on the protonation equilibra of the Cu(II)-peptides. Specifically, for Cu(II)(TRI-EH)32+ and Cu(II)(TRI-EHK24E)32+, as the pH was raised, an additional deprotonation process was observed as compared to Cu(II)(TRI-H)32+, which was attributed to the deprotonation of Glu22, forming a better H-bond acceptor. The interaction among residues on the 22nd, 24th, and 27th positions was thought to be an important factor that changes the H-bonding scheme around the Cu(His)3 center. For TRI-EHE27K, with the most positive charge in the series, Glu22 can form an H-bond with His23 (or a water bridging the two residues); meanwhile, Lys27 can form an interhelical salt-bridge with Glu22, weakening the H-bond interaction between Glu22 and His23 (or a water bridging the two residues). The other extreme is TRI-EHK24E with the largest negative charge. In this peptide, the putative H-bond between Glu22 and His23 (or a water molecule bridging the two residues) does not experience perturbations from other neighboring residues besides charge repulsion. Between these two situations is TRI-EH, which can have the H-bonding interaction between Glu22 and His23 (or a water bridging the two residues), together with interhelical salt-bridging interactions. These interactions are postulated to be the reason for the change of the protonation equilibra of Cu(II)-peptides. They can also lead to changes of Cu(II) affinities, structural perturbations of the Cu(I)(His)3 site, and differences of Cu(I) affinities at a particular pH across the series.
Based on the affinities of both Cu(I) and Cu(II) to the peptides, redox potentials were calculated from the Nernst equation. Similar to the previously reported construct Cu(II)/ (I)(TRI-H)32+/+, the redox potentials of these peptides are in the range of 400–600 mV, higher than those reported for the native T2Cu center in CuNiR,958 probably due to the high stability of the reduced-form of the peptide. The calculated potentials correlate very well with the charge mutations around the active site, forming two linear trends at pH 5.8 and pH 7.4, the distance between which is 100 mV. As a result, the mutation of the charged residues led to a ~200 mV variance in redox potential under different pH conditions. Furthermore, modulation of NiR activity was demonstrated. The more positively charged peptides, Cu(TRI-EHE27Q)3 and Cu(TRI-EHE27K)3 with higher redox potentials, exhibited lower rates than Cu(TRI-H)3, while the more negatively charged peptides with lower redox potentials showed higher rates. Similar to the parent peptide Cu(TRI-H)3, the rates of all of the mutated peptides were pH-dependent, which was associated with a single-proton rate-determining step. By changing the charged residues in a stepwise fashion, the NiR rates can be tuned by 4-fold at pH 5.8.
As reviewed previously, the systematic tuning of potentials of a specific redox site has been extensively studied for both heme and cupredoxin centers; however, similar investigations on T2Cu centers are lacking despite their functional importance and versatility in the native systems. This work represents the first systematic modulation of the second coordination sphere charged residues, leading to varied pH profiles, copper afinities, redox potentials, and NiR rates, demonstrating the critical role of ionizable residues in tuning the functional properties of a redox-active center, which brings us one step closer toward the goal of improving catalysis of this designed system.
3.3.3.3. A Di-iron Peptide: Due Ferri
DeGrado and coworkers have used retrostructural analysis of the active sites of several carboxylate-bridged di-iron proteins to design a family of artificial metalloproteins as models for enzymes such as RnR and bacterioferritin.973–978 Despite a low sequence identity (<5%) and overall complex protein folds, the active sites of these proteins are all found within a simple antiparallel D2-symmetric four-helix bundle.975 The Due Ferri (two-iron, DF) proteins, designed to understand how solvent accessibility, polarity, and electrostatic environment influence the properties, are comprised of a binuclear metal site with two chelating Glu residues, two His residues, and two bridging Glu residues. These primary ligands are buried in the interior of the scaffold with metal geometries stabilized by a network of hydrogen bonds to second-shell ligands. The parent model, DF1, is an antiparallel dimer of noncovalently associated HLH motifs, each 48 residues in length.975 The design was approached by first carefully defining the above-described dimetal coordinating site and second shell residues and then incorporating mainly hydrophobic core residues for packing and hydrophilic interfacial residues to define the topology. The design was first structurally characterized as a di-Zn(II) complex (but also binds Fe(II) and Co(II) in solution), confirming the presence of the dimetal cofactor near the center of the dimer.975 The subsequent apo-DF1 NMR structure demonstrated that the metal site is largely preorganized.979 Each metal is five-coordinate with a sixth vacant site lying on adjoining faces of the two metal ions to provide a potential binding site for small molecules. Complete characterization of DF1 was not possible due to low solubility and extreme stabilization (the scaffold had to be unfolded and refolded to bind metal). Further, Leu residues 13 and 13′ were found to block dimetal site access. DF2 was subsequently designed to increase solubility (more surface hydrophilic residues were incorporated, and, in a similar model DF2t, a longer interhelix loop was introduced for stabilization and minimization of aggregation) and to allow for access of small molecules to the metal site (by replacing Leu residues with Ala and Gly).976,978–982 The crystal structures of di-Mn(II)-L13A-DF1976 and di-Mn(II)-L13G-DF1981 confirm the presence of the designed cavity (which increases in size going from the larger Ala to Gly) filled with ordered water molecules. Notably, the asymmetric unit of di-Mn(II)-L13G-DF1 has four independent dimers, three of which have bridging water molecules and intermetal distances of 3.6 Å and one with two terminal water molecules and an intermetal distance of 4.2 Å.981 Superimposition of the structures revealed that two copies of the N-terminal helices have shifted in opposite directions, leading to the increased metal–metal bond length. Dubbed the sliding-helix mechanism, it is postulated that this mechanism could occur in native systems for signal transduction, but it would be difficult to observe this effect experimentally.
Having developed a construct with improved properties and a larger active site cavity, a combinatorial approach was undertaken to search for a functional assembly. To this end, DFtet, a noncovalent assembly of four separate helices that could easily be varied, was designed.983 One of the resulting heterotetramers, DFtet A2B2,983 with all Gly residues replacing the Leu and Ala residues in the active site cavity (G4-DFtet), could perform the rapid two-electron oxidation of 4-amino-phenol to benzoquinone monoamine with a kcat/KM of 25.7 M−1 s−1 and a rate enhancement of ~1000-fold relative to the background reaction (and no detectable intermediates).216 Further, this de novo design approach also demonstrates that the activity is sensitive to changes of the size of a methyl group in the active site cavity (substituting Gly for Ala decreased the rate up to 5-fold). The diferrous form of another resulting heterotetramer, DFtet AA′B2, reacts with oxygen to form a peroxo-bridged diferric species similar to an important reactive intermediate found in many native proteins.984 Given difficulties involving complex stoichiometry and unwanted ligand-exchange reactions with G4-DFtet, DF3 was designed on the basis of the original DF1 sequence with all Gly residues around the active site to accommodate the substrate and with modified loop residues to stabilize the resulting fold.215 Like DFtet, DF3 is a phenol oxidase for the substrates 4-aminophenol (kcat/KM of 23 M−1 s−1), 3,5-di-tert-butyl-catechol (3,5-DTBC, kcat/KM of 100 M−1 s−1), and p-phenylenediamine (kcat/KM of 14 M−1 s−1) (Table 1). No phenol oxidase activity was observed for o-phenylenediamine. These results are consistent with crystallographic analysis of the size of the cavity. The active site cleft created by the Gly mutations, at its narrowest, matched the width of the phenyl ring and, in wider sections, substitutions at the 3 and 5 positions could be accommodated. A single chain DF protein, DFsc (with Ala residues around the active site), was also designed, and the NMR structure of di-Zn(II)-DFsc was obtained.985,986 While rapid oxidation of the diferrous center was observed, it was due to an off-pathway iron-tyrosinate complex and not the important diferric intermediate observed for the other diferrous DF proteins.987,988 Substitution of the four Ala residues to Gly (G4-DFsc) led to the minimization of this complex and a scaffold, which could also catalyze 4-aminophenol oxidation on the same order of magnitude as DFsc was obtained.989 Most recently, the DeGrado group has shown that a single mutation (from Ile) provides an additional coordinating His residue at the dimetal center of G4-DFsc and results in a protein that can now catalyze the N-hydroxylation of arylamines such as p-anisidine, an activity not detected for G4-DFsc.989 Only three additional mutations outside of the coordination sphere were made to counterbalance the steric clashes and unfavorable electrostatic interactions introduced by the His residue. The design does not fold well in the absence of metal, but does adopt a fold similar to those of the other DF proteins in the presence of divalent metals. It is particularly striking that addition of a single metal-coordinating residue can result in complete loss of one activity, phenol oxidation, and generate another, N-hydroxylation. This work is an excellent example of how de novo design can be utilized to directly correlate structure with function by removing much of the complexity of native proteins.
3.3.3.4. Dirhodium Peptides
Combining nonbiological metal catalysts with polypeptides can generate new function while taking advantage of the natural chirality provided by amino acids. Ball and co-workers have developed an arsenal of dirhodium(II)–peptide complexes that react with enzyme-like selectivity.785 Dirhodium(II) complexes are known to catalyze a number of reactions and provide unique differentiated coordination environments. Under biological conditions, the strong equatorial ligands ensure stable metallopeptide complexes, while the kinetically inert axial ligands provide the open coordination sites necessary for catalytic activity.785
Having established the ability of dirhodium complexation to control secondary peptide structure,990,991 Ball and co-workers explored the catalytic activity of dirhodium–metallopeptides. Twenty-two metallopeptides that bind dirhodium(II) with carboxylate residues in the i and i+4 positions were screened as catalysts for the insertion of PhMe2SiH into methyl α-diazophenylacetate (Figure 49).992 Computations suggest, and the peptide library confirms, that mutations to positions i–1 and i+3 have the largest effect on enantioselectivity. At the i+3 position, sterically bulky ligand residues gave the best enantioselectivity, but the basis for selectivity at the i–1 position is not clear. The best catalyst, Rh2L212-isoB (L21 = KNDAAIDAK), produced silane in 92% ee and was further screened against 10 α-diazoesters, with the best enantioselec-tivities resulting from meta- and para-substituted substituents (90–99% ee). Pyrene excimer fluorescence identified Rh2L212-isoB as having antiparallel helices.993 Enantioselectivities from the catalyzed insertion of PhMe2SiH into methyl α-diazophenylacetate were improved by 5–18% for four Cbz-protected peptide sequences upon addition of triphenylphos-phite.994
Figure 49.
Tube and space-filling model for a dirhodium center bound to L12. Reproduced with permission from ref 992. Copyright 2010 American Chemical Society.
In the above enantioselective silane insertion reactions, the same enantiomer is always produced in excess. Accessing both enantiomers of a product using naturally derived chirality is challenging, necessitating the development of high-throughput screening methods.995,996 Utilizing the silane-insertion library for the enantioselective cyclopropanation of styrene, Rh2(L16)2 (L16 = KTDAALDLK) was quickly found to produce tert-butyl (1S,2R)-1,2-diphenylcyclopropane-1-carboxylate in 93% ee. Ninety-four unique [Rh2(peptide)(Oac)2] monopeptide complexes, with peptide ligands exhibiting a range of sterics and polarity, were prepared on polystyrene-polyethylene glycol resin beads. Trends in the production of the opposite enantiomer, methyl (1R,2S)-1,2-diphenylcyclopropane-1-carboxylate, highlighted successful mutations. These were then incorporated into a second, 96-peptide screen, from which eight sequences that gave greater than 45% ee were developed into [Rh2(peptide)2]bis-peptide catalysts. The best performer, [Rh2(KQDNANDTK)2-A], gave 83% and 92% ee for styrene cyclopropanation with methyl α-diazophenylacetate and tert-butyl α-diazophenylacetate, respectively. This “on bead” screening process significantly expedited the development of a new metalloenzyme.
Post-translational chemical modification of proteins is important for studying folding, function, protein–protein interactions, and localization in living cells. Most methods of modification rely on the reactivity of specific residues, but this can be problematic because proteins typically contain many copies of each residue. Alternatively, a precise residue within a complex environment may be targeted using molecular recognition to provide site-specific modification. Equipping a promiscuous dirhodium catalyst with the molecular recognition abilities of α-helices led to a general strategy for protein modification based on molecular shape rather than side-chain functionality.997,998 The well-established E3/K3 pair of α-helices self-assembles into a heterodimeric two-stranded assembly. Modification of the Lys-rich K3 peptide to include two coordinating glutamate residues allowed for the formation of the metallopeptide K3a,eRh2. The complementary peptide, E3gW, positions a Trp to flank the hydrophobic interface near the dirhodium center. Combination of these two peptides with a diazo reagent resulted in the covalent modification of the Trp residue (10 mol %, >95% conversion in 2 h). A >103 rate enhancement was observed as compared to control reactions using a free dirhodium reagent (100 mol %, 17% conversion in 20 h) or peptides in which the Trp residue and the dirhodium center are not optimally spaced.997 Additionally, K3a,eRh2 catalyzes modification of Tyr and Phe residues in E3gY and E3gF, respectively, but with slower rates than the Trp (20 mol %, 50% conversion in 5 h).997 This is the first reported example of Phe-specific post-translational modification, made possible by the interaction of two designed peptides. The scope of reactivity of K3a,eRh2 was expanded using a library of E3gX peptides with different amino acid residues (X) in the complementary position.998 Covalent modification was observed for over one-half of the amino acids: Cys, Gln, Asn, Arg, and Glu were modified in >65% conversion in 24 h, whereas Asp, His, Ser, and Lys showed conversion in lower yields, and Thr, Met, and alkyl side chains did not react.998
The specificity of the reaction was explored using a series of “matched” and “mismatched” coils. Six K3Rh2 metallopeptide variants and six E3W substrate variants were prepared resulting in 36 heterodimers (Figure 50).999 When combined, six catalyst/substrate pairs were matched both facially (catalyst and substrate on same side of coils) and axially (catalyst and substrate in same heptad along the axes of the coils), six had facial mismatches only, 12 had axial mismatches only, and 12 had both facial and axial mismatches. The mismatched substrates were less reactive (10–17% conversion when facially mismatched, 1–5% conversion when axially or axially/facially mismatched) than those that were matched (23–83% conversion), with the highest selectivities observed when the catalyst was located in the central heptad of the helix. Pairs of substrates, one matched and one mismatched, were combined with a catalyst in competition experiments. In five out of six cases, the selectivities improved further, typically in excess of 9:1. Overall, significant selectivities were observed for matched peptide pairs, demonstrating that site-specific catalysis overrides functional group reactivity. Like with natural enzymes, single-residue changes were observed to have dramatic effects on selectivity.
Figure 50.
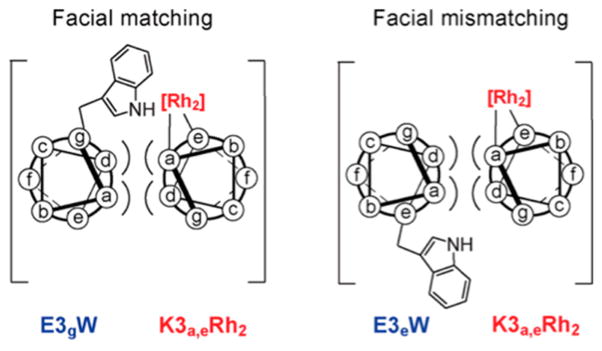
Conceptual illustration of axial and facial match/mismatch in coiled coils. Reproduced with permission from ref 999. Copyright 2012 American Chemical Society.
This successful strategy for protein modification based on molecular shape was applied to natural protein systems. Modification of the Trp residue on a helix fused to a recombinant maltose protein (MBP-E3g2W) can be catalyzed by a dirhodium metallopeptide at physiological pH within E. coli lysate. Lysozyme, which contains a Trp residue that is known to react with free Rh2(OAC)4, was unaffected, demonstrating the selectivity of the catalyst in the presence of a mixture of natural proteins.1000 Lysates from the expression of MBP-E3g2W and a recombinant S-transferase with an orthogonal peptide sequence at the C-terminus (GST-E3e2W) were combined with appropriate metallopeptides, and in each case only one product was observed, demonstrating selectivity even in the complex lysate environment.999
The oncogenic product c-Fos has a bZip domain that forms a coiled-coil with c-Jun, thus regulating transcription. The c-Jun variant Jun(Rh2) was designed to position a dirhodium center near a Gln residue on c-Fos during dimerization. Despite a significantly decreased affinity for c-Fos, Jun(Rh2) catalyzes the modification of the targeted Gln residue yielding 88% conversion at −15 °C.998 This example establishes the ability of templated modification to succeed, even in cases of transient assembly because the affinity between the catalyst and substrate is very low. Further, dirhodium-containing metallopeptides, with designs based on the MDM2-binding domain of p53, were found to inhibit MDM2.1001 Disruption of the MDM2-p53 interaction is one approach to restoring p53 function, which regulates cell cycle.
The protein-tyrosine kinase signaling protein Fyn has been implicated in tumor development and is an important therapeutic target. Fyn’s prototypical SH3 domain, which recognizes and binds weakly to short, Pro-rich PPII-helix sequences, contains several aromatic residues that are amenable to reaction with dirhodium catalysts. Out of a series of four designed peptides containing monodentate dirhodium ions, three were found to bind the Fyn SH3 domain with relatively high affinities (Kd = 0.24–0.76 and 0.65 μM, respectively). All three catalysts modified Trp42 of the SH3 domain effectively, showing that extensive optimization is not necessary when applying this post-translational modification method to natural proteins.999
Further stabilization of E3/K3 pairs was achieved by the inclusion of Lewis-basic side chains in E3gX, which coordinate to Rh(II) in K3a,eRh2.1002 When E3gX contained a His, Glu, or Met residue, Tm values were higher (66.1, 50.2, and 70.4 °C, respectively) than when it contained a noncoordinating Phe (E3gF, 39.5 °C). Cys led to decreased stability (Tm = 33.5 °C), which is consistent with models that show that Cys is too short to coordinate Rh(II) without disrupting the coiled-coil. The relative positions of dirhodium and the coordinating residues are important; K3g,dRh2 actually binds E3gH with lower affinity (Tm = 54.4 °C) than E3gF (Tm = 58.7 °C).1003 Further, properly positioned residues inhibit catalytic activity. E3gM and E3gH had significant inhibitory effects (IC50 = 3.7 and 0.5 μM, respectively) as compared to the control peptide E3gF (IC50 = 79 μM) in the modification of E3gW by K3a,eRh2 and a diazo reagent. The improperly positioned dirhodium in K3g,dRh2 was not as severely inhibited by E3gH (IC50 = 34 μM).
This model of organic–inorganic cooperativity was extended to design inhibitors for protein–protein interactions. The function of the cystic fibrosis transmembrane conductance regulator (CFTR) is affected by the binding of PDZ domains (a family of peptide-binding protein–protein interaction modules named for the first three members: PSD-95, Dlg, and ZO-1) in several proteins, including CAL (CFTR-associated ligand), making CAL inhibition a target. On the basis of sequences known to interact with the CAL PDZ domain, several short dirhodium metallopeptides were designed.1002 The peptide ERhVQSTRL is the best inhibitor for CAL PDZ reported to date with submicromolar affinity (Ki = 0.56 uM). The dirhodium moiety in this metallopeptide specifically targets coordination by the His301 residue on CAL, and mutation of this residue to an Ala (CALP-H301A) led to a 16-fold loss of affinity. By combining organic interactions and coordination chemistry, an inhibitor was quickly developed for a challenging protein target, demonstrating the potential of this approach for preparing inhibitors.
3.4. Summary
De novo design requires the construction of native-like, stable protein scaffolds with non-native sequences that exhibit desired properties and functionalities. Unlike protein redesign, which starts from native proteins, this “from scratch” design strategy considers the most fundamental interactions responsible for maintaining stable, native-like protein scaffolds. Hydrophobic interactions, H-bonding interactions, salt-bridges, van der Waals contacts, etc., contribute to the folding and topology of protein constructs. Early in the development of this field, significant progress was made in establishing stable constructs with common secondary structures, thus laying an excellent foundation for imparting functionality. The most common method to design stable constructs is to use hydrophobic interactions of amino acid side chains as a driving force to create self-assembled structures. In concert, the architecture of salt-bridges and van der Waals contacts directs the topology of the constructs. The concept of negative design, popularized by DeGrado and co-workers,10,643 has been highly employed to control the number of α-helical strands and parallel-versus-antiparallel orientations in the design of coiled coils. Alternatively, stable constructs can be built by attaching designed amphiphilic peptides onto preorganized templates to lower the entropic penalty of protein folding.
Starting from stable scaffolds, the next stage of de novo metalloprotein design involves the incorporation of metal cofactor-binding sites while retaining aggregate stability and a native folding state. Modifying the substitution pattern of hydrophobic residues by conversion into metal-binding residues with desired ligating atoms can potentially lead to perturbations of the protein structure. This can result in a decrease in protein stability, which illustrates the importance of choosing a stable construct as a starting point. The control of metal coordination through first and second coordination sphere interactions has been demonstrated in many cases. Recently, the field of de novo protein design began progressing toward the design of functional metalloenzymes, particularly those that aim to mimick native protein active sites using a minimalist approach. In the past few years, several successful examples have been reported, which possess the structural and functional characteristics of their native counterparts. In addition, conjugate systems of nonbiological metal complexes and de novo designed peptides have yielded new functions, some of which have enzyme-like selectivity. In summary, the de novo design approach allows one to employ knowledge learned from protein biochemistry, biophysics, coordination chemistry, and organometallic chemistry to achieve desired functions in proteins with a high level of precise control. It is truly a fantastic playground to showcase scientists’ creativity and ingenuity.
4. PERSPECTIVE
This Review focuses on the construction of metal sites in designed protein scaffolds to increase our understanding of heavy metal toxicity, to rationally modulate relevant properties, to create highly efficient metalloenzyme mimics, and to confer novel functions. The intrinsic complexity of proteins makes a full understanding of the structure–function relationship elusive, yet protein design has contributed significantly to what we have learned. Over the past three decades, we have witnessed a remarkable progress from constructing stable metalloproteins to imparting functionalities. The cases presented in this Review illustrate the depth of our understanding of the intricate interplay among various factors that dictate protein functions. Protein redesign utilizes the secondary and/or tertiary structure of native proteins, imparts desired functions into specific sites, and provides important details about how specific interactions around the metal or metal complex binding site influence functions. On the other hand, de novo design, a “bottom-up” approach, involves constructing metallopeptides from scratch to carry out predicted reactions. The minimalist approach often unveils the most important factors to building functional metalloenzymes.
Most protein design systems, especially de novo designed metalloproteins, focus on the first coordination sphere of metal cofactors. There are very few reported de novo designed metalloproteins in which the second coordination sphere of the metal site is taken into consideration with regard to modulating the properties of the metal center. One of the future directions of de novo protein design should be the rational design of the second coordination sphere. It is not uncommon to see in native proteins that specific residues not directly coordinated to the metal centers play important roles in catalysis or electron transfer by participating in H-bonding, electrostatic, and other interactions. These types of “remote” interactions should be the focus of the next level of design. Another area that has not received sufficient attention in the field of metalloprotein design is the building and optimizing of substrate binding sites. Because of the complexity of controlling metal structure and function, most effort thus far has been invested in taming the metal ions. However, regioselectivity, control of chirality, catalytic selectivity, and rate optimization are critically dependent on substrate recognition near the metal center. Additionally, workers in this field will need to evaluate more deeply the incorporation of dual functionality into protein constructs. Incorporating multiple metal centers within one protein is appealing, and native proteins with multiple metal-binding sites with cooperative fuctions are the targets for such designs. Little work has been completed emphasizing protein–protein or protein–nucleic acid recognition between designed metalloproteins. Because these types of interactions are crucial for the regulation of many complex biological processes, workers will need to learn how to dock, transfer electrons or substrates, and then release multicomponent protein systems to mimic more holistically complex biological processes. Finally, with the advent of chemical biology, future studies will require that metalloprotein designers learn how to express their new creations within cells to modify directly the behavior of organisms. Mastering the design of novel functional metalloprotein systems opens fascinating opportunities for application in biotechnology, pharmaceuticals, and diagnostics.
ABBREVIATIONS
- 2NA
2-naphthyl acetate
- 3SCCs
three-stranded coiled coils
- 3, 5-DTBC
3,5-di-tert-butyl-catechol
- ABTS
2,2′-azino-di(3-ethylbenzothiazoline-6-sulfonic acid)
- ALAD
δ-aminolevulinate dehydratase
- Ant
antennapedia homeodomain
- AP
amphiphilic membrane-associating redox maquette
- ATANP
(S)-2-amino-3-[1-(1,4,7-triazacyclononane)]-propanoic acid
- Avi
avidin
- Biot
biotin
- BPMCN
N,N′-bis(2-pyridylmethyl)-N,N′-dimethyl-trans-1,2-diaminocyclohexane
- BPMEN
N,N′-dimethyl-N,N′-bis(2-pyridylmethyl)ethane-1,2-diamine
- bPP
bovine pancreatic polypeptide
- bpy
bipyridyl
- BTC
butane-1,2,3-tricarboxylate
- CA
carbonic anhydrase
- CAL
CFTR-associated ligand
- CcO
cytochrome c oxidase
- CcP
cytochrome c peroxidase
- CD
circular dichroism
- CFTR
cystic fibrosis transmembrane conductance regulator
- CS
CoilSer
- CuNiR
copper nitrite reductase
- CV
cyclic voltammetry
- DAAO
D-amino acid oxidase
- DECP
diethyl 7-hydroxycoumarinyl
- DEPC
diethylpyrocarbonate
- DF
Due Ferri
- DFM
difluoromethionine
- DmsB
dimethyl sulfoxide reductase subunit B
- E. coli
Escherichia coli
- EDTA
ethylenediaminetetraacetate
- EDX
energy dispersed X-ray analysis
- en
ethylenediamine
- EPR
electron paramagnetic resonance
- EXAFS
extended X-ray absorption fine structure
- FdM
ferridoxin maquette
- Gln-Onp
Boc-glutamine 4-nitrophenyl ester
- GpA
glycophorin A
- H-bonding
hydrogen bonding
- HbpA
2-hydroxybiphenyl monooxygenase
- HCO
heme-copper oxidase
- Hcy
homocysteine
- HiPIP
high potential iron protein
- HLH
helix–loop–helix
- HPNP
hydroxypropyl-p-nitrophenol phosphate
- HRP
horseradish peroxidase
- LAAO
L-amino acid oxidase
- LCST
low critical solution temperature
- LiP
lignin peroxidase
- LMCT
ligand-to-metal charge transfer
- LmrR
lactococcal multidrug resistance regulator
- MADH
metholamine dehydrogenase
- MAO-N
monoamine oxidase
- Mb
myoglobin
- MCD
magnetic circular dichroism
- MMO
methane monooxygenase
- MnP
manganese peroxidase
- Mop
modular proteins
- MP
MiniPeroxidase
- Mp
3-maleimidopropionic acid
- N2OR
nitrous oxide reductase
- nbd
norbornadiene
- NHE
normal hydrogen electrode
- Nle
norleucine
- NMR
nuclear magnetic resonance
- NOR
nitric oxide reductase
- NTA
nitrilotriacetic acid
- OEC
oxygen evolving center
- OxM
oxomethionine
- PAC
perturbed angular correlation spectroscopy
- PDB
Protein Data Bank
- PDZ
PSD-95, Dlg, ZO-1 modules
- Pen
pennicillamine
- Phen
phenanthroline
- PNIPAM
poly(N-isopropylacrylamide)
- pNPA
p-nitrophenyl acetate
- pNPP
p-nitrophenyl phosphate
- PRIME
porphyrins in membrane
- PSI
Photosystem I
- PSII
Photosystem II
- RM
rubredoxin mimic
- RnR
ribonucleotide reductase
- Rop
repressor of primer
- rR
resonance Raman spectroscopy
- SALPN
1,3-bis(salicylideneamino)propane
- SAM
self-assembled monolayer
- Sav
streptavidin
- Sec
selenocysteine
- SeM
selenomethionine
- SOD
superoxide dismutase
- T1Cu
type 1 copper
- T2Cu
type 2 copper
- TASP
template-assembled synthetic proteins
- TFM
trifluoromethionine
- tHisF
imidazole glycerol phosphate synthase subunit from Thermotoga maritima
- TMB
2,2′,5,5′-tetramethylbenzidine
- Tren
tris(2-aminoethyl)amine
- Trx
thioredoxin
- UV–vis
UV–visible spectroscopy
- WT
wild type
- XAS
X-ray absorption spectroscopy
- ZF
zinc finger
Biographies

Fangting Yu received her B.S. in Chemistry in 2009 from the College of Chemistry and Molecular Engineering, Peking University, P. R. China. She did her undergraduate research in the National Lab of Rare Earth Material Chemistry and Application. Her work was focused on the influence of metal ions on the solubility of cholesterol in the aggregates of phospholipids and bile salts, during which time she was awarded the Undergraduate Student Innovation Training Grant from the Chinese Academy of Science. She also worked as a summer research assistant at the University of Hong Kong in 2008 synthesizing nickel nanoparticles. She then joined Prof. Pecoraro’s lab at the University of Michigan, Ann Arbor. She is currently a Ph.D. candidate, working primarily on de novo designed metallopeptides with type 2 copper centers as a model for copper nitrite reductase. She is also working on synthesizing manganese dimer complexes as a model for the Oxygen Evolving Center in Photosystem II.

Virginia M. Cangelosi received a B.A. in Chemistry in 2005 from Albion College, Albion, MI (with Clifford E. Harris). In 2010, she obtained her Ph.D. from the University of Oregon under the direction of Darren W. Johnson. She is currently an NIH Ruth L. Kirschstein fellow in the laboratory of Vincent L. Pecoraro at the University of Michigan. Her research interests include de novo protein design, enzyme kinetics, supramolecular chemistry, self-assembly, and toxic metals.

Melissa L. Zastrow received her B.S. in Chemistry from the University of Connecticut (2008) where she worked under Professors Isabelle Lagadic (2005–2007) and Christian Brückner (2007–2008). She received a Center for Environmental Kinetics Analysis NSF-REU Fellowship at Pennsylvania State University in the Department of Biochemistry and Molecular Biology in the summer of 2007 where she worked for Professor Ming Tien. She went on to earn her Ph.D. in Chemistry (2013) at the University of Michigan under the supervision of Professor Vincent L. Pecoraro. Her primary research focus was on the de novo design of dual-metal site three-stranded coiled coils for hydrolytic catalysis, including the design of zinc metalloenzymes. She is currently a postdoctoral researcher in Professor Stephen J. Lippard’s laboratory at Massachusetts Institute of Technology, where she is using fluorescence-based approaches for detecting and studying the functions of biological mobile zinc.

Matteo Tegoni graduated from the University of Parma, where he also obtained his Ph.D. in the field of the thermodynamics of formation of metal complexes in solution. In 2002 he was appointed Senior Research Associate in General and Inorganic Chemistry at the University of Parma, where he is a teacher of General and Inorganic Chemistry and Laboratory. He served as a member of the national (young chemists group) and regional boards of the Italian Chemical Society, and he is involved in activities of dissemination of science and chemistry for schools and citizenship. His current research interests are in the field of de novo designed metallopeptides and artificial metalloenzymes, metal-based supramolecular assemblies, and biologically relevant metal complexes.

Jefferson S. Plegaria was born in 1984 in the Rizal Province of the Philippines. He received his B.A. degree in Chemistry from La Salle University, Philadelphia, PA, in 2007. He then worked in a pediatric-oncology lab of Dr. John Maris at The Children’s Hospital of Philadelphia for two years, where he performed in vivo stage-one-drug studies using a neuroblastoma-xenograft model. In 2009, he began his Ph.D. degree with Professor Vincent L. Pecoraro at the University of Michigan in Ann Arbor, MI. He is currently studying the spectroscopic and electron transfer properties of a cupredoxin site that was incorporated in a de novo designed peptide using biophysical, electrochemical, and photophysical techniques. In addition, he is also solving the solution structure of a de novo designed peptide capable of binding to toxic heavy metals.

Alison G. Tebo was born and raised in San Diego, CA. She attended UCLA and received a B.S. in Biochemistry with a minor in Spanish. She began her Ph.D. at the University of Michigan in the Program in Chemical Biology in 2010. Her research in Dr. Vincent Pecoraro’s group focuses on the de novo design of iron electron transfer proteins, with an emphasis on chemical modifications to enforce asymmetry, append chromophores, and tune function.

Catherine S. Mocny received her B.A. in Chemistry from Kalamazoo College, Kalamazoo, MI, in 2010. She is currently a Ph.D. candidate of Chemistry at the University of Michigan, Ann Arbor, MI, studying de novo metalloprotein design with a focus on obtaining asymmetric peptide platforms.

Leela Ruckthong received her B.S. in Chemistry from Kasetsart University, Thailand in 2008. She has been awarded a full scholarship from the Ministry of Science, the Royal Thai Government, Thailand to pursue her Ph.D. degree in Biophysics, Department of Biophysics, University of Michigan where she has joined Prof. Dr. Vincent L. Pecoraro’s group. Her work focuses on heavy metal chemistry in de novo metalloprotein design using alternative chirality of D-amino acids.

Hira Qayyum obtained her B.S. in Chemistry from the University of Akron in 2012. She is currently a graduate student in Professor Vincent Pecoraro’s group at the University of Michigan, and her work involves the de novo design of metalloenzymes.

Vincent L. Pecoraro received his B.S. degree in biochemistry at the University of California, Los Angeles, in 1977 and his Ph.D. in chemistry under the supervision of Prof. Kenneth N. Raymond in 1981 at the University of California, Berkeley. After completing an NIH postdoctoral fellowship with Prof. W. W. Cleland in the Department of Biochemistry at the University of Wisconsin, Madison, Dr. Pecoraro joined the chemistry faculty at the University of Michigan, Ann Arbor, in 1984. He was promoted to associate professor in 1989 and professor in 1992 and then appointed as the John T. Groves Collegiate Professor in 2005. His research has focused on the role of metals in biology, in particular, defining the biological chemistry of manganese and vanadium. In recent years, his research group has become deeply involved in de novo metalloprotein design and the elucidation of heavy metal complexation by cysteine-rich proteins. An area of Prof. Pecoraro’s interest includes metallamacrocyclic chemistry, which was advanced significantly by his recognition of the metallacrown structural motif in 1989. Prof. Pecoraro has received numerous awards and honors including the G. D. Searle Biomedical Research Scholarship, an Alfred P. Sloan Fellowship, the Vanadis Award, an Alexander von Humboldt Award to Senior Research Scientists, and The Blaise Pascal International Chair for Research. He is a Fellow of the American Association for the Advancement of Science and the American Chemical Society. He has served as an Associate Editor of Inorganic Chemistry since 1994. Prof. Pecoraro is married to Prof. Peggy Carver of the College of Pharmacy at the University of Michigan, Ann Arbor. They enjoy traveling, eating well, fine art, and the company of their many good friends around the world.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Lu Y, Berry SM, Pfister TD. Chem Rev. 2001;101:3047. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 2.Lu Y. Inorg Chem. 2006;45:155. doi: 10.1021/ic052007t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marshall NM, Garner DK, Wilson TD, Gao Y-G, Robinson H, Nilges MJ, Lu Y. Nature. 2009;462:113. doi: 10.1038/nature08551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris KL, Lim S, Franklin SJ. Inorg Chem. 2006;45:10002. doi: 10.1021/ic060877k. [DOI] [PubMed] [Google Scholar]
- 5.Ringenberg MR, Ward TR. Chem Commun (Cambridge, UK) 2011;47:8470. doi: 10.1039/c1cc11592h. [DOI] [PubMed] [Google Scholar]
- 6.Ward TR. Acc Chem Res. 2011;44:47. doi: 10.1021/ar100099u. [DOI] [PubMed] [Google Scholar]
- 7.Steinreiber J, Ward TR. Coord Chem Rev. 2008;252:751. [Google Scholar]
- 8.Benson DE, Wisz MS, Hellinga HW. Proc Natl Acad Sci USA. 2000;97:6292. doi: 10.1073/pnas.97.12.6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hellinga HW. Fold Des. 1998;3:R1. doi: 10.1016/S1359-0278(98)00001-7. [DOI] [PubMed] [Google Scholar]
- 10.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. Annu Rev Biochem. 1999;68:779. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 11.Bryson JW, Betz SF, Lu HS, Suich DJ, Zhou HX, O’Neil KT, DeGrado WF. Science. 1995;270:935. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 12.Degrado WF, Wasserman ZR, Lear JD. Science. 1989;243:622. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 13.Cohen C, Parry DAD. Proteins: Struct, Funct, Genet. 1990;7:1. [Google Scholar]
- 14.Beasley JR, Hecht MH. J Biol Chem. 1997;272:2031. doi: 10.1074/jbc.272.4.2031. [DOI] [PubMed] [Google Scholar]
- 15.Richardson JS, Richardson DC, Tweedy NB, Gernert KM, Quinn TP, Hecht MH, Erickson BW, Yan Y, Mcclain RD, Donlan ME, Surles MC. Biophys J. 1992;63:1186. [PMC free article] [PubMed] [Google Scholar]
- 16.Baltzer L, Nilsson H, Nilsson J. Chem Rev. 2001;101:3153. doi: 10.1021/cr0000473. [DOI] [PubMed] [Google Scholar]
- 17.Saven JG. Chem Rev. 2001;101:3113. doi: 10.1021/cr000058w. [DOI] [PubMed] [Google Scholar]
- 18.Schneider JP, Lombardi A, DeGrado WF. Fold Des. 1998;3:R29. doi: 10.1016/S1359-0278(98)00011-X. [DOI] [PubMed] [Google Scholar]
- 19.Regan L. Trends Biochem Sci. 1995;20:280. doi: 10.1016/s0968-0004(00)89044-1. [DOI] [PubMed] [Google Scholar]
- 20.Baltzer L. Curr Opin Struct Biol. 1998;8:466. doi: 10.1016/s0959-440x(98)80124-3. [DOI] [PubMed] [Google Scholar]
- 21.Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reedy CJ, Gibney BR. Chem Rev. 2004;104:617. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 23.Zastrow ML, Pecoraro VL. Coord Chem Rev. 2013:1. doi: 10.1016/j.ccr.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samish I, MacDermaid CM, Perez-Aguilar JM, Saven JG. Annu Rev Phys Chem. 2011;62:129. doi: 10.1146/annurev-physchem-032210-103509. [DOI] [PubMed] [Google Scholar]
- 25.Barker PD. Curr Opin Struct Biol. 2003;13:490. doi: 10.1016/s0959-440x(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 26.Lu Y, Chakraborty S, Miner KD, Wilson TD, Mukherjee A, Yu Y, Liu J, Marshall NM, Champaign U. Comprehensive Inorganic Chemistry II. Elsevier Ltd; New York: 2013. p. 565. [Google Scholar]
- 27.Robson B, Vaithilingam A. Protein Folding Revisited. 1. Vol. 84. Elsevier Inc; New York: 2008. p. 161. [DOI] [PubMed] [Google Scholar]
- 28.Woolfson DN. Advances in Protein Chemistry. Vol. 70. Elsevier; New York: 2005. p. 79. [DOI] [PubMed] [Google Scholar]
- 29.Doerr AJ, McLendon GL. Inorg Chem. 2004;43:7916. doi: 10.1021/ic0490573. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Suzuki K, Kanaori K, Tajima K, Kashiwada A, Hiroaki H, Kohda D, Tanaka T. Protein Sci. 2000;9:1327. doi: 10.1110/ps.9.7.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiyokawa T, Kanaori K, Tajima K, Koike M, Mizuno T, Oku JJ-II, Tanaka T. J Pept Res. 2004;63:347. doi: 10.1111/j.1399-3011.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka T, Mizuno T, Fukui S, Hiroaki H, Oku J-I, Kanaori K, Tajima K, Shirakawa M. J Am Chem Soc. 2004;126:14023. doi: 10.1021/ja047945r. [DOI] [PubMed] [Google Scholar]
- 33.O’Neil KT, Degrado WF. Science. 1990;250:646. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 34.Lovejoy B, Choe S, Cascio D, Mcrorie DK, William F, Eisenberg D, Degrado WF. Science. 1993;259:1288. doi: 10.1126/science.8446897. [DOI] [PubMed] [Google Scholar]
- 35.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O’Halloran TV, Penner-Hahn JE, Degrado WF, Pecoraro VL. J Am Chem Soc. 1997;119:6195. [Google Scholar]
- 36.Ogihara NL, Weiss MS, Degrado WF, Eisenberg D. Protein Sci. 1997;6:80. doi: 10.1002/pro.5560060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. J Mol Biol. 1998;280:897. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 38.Farrer BT, Pecoraro VL. Proc Natl Acad Sci USA. 2003;100:3760. doi: 10.1073/pnas.0336055100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hellinga HW, Caradonna JP, Richards FM. J Mol Biol. 1991;222:787. doi: 10.1016/0022-2836(91)90511-4. [DOI] [PubMed] [Google Scholar]
- 40.Hellinga HW, Richards FM. J Mol Biol. 1991;222:763. doi: 10.1016/0022-2836(91)90510-d. [DOI] [PubMed] [Google Scholar]
- 41.Rohl CA, Strauss CEM, Misura KMS, Baker D. Methods Enzymol. 2004;383:66. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 42.Clarke ND, Yuan SM. Proteins: Struct, Funct, Genet. 1995;23:256. doi: 10.1002/prot.340230214. [DOI] [PubMed] [Google Scholar]
- 43.Lu Y, Valentine JS. Curr Opin Struct Biol. 1997;7:495. doi: 10.1016/s0959-440x(97)80112-1. [DOI] [PubMed] [Google Scholar]
- 44.Toscano MD, Woycechowsky KJ, Hilvert D. Angew Chem, Int Ed. 2007;46:3212. doi: 10.1002/anie.200604205. [DOI] [PubMed] [Google Scholar]
- 45.Ghirlanda G. Nature. 2008;453:164. doi: 10.1038/453164a. [DOI] [PubMed] [Google Scholar]
- 46.Kazlauskas RJ. Curr Opin Chem Biol. 2005;9:195. doi: 10.1016/j.cbpa.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 47.Khersonsky O, Roodveldt C, Tawfik DS. Curr Opin Chem Biol. 2006;10:498. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 48.Matthews J, Sunde M. IUBMB Life. 2002;54:351. doi: 10.1080/15216540216035. [DOI] [PubMed] [Google Scholar]
- 49.Kaptein R. Curr Opin Struct Biol. 1991;1:63. [Google Scholar]
- 50.Yamamoto KR. Annu Rev Genet. 1985;19:209. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
- 51.Payvar F, DeFranco D, Firestone GL, Edgar B, Wrange O, Okret S, Gustafsson JA, Yamamoto KR. Cell. 1983;35:381. doi: 10.1016/0092-8674(83)90171-x. [DOI] [PubMed] [Google Scholar]
- 52.Scheidereit C, Geisse S, Westphal H, Beato M. Nature. 1983;304:749. doi: 10.1038/304749a0. [DOI] [PubMed] [Google Scholar]
- 53.Chandler VL, Maler BA, Yamamoto KR. Cell. 1983;33:489. doi: 10.1016/0092-8674(83)90430-0. [DOI] [PubMed] [Google Scholar]
- 54.Sakai DD, Helms S, Carlstedt-Duke J, Gustafsson JA, Rottman FM, Yamamoto KR. Genes Dev. 1988;2:1144. doi: 10.1101/gad.2.9.1144. [DOI] [PubMed] [Google Scholar]
- 55.Evans R. Science. 1988;240:889. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hard T, Kellenbach E, Boelens R, Maler B, Dahlman K, Freedman L, Carlstedt-Duke J, Yamamoto K, Gustafsson J, Kaptein R. Science. 1990;249:157. doi: 10.1126/science.2115209. [DOI] [PubMed] [Google Scholar]
- 57.Schwabe JW, Chapman L, Finch JT, Rhodes D. Cell. 1993;75:567. doi: 10.1016/0092-8674(93)90390-c. [DOI] [PubMed] [Google Scholar]
- 58.Schwabe J, Neuhaus D, Rhodes D. Nature. 1990;348:458. doi: 10.1038/348458a0. [DOI] [PubMed] [Google Scholar]
- 59.Luisi B, Xu W, Otwinowski Z, Freedman LP, Yamamoto KR, Sigler PB. Nature. 1991;352:497. doi: 10.1038/352497a0. [DOI] [PubMed] [Google Scholar]
- 60.O’Connor TR, Graves RJ, de Murcia G, Castaing B, Laval J. J Biol Chem. 1993;268:9063. [PubMed] [Google Scholar]
- 61.Miyamoto I, Miura N, Niwa H, Miyazaki J, Tanaka K. J Biol Chem. 1992;267:12182. [PubMed] [Google Scholar]
- 62.Ikegami T, Kuraoka I, Saijo M, Kodo N, Kyogoku Y, Morikawa K, Tanaka K, Shirakawa M. Nat Struct Mol Biol. 1988;5:701. doi: 10.1038/1400. [DOI] [PubMed] [Google Scholar]
- 63.Buchko GW, Ni S, Thrall BD, Kennedy MA. Nucleic Acids Res. 1998;26:2779. doi: 10.1093/nar/26.11.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hartwig A, Asmuss M, Blessing H, Hoffmann S, Jahnke G, Khandelwal S, Pelzer A, Bürkle A. Food Chem Toxicol. 2002;40:1179. doi: 10.1016/s0278-6915(02)00043-1. [DOI] [PubMed] [Google Scholar]
- 65.Takinowaki H, Matsuda Y, Yoshida T, Kobayashi Y, Ohkubo T. Protein Sci. 2006;15:487. doi: 10.1110/ps.051786306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Janda I, Devedjiev Y, Derewenda U, Dauter Z, Bielnicki J, Cooper DR, Graf PCF, Joachimiak A, Jakob U, Derewenda ZS. Structure. 2004;12:1901. doi: 10.1016/j.str.2004.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Penner-Hahn J. Curr Opin Chem Biol. 2007;11:166. doi: 10.1016/j.cbpa.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 68.Collet J-F, D’Souza JC, Jakob U, Bardwell JCA. J Biol Chem. 2003;278:45325. doi: 10.1074/jbc.M307818200. [DOI] [PubMed] [Google Scholar]
- 69.Carballo E, Lai W, Blackshear P. Science. 1998;281:1001. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 70.Michalek JL, Besold AN, Michel SLJ. Dalton Trans. 2011;40:12619. doi: 10.1039/c1dt11071c. [DOI] [PubMed] [Google Scholar]
- 71.Cho Y, Gorina S, Jeffrey P, Pavletich N. Science. 1994;265:346. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 72.Hainaut P, Hollstein M. Adv Cancer Res. 2000;77:81. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 73.Lai WS, Carballo E, Thorn JM, Kennington Ea, Blackshear PJ. J Biol Chem. 2000;275:17827. doi: 10.1074/jbc.M001696200. [DOI] [PubMed] [Google Scholar]
- 74.Worthington MT, Amann BT, Nathans D, Berg JM. Proc Natl Acad Sci USA. 1996;93:13754. doi: 10.1073/pnas.93.24.13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Anzellotti AI, Farrell NP. Chem Soc Rev. 2008;37:1629. doi: 10.1039/b617121b. [DOI] [PubMed] [Google Scholar]
- 76.Lai WS, Blackshear PJ. J Biol Chem. 2001;276:23144. doi: 10.1074/jbc.M100680200. [DOI] [PubMed] [Google Scholar]
- 77.Lai W, Carballo E, Strum J, Kennington E, Phillips R, Blackshear P. Mol Cell Biol. 1999;19:4311. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.DiTargiani R, Lee S, Wassink S, Michel S. Biochemistry. 2006;45:13641. doi: 10.1021/bi060747n. [DOI] [PubMed] [Google Scholar]
- 79.Michel S, Guerrerio A, Berg J. Biochemistry. 2003;475:4626. doi: 10.1021/bi034073h. [DOI] [PubMed] [Google Scholar]
- 80.Worthington MT, Pelo JW, Sachedina MA, Applegate JL, Arseneau KO, Pizarro TT. J Biol Chem. 2002;277:48558. doi: 10.1074/jbc.M206505200. [DOI] [PubMed] [Google Scholar]
- 81.Brewer BY. J Biol Chem. 2004;279:27870. doi: 10.1074/jbc.M402551200. [DOI] [PubMed] [Google Scholar]
- 82.Klug A. Q Rev Biophys. 2010;43:1. doi: 10.1017/S0033583510000089. [DOI] [PubMed] [Google Scholar]
- 83.Laity JH, Lee BM, Wright PE. Curr Opin Struct Biol. 2001;11:39. doi: 10.1016/s0959-440x(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 84.Berg JM, Godwin HA. Annu Rev Biophys Biomol Struct. 1997;26:357. doi: 10.1146/annurev.biophys.26.1.357. [DOI] [PubMed] [Google Scholar]
- 85.Beerli RR, Barbas CF. Nat Biotechnol. 2002;20:135. doi: 10.1038/nbt0202-135. [DOI] [PubMed] [Google Scholar]
- 86.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Nat Rev Genet. 2010;11:636. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 87.Kröncke K, Klotz L-O. Antioxid Redox Signaling. 2009;11:1015. doi: 10.1089/ARS.2008.2269. [DOI] [PubMed] [Google Scholar]
- 88.Lee M, Gippert G, Soman K, Case D, Wright P. Science. 1989;245:635. doi: 10.1126/science.2503871. [DOI] [PubMed] [Google Scholar]
- 89.Klevit RE, Herriott JR, Horvath SJ. Proteins: Struct, Funct, Genet. 1990;7:215. doi: 10.1002/prot.340070303. [DOI] [PubMed] [Google Scholar]
- 90.Omichinski JG, Clore GM, Appella E, Sakaguchi K, Gronenborn AM. Biochemistry. 1990;29:9324. doi: 10.1021/bi00492a004. [DOI] [PubMed] [Google Scholar]
- 91.Negi S, Imanishi M, Matsumoto M, Sugiura Y. Chem- Eur J. 2008;14:3236. doi: 10.1002/chem.200701320. [DOI] [PubMed] [Google Scholar]
- 92.Besold AN, Oluyadi AA, Michel SLJ. Inorg Chem. 2013;52:4721. doi: 10.1021/ic4003516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berkovits HJ, Berg JM. Biochemistry. 1999;38:16826. doi: 10.1021/bi991433l. [DOI] [PubMed] [Google Scholar]
- 94.Berkovits-Cymet HJ, Amann BT, Berg JM. Biochemistry. 2004;43:898. doi: 10.1021/bi035159d. [DOI] [PubMed] [Google Scholar]
- 95.Kim JG, Armstrong RC, Agoston vD, Robinsky A, Wiese C, Nagle J, Hudson LD. J Neurosci Res. 1997;50:272. doi: 10.1002/(SICI)1097-4547(19971015)50:2<272::AID-JNR16>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 96.Vana AAC, Lucchinetti CFC, Le TTQ, Armstrong RRC. Glia. 2007;697:687. doi: 10.1002/glia.20492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Romm E, Nielsen JA, Kim JG, Hudson LD. J Neurochem. 2005;93:1444. doi: 10.1111/j.1471-4159.2005.03131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bellefroid EJ, Bourguignon C, Hollemann T, Ma Q, Anderson DJ, Kintner C, Pieler T. Cell. 1996;87:1191. doi: 10.1016/s0092-8674(00)81815-2. [DOI] [PubMed] [Google Scholar]
- 99.Blasie CA, Berg JM. Inorg Chem. 2000;39:348. doi: 10.1021/ic990913y. [DOI] [PubMed] [Google Scholar]
- 100.Nielsen JA, Berndt JA, Hudson LD, Armstrong RC. Mol Cell Neurosci. 2004;25:111. doi: 10.1016/j.mcn.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 101.Gamsjaeger R, Swanton MK, Kobus FJ, Lehtomaki E, Lowry Ja, Kwan AH, Matthews JM, Mackay JP. J Biol Chem. 2008;283:5158. doi: 10.1074/jbc.M703772200. [DOI] [PubMed] [Google Scholar]
- 102.Kim J, Hudson L. Mol Cell Biol. 1992;12:5632. doi: 10.1128/mcb.12.12.5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jiang Y, Yu VC, Buchholz F, O’Connel S, Rhodes SJ, Candloro C, Xia Y-R, Lusis AJ, Rosenfeld MG. J Biol Chem. 1996;271:10723. doi: 10.1074/jbc.271.18.10723. [DOI] [PubMed] [Google Scholar]
- 104.Yee KSY. J Biol Chem. 1998;273:5366. doi: 10.1074/jbc.273.9.5366. [DOI] [PubMed] [Google Scholar]
- 105.Lee SJ, Michel SLJ. Inorg Chem. 2010;49:1211. doi: 10.1021/ic9024298. [DOI] [PubMed] [Google Scholar]
- 106.Jantz D, Amann BT, Gatto GJ, Berg JM. Chem Rev. 2004;104:789. doi: 10.1021/cr020603o. [DOI] [PubMed] [Google Scholar]
- 107.Elrod-Erickson M, Rould MA, Nekludova L, Pabo CO. Structure. 1996;4:1171. doi: 10.1016/s0969-2126(96)00125-6. [DOI] [PubMed] [Google Scholar]
- 108.Pavletich N, Pabo C. Science. 1991;252:809. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 109.Oka S, Shiraishi Y, Yoshida T, Ohkubo T, Sugiura Y, Kobayashi Y. Biochemistry. 2004;43:16027. doi: 10.1021/bi048438p. [DOI] [PubMed] [Google Scholar]
- 110.Mandell JG, Barbas CF. Nucleic Acids Res. 2006;34:W516. doi: 10.1093/nar/gkl209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wolfe SA, Nekludova L, Pabo CO. Annu Rev Biophys Biomol Struct. 2000;29:183. doi: 10.1146/annurev.biophys.29.1.183. [DOI] [PubMed] [Google Scholar]
- 112.Desjarlais JR, Berg JM. Proc Natl Acad Sci USA. 1992;89:7345. doi: 10.1073/pnas.89.16.7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sera T. Adv Drug Delivery Rev. 2009;61:513. doi: 10.1016/j.addr.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 114.Davis D, Stokoe D. BMC Med. 2010;8:42. doi: 10.1186/1741-7015-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Berg JM, Shi Y. Science. 1996;271:1081. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 116.Coleman JE. Annu Rev Biochem. 1992;61:897. doi: 10.1146/annurev.bi.61.070192.004341. [DOI] [PubMed] [Google Scholar]
- 117.Harrison S. Nature. 1991;353:715. doi: 10.1038/353715a0. [DOI] [PubMed] [Google Scholar]
- 118.Klug A, Tocchini-Valentini G. Gene. 1993;135:83. doi: 10.1016/0378-1119(93)90053-6. [DOI] [PubMed] [Google Scholar]
- 119.Schwabe J, Klug A. Nat Struct Mol Biol. 1994;1:345. doi: 10.1038/nsb0694-345. [DOI] [PubMed] [Google Scholar]
- 120.Nardelli J, Gibson T, Charnay P. Nucleic Acids Res. 1992;20:4137. doi: 10.1093/nar/20.16.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sera T, Uranga C. Biochemistry. 2002;41:7074. doi: 10.1021/bi020095c. [DOI] [PubMed] [Google Scholar]
- 122.Greisman HA, Pabo CO. Science. 1997;275:657. doi: 10.1126/science.275.5300.657. [DOI] [PubMed] [Google Scholar]
- 123.Segal DJ, Dreier B, Beerli RR, Barbas CF. Proc Natl Acad Sci USA. 1999;96:2758. doi: 10.1073/pnas.96.6.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas CF. J Biol Chem. 2001;276:29466. doi: 10.1074/jbc.M102604200. [DOI] [PubMed] [Google Scholar]
- 125.Taylor WE, Suruki HK, Lin AH, Naraghi-Arani P, Igarashi RY, Younessian M, Katkus P, Vo NV. Biochemistry. 1995;34:3222. doi: 10.1021/bi00010a011. [DOI] [PubMed] [Google Scholar]
- 126.Wolfe SA, Grant RA, Elrod-Erickson M, Pabo CO. Structure. 2001;9:717. doi: 10.1016/s0969-2126(01)00632-3. [DOI] [PubMed] [Google Scholar]
- 127.Jamieson AC, Kim SH, Wells JA. Biochemistry. 1994;33:5689. doi: 10.1021/bi00185a004. [DOI] [PubMed] [Google Scholar]
- 128.Wu H, Yang WP, Barbas CF. Proc Natl Acad Sci USA. 1995;92:344. doi: 10.1073/pnas.92.2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Choo Y, Klug A. Proc Natl Acad Sci USA. 1994;91:11163. doi: 10.1073/pnas.91.23.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rebar E, Pabo C. Science. 1994;263:671. doi: 10.1126/science.8303274. [DOI] [PubMed] [Google Scholar]
- 131.Kono H, Imanishi M, Negi S, Tatsutani K, Sakaeda Y, Hashimoto A, Nakayama C, Futaki S, Sugiura Y. FEBS Lett. 2012;586:918. doi: 10.1016/j.febslet.2012.02.025. [DOI] [PubMed] [Google Scholar]
- 132.Hsu T, Gogos J, Kirsh S, Kafatos F. Science. 1992;257:1946. doi: 10.1126/science.1411512. [DOI] [PubMed] [Google Scholar]
- 133.Kadonaga JT, Carner KR, Masiarz FR, Tjian R. Cell. 1987;51:1079. doi: 10.1016/0092-8674(87)90594-0. [DOI] [PubMed] [Google Scholar]
- 134.Kadonaga JT, Jones KA, Tjian R. Trends Biochem Sci. 1986;11:20. [Google Scholar]
- 135.Nagaoka M, Doi Y, Kuwahara J, Sugiura Y. J Am Chem Soc. 2002;124:6526. doi: 10.1021/ja025856d. [DOI] [PubMed] [Google Scholar]
- 136.Dynan WS, Tjian R. Cell. 1983;32:669. doi: 10.1016/0092-8674(83)90053-3. [DOI] [PubMed] [Google Scholar]
- 137.Gidoni D, Dynan W, Tjian R. Nature. 1984;312:401. doi: 10.1038/312409a0. [DOI] [PubMed] [Google Scholar]
- 138.Shiraishi Y, Imanishi M, Morisaki T, Sugiura Y. Biochemistry. 2005;44:2523. doi: 10.1021/bi047797h. [DOI] [PubMed] [Google Scholar]
- 139.Hirata T, Nomura W, Imanishi M, Sugiura Y. Bioorg Med Chem Lett. 2005;15:2197. doi: 10.1016/j.bmcl.2005.03.040. [DOI] [PubMed] [Google Scholar]
- 140.Liu Q, Segal D, Ghiara J, Barbas CF. Proc Natl Acad Sci USA. 1997;94:5525. doi: 10.1073/pnas.94.11.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kamiuchi T, Abe E, Imanishi M, Kaji T, Nagaoka M, Sugiura Y. Biochemistry. 1998;37:13827. doi: 10.1021/bi9811112. [DOI] [PubMed] [Google Scholar]
- 142.Lund CV, Popkov M, Magnenat L, Barbas CF. Mol Cell Biol. 2005;25:9082. doi: 10.1128/MCB.25.20.9082-9091.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pomerantz JJL, Wolfe SSA, Pabo COC. Biochemistry. 1998;37:1. doi: 10.1021/bi972464o. [DOI] [PubMed] [Google Scholar]
- 144.Wolfe SSA, Grant RAR, Pabo COC. Biochemistry. 2003;42:13401. doi: 10.1021/bi034830b. [DOI] [PubMed] [Google Scholar]
- 145.Nagaoka M, Nomura W, Shiraishi Y, Sugiura Y. Biochem Biophys Res Commun. 2001;282:1001. doi: 10.1006/bbrc.2001.4672. [DOI] [PubMed] [Google Scholar]
- 146.Yan W, Imanishi M, Futaki S, Sugiura Y. Biochemistry. 2007;46:8517. doi: 10.1021/bi7006417. [DOI] [PubMed] [Google Scholar]
- 147.Merutka G, Shalongo W, Stellwagen E. Biochemistry. 1991;30:4245. doi: 10.1021/bi00231a020. [DOI] [PubMed] [Google Scholar]
- 148.Imanishi M, Yan W, Morisaki T, Sugiura Y. Biochem Biophys Res Commun. 2005;333:167. doi: 10.1016/j.bbrc.2005.05.090. [DOI] [PubMed] [Google Scholar]
- 149.Nomura W, Nagaoka M, Shiraishi Y, Sugiura Y. Biochem Biophys Res Commun. 2003;300:87. doi: 10.1016/s0006-291x(02)02792-4. [DOI] [PubMed] [Google Scholar]
- 150.Nomura W, Sugiura Y. Biochemistry. 2003;42:14805. doi: 10.1021/bi035446h. [DOI] [PubMed] [Google Scholar]
- 151.Imanishi M, Hori Y, Nagaoka M, Sugiura Y. Biochemistry. 2000;39:4383. doi: 10.1021/bi992989b. [DOI] [PubMed] [Google Scholar]
- 152.Imanishi M, Sugiura Y. Biochemistry. 2002;41:1328. doi: 10.1021/bi011761x. [DOI] [PubMed] [Google Scholar]
- 153.Dahiyat BI. Science. 1997;278:82. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 154.Dahiyat BI, Sarisky CA, Mayo SL. J Mol Biol. 1997;273:789. doi: 10.1006/jmbi.1997.1341. [DOI] [PubMed] [Google Scholar]
- 155.Struthers MD, Cheng RP, Imperiali B. J Am Chem Soc. 1996;118:3073. [Google Scholar]
- 156.Seelig B, Szostak JW. Nature. 2007;448:828. doi: 10.1038/nature06032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Green A, Sarkar B. Biochem J. 1998;333:85. doi: 10.1042/bj3330085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Christy BA, Lau LF, Nathans D. Proc Natl Acad Sci USA. 1988;85:7857. doi: 10.1073/pnas.85.21.7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Christy B, Nathans D. Proc Natl Acad Sci USA. 1989;86:8737. doi: 10.1073/pnas.86.22.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hori Y, Suzuki K, Okuno Y, Nagaoka M, Futaki S, Sugiura Y. J Am Chem Soc. 2000;122:7648. doi: 10.1093/nass/44.1.295. [DOI] [PubMed] [Google Scholar]
- 161.Nagaoka M, Sugiura Y. Biochemistry. 1996;35:8761. doi: 10.1021/bi952530r. [DOI] [PubMed] [Google Scholar]
- 162.Nagaoka M, Kuwahara J, Sugiura Y. Biochem Biophys Res Commun. 1993;194:1515. doi: 10.1006/bbrc.1993.1996. [DOI] [PubMed] [Google Scholar]
- 163.Kuwahara J, Yonezawa A, Futamura M, Sugiura Y. Biochemistry. 1993;32:5994. doi: 10.1021/bi00074a010. [DOI] [PubMed] [Google Scholar]
- 164.Yokono M, Saegusa N, Matsushita K, Sugiura Y. Biochemistry. 1998;37:6824. doi: 10.1021/bi9727646. [DOI] [PubMed] [Google Scholar]
- 165.Berg JM. Proc Natl Acad Sci USA. 1992;89:11109. doi: 10.1073/pnas.89.23.11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Suske G. Gene. 1999;238:291. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 167.Kriwacki RW. J Biol Chem. 1997;272:7801. doi: 10.1074/jbc.272.12.7801. [DOI] [PubMed] [Google Scholar]
- 168.Nomura A, Sugiura Y. Inorg Chem. 2002;41:3693. doi: 10.1021/ic025557p. [DOI] [PubMed] [Google Scholar]
- 169.Nomura A, Sugiura Y. Inorg Chem. 2004;43:1708. doi: 10.1021/ic034931y. [DOI] [PubMed] [Google Scholar]
- 170.Klemba M, Gardner KH, Marino S, Clarke ND, Regan L. Nat Struct Biol. 1995;2:368. doi: 10.1038/nsb0595-368. [DOI] [PubMed] [Google Scholar]
- 171.Wisz MS, Garrett CZ, Hellinga HW. Biochemistry. 1998;37:8269. doi: 10.1021/bi980718f. [DOI] [PubMed] [Google Scholar]
- 172.Hori Y, Sugiura Y. J Am Chem Soc. 2002;124:9362. doi: 10.1021/ja026577t. [DOI] [PubMed] [Google Scholar]
- 173.Hori Y, Sugiura Y. Biochemistry. 2004;43:3068. doi: 10.1021/bi035742u. [DOI] [PubMed] [Google Scholar]
- 174.Qian YQ, Resendez-Perez D, Gehring WJ, Wüthrich K. Proc Natl Acad Sci USA. 1994;91:4091. doi: 10.1073/pnas.91.9.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Vallee BL, Auld DS. Proc Natl Acad Sci USA. 1990;87:220. doi: 10.1073/pnas.87.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tripp BC, Smith K, Ferry JG. J Biol Chem. 2001;276:48615. doi: 10.1074/jbc.R100045200. [DOI] [PubMed] [Google Scholar]
- 177.Smith KS, Ferry JG. FEMS Microbiol Rev. 2000;24:335. doi: 10.1111/j.1574-6976.2000.tb00546.x. [DOI] [PubMed] [Google Scholar]
- 178.Liljas A, Kannan KK, Bergstén PC, Waara I, Fridborg K, Strandberg B, Carlbom U, Järup L, Lövgren S, Petef M. Nature (London), New Biol. 1972;235:131. doi: 10.1038/newbio235131a0. [DOI] [PubMed] [Google Scholar]
- 179.Kannan KK, Notstrand B, Fridborg K, Lövgren S, Ohlsson A, Petef M. Proc Natl Acad Sci USA. 1975;72:51. doi: 10.1073/pnas.72.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Alberts IL, Nadassy K, Wodak SJ. Protein Sci. 1998;7:1700. doi: 10.1002/pro.5560070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Håkansson K, Carlsson M, Svensson LA, Liljas A. J Mol Biol. 1992;227:1192. doi: 10.1016/0022-2836(92)90531-n. [DOI] [PubMed] [Google Scholar]
- 182.Christianson DW, Fierke CA. Acc Chem Res. 1996;29:331. [Google Scholar]
- 183.Kiefer LL, Fierke CA. Biochemistry. 1994;33:15233. doi: 10.1021/bi00255a003. [DOI] [PubMed] [Google Scholar]
- 184.Kiefer LL, Ippolito JA, Fierke CA, Christianson DW. J Am Chem Soc. 1993;115:12581. [Google Scholar]
- 185.Alexander RS, Kiefer LL, Fierke CA, Christianson DW. Biochemistry. 1993;32:1510. doi: 10.1021/bi00057a015. [DOI] [PubMed] [Google Scholar]
- 186.Lesburg CA, Huang C, Christianson DW, Fierke CA. Biochemistry. 1997;36:15780. doi: 10.1021/bi971296x. [DOI] [PubMed] [Google Scholar]
- 187.Kiefer LL, Krebs JF, Paterno SA, Fierke CA. Biochemistry. 1993;32:9896. doi: 10.1021/bi00089a004. [DOI] [PubMed] [Google Scholar]
- 188.Krebs JF, Fierke CA, Alexander RS, Christianson DW. Biochemistry. 1991;30:9153. doi: 10.1021/bi00102a005. [DOI] [PubMed] [Google Scholar]
- 189.Kiefer LL, Paterno SA, Fierke CA. J Am Chem Soc. 1995;117:6831. [Google Scholar]
- 190.Lesburg CA, Christianson DW. J Am Chem Soc. 1995;117:6838. [Google Scholar]
- 191.Ippolito JA, Baird TT, McGee SA, Christianson DW, Fierke CA. Proc Natl Acad Sci USA. 1995;92:5017. doi: 10.1073/pnas.92.11.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fierke CA, Calderone TL, Krebs JF. Biochemistry. 1991;30:11054. doi: 10.1021/bi00110a007. [DOI] [PubMed] [Google Scholar]
- 193.Avvaru BS, Kim CU, Sippel KH, Gruner SM, Agbandje-McKenna M, Silverman DN, McKenna R. Biochemistry. 2010;49:249. doi: 10.1021/bi902007b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ippolito JA, Christianson DW. Biochemistry. 1993;32:9901. doi: 10.1021/bi00089a005. [DOI] [PubMed] [Google Scholar]
- 195.Carter ND, Dodgson SJ, Gros G, Tashian RE. The Carbonic Anhydrases: Cellular Physiology and Molecular Genetics. Springer; New York: 1991. p. 379. [Google Scholar]
- 196.Elleby B, Sjoblom B, Lindskog S. Eur J Biochem. 1999;262:516. doi: 10.1046/j.1432-1327.1999.00400.x. [DOI] [PubMed] [Google Scholar]
- 197.Gould SM, Tawfik DS. Biochemistry. 2005;44:5444. doi: 10.1021/bi0475471. [DOI] [PubMed] [Google Scholar]
- 198.Höst G, Mårtensson L-G, Jonsson B-H. Biochim Biophys Acta. 2006;1764:1601. doi: 10.1016/j.bbapap.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 199.Höst GE, Jonsson B-H. Biochim Biophys Acta. 2008;1784:811. doi: 10.1016/j.bbapap.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 200.Nomura A, Sugiura Y. J Am Chem Soc. 2004;126:15374. doi: 10.1021/ja045663l. [DOI] [PubMed] [Google Scholar]
- 201.Negi S, Umeda Y, Masuyama S, Kano K, Sugiura Y. Bioorg Med Chem Lett. 2009;19:2789. doi: 10.1016/j.bmcl.2009.03.088. [DOI] [PubMed] [Google Scholar]
- 202.Porteus MH, Baltimore D. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 203.Nakatsukasa T, Shiraishi Y, Negi S, Imanishi M, Futaki S, Sugiura Y. Biochem Biophys Res Commun. 2005;330:247. doi: 10.1016/j.bbrc.2005.02.164. [DOI] [PubMed] [Google Scholar]
- 204.Khare SD, Kipnis Y, Greisen P, Takeuchi R, Ashani Y, Goldsmith M, Song Y, Gallaher JL, Silman I, Leader H, Sussman JL, Stoddard BL, Tawfik DS, Baker D. Nat Chem Biol. 2012;8:294. doi: 10.1038/nchembio.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zanghellini A, Jiang L, Wollacott AM, Cheng G, Meiler J, Althoff EA, Röthlisberger D, Baker D. Protein Sci. 2006;15:2785. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Jiang L, Althoff EA, Clemente FR, Doyle L, Röthlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, Hilvert D, Houk KN, Stoddard BL, Baker D. Science. 2008;319:1387. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Röthlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D. Nature. 2008;453:190. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 208.Park H-S, Nam S, Lee JK, Yoon CN, Mannervik B, Benkovic SJ, Kim H-S. Science. 2006;311:535. doi: 10.1126/science.1118953. [DOI] [PubMed] [Google Scholar]
- 209.Otten LG, Quax WJ. Biomol Eng. 2005;22:1. doi: 10.1016/j.bioeng.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 210.Mills JH, Khare SD, Bolduc JM, Forouhar F, Mulligan VK, Lew S, Seetharaman J, Tong L, Stoddard BL, Baker D. J Am Chem Soc. 2013;135:13393. doi: 10.1021/ja403503m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Der BS, Edwards DR, Kuhlman B. Biochemistry. 2012;51:3933. doi: 10.1021/bi201881p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zastrow ML, Pecoraro VL. J Am Chem Soc. 2013;135:5895. doi: 10.1021/ja401537t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zastrow ML, Peacock AFA, Stuckey JA, Pecoraro VL. Nat Chem. 2012;4:118. doi: 10.1038/nchem.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, Pecoraro VL. Proc Natl Acad Sci USA. 2012;109:21234. doi: 10.1073/pnas.1212893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Faiella M, Andreozzi C, de Rosales RTM, Pavone V, Maglio O, Nastri F, DeGrado WF, Lombardi A. Nat Chem Biol. 2009;5:882. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kaplan J, DeGrado WF. Proc Natl Acad Sci USA. 2004;101:11566. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nastri F, Lista L, Ringhieri P, Vitale R, Faiella M, Andreozzi C, Travascio P, Maglio O, Lombardi A, Pavone V. Chem-Eur J. 2011;17:4444. doi: 10.1002/chem.201003485. [DOI] [PubMed] [Google Scholar]
- 218.Faiella M, Maglio O, Nastri F, Lombardi A, Lista L, Hagen WR, Pavone V. Chem-Eur J. 2012;18:15960. doi: 10.1002/chem.201201404. [DOI] [PubMed] [Google Scholar]
- 219.Maines MD. Annu Rev Pharmacol Toxicol. 1997;37:517. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- 220.Yoshikawa S. Adv Protein Chem. 2002;60:341. doi: 10.1016/s0065-3233(02)60057-9. [DOI] [PubMed] [Google Scholar]
- 221.Narula J, Pandey P, Arbustini E, Haider N, Narula N, Kolodgie FD, Dal Bello B, Semigran MJ, Bielsa-Masdeu A, Dec GW, Israels S, Ballester M, Virmani R, Saxena S, Kharbanda S. Proc Natl Acad Sci USA. 1999;96:8144. doi: 10.1073/pnas.96.14.8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rodgers KR. Curr Opin Chem Biol. 1999;3:158. doi: 10.1016/S1367-5931(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 223.Chan MK. Curr Opin Chem Biol. 2001;5:216. doi: 10.1016/s1367-5931(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 224.Anderson GJ, Vulpe CD. Cell Mol Life Sci. 2009;66:3241. doi: 10.1007/s00018-009-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Martin AC, Orengo CA, Hutchinson EG, Jones S, Karmirantzou M, Laskowski RA, Mitchell JB, Taroni C, Thornton JM. Structure. 1998;6:875. doi: 10.1016/s0969-2126(98)00089-6. [DOI] [PubMed] [Google Scholar]
- 226.Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. J Cereb Blood Flow Metab. 2003;23:629. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- 227.Cordova JM, Noack PL, Hilcove SA, Lear JD, Ghirlanda G. J Am Chem Soc. 2007;129:512. doi: 10.1021/ja057495i. [DOI] [PubMed] [Google Scholar]
- 228.Yeung BKS, Wang X, Sigman JA, Petillo PA, Lu Y. Chem Biol. 1997;4:215. doi: 10.1016/s1074-5521(97)90291-x. [DOI] [PubMed] [Google Scholar]
- 229.Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. Proc Natl Acad Sci USA. 2003;100:3629. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhao X, Yeung N, Russell BS, Garner DK, Lu Y. J Am Chem Soc. 2006;128:6766. doi: 10.1021/ja058822p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Liu X, Yu Y, Hu C, Zhang W, Lu Y, Wang J. Angew Chem, Int Ed. 2012;51:4312. doi: 10.1002/anie.201108756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Yeung N, Lin Y-W, Gao Y-G, Zhao X, Russell BS, Lei L, Miner KD, Robinson H, Lu Y. Nature. 2009;462:1079. doi: 10.1038/nature08620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lin Y-W, Yeung N, Gao Y-G, Miner KD, Tian S, Robinson H, Lu Y. Proc Natl Acad Sci USA. 2010;107:8581. doi: 10.1073/pnas.1000526107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kitagishi H, Oohora K, Yamaguchi H, Sato H, Matsuo T, Harada A, Hayashi T. J Am Chem Soc. 2007;129:10326. doi: 10.1021/ja073295q. [DOI] [PubMed] [Google Scholar]
- 235.Kitagishi H, Kakikura Y, Yamaguchi H, Oohora K, Harada A, Hayashi T. Angew Chem, Int Ed. 2009;48:1271. doi: 10.1002/anie.200804006. [DOI] [PubMed] [Google Scholar]
- 236.Onoda A, Ueya Y, Sakamoto T, Uematsu T, Hayashi T. Chem Commun (Cambridge, UK) 2010;46:9107. doi: 10.1039/c0cc03430d. [DOI] [PubMed] [Google Scholar]
- 237.Onoda A, Kakikura Y, Uematsu T, Kuwabata S, Hayashi T. Angew Chem, Int Ed. 2012;51:2628. doi: 10.1002/anie.201105186. [DOI] [PubMed] [Google Scholar]
- 238.Onoda A, Himiyama T, Ohkubo K, Fukuzumi S, Hayashi T. Chem Commun (Cambridge, UK) 2012;48:8054. doi: 10.1039/c2cc33046f. [DOI] [PubMed] [Google Scholar]
- 239.Oohora K, Onoda A, Kitagishi H, Yamaguchi H, Harada A, Hayashi T. Chem Sci. 2011;2:1033. [Google Scholar]
- 240.Oohora K, Burazerovic S, Onoda A, Wilson YM, Ward TR, Hayashi T. Angew Chem, Int Ed. 2012;51:3818. doi: 10.1002/anie.201107067. [DOI] [PubMed] [Google Scholar]
- 241.Asakura T, Sono M. J Biol Chem. 1974;249:7087. [PubMed] [Google Scholar]
- 242.Singh UP, Obayashi E, Takahashi S, Iizuka T, Shoun H, Shiro Y. Biochim Biophys Acta. 1998;1384:103. doi: 10.1016/s0167-4838(98)00006-5. [DOI] [PubMed] [Google Scholar]
- 243.De Sandro V, Dupuy C, Kaniewski J, Ohayon R, Deme D, Virion A, Pommier J. Eur J Biochem. 1991;201:507. doi: 10.1111/j.1432-1033.1991.tb16310.x. [DOI] [PubMed] [Google Scholar]
- 244.Banci L, Bertini I, Branchini BR, Hajieva P, Spyroulias GA, Turano P. JBIC, J Biol Inorg Chem. 2001;6:490. doi: 10.1007/s007750100217. [DOI] [PubMed] [Google Scholar]
- 245.Sato H, Watanabe M, Hisaeda Y, Hayashi T. J Am Chem Soc. 2005;127:56. doi: 10.1021/ja044984u. [DOI] [PubMed] [Google Scholar]
- 246.Matsuo T, Nagai H, Hisaeda Y, Hayashi T. Chem Commun (Cambridge, UK) 2006:3131. doi: 10.1039/b606060a. [DOI] [PubMed] [Google Scholar]
- 247.Matsuo T, Ikegami T, Sato H, Hisaeda Y, Hayashi T. J Inorg Biochem. 2006;100:1265. doi: 10.1016/j.jinorgbio.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 248.Harada K, Makino M, Sugimoto H, Hirota S, Matsuo T, Shiro Y, Hisaeda Y, Hayashi T. Biochemistry. 2007;46:9406. doi: 10.1021/bi7007068. [DOI] [PubMed] [Google Scholar]
- 249.Matsuo T, Fukumoto K, Watanabe T, Hayashi T. Chem Asian J. 2011;6:2491. doi: 10.1002/asia.201100107. [DOI] [PubMed] [Google Scholar]
- 250.Hayashi T, Harada K, Sakurai K, Shimada H, Hirota S. J Am Chem Soc. 2009;131:1398. doi: 10.1021/ja807420k. [DOI] [PubMed] [Google Scholar]
- 251.Harada K, Sakurai K, Ikemura K, Ogura T, Hirota S, Shimada H, Hayashi T. J Am Chem Soc. 2008;130:432. doi: 10.1021/ja077902l. [DOI] [PubMed] [Google Scholar]
- 252.Sato H, Hayashi T, Ando T, Hisaeda Y, Ueno T, Watanabe Y. J Am Chem Soc. 2004;126:436. doi: 10.1021/ja038798k. [DOI] [PubMed] [Google Scholar]
- 253.Matsuo T, Dejima H, Hirota S, Murata D, Sato H, Ikegami T, Hori H, Hisaeda Y, Hayashi T. J Am Chem Soc. 2004;126:16007. doi: 10.1021/ja045880m. [DOI] [PubMed] [Google Scholar]
- 254.Hayashi T, Murata D, Makino M, Sugimoto H, Matsuo T, Sato H, Shiro Y, Hisaeda Y. Inorg Chem. 2006;45:10530. doi: 10.1021/ic061130x. [DOI] [PubMed] [Google Scholar]
- 255.Matsuo T, Ito K, Nakashima Y, Hisaeda Y, Hayashi T. J Inorg Biochem. 2008;102:166. doi: 10.1016/j.jinorgbio.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 256.Matsuo T, Murata D, Hisaeda Y, Hori H, Hayashi T. J Am Chem Soc. 2007;129:12906. doi: 10.1021/ja074685f. [DOI] [PubMed] [Google Scholar]
- 257.Matsuo T, Hayashi A, Abe M, Matsuda T, Hisaeda Y, Hayashi T. J Am Chem Soc. 2009;131:15124. doi: 10.1021/ja907428e. [DOI] [PubMed] [Google Scholar]
- 258.Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM. Nat Chem Biol. 2013;9:485. doi: 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- 260.Zhao X, Yeung N, Wang Z, Guo Z, Lu Y. Biochemistry. 2005;44:1210. doi: 10.1021/bi0479151. [DOI] [PubMed] [Google Scholar]
- 261.Korendovych IV, Senes A, Kim YH, Lear JD, Fry HC, Therien MJ, Blasie JK, Walker FA, Degrado WF. J Am Chem Soc. 2010;132:15516. doi: 10.1021/ja107487b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Reedy CJ, Kennedy ML, Gibney BR. Chem Commun (Cambridge, UK) 2003;1:570. doi: 10.1039/b211488g. [DOI] [PubMed] [Google Scholar]
- 263.Privett HK, Reedy CJ, Kennedy ML, Gibney BR. J Am Chem Soc. 2002;124:6828. doi: 10.1021/ja025534+. [DOI] [PubMed] [Google Scholar]
- 264.Zhuang J, Amoroso JH, Kinloch R, Dawson JH, Baldwin MJ, Gibney BR. Inorg Chem. 2004;43:8218. doi: 10.1021/ic048502r. [DOI] [PubMed] [Google Scholar]
- 265.Zhuang J, Amoroso JH, Kinloch R, Dawson JH, Baldwin MJ, Gibney BR. Inorg Chem. 2006;45:4685. doi: 10.1021/ic060072c. [DOI] [PubMed] [Google Scholar]
- 266.Zhang L, Anderson JLR, Ahmed I, Norman JA, Negron C, Mutter AC, Dutton PL, Koder RL. Biochemistry. 2011;50:10254. doi: 10.1021/bi201242a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Rau HK, DeJonge N, Haehnel W. Proc Natl Acad Sci USA. 1998;95:11526. doi: 10.1073/pnas.95.20.11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Lombardi A, Nastri F, Marasco D, Maglio O, De Sanctis G, Sinibaldi F, Santucci R, Coletta M, Pavone V. Chem—Eur J. 2003;9:5643. doi: 10.1002/chem.200304831. [DOI] [PubMed] [Google Scholar]
- 269.Das A, Trammell SA, Hecht MH. Biophys Chem. 2006;123:102. doi: 10.1016/j.bpc.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 270.Howard JB, Rees DC. Adv Protein Chem. 1991;42:199. doi: 10.1016/s0065-3233(08)60537-9. [DOI] [PubMed] [Google Scholar]
- 271.Farinas E, Regan L. Protein Sci. 1998;7:1939. doi: 10.1002/pro.5560070909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Baldwin EP, Hajiseyedjavadi O, Baase Wa, Matthews BW. Science. 1993;262:1715. doi: 10.1126/science.8259514. [DOI] [PubMed] [Google Scholar]
- 273.Lim WA, Hodel A, Sauer RT, Richards FM. Proc Natl Acad Sci USA. 1994;91:423. doi: 10.1073/pnas.91.1.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Benson DE, Wisz MS, Liu W, Hellinga HW. Biochemistry. 1998;2960:7070. doi: 10.1021/bi980583d. [DOI] [PubMed] [Google Scholar]
- 275.Coldren CD, Hellinga HW, Caradonna JP. Proc Natl Acad Sci USA. 1997;94:6635. doi: 10.1073/pnas.94.13.6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Hill CL, Renaud J, Holm RH, Mortenson LE. J Am Chem Soc. 1977;99:2549. doi: 10.1021/ja00450a024. [DOI] [PubMed] [Google Scholar]
- 277.Meyer TE, Przysiecki CT, Watkins JA, Bhattacharyya A, Simondsen RP, Cusanovich MA, Tollin G. Proc Natl Acad Sci U SA. 1983;80:6740. doi: 10.1073/pnas.80.22.6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Hoppe A, Pandelia M, Gärtner W, Lubitz W. Biochim Biophys Acta. 2011;1807:1414. doi: 10.1016/j.bbabio.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 279.Arciero DM, Lipscomb JD, Huynh BH, Kent TA, Münck E. J Biol Chem. 1983;258:14981. [PubMed] [Google Scholar]
- 280.Sugimoto K, Senda T, Aoshima H, Masai E, Fukuda M, Mitsui Y. Structure. 1999;7:953. doi: 10.1016/s0969-2126(99)80122-1. [DOI] [PubMed] [Google Scholar]
- 281.Kita A, Kita S, Fujisawa I, Inaka K, Ishida T, Horiike K, Nozaki M, Miki K. Structure. 1999;7:25. doi: 10.1016/s0969-2126(99)80006-9. [DOI] [PubMed] [Google Scholar]
- 282.Sieker LC, Holmes M, Le Trong I, Turley S, Liu MY, LeGall J, Stenkamp RE. JBIC, J Biol Inorg Chem. 2000;5:505. doi: 10.1007/pl00021450. [DOI] [PubMed] [Google Scholar]
- 283.Smith P, Zhou B, Ho N, Yuan Y-C, Su L, Tsai S-C, Yen Y. Biochemistry. 2009;48:11134. doi: 10.1021/bi9001425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zhang Z, Ren J, Clifton IJ, Schofield CJ. Chem Biol. 2004;11:1383. doi: 10.1016/j.chembiol.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 285.Muñoz IG, Moran JF, Becana M, Montoya G. Protein Sci. 2005;14:387. doi: 10.1110/ps.04979505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Erlandsen H, Fusetti F, Martinez A, Hough E, Flatmark T, Stevens RC. Nat Struct Biol. 1997;4:995. doi: 10.1038/nsb1297-995. [DOI] [PubMed] [Google Scholar]
- 287.Fusetti F, Erlandsen H, Flatmark T, Stevens RC. J Biol Chem. 1998;273:16962. doi: 10.1074/jbc.273.27.16962. [DOI] [PubMed] [Google Scholar]
- 288.Zhang Z, Ren JS, Harlos K, McKinnon CH, Clifton IJ, Schofield CJ. FEBS Lett. 2002;517:7. doi: 10.1016/s0014-5793(02)02520-6. [DOI] [PubMed] [Google Scholar]
- 289.Lehmann TE, Ming LJ, Rosen ME, Que L. Biochemistry. 1997;36:2807. doi: 10.1021/bi962748t. [DOI] [PubMed] [Google Scholar]
- 290.Takahashi S, Sam JW, Peisach J, Rousseau DL. J Am Chem Soc. 1994;116:4408. [Google Scholar]
- 291.Dumas P, Bergdoll M, Cagnon C, Masson JM. EMBO J. 1994;13:2483. doi: 10.1002/j.1460-2075.1994.tb06535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Kovaleva EG, Lipscomb JD. Nat Chem Biol. 2008;4:186. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Ryle MJ, Hausinger RP. Curr Opin Chem Biol. 2002;6:193. doi: 10.1016/s1367-5931(02)00302-2. [DOI] [PubMed] [Google Scholar]
- 294.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 295.Miller A-F. FEBS Lett. 2012;586:585. doi: 10.1016/j.febslet.2011.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Wuerges J, Lee J-W, Yim Y-I, Yim H-S, Kang S-O, Djinovic Carugo K. Proc Natl Acad Sci USA. 2004;101:8569. doi: 10.1073/pnas.0308514101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Zhang N-N, He Y-X, Li W-F, Teng Y-B, Yu J, Chen Y, Zhou C-Z. Proteins: Struct, Funct, Genet. 2010;78:1999. doi: 10.1002/prot.22709. [DOI] [PubMed] [Google Scholar]
- 298.Stallings WC, Pattridge KA, Strong RK, Ludwig ML. J Biol Chem. 1984;259:10695. [PubMed] [Google Scholar]
- 299.Stallings WC, Powers TB, Pattridge KA, Fee JA, Ludwig ML. Proc Natl Acad Sci USA. 1983;80:3884. doi: 10.1073/pnas.80.13.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Yamakura F, Suzuki K. J Biochem. 1980;88:191. [PubMed] [Google Scholar]
- 301.Brock CJ, Harris JI. Biochem Soc Trans. 1977;5:1537. doi: 10.1042/bst0051537. [DOI] [PubMed] [Google Scholar]
- 302.Ose DE, Fridovich I. Arch Biochem Biophys. 1979;194:360. doi: 10.1016/0003-9861(79)90628-3. [DOI] [PubMed] [Google Scholar]
- 303.Vance CK, Miller A-F. J Am Chem Soc. 1998;120:461. [Google Scholar]
- 304.Miller A-F. Acc Chem Res. 2008;41:501. doi: 10.1021/ar700237u. [DOI] [PubMed] [Google Scholar]
- 305.Stallings WC, Metzger AL, Pattridge KA, Fee JA, Ludwig ML. Free Radical Res Commun. 1991;12–13:259. doi: 10.3109/10715769109145794. [DOI] [PubMed] [Google Scholar]
- 306.Pinto AL, Hellinga HW, Caradonna JP. Proc Natl Acad Sci USA. 1997;94:5562. doi: 10.1073/pnas.94.11.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Richardson DJ, Watmough NJ. Curr Opin Chem Biol. 1999;3:207. doi: 10.1016/S1367-5931(99)80034-9. [DOI] [PubMed] [Google Scholar]
- 308.Watmough NJ, Butland G, Cheesman MR, Moir JW, Richardson DJ, Spiro S. Biochim Biophys Acta. 1999;1411:456. doi: 10.1016/s0005-2728(99)00032-8. [DOI] [PubMed] [Google Scholar]
- 309.Sigman JA, Kwok BC, Lu Y. J Am Chem Soc. 2000;122:8192. [Google Scholar]
- 310.Flock U, Lachmann P, Reimann J, Watmough NJ, Adelroth P. J Inorg Biochem. 2009;103:845. doi: 10.1016/j.jinorgbio.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 311.Butland G, Spiro S, Watmough NJ, Richardson DJ. J Bacteriol. 2001;183:189. doi: 10.1128/JB.183.1.189-199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Stenkamp RE. Chem Rev. 1994;94:715. [Google Scholar]
- 313.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Nature. 1993;366:537. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 314.de Maré F, Nordlund P, Gupta N, Shenvi NV, Cui X, Kurtz DM. Inorg Chim Acta. 1997;263:255. [Google Scholar]
- 315.Ormö M, DeMaré F, Regnström K, Aberg A, Sahlin M, Ling J, Loehr TM, Sanders-Loehr J, Sjöberg BM. J Biol Chem. 1992;267:8711. [PubMed] [Google Scholar]
- 316.Larsson A, Sjöberg BM. EMBO J. 1986;5:2037. doi: 10.1002/j.1460-2075.1986.tb04461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Nordlund P, Sjöberg BM, Eklund H. Nature. 1990;345:593. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 318.Sano Y, Onoda A, Sakurai R, Kitagishi H, Hayashi T. J Inorg Biochem. 2011;105:702. doi: 10.1016/j.jinorgbio.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 319.Sano Y, Onoda A, Hayashi T. Chem Commun. 2011;47:8229. doi: 10.1039/c1cc11157d. [DOI] [PubMed] [Google Scholar]
- 320.Volbeda A, Charon MH, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC. Nature. 1995;373:580. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- 321.Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC. Science. 1998;282:1853. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- 322.Nicolet Y, Piras C, Legrand P, Hatchikian CE, Fontecilla-Camps JC. Structure. 1999;7:13. doi: 10.1016/s0969-2126(99)80005-7. [DOI] [PubMed] [Google Scholar]
- 323.Mulder DW, Boyd ES, Sarma R, Lange RK, Endrizzi JA, Broderick JB, Peters JW. Nature. 2010;465:248. doi: 10.1038/nature08993. [DOI] [PubMed] [Google Scholar]
- 324.Berggren G, Adamska A, Lambertz C, Simmons TR, Esselborn J, Atta M, Gambarelli S, Mouesca J-M, Reijerse E, Lubitz W, Happe T, Artero V, Fontecave M. Nature. 2013;499:66. doi: 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Malkin R, Malmström BG. Adv Enzymol Relat Areas Mol Biol. 1970;33:177. doi: 10.1002/9780470122785.ch4. [DOI] [PubMed] [Google Scholar]
- 326.Lu Y. Compr Coord Chem II. 2003;8:91. [Google Scholar]
- 327.Messerschmidt A, Huber R, Poulos T, Wieghardt K. Handbook of Metalloproteins: Cupredoxins (Type-1 Copper Proteins) Wiley; New York: 2001. p. 1153. [Google Scholar]
- 328.Hart P, Nersissian A, George S. Encyclopedia of Inorganic Bioinorganic Chemistry. Wiley; New York: 2011. [Google Scholar]
- 329.Gray HB, Malmström BG, Williams RJP. JBIC, J Biol Inorg Chem. 2000;5:551. doi: 10.1007/s007750000146. [DOI] [PubMed] [Google Scholar]
- 330.Solomon E, Gorelsky S, Dey A. Wiley Intersci. 2006;405:1415. [Google Scholar]
- 331.Lowery M, Solomon E. Inorg Chim Acta. 1992;200:233. [Google Scholar]
- 332.Visser JW, Amesz J, Van Gelder BF. Biochim Biophys Acta. 1974;333:279. doi: 10.1016/0005-2728(74)90011-5. [DOI] [PubMed] [Google Scholar]
- 333.Gewirth AA, Solomon EI. J Am Chem Soc. 1988;110:3811. [Google Scholar]
- 334.Sykes AG. Adv Inorg Chem. 1991;36:377. [Google Scholar]
- 335.Gray HB. Metals Ions in Biology. Wiley; New York: 1980. p. 1. [Google Scholar]
- 336.Solomon EI, Hare JW, Gray HB. Proc Natl Acad Sci USA. 1976;73:1389. doi: 10.1073/pnas.73.5.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Canters GW, Gilardi G. FEBS Lett. 1993;325:39. doi: 10.1016/0014-5793(93)81410-2. [DOI] [PubMed] [Google Scholar]
- 338.Ainscough EW, Bingham AG, Brodie AM, Ellis WR, Gray HB, Loehr TM, Plowman JE, Norris GE, Baker EN. Biochemistry. 1987;26:71. doi: 10.1021/bi00375a011. [DOI] [PubMed] [Google Scholar]
- 339.Colman P, Freeman H, Guss J, Murata M, Norris V, Ramshaw A, Venkatappa M. Nature. 1978;272:319. [Google Scholar]
- 340.Guss JM, Freeman HC. J Mol Biol. 1983;169:521. doi: 10.1016/s0022-2836(83)80064-3. [DOI] [PubMed] [Google Scholar]
- 341.Guss JM, Bartunik HD, Freeman HC. Acta Crystallogr, Sect B. 1992;48:790. doi: 10.1107/s0108768192004270. [DOI] [PubMed] [Google Scholar]
- 342.Solomon EI. J Inorg Biochem. 1992;47:29. [Google Scholar]
- 343.Solomon EI, Lowery M. Science. 1993;259:1575. doi: 10.1126/science.8384374. [DOI] [PubMed] [Google Scholar]
- 344.Solomon EI, Lowery M, Lacroix L, Root D. Methods Enzymol. 1993;226:1. doi: 10.1016/0076-6879(93)26003-r. [DOI] [PubMed] [Google Scholar]
- 345.Guckert JA, Lowery MD, Solomon EI. J Am Chem Soc. 1995;117:2817. [Google Scholar]
- 346.Solomon EI, Penfield KW, Gewirth AA. Inorg Chim Acta. 1996;243:67. [Google Scholar]
- 347.Lacroix LB, Shadle SE, Wang Y, Averill BA, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1996:7755. [Google Scholar]
- 348.LaCroix L, Randall D. J Am Chem Soc. 1998:9621. [Google Scholar]
- 349.Solomon EI, Randall DW, Glaser T. Coord Chem Rev. 2000;200–202:595. [Google Scholar]
- 350.Hannan JP, Busch JLH, Breton J, James R, Thomson AJ, Moore GR, Davy SL. JBIC, J Biol Inorg Chem. 2000;5:432. doi: 10.1007/pl00021445. [DOI] [PubMed] [Google Scholar]
- 351.Solomon EI, Szilagyi RK, DeBeer George S, Basumallick L. Chem Rev. 2004;104:419. doi: 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]
- 352.Solomon EI. Inorg Chem. 2006;45:8012. doi: 10.1021/ic060450d. [DOI] [PubMed] [Google Scholar]
- 353.Solomon EI, Hadt RG. Coord Chem Rev. 2011;255:774. [Google Scholar]
- 354.Cox JC, Boxer DH. Biochem J. 1978;174:497. doi: 10.1042/bj1740497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Cox JC, Aasa R, Maimstrom BG, Press ENB. FEBS Lett. 1978;93:157. doi: 10.1016/0014-5793(78)80826-6. [DOI] [PubMed] [Google Scholar]
- 356.Peisach J, Levine WG, Blumberg WE. J Biol Chem. 1967;242:2847. [PubMed] [Google Scholar]
- 357.Malmström BG, Reinhammar B, Vänngård T. Biochim Biophys Acta—Bioenerg. 1970;205:48. doi: 10.1016/0005-2728(70)90060-5. [DOI] [PubMed] [Google Scholar]
- 358.Kakutani T, Watanabe H, Arima K, Beppu T. J Biochem. 1981;89:463. doi: 10.1093/oxfordjournals.jbchem.a133221. [DOI] [PubMed] [Google Scholar]
- 359.Arciero DM, Pierce BS, Hendrich MP, Hooper AB. Biochemistry. 2002;41:1703. doi: 10.1021/bi015908w. [DOI] [PubMed] [Google Scholar]
- 360.Lieberman RL, Arciero DM, Hooper AB, Rosenzweig AC. Biochemistry. 2001;40:5674. doi: 10.1021/bi0102611. [DOI] [PubMed] [Google Scholar]
- 361.Basumallick L, Sarangi R, DeBeer George S, Elmore B, Hooper AB, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2005;127:3531. doi: 10.1021/ja044412+. [DOI] [PubMed] [Google Scholar]
- 362.Malmström BG. Chem Rev. 1990;90:1247. [Google Scholar]
- 363.Babcock G, Wikström M. Nature. 1992;356:302. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 364.Musser SM, Stowell MH, Chan SI. FEBS Lett. 1993;327:131. doi: 10.1016/0014-5793(93)80156-o. [DOI] [PubMed] [Google Scholar]
- 365.Pascher T, Karlsson BG, Nordling M, Malmström BG, Vänngård T. Eur J Biochem. 1993;212:289. doi: 10.1111/j.1432-1033.1993.tb17661.x. [DOI] [PubMed] [Google Scholar]
- 366.Lappalainen P, Saraste M. Biochim Biophys Acta. 1994;1187:222. [Google Scholar]
- 367.Beinert H. Eur J Biochem. 1997;245:521. doi: 10.1111/j.1432-1033.1997.t01-1-00521.x. [DOI] [PubMed] [Google Scholar]
- 368.Solomon EI, Xie X, Dey A. Chem Soc Rev. 2008;37:623. doi: 10.1039/b714577m. [DOI] [PubMed] [Google Scholar]
- 369.Ramirez BE. Proc Natl Acad Sci USA. 1995;92:11949. doi: 10.1073/pnas.92.26.11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Kroneck P, Riester J, Zumft W, Antholine W. Biol Met. 1990;3:103. doi: 10.1007/BF01179514. [DOI] [PubMed] [Google Scholar]
- 371.Zumft WG, Kroneck P. Adv Inorg Biochem. 1996;11:193. [Google Scholar]
- 372.Hay M, Ang M, Gamelin D, Solomon E, Antholine WE, Ralle M, Blackburn NJ, Massey PD, Wang X, Kwon AH, Lu Y. Inorg Chem. 1998:191. [Google Scholar]
- 373.Gamelin DR, Randall DW, Hay MT, Houser RP, Mulder TC, Canters GW, Vries S, De Tolman WB, Lu Y, Solomon EI. J Am Chem Soc. 1998:5246. [Google Scholar]
- 374.Szilagyi RK, Solomon EI. Curr Opin Chem Biol. 2002;6:250. doi: 10.1016/s1367-5931(02)00304-6. [DOI] [PubMed] [Google Scholar]
- 375.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 376.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 377.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature. 1995;376:660. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 378.Ostermeier C, Harrenga A, Ermler U, Michel H. Proc Natl Acad Sci USA. 1997;94:10547. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Blackburn NJ, Barr ME, Woodruff WH, van der Ooost J, de Vries S. Biochemistry. 1994;33:10401. doi: 10.1021/bi00200a022. [DOI] [PubMed] [Google Scholar]
- 380.Blackburn NJ, de Vries S, Barr ME, Houser RP, Tolman WB, Sanders D, Fee JA. J Am Chem Soc. 1997;119:6135. [Google Scholar]
- 381.Kroneck PM, Antholine WA, Riester J, Zumft WG. FEBS Lett. 1988;242:70. doi: 10.1016/0014-5793(88)80987-6. [DOI] [PubMed] [Google Scholar]
- 382.Kroneck PM, Antholine WA, Riester J, Zumft WG. FEBS Lett. 1989;248:212. doi: 10.1016/0014-5793(89)80464-8. [DOI] [PubMed] [Google Scholar]
- 383.Kroneck PM, Antholine WE, Kastrau DH, Buse G, Steffens GC, Zumft WG. FEBS Lett. 1990;268:274. doi: 10.1016/0014-5793(90)81026-k. [DOI] [PubMed] [Google Scholar]
- 384.Riester J, Zumft WGG, Kroneck PMM. Eur J Biochem. 1989;178:751. doi: 10.1111/j.1432-1033.1989.tb14506.x. [DOI] [PubMed] [Google Scholar]
- 385.Chan SI, Li PM. Biochemistry. 1990;29:1. doi: 10.1021/bi00453a001. [DOI] [PubMed] [Google Scholar]
- 386.Morgan JE, Wikström M. Biochemistry. 1991;30:948. doi: 10.1021/bi00218a010. [DOI] [PubMed] [Google Scholar]
- 387.Immoos C, Hill MG, Sanders D, Fee JA, Slutter CE, Richards JH, Gray HB. JBIC, J Biol Inorg Chem. 1996;1:529. [Google Scholar]
- 388.Berry SM, Gieselman MD, Nilges MJ, van der Donk WA, Lu Y. J Am Chem Soc. 2002;124:2084. doi: 10.1021/ja0169163. [DOI] [PubMed] [Google Scholar]
- 389.Ralle M, Berry SM, Nilges MJ, Gieselman MD, van der Donk Wa, Lu Y, Blackburn NJ. J Am Chem Soc. 2004;126:7244. doi: 10.1021/ja031821h. [DOI] [PubMed] [Google Scholar]
- 390.Berry SM, Ralle M, Low DW, Blackburn NJ, Lu Y. J Am Chem Soc. 2003;125:8760. doi: 10.1021/ja029699u. [DOI] [PubMed] [Google Scholar]
- 391.Garner DK, Vaughan MD, Hwang HJ, Savelieff MG, Berry SM, Honek JF, Lu Y. J Am Chem Soc. 2006;128:15608. doi: 10.1021/ja062732i. [DOI] [PubMed] [Google Scholar]
- 392.Clark KM, Yu Y, Marshall NM, Sieracki NA, Nilges MJ, Blackburn NJ, van der Donk Wa, Lu Y. J Am Chem Soc. 2010;132:10093. doi: 10.1021/ja102632p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Farver O, Marshall NM, Wherland S, Lu Y, Pecht I. Proc Natl Acad Sci USA. 2013;110:10536. doi: 10.1073/pnas.1215081110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Van der Oost J, Lappalainen P, Musacchio A, Warne A, Lemieux L, Rumbley J, Gennis RB, Aasa R, Pascher T, Malmström BG. EMBO J. 1992;11:3209. doi: 10.1002/j.1460-2075.1992.tb05398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Hay M, Richards JH, Lu Y. Proc Natl Acad Sci USA. 1996;93:461. doi: 10.1073/pnas.93.1.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Hwang HJ, Lu Y. Proc Natl Acad Sci USA. 2004;101:12842. doi: 10.1073/pnas.0403473101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Hwang HJ, Berry SM, Nilges MJ, Lu Y. J Am Chem Soc. 2005;127:7274. doi: 10.1021/ja0501114. [DOI] [PubMed] [Google Scholar]
- 398.Farver O, Lu Y, Ang MC, Pecht I. Proc Natl Acad Sci USA. 1999;96:899. doi: 10.1073/pnas.96.3.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.New SY, Marshall NM, Hor TSA, Xue F, Lu Y. Chem Commun (Cambridge, UK) 2012;48:4217. doi: 10.1039/c2cc30901g. [DOI] [PubMed] [Google Scholar]
- 400.Dennison C, Vijgenboom E, de Vries S, van der Oost J, Canters GW. FEBS Lett. 1995;365:92. doi: 10.1016/0014-5793(95)00429-d. [DOI] [PubMed] [Google Scholar]
- 401.Jones LH, Liu A, Davidson VL. J Biol Chem. 2003;278:47269. doi: 10.1074/jbc.M308863200. [DOI] [PubMed] [Google Scholar]
- 402.Kitagawa Y, Tanaka N, Hata Y, Kusunoki M, Lee GP, Katsube Y, Asada K, Aibara S, Morita Y. J Biochem. 1991;109:477. doi: 10.1093/oxfordjournals.jbchem.a123407. [DOI] [PubMed] [Google Scholar]
- 403.Djinović Carugo K, Battistoni A, Carrì MT, Polticelli F, Desideri A, Rotilio G, Coda A, Wilson KS, Bolognesi M. Acta Crystallogr, Sect D. 1996;D52:176. doi: 10.1107/S0907444995007608. [DOI] [PubMed] [Google Scholar]
- 404.Miller A. Compr Coord Chem II. 2003;8:479. [Google Scholar]
- 405.Lu Y, Gralla EB, Roe JA, Valentine JS. J Am Chem Soc. 1992;114:3560. [Google Scholar]
- 406.Lu Y, LaCroix LB, Lowery MD, Solomon EI, Bender CJ, Peisach J, Roe JA, Gralla EB, Valentine JS. J Am Chem Soc. 1993;115:5907. [Google Scholar]
- 407.Han J, Loehr TM, Lu Y, Valentine JS, Averill BA, Sanders-Loehr J. J Am Chem Soc. 1993;115:4256. [Google Scholar]
- 408.Lu Y, Roe JA, Bender CJ, Peisach J, Banci L, Bertini I, Gralla EB, Valentine JS. Inorg Chem. 1996;35:1692. doi: 10.1021/ic9513189. [DOI] [PubMed] [Google Scholar]
- 409.Banci L, Bertini I, Borsari M, Viezzoli MS, Hallewell RA. Eur J Biochem. 1995;232:220. doi: 10.1111/j.1432-1033.1995.tb20802.x. [DOI] [PubMed] [Google Scholar]
- 410.Tainer JA, Getzoff ED, Beem KM, Richardson JS, Richardson DC. J Mol Biol. 1982;160:181. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 411.Hellinga HW. J Am Chem Soc. 1998;120:10055. [Google Scholar]
- 412.Katti SK, LeMaster DM, Eklund H. J Mol Biol. 1990;212:167. doi: 10.1016/0022-2836(90)90313-B. [DOI] [PubMed] [Google Scholar]
- 413.Korszun ZR. J Mol Biol. 1987;196:413. doi: 10.1016/0022-2836(87)90701-7. [DOI] [PubMed] [Google Scholar]
- 414.Adman ET, Stenkamp RE, Sieker LC, Jensen LH. J Mol Biol. 1978;123:35. doi: 10.1016/0022-2836(78)90375-3. [DOI] [PubMed] [Google Scholar]
- 415.Baker EN. J Mol Biol. 1988;203:1071. doi: 10.1016/0022-2836(88)90129-5. [DOI] [PubMed] [Google Scholar]
- 416.Nar H, Messerschmidt A, Huber R, van de Kamp M, Canters GW. J Mol Biol. 1991;221:765. doi: 10.1016/0022-2836(91)80173-r. [DOI] [PubMed] [Google Scholar]
- 417.Vijgenboom E, Busch JE, Canters GW. Microbiology. 1997;143:2853. doi: 10.1099/00221287-143-9-2853. [DOI] [PubMed] [Google Scholar]
- 418.Piccioli M, Luchinat C, Mizoguchi TJ, Ramirez BE, Gray HB, Richards JH. Inorg Chem. 1995;34:737. [Google Scholar]
- 419.Faham S, Mizoguchi TJ, Adman ET, Gray HB, Richards JH, Rees DC. JBIC, J Biol Inorg Chem. 1997;2:464. [Google Scholar]
- 420.DeBeer S, Kiser CN, Mines GA, Richards JH, Gray HB, Solomon EI, Hedman B, Hodgson KO. Inorg Chem. 1999;38:433. doi: 10.1021/ic9804622. [DOI] [PubMed] [Google Scholar]
- 421.Lancaster KM, DeBeer George S, Yokoyama K, Richards JH, Gray HB. Nat Chem. 2009;1:711. doi: 10.1038/nchem.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Lancaster KM, Yokoyama K, Richards JH, Winkler JR, Gray HB. Inorg Chem. 2009;48:1278. doi: 10.1021/ic802322e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Lancaster KM, Sproules S, Palmer JH, Richards JH, Gray HB. J Am Chem Soc. 2010;132:14590. doi: 10.1021/ja105731x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Lancaster KM, Farver O, Wherland S, Crane EJ, Richards JH, Pecht I, Gray HB. J Am Chem Soc. 2011;133:4865. doi: 10.1021/ja1093919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Karlsson BG, Aasa R, Malmström BG, Lundberg LG. FEBS Lett. 1989;253:99. [Google Scholar]
- 426.Karlsson BG, Nordling M, Pascher T, Tsai L, Sjölin L, Lundberg LG. Protein Eng. 1991;4:343. doi: 10.1093/protein/4.3.343. [DOI] [PubMed] [Google Scholar]
- 427.Tsai LC, Bonander N, Harata K, Karlsson G, Vänngård T, Langer V, Sjölin L. Acta Crystallogr, Sect D. 1996;52:950. doi: 10.1107/S0907444996004982. [DOI] [PubMed] [Google Scholar]
- 428.Kroes SJ, Hoitink CWG, Andrew CR, Ai J, Sanders-Loehr J, Messerschmidt A, Hagen WR, Canters GW. Eur J Biochem. 1996;240:342. doi: 10.1111/j.1432-1033.1996.0342h.x. [DOI] [PubMed] [Google Scholar]
- 429.Bonander N, Karlsson BG, Vänngård T. Biochemistry. 1996;35:2429. doi: 10.1021/bi9522110. [DOI] [PubMed] [Google Scholar]
- 430.Salgado J, Kroes SJ, Berg A, Moratal JM, Canters GW. J Biol Chem. 1998;273:177. doi: 10.1074/jbc.273.1.177. [DOI] [PubMed] [Google Scholar]
- 431.Messerschmidt A, Prade L, Kroes SJ, Sanders-Loehr J, Huber R, Canters GW. Proc Natl Acad Sci USA. 1998;95:3443. doi: 10.1073/pnas.95.7.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Mizoguchi TTJ, Di Bilio AJ, Gray HB, Richards JH, Di Bilio A. J Am Chem Soc. 1992;114:10076. [Google Scholar]
- 433.Gray HB, Malmström BG. Comments Inorg Chem. 1983;2:203. [Google Scholar]
- 434.Ryde U, Olsson MHM, Roos BO, Borin AC. Theor Chem Acc. 2001;105:452. [Google Scholar]
- 435.Sailasuta N, Anson FC, Gray HB. J Am Chem Soc. 1979;101:455. [Google Scholar]
- 436.Muir TW, Sondhi D, Cole PA. Proc Natl Acad Sci USA. 1998;95:6705. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Hall J, Kanbi L, Strange R, Hasnain S. Biochemistry. 1999:12675. doi: 10.1021/bi990983g. [DOI] [PubMed] [Google Scholar]
- 438.Xu F, Palmer AE, Yaver S, Berka RM, Gambetta GA, Brown SH, Solomon EI. J Biol Chem. 1999;274:12372. doi: 10.1074/jbc.274.18.12372. [DOI] [PubMed] [Google Scholar]
- 439.Xu F, Berka RM, Wahleithner JA, Nelson BA, Shuster JR, Brown SH, Palmer AE, Solomon EI. Biochem J. 1998;334:63. doi: 10.1042/bj3340063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Nersissian AM, Immoos C, Hill MG, Hart PJ, Williams G, Herrmann RG, Valentine JS. Protein Sci. 1998;7:1915. doi: 10.1002/pro.5560070907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Berry SM, Bladholm EL, Mostad EJ, Schenewerk AR. JBIC, J Biol Inorg Chem. 2011;16:473. doi: 10.1007/s00775-010-0746-7. [DOI] [PubMed] [Google Scholar]
- 442.Lappin A, Lewis C, Ingledew W. Inorg Chem. 1985:1446. [Google Scholar]
- 443.Dennison C. Coord Chem Rev. 2005;249:3025. [Google Scholar]
- 444.Kanbi LD, Antonyuk S, Hough MA, Hall JF, Dodd FE, Hasnain SS. J Mol Biol. 2002;320:263. doi: 10.1016/S0022-2836(02)00443-6. [DOI] [PubMed] [Google Scholar]
- 445.Grossmann JG, Ingledew WJ, Harvey I, Strange RW, Hasnain SS. Biochemistry. 1995;34:8406. doi: 10.1021/bi00026a023. [DOI] [PubMed] [Google Scholar]
- 446.Alcaraz LA, Gómez J, Ramírez P, Calvente JJ, Andreu R, Donaire A. Bioinorg Chem Appl. 2007;2007:54232. doi: 10.1155/2007/54232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Romero A, Hoitink CW, Nar H, Huber R, Messerschmidt A, Canters GW. J Mol Biol. 1993;229:1007. doi: 10.1006/jmbi.1993.1101. [DOI] [PubMed] [Google Scholar]
- 448.Hadt RG, Sun N, Marshall NM, Hodgson KO, Hedman B, Lu Y, Solomon EI. J Am Chem Soc. 2012;134:16701. doi: 10.1021/ja306438n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Marcus RA, Sutin N. Biochim Biophys Acta. 1985;811:265. [Google Scholar]
- 450.Marcus RA. Angew Chem, Int Ed Engl. 1993;32:1111. [Google Scholar]
- 451.Rosenberger N, Studer A, Takatani N, Nakajima H, Watanabe Y. Angew Chem, Int Ed. 2009;48:1946. doi: 10.1002/anie.200804440. [DOI] [PubMed] [Google Scholar]
- 452.Hoffman AS, Stayton PS. Macromol Symp. 2004;207:139. [Google Scholar]
- 453.Heskins M, Guillet JE. J Macromol Sci, Part A. 1968;2:1441. [Google Scholar]
- 454.Schild HG. Prog Polym Sci. 1992;17:163. [Google Scholar]
- 455.Hay MT, Milberg RM, Lu Y. J Am Chem Soc. 1996;118:11976. [Google Scholar]
- 456.Wang X, Ang MC, Lu Y. J Am Chem Soc. 1999;121:2947. [Google Scholar]
- 457.Wilson TD, Savelieff MG, Nilges MJ, Marshall NM, Lu Y. J Am Chem Soc. 2011;133:20778. doi: 10.1021/ja205281t. [DOI] [PubMed] [Google Scholar]
- 458.Wang X, Berry SM, Xia Y, Lu Y. J Am Chem Soc. 1999;121:7449. [Google Scholar]
- 459.Berry SM, Wang X, Lu Y. J Inorg Biochem. 2000;78:89. doi: 10.1016/s0162-0134(99)00214-7. [DOI] [PubMed] [Google Scholar]
- 460.Hwang HJ, Nagraj N, Lu Y. Inorg Chem. 2006;45:102. doi: 10.1021/ic051375u. [DOI] [PubMed] [Google Scholar]
- 461.Kelly M, Lappalainen P, Talbo G, Haltia T, van der Oost J, Saraste M. J Biol Chem. 1993;268:16781. [PubMed] [Google Scholar]
- 462.Brown K, Tegoni M, Prudêncio M, Pereira AS, Besson S, Moura JJ, Moura I, Cambillau C. Nat Struct Biol. 2000;7:191. doi: 10.1038/73288. [DOI] [PubMed] [Google Scholar]
- 463.Coyle CL, Zumft WG, Kroneck PMH, Korner H, Jakob W. Eur J Biochem. 1985;153:459. doi: 10.1111/j.1432-1033.1985.tb09324.x. [DOI] [PubMed] [Google Scholar]
- 464.Farrar JA, Thomson AJ, Cheesman MR, Dooley DM, Zumft WG. FEBS Lett. 1991;294:11. doi: 10.1016/0014-5793(91)81331-2. [DOI] [PubMed] [Google Scholar]
- 465.Lappalainen P, Aasa R, Malmström BG, Saraste M. J Biol Chem. 1993;268:26416. [PubMed] [Google Scholar]
- 466.Chacón KN, Blackburn NJ. J Am Chem Soc. 2012;134:16401. doi: 10.1021/ja307276z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Robinson H, Ang MMC, Gao YG, Hay MMT, Lu Y, Wang AH. Biochemistry. 1999;38:5677. doi: 10.1021/bi9901634. [DOI] [PubMed] [Google Scholar]
- 468.Farrar JA, Lappalainen P, Zumft WG, Saraste M, Thomson AJ. Eur J Biochem. 1995;232:294. doi: 10.1111/j.1432-1033.1995.tb20811.x. [DOI] [PubMed] [Google Scholar]
- 469.Farrar JA, Neese F, Lappalainen P, Kroneck PMH, Saraste M, Zumft WG, Thomson AJ. J Am Chem Soc. 1996;118:11501. [Google Scholar]
- 470.Anthony C. Methods Enzymol. 1990;188:284. doi: 10.1016/0076-6879(90)88046-d. [DOI] [PubMed] [Google Scholar]
- 471.Davidson VL. Biosensor Design and Application; ACS Symposium Series; Washington, DC: American Chemical Society; 1992. p. 1. [Google Scholar]
- 472.Gray KA, Knaff DB, Husain M, Davidson VL. FEBS Lett. 1986;207:239. doi: 10.1016/0014-5793(86)81496-x. [DOI] [PubMed] [Google Scholar]
- 473.Andrew CR, Han J, de Vries S, van der Oost J, Averill BA, Loehr TM, Sanders-Loehr J. J Am Chem Soc. 1994;116:10805. [Google Scholar]
- 474.Babcock GT, Wikström M. Nature. 1992;356:301. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 475.Ferguson-Miller S, Babcock GT. Chem Rev. 1996;96:2889. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 476.García-Horsman JA, Barquera B, Rumbley J, Ma J, Gennis RB. J Bacteriol. 1994;176:5587. doi: 10.1128/jb.176.18.5587-5600.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Michel H, Behr J, Harrenga A, Kannt A. Annu Rev Biophys Biomol Struct. 1998;27:329. doi: 10.1146/annurev.biophys.27.1.329. [DOI] [PubMed] [Google Scholar]
- 478.Poulos TL, Li H, Raman C. Curr Opin Chem Biol. 1999;3:131. doi: 10.1016/s1367-5931(99)80024-6. [DOI] [PubMed] [Google Scholar]
- 479.Namslauer A, Brzezinski P. FEBS Lett. 2004;567:103. doi: 10.1016/j.febslet.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 480.Svensson-Ek M, Abramson J, Larsson G, Törnroth S, Brzezinski P, Iwata S. J Mol Biol. 2002;321:329. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 481.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Science. 1998;280:1723. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 482.Buse G, Soulimane T, Dewor M, Meyer HE, Blüggel M. Protein Sci. 1999;8:985. doi: 10.1110/ps.8.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Bu Y, Cukier RI. J Phys Chem B. 2005;109:22013. doi: 10.1021/jp053046t. [DOI] [PubMed] [Google Scholar]
- 484.Kaila VRI, Johansson MP, Sundholm D, Laakkonen L, Wikström M. Biochim Biophys Acta. 2009;1787:221. doi: 10.1016/j.bbabio.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 485.Miner KD, Mukherjee A, Gao Y-G, Null EL, Petrik ID, Zhao X, Yeung N, Robinson H, Lu Y. Angew Chem, Int Ed. 2012;51:5589. doi: 10.1002/anie.201201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Buschmann S, Warkentin E, Xie H, Langer JD, Ermler U, Michel H. Science. 2010;329:327. doi: 10.1126/science.1187303. [DOI] [PubMed] [Google Scholar]
- 487.Wade RS, Castro CE. Chem Res Toxicol. 1996;9:1382. doi: 10.1021/tx9600457. [DOI] [PubMed] [Google Scholar]
- 488.Copeland DM, West AH, Richter-Addo GB. Proteins: Struct, Funct, Genet. 2003;53:182. doi: 10.1002/prot.10495. [DOI] [PubMed] [Google Scholar]
- 489.Reynolds MF, Parks RB, Burstyn JN, Shelver D, Thorsteinsson MV, Kerby RL, Roberts GP, Vogel KM, Spiro TG. Biochemistry. 2000;39:388. doi: 10.1021/bi991378g. [DOI] [PubMed] [Google Scholar]
- 490.Ludwig ML, Metzger AL, Pattridge KA, Stallings WC. J Mol Biol. 1991;219:335. doi: 10.1016/0022-2836(91)90569-r. [DOI] [PubMed] [Google Scholar]
- 491.Edwards RA, Baker HM, Whittaker MM, Whittaker JW, Jameson GB, Baker EN. JBIC, J Biol Inorg Chem. 1998;3:161. [Google Scholar]
- 492.Wagner UG, Werber MM, Beck Y, Hartman JR, Frolow F, Sussman JL. J Mol Biol. 1989;206:787. doi: 10.1016/0022-2836(89)90586-x. [DOI] [PubMed] [Google Scholar]
- 493.Wagner UG, Pattridge KA, Ludwig ML, Stallings WC, Werber MM, Oefner C, Frolow F, Sussman JL. Protein Sci. 1993;2:814. doi: 10.1002/pro.5560020511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Borgstahl GEO, Parge HE, Hickey MJ, Beyer WF, Hallewell RA, Tainer JA. Cell. 1992;71:107. doi: 10.1016/0092-8674(92)90270-m. [DOI] [PubMed] [Google Scholar]
- 495.Barynin VV, Whittaker MM, Antonyuk SV, Lamzin VS, Harrison PM, Artymiuk PJ, Whittaker JW. Structure. 2001;9:725. doi: 10.1016/s0969-2126(01)00628-1. [DOI] [PubMed] [Google Scholar]
- 496.Kono Y, Fridovich I. J Biol Chem. 1983;258:6015. [PubMed] [Google Scholar]
- 497.Barber J, Kühlbrandt W. Curr Opin Struct Biol. 1999;9:469. doi: 10.1016/S0959-440X(99)80066-9. [DOI] [PubMed] [Google Scholar]
- 498.Barber J. Curr Opin Struct Biol. 2002;12:523. doi: 10.1016/s0959-440x(02)00357-3. [DOI] [PubMed] [Google Scholar]
- 499.McEvoy JP, Brudvig GW. Chem Rev. 2006;106:4455. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 500.Barber J. Inorg Chem. 2008;47:1700. doi: 10.1021/ic701835r. [DOI] [PubMed] [Google Scholar]
- 501.Umena Y, Kawakami K, Shen J-R, Kamiya N. Nature. 2011;473:55. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 502.Tomter AB, Zoppellaro G, Andersen NH, Hersleth H-P, Hammerstad M, Røhr ÅK, Sandvik GK, Strand KR, Nilsson GE, Bell CB, Barra A-L, Blasco E, Le Pape L, Solomon EI, Andersson KK. Coord Chem Rev. 2013;257:3. [Google Scholar]
- 503.Sharma CB, Lehle L, Tanner W. Eur J Biochem. 1981;116:101. doi: 10.1111/j.1432-1033.1981.tb05306.x. [DOI] [PubMed] [Google Scholar]
- 504.Powell JT, Brew K. J Biol Chem. 1976;251:3645. [PubMed] [Google Scholar]
- 505.Jiao Y, Shashkina E, Shashkin P, Hansson A, Katz A. Biochim Biophys Acta. 1999;1427:1. doi: 10.1016/s0304-4165(98)00142-1. [DOI] [PubMed] [Google Scholar]
- 506.Fernández-Gacio A, Codina A, Fastrez J, Riant O, Soumillion P. ChemBioChem. 2006;7:1013. doi: 10.1002/cbic.200600127. [DOI] [PubMed] [Google Scholar]
- 507.Okrasa K, Kazlauskas RJ. Chem—Eur J. 2006;12:1587. doi: 10.1002/chem.200501413. [DOI] [PubMed] [Google Scholar]
- 508.Ugarova NN, Lebedeva OV, Berezin IV. J Mol Catal. 1981;13:215. [Google Scholar]
- 509.Finzel BC, Poulos TL, Kraut J. J Biol Chem. 1984;259:13027. [PubMed] [Google Scholar]
- 510.Poulos TL, Edwards SL, Wariishi H, Gold MH. J Biol Chem. 1993;268:4429. doi: 10.2210/pdb1lga/pdb. [DOI] [PubMed] [Google Scholar]
- 511.Sundaramoorthy M, Kishi K, Gold MH, Poulos TL. J Biol Chem. 1994;269:32759. [PubMed] [Google Scholar]
- 512.Kuan IC, Johnson KA, Tien M. J Biol Chem. 1993;268:20064. [PubMed] [Google Scholar]
- 513.Kishi K, Kusters-van Someren M, Mayfield MB, Sun J, Loehr TM, Gold MH. Biochemistry. 1996;35:8986. doi: 10.1021/bi960679c. [DOI] [PubMed] [Google Scholar]
- 514.Banci L. J Biotechnol. 1997;53:253. doi: 10.1016/s0168-1656(97)01677-5. [DOI] [PubMed] [Google Scholar]
- 515.Wang X, Lu Y. Biochemistry. 1999;38:9146. doi: 10.1021/bi990235r. [DOI] [PubMed] [Google Scholar]
- 516.Gengenbach A, Syn S, Wang X, Lu Y. Biochemistry. 1999;38:11425. doi: 10.1021/bi990666+. [DOI] [PubMed] [Google Scholar]
- 517.Erman JE, Vitello LB, Mauro JM, Kraut J. Biochemistry. 1989;28:7992. doi: 10.1021/bi00446a004. [DOI] [PubMed] [Google Scholar]
- 518.Roe JA, Goodin DB. J Biol Chem. 1993;268:20037. [PubMed] [Google Scholar]
- 519.English AM, Tsaprailis G. Adv Inorg Chem. 1995;43:79. [Google Scholar]
- 520.Glenn JK, Gold MH. Arch Biochem Biophys. 1985;242:329. doi: 10.1016/0003-9861(85)90217-6. [DOI] [PubMed] [Google Scholar]
- 521.Pfister TD, Mirarefi AY, Gengenbach AJ, Zhao X, Danstrom C, Conatser N, Gao Y-G, Robinson H, Zukoski CF, Wang AH-J, Lu Y. JBIC, J Biol Inorg Chem. 2007;12:126. doi: 10.1007/s00775-006-0171-0. [DOI] [PubMed] [Google Scholar]
- 522.Wilcox SK, Putnam CD, Sastry M, Blankenship J, Chazin WJ, McRee DE, Goodin DB. Biochemistry. 1998;37:16853. doi: 10.1021/bi9815039. [DOI] [PubMed] [Google Scholar]
- 523.McEvoy JP, Gascon JA, Batista VS, Brudvig GW. Photochem Photobiol Sci. 2005;4:940. doi: 10.1039/b506755c. [DOI] [PubMed] [Google Scholar]
- 524.Pecoraro VL, Hsieh W-Y. Inorg Chem. 2008;47:1765. doi: 10.1021/ic7017488. [DOI] [PubMed] [Google Scholar]
- 525.Meelich K, Zaleski CM, Pecoraro VL. Philos Trans R Soc, B. 2008;363:1271. doi: 10.1098/rstb.2007.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Thielges M, Uyeda G, Cámara-Artigas A, Kálmán L, Williams JC, Allen JP, Ca A, Ka L. Biochemistry. 2005;44:7389. doi: 10.1021/bi050377n. [DOI] [PubMed] [Google Scholar]
- 527.Michel H, Deisenhofer J. Biochemistry. 1988;27:1. [Google Scholar]
- 528.Komiya H, Yeates TO, Rees DC, Allen JP, Feher G. Proc Natl Acad Sci USA. 1988;85:9012. doi: 10.1073/pnas.85.23.9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303:1831. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 530.Jing Q, Okrasa K, Kazlauskas RJ. Chem—Eur J. 2009;15:1370. doi: 10.1002/chem.200801673. [DOI] [PubMed] [Google Scholar]
- 531.Jing Q, Kazlauskas RJ. ChemCatChem. 2010;2:953. [Google Scholar]
- 532.Ward TR. Comprehensive Inorganic Chemistry II. Elsevier Ltd; New York: 2013. p. 737. [Google Scholar]
- 533.Wilson ME, Whitesides GM. J Am Chem Soc. 1978;100:306. [Google Scholar]
- 534.Creus M, Ward TR. In: Progress in Inorganic Chemistry. Karlin KD, editor. John Wiley & Sons, Inc; New York: 2011. p. 203. [Google Scholar]
- 535.Wilchek M, Bayer EA. Methods in Enzymology. Vol. 184. Elsevier; New York: 1990. p. 5. [DOI] [PubMed] [Google Scholar]
- 536.Green NM. Advances in Protein Chemistry. Vol. 29. Elsevier; New York: 1975. p. 85. [DOI] [PubMed] [Google Scholar]
- 537.Weber PC, Wendoloski JJ, Pantoliano MW, Salemme FR. J Am Chem Soc. 1992;114:3197. [Google Scholar]
- 538.Panek J, Ward T, Jezierska-Mazzarello A, Novič M. J Comput-Aided Mol Des. 2010;24:719. doi: 10.1007/s10822-010-9369-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Ward TR, Collot J, Gradinaru J, Loosli A, Skander M, Letondor C, Joseph E, Klein G. Chimia. 2003;57:586. [Google Scholar]
- 540.Collot J, Gradinaru J, Humbert N, Skander M, Zocchi A, Ward TR. J Am Chem Soc. 2003;125:9030. doi: 10.1021/ja035545i. [DOI] [PubMed] [Google Scholar]
- 541.Skander M, Humbert N, Collot J, Gradinaru J, Klein G, Loosli A, Sauser J, Zocchi A, Gilardoni F, Ward TR. J Am Chem Soc. 2004;126:14411. doi: 10.1021/ja0476718. [DOI] [PubMed] [Google Scholar]
- 542.Thomas CM, Ward TR. Chem Soc Rev. 2005;34:337. doi: 10.1039/b314695m. [DOI] [PubMed] [Google Scholar]
- 543.Klein G, Humbert N, Gradinaru J, Ivanova A, Gilardoni F, Rusbandi UE, Ward TR. Angew Chem, Int Ed. 2005;44:7764. doi: 10.1002/anie.200502000. [DOI] [PubMed] [Google Scholar]
- 544.Loosli A, Rusbandi UE, Gradinaru J, Bernauer K, Schlaepfer CW, Meyer M, Mazurek S, Novic M, Ward TR. Inorg Chem. 2006;45:660. doi: 10.1021/ic051405t. [DOI] [PubMed] [Google Scholar]
- 545.Collot J, Humbert N, Skander M, Klein G, Ward TR. J Organomet Chem. 2004;689:4868. [Google Scholar]
- 546.Skander M, Malan C, Ivanova A, Ward TR. Chem Commun (Cambridge, UK) 2005:4815. doi: 10.1039/b509015f. [DOI] [PubMed] [Google Scholar]
- 547.Rusbandi UE, Skander M, Ivanova A, Malan C, Ward TR. C R Chim. 2007;10:678. [Google Scholar]
- 548.Rusbandi UE, Lo C, Skander M, Ivanova A, Creus M, Humbert N, Ward TR. Adv Synth Catal. 2007;349:1923. [Google Scholar]
- 549.Pordea A, Ward T. Synlett. 2009;2009:3225. [Google Scholar]
- 550.Sardo A, Wohlschlager T, Lo C, Zoller H, Ward TR, Creus M. Protein Expression Purif. 2011;77:131. doi: 10.1016/j.pep.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 551.Letondor C, Humbert N, Ward TR. Proc Natl Acad Sci USA. 2005;102:4683. doi: 10.1073/pnas.0409684102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Letondor C, Pordea A, Humbert N, Ivanova A, Mazurek S, Novic M, Ward TR. J Am Chem Soc. 2006;128:8320. doi: 10.1021/ja061580o. [DOI] [PubMed] [Google Scholar]
- 553.Creus M, Pordea A, Rossel T, Sardo A, Letondor C, Ivanova A, Letrong I, Stenkamp RE, Ward TR. Angew Chem, Int Ed. 2008;47:1400. doi: 10.1002/anie.200704865. [DOI] [PubMed] [Google Scholar]
- 554.Dürrenberger M, Heinisch T, Wilson YM, Rossel T, Nogueira E, Knörr L, Mutschler A, Kersten K, Zimbron MJ, Pierron J, Schirmer T, Ward TR. Angew Chem, Int Ed. 2011;50:3026. doi: 10.1002/anie.201007820. [DOI] [PubMed] [Google Scholar]
- 555.Köhler V, Wilson YM, Dürrenberger M, Ghislieri D, Churakova E, Quinto T, Knörr L, Häussinger D, Hollmann F, Turner NJ, Ward TR. Nat Chem. 2013;5:93. doi: 10.1038/nchem.1498. [DOI] [PubMed] [Google Scholar]
- 556.Thomas CM, Letondor C, Humbert N, Ward TR. J Organomet Chem. 2005;690:4488. [Google Scholar]
- 557.Pordea A, Mathis D, Ward TR. J Organomet Chem. 2009;694:930. [Google Scholar]
- 558.Pierron J, Malan C, Creus M, Gradinaru J, Hafner I, Ivanova A, Sardo A, Ward TR. Angew Chem, Int Ed. 2008;120:713. doi: 10.1002/anie.200703159. [DOI] [PubMed] [Google Scholar]
- 559.Hyster TK, Knörr L, Ward TR, Rovis T. Science. 2012;338:500. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Pordea A, Creus M, Panek J, Duboc C, Mathis D, Novic M, Ward TR. J Am Chem Soc. 2008;130:8085. doi: 10.1021/ja8017219. [DOI] [PubMed] [Google Scholar]
- 561.Köhler V, Mao J, Heinisch T, Pordea A, Sardo A, Wilson YM, Knörr L, Creus M, Prost J-C, Schirmer T, Ward TR. Angew Chem, Int Ed. 2011;50:10863. doi: 10.1002/anie.201103632. [DOI] [PubMed] [Google Scholar]
- 562.Mahy J-P, Raffy Q, Allard M, Ricoux R. Biochimie. 2009;91:1321. doi: 10.1016/j.biochi.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 563.Ricoux R, Sauriat-Dorizon H, Girgenti E, Blanchard D, Mahy J-P. J Immunol Methods. 2002;269:39. doi: 10.1016/s0022-1759(02)00223-5. [DOI] [PubMed] [Google Scholar]
- 564.Ricoux R, Lukowska E, Pezzotti F, Mahy J-P. Eur J Biochem. 2004;271:1277. doi: 10.1111/j.1432-1033.2004.04032.x. [DOI] [PubMed] [Google Scholar]
- 565.Ricoux R, Girgenti E, Sauriat-Dorizon H, Blanchard D, Mahy J-P. J Protein Chem. 2002;21:473. doi: 10.1023/a:1021351120772. [DOI] [PubMed] [Google Scholar]
- 566.Quilez R, de Lauzon S, Desfosses B, Mansuy D, Mahy JP. FEBS Lett. 1996;395:73. doi: 10.1016/0014-5793(96)01006-x. [DOI] [PubMed] [Google Scholar]
- 567.Raffy Q, Ricoux R, Mahy J-P. Tetrahedron Lett. 2008;49:1865. [Google Scholar]
- 568.Raffy Q, Ricoux R, Sansiaume E, Pethe S, Mahy J-P. J Mol Catal A: Chem. 2010;317:19. [Google Scholar]
- 569.Ricoux R, Dubuc R, Dupont C, Marechal J, Martin A, Sellier M, Mahy J-P. Bioconjugate Chem. 2008;19:899. doi: 10.1021/bc700435a. [DOI] [PubMed] [Google Scholar]
- 570.Ricoux R, Allard M, Dubuc R, Dupont C, Maréchal J-D, Mahy J-P. Org Biomol Chem. 2009;7:3208. doi: 10.1039/b907534h. [DOI] [PubMed] [Google Scholar]
- 571.Allard M, Dupont C, Muñoz Robles V, Doucet N, Lledós A, Maréchal J-D, Urvoas A, Mahy J-P, Ricoux R. ChemBioChem. 2012;13:240. doi: 10.1002/cbic.201100659. [DOI] [PubMed] [Google Scholar]
- 572.Rousselot-Pailley P, Bochot C, Marchi-Delapierre C, Jorge-Robin A, Martin L, Fontecilla-Camps JC, Cavazza C, Ménage S. ChemBioChem. 2009;10:545. doi: 10.1002/cbic.200800595. [DOI] [PubMed] [Google Scholar]
- 573.Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. J Mol Biol. 2005;353:38. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 574.Heddle J, Scott DJ, Unzai S, Park S-Y, Tame JRH. J Biol Chem. 2003;278:50322. doi: 10.1074/jbc.M307941200. [DOI] [PubMed] [Google Scholar]
- 575.De Pina K, Navarro C, McWalter L, Boxer DH, Price NC, Kelly SM, Mandrand-Berthelot MA, Wu LF. Eur J Biochem. 1995;227:857. doi: 10.1111/j.1432-1033.1995.tb20211.x. [DOI] [PubMed] [Google Scholar]
- 576.Yohannes E, Barnhart DM, Slonczewski JL. J Bacteriol. 2003;186:192. doi: 10.1128/JB.186.1.192-199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Salins L, Goldsmith E, Ensor M, Daunert S. Anal Bioanal Chem. 2002;372:174. doi: 10.1007/s00216-001-1169-7. [DOI] [PubMed] [Google Scholar]
- 578.Cherrier MV, Martin L, Cavazza C, Jacquamet L, Lemaire D, Gaillard J, Fontecilla-Camps JC. J Am Chem Soc. 2005;127:10075. doi: 10.1021/ja0518530. [DOI] [PubMed] [Google Scholar]
- 579.Cherrier MV, Cavazza C, Bochot C, Lemaire D, Fontecilla-Camps JC. Biochemistry. 2008;47:9937. doi: 10.1021/bi801051y. [DOI] [PubMed] [Google Scholar]
- 580.Cavazza C, Bochot C, Rousselot-Pailley P, Carpentier P, Cherrier MV, Martin L, Marchi-Delapierre C, Fontecilla-Camps JC, Ménage S. Nat Chem. 2010;2:1069. doi: 10.1038/nchem.841. [DOI] [PubMed] [Google Scholar]
- 581.Cherrier MV, Girgenti E, Amara P, Iannello M, Marchi-Delapierre C, Fontecilla-Camps JC, Ménage S, Cavazza C. JBIC, J Biol Inorg Chem. 2012;17:817. doi: 10.1007/s00775-012-0899-7. [DOI] [PubMed] [Google Scholar]
- 582.Esmieu C, Cherrier MV, Amara P, Girgenti E, Marchi-Delapierre C, Oddon F, Iannello M, Jorge-Robin A, Cavazza C, Ménage S. Angew Chem, Int Ed Engl. 2013;52:3922. doi: 10.1002/anie.201209021. [DOI] [PubMed] [Google Scholar]
- 583.Satake Y, Abe S, Okazaki S, Ban N, Hikage T, Ueno T, Nakajima H, Suzuki A, Yamane T, Nishiyama H, Watanabe Y. Organometallics. 2007;26:4904. [Google Scholar]
- 584.Ohashi M, Koshiyama T, Ueno T, Yanase M, Fujii H, Watanabe Y. Angew Chem, Int Ed. 2003;42:1005. doi: 10.1002/anie.200390256. [DOI] [PubMed] [Google Scholar]
- 585.Ueno T, Ohashi M, Kono M, Kondo K, Suzuki A, Yamane T, Watanabe Y. Inorg Chem. 2004;43:2852. doi: 10.1021/ic0498539. [DOI] [PubMed] [Google Scholar]
- 586.Ueno T, Koshiyama T, Ohashi M, Kondo K, Kono M, Suzuki A, Yamane T, Watanabe Y. J Am Chem Soc. 2005;127:6556. doi: 10.1021/ja045995q. [DOI] [PubMed] [Google Scholar]
- 587.Ueno T, Koshiyama T, Abe S, Yokoi N, Ohashi M, Nakajima H, Watanabe Y. J Organomet Chem. 2007;692:142. [Google Scholar]
- 588.Wegner SV, Boyaci H, Chen H, Jensen MP, He C. Angew Chem, Int Ed. 2009;48:2339. doi: 10.1002/anie.200805262. [DOI] [PubMed] [Google Scholar]
- 589.Schreiter ER, Sintchak MD, Guo Y, Chivers PT, Sauer RT, Drennan CL. Nat Struct Biol. 2003;10:794. doi: 10.1038/nsb985. [DOI] [PubMed] [Google Scholar]
- 590.Carey JR, Ma SK, Pfister TD, Garner DK, Kim HK, Abramite JA, Wang Z, Guo Z, Lu Y. J Am Chem Soc. 2004;126:10812. doi: 10.1021/ja046908x. [DOI] [PubMed] [Google Scholar]
- 591.Zhang J-L, Garner DK, Liang L, Chen Q, Lu Y. Chem Commun (Cambridge, UK) 2008:1665. doi: 10.1039/b718915j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Garner DK, Liang L, Barrios DA, Zhang J, Lu Y. ACS Catal. 2011;1:1083. doi: 10.1021/cs200258e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Otto S, Engberts JBFN. J Am Chem Soc. 1999;121:6798. [Google Scholar]
- 594.Otto S, Boccaletti G, Engberts JBFN. J Am Chem Soc. 1998;120:4238. [Google Scholar]
- 595.Boersma AJ, de Bruin B, Feringa BL, Roelfes G. Chem Commun (Cambridge, UK) 2012;48:2394. doi: 10.1039/c2cc17350f. [DOI] [PubMed] [Google Scholar]
- 596.Boersma AJ, Megens RP, Feringa BL, Roelfes G. Chem Soc Rev. 2010;39:2083. doi: 10.1039/b811349c. [DOI] [PubMed] [Google Scholar]
- 597.García-Fernández A, Roelfes G. Met Ions Life Sci. 2012;10:249. doi: 10.1007/978-94-007-2172-2_9. [DOI] [PubMed] [Google Scholar]
- 598.Reetz MT, Jiao N. Angew Chem, Int Ed. 2006;118:2476. [Google Scholar]
- 599.Bos J, Fusetti F, Driessen AJM, Roelfes G. Angew Chem, Int Ed. 2012;51:7472. doi: 10.1002/anie.201202070. [DOI] [PubMed] [Google Scholar]
- 600.Madoori PK, Agustiandari H, Driessen AJM, Thunnissen A-MWH. EMBO J. 2009;28:156. doi: 10.1038/emboj.2008.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Agustiandari H, Lubelski J, van den Berg van Saparoea HB, Kuipers OP, Driessen AJM. J Bacteriol. 2008;190:759. doi: 10.1128/JB.01151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Beismann-Driemeyer S, Sterner R. J Biol Chem. 2001;276:20387. doi: 10.1074/jbc.M102012200. [DOI] [PubMed] [Google Scholar]
- 603.Podtetenieff J, Taglieber A, Bill E, Reijerse EJ, Reetz MT. Angew Chem, Int Ed. 2010;49:5151. doi: 10.1002/anie.201002106. [DOI] [PubMed] [Google Scholar]
- 604.Roelfes G, Feringa BL. Angew Chem, Int Ed. 2005;44:3230. doi: 10.1002/anie.200500298. [DOI] [PubMed] [Google Scholar]
- 605.Bettio A, Beck-Sickinger AG. Biopolymers. 2001;60:420. doi: 10.1002/1097-0282(2001)60:6<420::AID-BIP10183>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 606.Lerch M, Gafner V, Bader R, Christen B, Folkers G, Zerbe O. J Mol Biol. 2002;322:1117. doi: 10.1016/s0022-2836(02)00889-6. [DOI] [PubMed] [Google Scholar]
- 607.Li XA, Sutcliffe MJ, Schwartz TW, Dobson CM. Biochemistry. 1992;31:1245. doi: 10.1021/bi00119a038. [DOI] [PubMed] [Google Scholar]
- 608.Coquiére D, Bos J, Beld J, Roelfes G. Angew Chem, Int Ed. 2009;48:5159. doi: 10.1002/anie.200901134. [DOI] [PubMed] [Google Scholar]
- 609.Dickerson MB, Sandhage KH, Naik RR. Chem Rev. 2008;108:4935. doi: 10.1021/cr8002328. [DOI] [PubMed] [Google Scholar]
- 610.Theil EC, Behera RK, Tosha T. Coord Chem Rev. 2013;257:579. doi: 10.1016/j.ccr.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 611.Wong KKW, Mann S. Adv Mater. 1996;8:928. [Google Scholar]
- 612.Domínguez-Vera JM, Colacio E. Inorg Chem. 2003;42:1828. doi: 10.1021/ic034783b. [DOI] [PubMed] [Google Scholar]
- 613.Yamashita I, Hayashi J, Hara M. Chem Lett. 2004;33:1158. [Google Scholar]
- 614.Zhang L, Swift J, Butts CA, Yerubandi V, Dmochowski IJ. J Inorg Biochem. 2007;101:1719. doi: 10.1016/j.jinorgbio.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 615.Butts CA, Swift J, Kang S-G, Di Costanzo L, Christianson DW, Saven JG, Dmochowski IJ. Biochemistry. 2008;47:12729. doi: 10.1021/bi8016735. [DOI] [PubMed] [Google Scholar]
- 616.Aime S, Frullano L, Geninatti Crich S. Angew Chem, Int Ed. 2002;41:1017. doi: 10.1002/1521-3773(20020315)41:6<1017::aid-anie1017>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 617.Domínguez-Vera JM. J Inorg Biochem. 2004;98:469. doi: 10.1016/j.jinorgbio.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 618.Yang Z, Wang X, Diao H, Zhang J, Li H, Sun H, Guo Z. Chem Commun. 2007;7345:3453. doi: 10.1039/b705326f. [DOI] [PubMed] [Google Scholar]
- 619.Niemeyer J, Abe S, Hikage T, Ueno T, Erker G, Watanabe Y. Chem Commun. 2008;1:6519. doi: 10.1039/b813181c. [DOI] [PubMed] [Google Scholar]
- 620.Takezawa Y, Böckmann P, Sugi N, Wang Z, Abe S, Murakami T, Hikage T, Erker G, Watanabe Y, Kitagawa S, Ueno T. Dalton Trans. 2011;40:2190. doi: 10.1039/c0dt00955e. [DOI] [PubMed] [Google Scholar]
- 621.Ueno T, Abe M, Hirata K, Abe S, Suzuki M, Shimizu N, Yamamoto M, Takata M, Watanabe Y. J Am Chem Soc. 2009;131:5094. doi: 10.1021/ja806688s. [DOI] [PubMed] [Google Scholar]
- 622.Ueno T, Suzuki M, Goto T, Matsumoto T, Nagayama K, Watanabe Y. Angew Chem, Int Ed. 2004;43:2527. doi: 10.1002/anie.200353436. [DOI] [PubMed] [Google Scholar]
- 623.Suzuki M, Abe M, Ueno T, Abe S, Goto T, Toda Y, Akita T, Yamada Y, Watanabe Y. Chem Commun (Cambridge, UK) 2009:4871. doi: 10.1039/b908742g. [DOI] [PubMed] [Google Scholar]
- 624.Abe S, Hirata K, Ueno T, Morino K, Shimizu N, Yamamoto M, Takata M, Yashima E, Watanabe Y. J Am Chem Soc. 2009;131:6958. doi: 10.1021/ja901234j. [DOI] [PubMed] [Google Scholar]
- 625.Abe S, Niemeyer J, Abe M, Takezawa Y, Ueno T, Hikage T, Erker G, Watanabe Y. J Am Chem Soc. 2008;130:10512. doi: 10.1021/ja802463a. [DOI] [PubMed] [Google Scholar]
- 626.Abe S, Hikage T, Watanabe Y, Kitagawa S, Ueno T. Inorg Chem. 2010;49:6967. doi: 10.1021/ic1003758. [DOI] [PubMed] [Google Scholar]
- 627.Salgado EN, Faraone-Mennella J, Tezcan FA. J Am Chem Soc. 2007;129:13374. doi: 10.1021/ja075261o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628.Salgado EN, Lewis RA, Faraone-Mennella J, Tezcan FA. J Am Chem Soc. 2008;130:6082. doi: 10.1021/ja8012177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629.Salgado EN, Lewis RA, Mossin S, Rheingold AL, Tezcan FA. Inorg Chem. 2009;48:2726. doi: 10.1021/ic9001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630.Brodin JD, Medina-Morales A, Ni T, Salgado EN, Ambroggio XI, Tezcan FA. J Am Chem Soc. 2010;132:8610. doi: 10.1021/ja910844n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 631.Salgado EN, Brodin JD, To MM, Tezcan FA. Inorg Chem. 2011;50:6323. doi: 10.1021/ic200746m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Brodin JD, Ambroggio XI, Tang C, Parent KN, Baker TS, Tezcan FA. Nat Chem. 2012;4:375. doi: 10.1038/nchem.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Huard DJE, Kane KM, Tezcan FA. Nat Chem Biol. 2013;9:169. doi: 10.1038/nchembio.1163. [DOI] [PubMed] [Google Scholar]
- 634.Medina-Morales A, Perez A, Brodin JD, Tezcan FA. J Am Chem Soc. 2013;135:12013. doi: 10.1021/ja405318d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Ni TW, Tezcan FA. Angew Chem, Int Ed. 2010;49:7014. doi: 10.1002/anie.201001487. [DOI] [PubMed] [Google Scholar]
- 636.Salgado EN, Ambroggio XI, Brodin JD, Lewis RA, Kuhlman B, Tezcan FA. Proc Natl Acad Sci USA. 2010;107:1827. doi: 10.1073/pnas.0906852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 637.Koshiyama T, Yokoi N, Ueno T, Kanamaru S, Nagano S, Shiro Y, Arisaka F, Watanabe Y. Small. 2008;4:50. doi: 10.1002/smll.200700855. [DOI] [PubMed] [Google Scholar]
- 638.Yokoi N, Inaba H, Terauchi M, Stieg AZ, Sanghamitra NJM, Koshiyama T, Yutani K, Kanamaru S, Arisaka F, Hikage T, Suzuki A, Yamane T, Gimzewski JK, Watanabe Y, Kitagawa S, Ueno T. Small. 2010;6:1873. doi: 10.1002/smll.201000772. [DOI] [PubMed] [Google Scholar]
- 639.Yokoi N, Miura Y, Huang C-Y, Takatani N, Inaba H, Koshiyama T, Kanamaru S, Arisaka F, Watanabe Y, Kitagawa S, Ueno T. Chem Commun (Cambridge, UK) 2011;47:2074. doi: 10.1039/c0cc03015e. [DOI] [PubMed] [Google Scholar]
- 640.Inaba H, Kanamaru S, Arisaka F, Kitagawa S, Ueno T. Dalton Trans. 2012;41:11424. doi: 10.1039/c2dt31030a. [DOI] [PubMed] [Google Scholar]
- 641.Feynman R. Phys Today. 1989;42:88. [Google Scholar]
- 642.Hecht MH, Richardson JS, Richardson DC, Ogden RC. Science. 1990;249:884. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 643.Hill RB, Raleigh DP, Lombardi A, DeGrado WF. Acc Chem Res. 2000;33:745. doi: 10.1021/ar970004h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Onuchic JN, Wolynes PG. Curr Opin Struct Biol. 2004;14:70. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 645.Regan L, DeGrado WF. Science. 1988;241:976. doi: 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- 646.Rose GD, Gierasch LM, Smith JA. Advances in Protein Chemistry. Vol. 37. Academic Press, Inc; New York: 1985. p. 1. [DOI] [PubMed] [Google Scholar]
- 647.Hecht MH. Proc Natl Acad Sci USA. 1994;91:8729. doi: 10.1073/pnas.91.19.8729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 648.Nesloney CL, Kelly JW. Bioorg Med Chem. 1996;4:739. doi: 10.1016/0968-0896(96)00051-x. [DOI] [PubMed] [Google Scholar]
- 649.Smith CK, Regan L. Acc Chem Res. 1997;30:153. [Google Scholar]
- 650.Hughes RM, Waters ML. Curr Opin Struct Biol. 2006;16:514. doi: 10.1016/j.sbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 651.Pantoja-Uceda D, Santiveri CM, Jiménez MA. Methods Mol Biol. 2006;340:27. doi: 10.1385/1-59745-116-9:27. [DOI] [PubMed] [Google Scholar]
- 652.Kim DE, Chivian D, Baker D. Nucleic Acids Res. 2004;32:W526. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Science. 2003;302:1364. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 654.Bradley P, Misura KMS, Baker D. Science. 2005;309:1868. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 655.Saven JG. Curr Opin Struct Biol. 2002;12:453. doi: 10.1016/s0959-440x(02)00347-0. [DOI] [PubMed] [Google Scholar]
- 656.Park S, Yang X, Saven JG. Curr Opin Struct Biol. 2004;14:487. doi: 10.1016/j.sbi.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 657.Saven JG. Curr Opin Chem Biol. 2011;15:452. doi: 10.1016/j.cbpa.2011.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 658.Hecht MH, Das A, Go A, Bradley LH, Wei Y. Protein Sci. 2004;13:1711. doi: 10.1110/ps.04690804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Moffet DA, Hecht MH. Chem Rev. 2001;101:3191. doi: 10.1021/cr000051e. [DOI] [PubMed] [Google Scholar]
- 660.Smith BA, Hecht MH. Curr Opin Chem Biol. 2011;15:421. doi: 10.1016/j.cbpa.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 661.Bradley LH, Thumfort PP, Hecht MH. Methods Mol Biol. 2006;340:53. doi: 10.1385/1-59745-116-9:53. [DOI] [PubMed] [Google Scholar]
- 662.Butterfoss GL, Kuhlman B. Annu Rev Biophys Biomol Struct. 2006;35:49. doi: 10.1146/annurev.biophys.35.040405.102046. [DOI] [PubMed] [Google Scholar]
- 663.Floudas CA, Fung HK, McAllister SR, Mönnigmann M, Rajgaria R. Chem Eng Sci. 2006;61:966. [Google Scholar]
- 664.Kang S, Saven JG. Curr Opin Chem Biol. 2007;11:329. doi: 10.1016/j.cbpa.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 665.Langer HG. J Inorg Nucl Chem. 1964;26:59. [Google Scholar]
- 666.Baumann EW, Wallace RM. Anal Chem. 1969;41:2072. [Google Scholar]
- 667.Caudle MT, Pecoraro VL. J Am Chem Soc. 1997;119:3415. [Google Scholar]
- 668.Hamstra BJ, Colpas GJ, Pecoraro VL. Inorg Chem. 1998;37:949. doi: 10.1021/ic970284x. [DOI] [PubMed] [Google Scholar]
- 669.Peacock AFA. Curr Opin Chem Biol. 2013;17:934. doi: 10.1016/j.cbpa.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 670.Neustadt J, Pieczenik S. Integr Med. 2007;6:26. [Google Scholar]
- 671.Ibrahim D, Froberg B, Wolf A, Rusyniak DE. Clin Lab Med. 2006;26:67. doi: 10.1016/j.cll.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 672.Busenlehner LS, Pennella MA, Giedroc DP. FEMS Microbiol Rev. 2003;27:131. doi: 10.1016/S0168-6445(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 673.Brown NL, Stoyanov JV, Kidd SP, Hobman JL. FEMS Microbiol Rev. 2003;27:145. doi: 10.1016/S0168-6445(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 674.Utschig LM, Bryson JW, O’Halloran TV. Science. 1995;268:380. doi: 10.1126/science.7716541. [DOI] [PubMed] [Google Scholar]
- 675.Summers AO. J Bacteriol. 1992;174:3097. doi: 10.1128/jb.174.10.3097-3101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Wright JG, Tsang HT, Penner-Hahn JE, O’Halloran TV. J Am Chem Soc. 1990;112:2434. [Google Scholar]
- 677.Helmann JD, Ballard BT, Walsh CT. Science. 1990;247:946. doi: 10.1126/science.2305262. [DOI] [PubMed] [Google Scholar]
- 678.Shewchuk LM, Verdine GL, Walsh CT. Biochemistry. 1989;28:2331. doi: 10.1021/bi00431a052. [DOI] [PubMed] [Google Scholar]
- 679.Ralston DM, O’Halloran TV. Proc Natl Acad Sci USA. 1990;87:3846. doi: 10.1073/pnas.87.10.3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 680.Chen PR, He C. Curr Opin Chem Biol. 2008;12:214. doi: 10.1016/j.cbpa.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 681.Novick RP, Roth C. J Bacteriol. 1968;95:1335. doi: 10.1128/jb.95.4.1335-1342.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Ye J, Kandegedara A, Martin P, Rosen BP. J Bacteriol. 2005;187:4214. doi: 10.1128/JB.187.12.4214-4221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 683.Rensing C. J Biol Chem. 1998;273:32614. doi: 10.1074/jbc.273.49.32614. [DOI] [PubMed] [Google Scholar]
- 684.Diner B. Toxicity, Mercury. 2006:1. [Google Scholar]
- 685.Endo G, Silver S. J Bacteriol. 1995;177:4437. doi: 10.1128/jb.177.15.4437-4441.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 686.Shi W, Dong J, Scott RA, Ksenzenko MY, Rosen BP. J Biol Chem. 1996;271:9291. doi: 10.1074/jbc.271.16.9291. [DOI] [PubMed] [Google Scholar]
- 687.Zhou T, Radaev S, Rosen BP, Gatti DL. EMBO J. 2000;19:4838. doi: 10.1093/emboj/19.17.4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Martin P, DeMel S, Shi J, Gladysheva T, Gatti DL, Rosen BP, Edwards BF. Structure. 2001;9:1071. doi: 10.1016/s0969-2126(01)00672-4. [DOI] [PubMed] [Google Scholar]
- 689.Messens J, Martins JC, Van Belle K, Brosens E, Desmyter A, De Gieter M, Wieruszeski J-M, Willem R, Wyns L, Zegers I. Proc Natl Acad Sci USA. 2002;99:8506. doi: 10.1073/pnas.132142799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690.Lin Y, Walmsley AR, Rosen BP. Proc Natl Acad Sci USA. 2006;103:15617. doi: 10.1073/pnas.0603974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 691.Rosen BP. FEBS Lett. 2002;529:86. doi: 10.1016/s0014-5793(02)03186-1. [DOI] [PubMed] [Google Scholar]
- 692.Chen P, Greenberg B, Taghavi S, Romano C, van der Lelie D, He C. Angew Chem, Int Ed. 2005;44:2715. doi: 10.1002/anie.200462443. [DOI] [PubMed] [Google Scholar]
- 693.Chen PR, Wasinger EC, Zhao J, van der Lelie D, Chen LX, He C. J Am Chem Soc. 2007;129:12350. doi: 10.1021/ja0733890. [DOI] [PubMed] [Google Scholar]
- 694.Erskine PT, Norton E, Cooper JB, Lambert R, Coker A, Lewis G, Spencer P, Sarwar M, Wood SP, Warren MJ, Shoolingin-Jordan PM. Biochemistry. 1999;38:4266. doi: 10.1021/bi982137w. [DOI] [PubMed] [Google Scholar]
- 695.Jaffe EK, Martins J, Li J, Kervinen J, Dunbrack RL. J Biol Chem. 2001;276:1531. doi: 10.1074/jbc.M007663200. [DOI] [PubMed] [Google Scholar]
- 696.Kelada SN, Shelton E, Kaufmann RB, Khoury MJ. Am J Epidemiol. 2001;154:1. doi: 10.1093/aje/154.1.1. [DOI] [PubMed] [Google Scholar]
- 697.Magyar JS, Weng T-C, Stern CM, Dye DF, Rous BW, Payne JC, Bridgewater BM, Mijovilovich A, Parkin G, Zaleski JM, Penner-Hahn JE, Godwin HA. J Am Chem Soc. 2005;127:9495. doi: 10.1021/ja0424530. [DOI] [PubMed] [Google Scholar]
- 698.Bridgewater BM, Parkin G. J Am Chem Soc. 2000;122:7140. [Google Scholar]
- 699.Molecular Simulations, Inc. 2000 [Google Scholar]
- 700.Dill KA, Bromberg S, Kaizhi Y, Fiebig KM, Yee DP, Thomas PD, Chan HS, Yue K. Protein Sci. 1995;4:561. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 701.Berne BJ, Weeks JD, Zhou R. Annu Rev Phys Chem. 2009;60:85. doi: 10.1146/annurev.physchem.58.032806.104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 702.Agashe VR, Shastry MC, Udgaonkar JB. Nature. 1995;377:754. doi: 10.1038/377754a0. [DOI] [PubMed] [Google Scholar]
- 703.Gutin AM, Abkevich VI, Shakhnovich EI. Biochemistry. 1995;34:3066. doi: 10.1021/bi00009a038. [DOI] [PubMed] [Google Scholar]
- 704.Ghosh D, Pecoraro VL. Inorg Chem. 2004;43:7902. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]
- 705.Peacock AFA, Iranzo O, Pecoraro VL. Dalton Trans. 2009;9226:2271. doi: 10.1039/b818306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 706.Su JY, Hodges RS, Kay CM. Biochemistry. 1994;33:15501. doi: 10.1021/bi00255a032. [DOI] [PubMed] [Google Scholar]
- 707.Ghosh D, Lee K-H, Demeler B, Pecoraro VL. Biochemistry. 2005;44:10732. doi: 10.1021/bi0506674. [DOI] [PubMed] [Google Scholar]
- 708.Ptitsyn OB, Pain RH, Semisotnov GV, Zerovnik E, Razgulyaev OI. FEBS Lett. 1990;262:20. doi: 10.1016/0014-5793(90)80143-7. [DOI] [PubMed] [Google Scholar]
- 709.Ptitsyn OB. Protein Folding. W. H. Freeman and Co; New York: 1992. p. 243. [Google Scholar]
- 710.Ptitsyn OB. Adv Protein Chem. 1995;47:83. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 711.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Proc Natl Acad Sci USA. 2007;104:11969. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 712.Zampella G, Neupane KP, De Gioia L, Pecoraro VL. Chem—Eur J. 2012;18:2040. doi: 10.1002/chem.201102786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 713.Allen DW, Coppola JC, Kennard O, Mann FG, Motherwell WDS, Watson DG. J Chem Soc C. 1970:810. [Google Scholar]
- 714.Pappalardo G, Chakravorty R, Irgolic KJ, Meyers EA. Acta Crystallogr, Sect C. 1983;39:1618. [Google Scholar]
- 715.Shaikh TA, Bakus RC, Parkin S, Atwood DA. J Organomet Chem. 2006;691:1825. [Google Scholar]
- 716.Shaikh TA, Parkin S, Atwood DA. J Organomet Chem. 2006;691:4167. [Google Scholar]
- 717.Carter TG, Healey ER, Pitt MA, Johnson DW. Inorg Chem. 2005;44:9634. doi: 10.1021/ic051522o. [DOI] [PubMed] [Google Scholar]
- 718.Bally R. Acta Crystallogr. 1967;23:295. [Google Scholar]
- 719.Whitfield HJ. J Chem Soc A. 1970;11:1800. [Google Scholar]
- 720.Betz S, Fairman R, O’Neil K, Lear J, Degrado W. Philos Trans R Soc, B. 1995;348:81. doi: 10.1098/rstb.1995.0048. [DOI] [PubMed] [Google Scholar]
- 721.Chakraborty S, Touw DS, Peacock AFA, Stuckey J, Pecoraro VL. J Am Chem Soc. 2010;132:13240. doi: 10.1021/ja101812c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 722.Matzapetakis M, Ghosh D, Weng T-C, Penner-Hahn JE, Pecoraro VL. JBIC, J Biol Inorg Chem. 2006;11:876. doi: 10.1007/s00775-006-0140-7. [DOI] [PubMed] [Google Scholar]
- 723.Farrer BT, Harris NP, Valchus KE, Pecoraro VL. Biochemistry. 2001;40:14696. doi: 10.1021/bi015649a. [DOI] [PubMed] [Google Scholar]
- 724.Iranzo O, Thulstrup PW, Ryu S-B, Hemmingsen L, Pecoraro VL. Chem—Eur J. 2007;13:9178. doi: 10.1002/chem.200701208. [DOI] [PubMed] [Google Scholar]
- 725.Pecoraro VL, Peacock AFA, Iranzo O, Luczkowski M. ACS Symposium Series. American Chemical Society; Washington, DC: 2009. p. 183. [Google Scholar]
- 726.Lee K-H, Matzapetakis M, Mitra S, Marsh ENG, Pecoraro VL. J Am Chem Soc. 2004;126:9178. doi: 10.1021/ja048839s. [DOI] [PubMed] [Google Scholar]
- 727.Lee K-H, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL. Angew Chem, Int Ed. 2006;45:2864. doi: 10.1002/anie.200504548. [DOI] [PubMed] [Google Scholar]
- 728.Iranzo O, Jakusch T, Lee K-H, Hemmingsen L, Pecoraro VL. Chem—Eur J. 2009;15:3761. doi: 10.1002/chem.200802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 729.Hemmingsen L, Sas KN, Danielsen E. Chem Rev. 2004;104:4027. doi: 10.1021/cr030030v. [DOI] [PubMed] [Google Scholar]
- 730.Neupane KP, Pecoraro VL. Angew Chem, Int Ed. 2010;49:8177. doi: 10.1002/anie.201004429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Neupane KP, Pecoraro VL. J Inorg Biochem. 2011;105:1030. doi: 10.1016/j.jinorgbio.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 732.Peacock AFA, Stuckey JA, Pecoraro VL. Angew Chem, Int Ed. 2009;48:7371. doi: 10.1002/anie.200902166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733.Peacock AFA, Hemmingsen L, Pecoraro VL. Proc Natl Acad Sci USA. 2008;105:16566. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.Hohsaka T, Sisido M. Curr Opin Chem Biol. 2002;6:809. doi: 10.1016/s1367-5931(02)00376-9. [DOI] [PubMed] [Google Scholar]
- 735.Hodgson DRW, Sanderson JM. Chem Soc Rev. 2004;33:422. doi: 10.1039/b312953p. [DOI] [PubMed] [Google Scholar]
- 736.Yoder NC, Kumar K. Chem Soc Rev. 2002;31:335. doi: 10.1039/b201097f. [DOI] [PubMed] [Google Scholar]
- 737.Wang A, Nairn NW, Marelli M, Grabstein K. In: Protein Engineering. Kaumaya PP, editor. InTech; Croatia: 2012. p. 253. [Google Scholar]
- 738.Erskine PT, Senior N, Awan S, Lambert R, Lewis G, Tickle IJ, Sarwar M, Spencer P, Thomas P, Warren MJ, Shoolingin-Jordan PM, Wood SP, Cooper JB. Nat Struct Biol. 1997;4:1025. doi: 10.1038/nsb1297-1025. [DOI] [PubMed] [Google Scholar]
- 739.Erskine PT, Duke EMH, Tickle IJ, Senior NM, Warren MJ, Cooper JB. Acta Crystallogr, Sect D. 2000;56:421. doi: 10.1107/s0907444900000597. [DOI] [PubMed] [Google Scholar]
- 740.MacPherson IS, Murphy MEP. Cell Mol Life Sci. 2007;64:2887. doi: 10.1007/s00018-007-7310-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Murphy MEP, Turley S, Kukimoto M, Nishiyama M, Horinouchi S, Sasaki H, Tanakura M, Adman ET. Biochemistry. 1995;34:12107. doi: 10.1021/bi00038a003. [DOI] [PubMed] [Google Scholar]
- 742.Godden J, Turley S, Teller D, Adman E, Liu M, Payne W, LeGall J. Science. 1991;253:438. doi: 10.1126/science.1862344. [DOI] [PubMed] [Google Scholar]
- 743.Iranzo O, Cabello C, Pecoraro VL. Angew Chem, Int Ed. 2007;46:6688. doi: 10.1002/anie.200701729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Iranzo O, Chakraborty S, Hemmingsen L, Pecoraro VL. J Am Chem Soc. 2011;133:239. doi: 10.1021/ja104433n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745.Touw DS. Structural and Spectroscopic Studies of Heavy Metal Binding to De Novo Designed Coiled Coil Peptides. University of Michigan; Ann Arbor, MI: 2007. [Google Scholar]
- 746.DeSilva TM, Veglia G, Porcelli F, Prantner AM, Opella SJ. Biopolymers. 2002;64:189. doi: 10.1002/bip.10149. [DOI] [PubMed] [Google Scholar]
- 747.Veglia G, Porcelli F, DeSilva T, Prantner A, Opella SJ. J Am Chem Soc. 2000;122:2389. [Google Scholar]
- 748.Opella SJ, DeSilva TM, Veglia G. Curr Opin Chem Biol. 2002;6:217. doi: 10.1016/s1367-5931(02)00314-9. [DOI] [PubMed] [Google Scholar]
- 749.Pires S, Habjanič J, Sezer M, Soares CM, Hemmingsen L, Iranzo O. Inorg Chem. 2012;51:11339. doi: 10.1021/ic3008014. [DOI] [PubMed] [Google Scholar]
- 750.Chakraborty S, Kravitz JY, Thulstrup PW, Hemmingsen L, DeGrado WF, Pecoraro VL. Angew Chem, Int Ed. 2011;50:2049. doi: 10.1002/anie.201006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 751.Walsh STR, Cheng H, Bryson JW, Roder H, DeGrado WF. Proc Natl Acad Sci USA. 1999;96:5486. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 752.Bryson JW, Desjarlais JR, Handel TM, DeGrado WF. Protein Sci. 1998;7:1404. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Salemme FR. Prog Biophys Mol Biol. 1983;42:95. doi: 10.1016/0079-6107(83)90005-6. [DOI] [PubMed] [Google Scholar]
- 754.Richardson JS. Adv Protein Chem. 1981:167. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 755.Chothia C. Annu Rev Biochem. 1984;53:537. doi: 10.1146/annurev.bi.53.070184.002541. [DOI] [PubMed] [Google Scholar]
- 756.Somers WS, Philips SEV. Nature. 1992;359:387. doi: 10.1038/359387a0. [DOI] [PubMed] [Google Scholar]
- 757.Puglisi JD, Chen L, Blanchard S, Frankel AD. Science. 1995;270:1200. doi: 10.1126/science.270.5239.1200. [DOI] [PubMed] [Google Scholar]
- 758.Derrick JP, Wigley DB. Nature. 1992;259:752. doi: 10.1038/359752a0. [DOI] [PubMed] [Google Scholar]
- 759.Pessi A, Bianchi E, Crameri A, Venturini S, Tramontano A, Sollazzo M. Nature. 1993;362:367. doi: 10.1038/362367a0. [DOI] [PubMed] [Google Scholar]
- 760.Satow Y, Cohen GH, Padlan EA, Davies DR. J Mol Biol. 1986;190:593. doi: 10.1016/0022-2836(86)90245-7. [DOI] [PubMed] [Google Scholar]
- 761.Bianchi E, Venturini S, Pessi A, Tramontano A, Sollazzo M. J Mol Biol. 1994;236:649. doi: 10.1006/jmbi.1994.1174. [DOI] [PubMed] [Google Scholar]
- 762.Zappacosta F, Ingallinella P, Scaloni A, Pessi A, Bianchi E, Sollazzo M, Tramontano A, Marino G, Pucci P. Protein Sci. 1997;6:1901. doi: 10.1002/pro.5560060911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 763.Zappacosta F, Pessi A, Bianchi E, Venturini S, Sollazzo M, Tramontano A, Marino G, Pucci P. Protein Sci. 1996;5:802. doi: 10.1002/pro.5560050502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 764.Guy PA, Anderegg RJ, Lim A, Saderholm MJ, Yan Y, Erickson BW. J Am Soc Mass Spectrom. 1999;10:969. doi: 10.1016/S1044-0305(99)00069-0. [DOI] [PubMed] [Google Scholar]
- 765.Lim A, Guy PA, Makhov AM, Saderholm MJ, Kroll M, Yan Y, Anderegg RJ, Griffith JD, Erickson BW. Lett Pept Sci. 1999;6:3. [Google Scholar]
- 766.Richardson JS, Richardson DC. In: Prediction of Protein Structure and the Principles of Protein Conformation. Fasman GD, editor. Springer US; Boston, MA: 1989. p. 1. [Google Scholar]
- 767.Yan Y, Tropsha A, Hermans J, Erickson BW. Proc Natl Acad Sci USA. 1993;90:7898. doi: 10.1073/pnas.90.16.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 768.Yang Y, Erickson BW, Tropsha A. J Am Chem Soc. 1995;117:7592. [Google Scholar]
- 769.Lim A, Makhov AM, Bond J, Inouye H, Connors LH, Griffith JD, Erickson BW, Kirschner DA, Costello CE. J Struct Biol. 2000;130:363. doi: 10.1006/jsbi.2000.4272. [DOI] [PubMed] [Google Scholar]
- 770.Inouye H, Bond JE, Deverin SP, Lim A, Costello CE, Kirschner DA. Biophys J. 2002;83:1716. doi: 10.1016/S0006-3495(02)73939-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 771.Imperiali B, Fisher SL. J Am Chem Soc. 1991;113:8527. [Google Scholar]
- 772.Cheng RP, Fisher SL, Imperiali B. J Am Chem Soc. 1996;118:11349. [Google Scholar]
- 773.Loving G, Imperiali B. J Am Chem Soc. 2008;130:13630. doi: 10.1021/ja804754y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 774.Vogel EM, Imperiali B. Protein Sci. 2007;16:550. doi: 10.1110/ps.062549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 775.Reynolds AM, Sculimbrene BR, Imperiali B. Bioconjugate Chem. 2008;19:588. doi: 10.1021/bc700426c. [DOI] [PubMed] [Google Scholar]
- 776.Regan L, Clarke ND. Biochemistry. 1990;29:10878. doi: 10.1021/bi00501a003. [DOI] [PubMed] [Google Scholar]
- 777.Klemba M, Regan L. Biochemistry. 1995;34:10094. doi: 10.1021/bi00031a034. [DOI] [PubMed] [Google Scholar]
- 778.Michael SF, Kilfoil VJ, Schmidt MH, Amann BT, Berg JM. Proc Natl Acad Sci USA. 1992;89:4796. doi: 10.1073/pnas.89.11.4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 779.Miller J, McLachlan AD, Klug A. EMBO J. 1985;4:1609. doi: 10.1002/j.1460-2075.1985.tb03825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 780.Sénèque O, Bourlès E, Lebrun V, Bonnet E, Dumy P, Latour J-M. Angew Chem, Int Ed. 2008;47:6888. doi: 10.1002/anie.200800677. [DOI] [PubMed] [Google Scholar]
- 781.Sénèque O, Bonnet E, Joumas FL, Latour J-M. Chem—Eur J. 2009;15:4798. doi: 10.1002/chem.200900147. [DOI] [PubMed] [Google Scholar]
- 782.Sénèque O, Latour J-M. J Am Chem Soc. 2010;132:17760. doi: 10.1021/ja104992h. [DOI] [PubMed] [Google Scholar]
- 783.Krizek BA, Merkle DL, Berg JM. Inorg Chem. 1993;32:937. [Google Scholar]
- 784.Ghosh D, Pecoraro VL. Curr Opin Chem Biol. 2005;9:97. doi: 10.1016/j.cbpa.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 785.Ball ZT. Acc Chem Res. 2013;46:560. doi: 10.1021/ar300261h. [DOI] [PubMed] [Google Scholar]
- 786.Ghadiri MR, Soares C, Choi C. J Am Chem Soc. 1992;114:825. [Google Scholar]
- 787.Tsurkan MV, Ogawa MY. Inorg Chem. 2007;46:6849. doi: 10.1021/ic700958h. [DOI] [PubMed] [Google Scholar]
- 788.Suzuki K, Hiroaki H, Kohda D, Nakamura H, Tanaka T. J Am Chem Soc. 1998;120:13008. [Google Scholar]
- 789.Li X, Suzuki K, Kanaori K, Tajima K, Kashiwada A, Hiroaki H, Kohda D, Tanaka T. Protein Sci. 2000;9:1327. doi: 10.1110/ps.9.7.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 790.Ghadiri MR, Choi C. J Am Chem Soc. 1990;112:1630. [Google Scholar]
- 791.Ghadiri MR, Fernholz AK. J Am Chem Soc. 1990;112:9633. [Google Scholar]
- 792.Ghadiri MR, Case MA. Angew Chem, Int Ed Engl. 1993;32:1594. [Google Scholar]
- 793.Ghadiri MR, Soares C, Choi C. J Am Chem Soc. 1992;114:4000. [Google Scholar]
- 794.Hong J, Kharenko OA, Ogawa MY. Inorg Chem. 2006;45:9974. doi: 10.1021/ic060222j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 795.Kharenko OA, Ogawa MY. J Inorg Biochem. 2004;98:1971. doi: 10.1016/j.jinorgbio.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 796.Kharenko OA, Kennedy DC, Demeler B, Maroney MJ, Ogawa MY. J Am Chem Soc. 2005;127:7678. doi: 10.1021/ja042757m. [DOI] [PubMed] [Google Scholar]
- 797.Suzuki K, Hiroaki H, Kohda D, Tanaka T. Protein Eng, Des Sel. 1998;11:1051. doi: 10.1093/protein/11.11.1051. [DOI] [PubMed] [Google Scholar]
- 798.Murase S, Ishino S, Ishino Y, Tanaka T. JBIC, J Biol Inorg Chem. 2012;17:791. doi: 10.1007/s00775-012-0896-x. [DOI] [PubMed] [Google Scholar]
- 799.Dublin SN, Conticello VP. J Am Chem Soc. 2008;130:49. doi: 10.1021/ja0775016. [DOI] [PubMed] [Google Scholar]
- 800.Harbury PB, Kim PS, Alber T. Nature. 1994;371:80. doi: 10.1038/371080a0. [DOI] [PubMed] [Google Scholar]
- 801.Zimenkov Y, Dublin SN, Ni R, Tu RS, Breedveld V, Apkarian RP, Conticello VP. J Am Chem Soc. 2006;128:6770. doi: 10.1021/ja0605974. [DOI] [PubMed] [Google Scholar]
- 802.Cal M, Jaremko Ł, Jaremko M, Stefanowicz P. New J Chem. 2013;37:3770. [Google Scholar]
- 803.Halfen JA, Bodwin JJ, Pecoraro VL. Inorg Chem. 1998;37:5416. doi: 10.1021/ic9807386. [DOI] [PubMed] [Google Scholar]
- 804.Bodwin JJ, Cutland AD, Malkani RG, Pecoraro VL. Coord Chem Rev. 2001;216–217:489. [Google Scholar]
- 805.Tegoni M, Remelli M. Coord Chem Rev. 2012;256:289. [Google Scholar]
- 806.Kaziro Y, Itoh H, Kozasa T, Nakafuku M, Satoh T. Annu Rev Biochem. 1991;60:349. doi: 10.1146/annurev.bi.60.070191.002025. [DOI] [PubMed] [Google Scholar]
- 807.Gerstein M, Echols N. Curr Opin Chem Biol. 2004;8:14. doi: 10.1016/j.cbpa.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 808.Vale RD, Milligan RA. Science. 2000;288:88. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- 809.Robinson VL, Buckler DR, Stock AM. Nat Struct Biol. 2000;7:626. doi: 10.1038/77915. [DOI] [PubMed] [Google Scholar]
- 810.Beckett D. Proc Natl Acad Sci USA. 2009;106:22035. doi: 10.1073/pnas.0912300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 811.Champoux JJ. Annu Rev Biochem. 2001;70:369. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 812.Page MJ, Cera EDi. Physiol Rev. 2006;86:1049. doi: 10.1152/physrev.00008.2006. [DOI] [PubMed] [Google Scholar]
- 813.Reyes-Caballero H, Campanello GC, Giedroc DP. Biophys Chem. 2011;156:103. doi: 10.1016/j.bpc.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 814.Fan SW, George RA, Haworth NL, Feng LL, Liu JY, Wouters MA. Protein Sci. 2009;18:1745. doi: 10.1002/pro.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 815.Kelly JW. Curr Opin Struct Biol. 1998;8:101. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 816.Carrell RW, Gooptut B. Curr Opin Struct Biol. 1998;8:799. doi: 10.1016/s0959-440x(98)80101-2. [DOI] [PubMed] [Google Scholar]
- 817.Cerasoli E, Sharpe BK, Woolfson DN. J Am Chem Soc. 2005;127:15008. doi: 10.1021/ja0543604. [DOI] [PubMed] [Google Scholar]
- 818.Ambroggio XI, Kuhlman B. J Am Chem Soc. 2006;128:1154. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 819.Pagel K, Seri T, von Berlepsch H, Griebel J, Kirmse R, Böttcher C, Koksch B. ChemBioChem. 2008;9:531. doi: 10.1002/cbic.200700656. [DOI] [PubMed] [Google Scholar]
- 820.Der BS, Machius M, Miley MJ, Mills JL, Szyperski T, Kuhlman B. J Am Chem Soc. 2012;134:375. doi: 10.1021/ja208015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 821.Gomez-Tagle P, Vargas-Zúñiga I, Taran O, Yatsimirsky AK. J Org Chem. 2006;71:9713. doi: 10.1021/jo061780i. [DOI] [PubMed] [Google Scholar]
- 822.Zastrow ML, Pecoraro VL. Biochemistry. 2014 doi: 10.1021/bi4016617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 823.Kimura E, Shiota T, Koike T, Shiro M, Kodama M. J Am Chem Soc. 1990;112:5805. [Google Scholar]
- 824.Koerner TB, Brown RS. Can J Chem. 2002;80:183. [Google Scholar]
- 825.Bazzicalupi C, Bencini A, Bianchi A, Fusi V, Giorgi C, Paoletti P, Valtancoli B, Zanchi D. Inorg Chem. 1997;36:2784. doi: 10.1021/ic961521j. [DOI] [PubMed] [Google Scholar]
- 826.Verpoorte JA, Mehta S, Edsall JT. J Biol Chem. 1967;242:4221. [PubMed] [Google Scholar]
- 827.Innocenti A, Scozzafava A, Parkkila S, Puccetti L, De Simone G, Supuran CT. Bioorg Med Chem Lett. 2008;18:2267. doi: 10.1016/j.bmcl.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 828.Liang Z, Xue Y, Behravan G, Jonsson BH, Lindskog S. Eur J Biochem. 1993;211:821. doi: 10.1111/j.1432-1033.1993.tb17614.x. [DOI] [PubMed] [Google Scholar]
- 829.Krebs JF, Ippolito JA, Christianson DW, Fierke CA. J Biol Chem. 1993;268:27458. [PubMed] [Google Scholar]
- 830.Woolley P. Nature. 1975;258:677. doi: 10.1038/258677a0. [DOI] [PubMed] [Google Scholar]
- 831.Huguet J, Brown RS. J Am Chem Soc. 1980;102:7571. [Google Scholar]
- 832.Brown RS, Curtis NJ, Huguet J. J Am Chem Soc. 1981;103:6953. [Google Scholar]
- 833.Brown RS, Salmon D, Curtis NJ, Kusuma S. J Am Chem Soc. 1982;104:3188. [Google Scholar]
- 834.Slebocka-Tilk H, Cocho JL, Frackman Z, Brown RS. J Am Chem Soc. 1984;106:2421. [Google Scholar]
- 835.Zhang X, van Eldik R, Koike T, Kimura E. Inorg Chem. 1993;32:5749. [Google Scholar]
- 836.Zhang X, van Eldik R. Inorg Chem. 1995;34:5606. [Google Scholar]
- 837.Nakata K, Shimomura N, Shiina N, Izumi M, Ichikawa K, Shiro M. J Inorg Biochem. 2002;89:255. doi: 10.1016/s0162-0134(01)00419-6. [DOI] [PubMed] [Google Scholar]
- 838.Holm RH, Kennepohl P, Solomon EI. Chem Rev. 1996;96:2239. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 839.Müller HN, Skerra A. Biochemistry. 1994;33:14126. doi: 10.1021/bi00251a023. [DOI] [PubMed] [Google Scholar]
- 840.Vita C, Roumestand C, Toma F, Ménez A. Proc Natl Acad Sci USA. 1995;92:6404. doi: 10.1073/pnas.92.14.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 841.Stewart JD, Roberts VA, Crowder MW, Getzoff ED, Benkovic SJ. J Am Chem Soc. 1994;116:415. [Google Scholar]
- 842.Wade WS, Koh JS, Han N, Hoekstra DM, Lerner RA. J Am Chem Soc. 1993;115:4449. [Google Scholar]
- 843.Handel T, Williams SA, DeGrado WF. Science. 1993;261:879. doi: 10.1126/science.8346440. [DOI] [PubMed] [Google Scholar]
- 844.Hitomi Y, Outten CE, O’Halloran TV. J Am Chem Soc. 2001;123:8614. doi: 10.1021/ja016146v. [DOI] [PubMed] [Google Scholar]
- 845.Fierke CA, Thompson RB. BioMetals. 2001;14:205. doi: 10.1023/a:1012980628412. [DOI] [PubMed] [Google Scholar]
- 846.Song H, Wilson DL, Farquhar ER, Lewis EA, Emerson JP. Inorg Chem. 2012;51:11098. doi: 10.1021/ic301645j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847.Weston J. Chem Rev. 2005;105:2151. doi: 10.1021/cr020057z. [DOI] [PubMed] [Google Scholar]
- 848.Wilcox DE. Chem Rev. 1996;96:2435. doi: 10.1021/cr950043b. [DOI] [PubMed] [Google Scholar]
- 849.Jedrzejas MJ, Setlow P. Chem Rev. 2001;101:607. doi: 10.1021/cr000253a. [DOI] [PubMed] [Google Scholar]
- 850.Lowther WT, Matthews BW. Chem Rev. 2002;102:4581. doi: 10.1021/cr0101757. [DOI] [PubMed] [Google Scholar]
- 851.Dupureur CM. Curr Opin Chem Biol. 2008;12:250. doi: 10.1016/j.cbpa.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 852.Dupureur CM. Metallomics. 2010;2:609. doi: 10.1039/c0mt00013b. [DOI] [PubMed] [Google Scholar]
- 853.Brown RS, Lu Z-L, Liu CT, Tsang WY, Edwards DR, Neverov AA. J Phys Org Chem. 2010;23:1. [Google Scholar]
- 854.Lönnberg H. Org Biomol Chem. 2011;9:1687. doi: 10.1039/c0ob00486c. [DOI] [PubMed] [Google Scholar]
- 855.Liu C, Wang L. Dalton Trans. 2009:227. doi: 10.1039/b811616d. [DOI] [PubMed] [Google Scholar]
- 856.Morrow JR, Amyes TL, Richard JP. Acc Chem Res. 2008;41:539. doi: 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 857.Mancin F, Scrimin P, Tecilla P, Tonellato U. Chem Commun (Cambridge, UK) 2005:2540. doi: 10.1039/b418164f. [DOI] [PubMed] [Google Scholar]
- 858.Yatsimirsky A. Coord Chem Rev. 2005;249:1997. [Google Scholar]
- 859.Liu C, Wang M, Zhang T, Sun H. Coord Chem Rev. 2004;248:147. [Google Scholar]
- 860.Krämer R. Coord Chem Rev. 1999;182:243. [Google Scholar]
- 861.Chin J. Acc Chem Res. 1991;24:145. [Google Scholar]
- 862.Vichard C, Kaden TA. Inorg Chim Acta. 2002;337:173. [Google Scholar]
- 863.Rossi P, Felluga F, Tecilla P, Formaggio F, Crisma M, Toniolo C, Scrimin P. J Am Chem Soc. 1999;121:6948. [Google Scholar]
- 864.Rossi P, Felluga F, Tecilla P, Formaggio F, Crisma M, Toniolo C, Scrimin P. Biopolymers. 2000;55:496. doi: 10.1002/1097-0282(2000)55:6<496::AID-BIP1025>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 865.Sissi C, Rossi P, Felluga F, Gormaggio F, Palumbo M, Tecilla P, Toniolo C, Scrimin P. J Am Chem Soc. 2001;123:3169. doi: 10.1021/ja005675g. [DOI] [PubMed] [Google Scholar]
- 866.Scarso A, Scheffer U, Göbel M, Broxterman QB, Kaptein B, Formaggio F, Toniolo C, Scrimin P. Proc Natl Acad Sci USA. 2002;99:5144. doi: 10.1073/pnas.072642699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 867.Rossi P, Tecilla P, Baltzer L, Scrimin P. Chem—Eur J. 2004;10:4163. doi: 10.1002/chem.200400160. [DOI] [PubMed] [Google Scholar]
- 868.Olofsson S, Johansson G, Baltzer L. J Chem Soc, Perkin Trans 2. 1995:2047. [Google Scholar]
- 869.Olofsson S, Baltzer L. Fold Des. 1996;1:347. doi: 10.1016/S1359-0278(96)00050-8. [DOI] [PubMed] [Google Scholar]
- 870.Broo KS, Brive L, Ahlberg P, Baltzer L. J Am Chem Soc. 1997;7863:11362. [Google Scholar]
- 871.Sasaki T, Kaiser ET. J Am Chem Soc. 1989;111:380. [Google Scholar]
- 872.Choma CT, Lear JD, Nelson MJ, Dutton PL, Robertson DE, DeGrado WF. J Am Chem Soc. 1994;116:856. [Google Scholar]
- 873.Shifman JM, Moser CC, Kalsbeck WA, Bocian DF, Dutton PL. Biochemistry. 1998;37:16815. doi: 10.1021/bi9816857. [DOI] [PubMed] [Google Scholar]
- 874.Chen X, Moser CC, Pilloud DL, Dutton PL. J Phys Chem B. 1998;102:6425. [Google Scholar]
- 875.Pilloud DL, Rabanal F, Gibney BR, Farid RS, Dutton PL, Moser CC. J Phys Chem B. 1998;102:1926. [Google Scholar]
- 876.Chen X, Moser CC, Pilloud DL, Gibney BR, Dutton PL. J Phys Chem B. 1999;103:9029. [Google Scholar]
- 877.Chen X, Discher BM, Pilloud DL, Gibney BR, Moser CC, Dutton PL. J Phys Chem B. 2002;106:617. [Google Scholar]
- 878.Ye S, Strzalka J, Chen X, Moser CC, Dutton PL, Blasie JK. Langmuir. 2003;19:1515. [Google Scholar]
- 879.Topoglidis E, Discher BM, Moser CC, Dutton PL, Durrant JR. ChemBioChem. 2003;4:1332. doi: 10.1002/cbic.200300707. [DOI] [PubMed] [Google Scholar]
- 880.Ye S, Discher BM, Strzalka J, Xu T, Wu SP, Noy D, Kuzmenko I, Gog T, Therien MJ, Dutton PL, Blasie JK. Nano Lett. 2005;5:1658. doi: 10.1021/nl050542k. [DOI] [PubMed] [Google Scholar]
- 881.Discher BM, Koder RL, Moser CC, Dutton PL. Curr Opin Chem Biol. 2003;7:741. doi: 10.1016/j.cbpa.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 882.Ye S, Strzalka JW, Discher BM, Noy D, Zheng S, Dutton PL, Blasie JK. Langmuir. 2004;20:5897. doi: 10.1021/la0363884. [DOI] [PubMed] [Google Scholar]
- 883.Discher BM, Noy D, Strzalka J, Ye S, Moser CC, Lear JD, Blasie JK, Dutton PL. Biochemistry. 2005;44:12329. doi: 10.1021/bi050695m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 884.Ghirlanda G. Curr Opin Chem Biol. 2009;13:643. doi: 10.1016/j.cbpa.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 885.Gibney BR, Isogai Y, Rabanal F, Reddy KS, Grosset AM, Moser CC, Dutton PL. Biochemistry. 2000;39:11041. doi: 10.1021/bi000925r. [DOI] [PubMed] [Google Scholar]
- 886.Ghirlanda G, Osyczka A, Liu W, Antolovich M, Smith KM, Dutton PL, Wand AJ, DeGrado WF. J Am Chem Soc. 2004;126:8141. doi: 10.1021/ja039935g. [DOI] [PubMed] [Google Scholar]
- 887.Cochran FV, Wu SP, Wang W, Nanda V, Saven JG, Therien MJ, DeGrado WF. J Am Chem Soc. 2005;127:1346. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- 888.Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, DeGrado WF. J Am Chem Soc. 2007;129:10732. doi: 10.1021/ja071199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 889.Fry HC, Lehmann A, Saven JG, DeGrado WF, Therien MJ. J Am Chem Soc. 2010;132:3997. doi: 10.1021/ja907407m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 890.Robertson DE, Farid RS, Moser CC, Urbauer JL, Mulholland SE, Pidikiti R, Lear JD, Wand AJ, DeGrado WF, Dutton PL. Nature. 1994;368:425. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 891.Rabanal F, DeGrado WF, Dutton PL. J Am Chem Soc. 1996;118:473. [Google Scholar]
- 892.Zhuang J, Reddi AR, Wang Z, Khodaverdian B, Hegg EL, Gibney BR. Biochemistry. 2006;45:12530. doi: 10.1021/bi060565t. [DOI] [PubMed] [Google Scholar]
- 893.Rau HK, Haehnel W. J Am Chem Soc. 1998;120:468. [Google Scholar]
- 894.Rau H, DeJonge N, Haehnel W. Angew Chem, Int Ed. 2000;39:250. [PubMed] [Google Scholar]
- 895.Mutter M, Altmann E, Altmann K-H, Hersperger R, Koziej P, Nebel K, Tuchsecherer G, Vuilleumier S, Gremlich H-U, Muller K. Helv Chim Acta. 1988;71:835. [Google Scholar]
- 896.Mutter M, Tuchscherer GG, Miller C, Altmann KH, Carey RI, Wyss DF, Labhardt AM, Rivier JE. J Am Chem Soc. 1992;114:1463. [Google Scholar]
- 897.Mutter M, Tuchscherer G. Cell Mol Life Sci. 1997;53:851. doi: 10.1007/s000180050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 898.Fontecave M. Nat Chem Biol. 2006;2:171. doi: 10.1038/nchembio0406-171. [DOI] [PubMed] [Google Scholar]
- 899.Beinert H. JBIC, J Biol Inorg Chem. 2000;5:2. doi: 10.1007/s007750050002. [DOI] [PubMed] [Google Scholar]
- 900.Beinert H. Science. 1997;277:653. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 901.Beinert H, Kiley PJ. Curr Opin Chem Biol. 1999;3:152. doi: 10.1016/S1367-5931(99)80027-1. [DOI] [PubMed] [Google Scholar]
- 902.Kiley PJ, Beinert H. Curr Opin Microbiol. 2003;6:181. doi: 10.1016/s1369-5274(03)00039-0. [DOI] [PubMed] [Google Scholar]
- 903.Venkateswara Rao P, Holm RH. Chem Rev. 2004;104:527. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 904.Lombardi A, Marasco D, Maglio O, Costanzo L, Di Nastri F, Pavone V. Proc Natl Acad Sci USA. 2000;97:11922. doi: 10.1073/pnas.97.22.11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 905.Nanda V, Rosenblatt MM, Osyczka A, Kono H, Getahun Z, Dutton PL, Saven JG, Degrado WF. J Am Chem Soc. 2005;127:5804. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 906.Stephens PJ, Jollie DR, Warshel A. Chem Rev. 1996;96:2491. doi: 10.1021/cr950045w. [DOI] [PubMed] [Google Scholar]
- 907.Eidsness MK, Burden AE, Richie KA, Kurtz DM, Scott RA, Smith ET, Ichiye T, Beard B, Min T, Kang C. Biochemistry. 1999;38:14803. doi: 10.1021/bi991661f. [DOI] [PubMed] [Google Scholar]
- 908.Jacques A, Clémancey M, Blondin G, Fourmond V, Latour J-M, Sénèque O. Chem Commun (Cambridge, UK) 2013;49:2915. doi: 10.1039/c3cc40517f. [DOI] [PubMed] [Google Scholar]
- 909.Dey A, Jenney FE, Adams MWW, Babini E, Takahashi Y, Fukuyama K, Hodgson KO, Hedman B, Solomon EI. Science. 2007;318:1464. doi: 10.1126/science.1147753. [DOI] [PubMed] [Google Scholar]
- 910.Gibney BR, Mulholland SE, Rabanal F, Dutton PL. Proc Natl Acad Sci USA. 1996;93:15041. doi: 10.1073/pnas.93.26.15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 911.Antonkine ML, Koay MS, Epel B, Breitenstein C, Gopta O, Gärtner W, Bill E, Lubitz W. Biochim Biophys Acta. 2009;1787:995. doi: 10.1016/j.bbabio.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 912.Scott MP, Biggins J. Protein Sci. 1997;6:340. doi: 10.1002/pro.5560060209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 913.Grzyb J, Xu F, Nanda V, Luczkowska R, Reijerse E, Lubitz W, Noy D. Biochim Biophys Acta. 2012;1817:1256. doi: 10.1016/j.bbabio.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 914.Roy A, Sarrou I, Vaughn MD, Astashkin AV, Ghirlanda G. Biochemistry. 2013 doi: 10.1021/bi401199s. [DOI] [PubMed] [Google Scholar]
- 915.Sweeney WV, Rabinowitz JC. Annu Rev Biochem. 1980;49:139. doi: 10.1146/annurev.bi.49.070180.001035. [DOI] [PubMed] [Google Scholar]
- 916.Faiella M, Roy A, Sommer D, Ghirlanda G. Biopolymers. 2013;100:558. doi: 10.1002/bip.22420. [DOI] [PubMed] [Google Scholar]
- 917.Schnepf R, Hörth P, Bill E, Wieghardt K, Hildebrandt P, Haehnel W. J Am Chem Soc. 2001;123:2186. doi: 10.1021/ja001880k. [DOI] [PubMed] [Google Scholar]
- 918.Schnepf R, Haehnel W, Wieghardt K, Hildebrandt P. J Am Chem Soc. 2004;126:14389. doi: 10.1021/ja0484294. [DOI] [PubMed] [Google Scholar]
- 919.Banner DW, Kokkinidis M, Tsernoglou D. J Mol Biol. 1987;196:657. doi: 10.1016/0022-2836(87)90039-8. [DOI] [PubMed] [Google Scholar]
- 920.Wallace-Williams SE, James CA, de Vries S, Saraste M, Lappalainen P, van der Oost J, Fabian M, Palmer G, Woodruff WH. J Am Chem Soc. 1996;118:3986. [Google Scholar]
- 921.Andrew CR, Fraczkiewicz R, Czernuszewicz RS, Lappalainen P, Saraste M, Sanders-Loehr J. J Am Chem Soc. 1996;118:10436. [Google Scholar]
- 922.Andrew CR, Sanders-Loehr J. Acc Chem Res. 1996;29:365. [Google Scholar]
- 923.Shiga D, Nakane D, Inomata T, Funahashi Y, Masuda H, Kikuchi A, Oda M, Noda M, Uchiyama S, Fukui K, Kanaori K, Tajima K, Takano Y, Nakamura H, Tanaka T. J Am Chem Soc. 2010;132:18191. doi: 10.1021/ja106263y. [DOI] [PubMed] [Google Scholar]
- 924.Shiga D, Hamano Y, Kamei M, Funahashi Y, Masuda H, Sakaguchi M, Ogura T, Tanaka T. JBIC, J Biol Inorg Chem. 2012;17:1025. doi: 10.1007/s00775-012-0916-x. [DOI] [PubMed] [Google Scholar]
- 925.Li H, Webb SP, Ivanic J, Jensen JH. J Am Chem Soc. 2004;126:8010. doi: 10.1021/ja049345y. [DOI] [PubMed] [Google Scholar]
- 926.Koch M, Velarde M, Harrison MD, Echt S, Fischer M, Messerschmidt A, Dennison C. J Am Chem Soc. 2005;127:158. doi: 10.1021/ja046184p. [DOI] [PubMed] [Google Scholar]
- 927.Shiga D, Funahashi Y, Masuda H, Kikuchi A, Noda M, Uchiyama S, Fukui K, Kanaori K, Tajima K, Takano Y, Nakamura H, Kamei M, Tanaka T. Biochemistry. 2012;51:7901. doi: 10.1021/bi3007884. [DOI] [PubMed] [Google Scholar]
- 928.Nastri F, Lombardi A, Morelli G, Maglio O, Auria GD, Pedone C, Pavone V, Mazzeo M, Paolillo L. Chem—Eur J. 1997;3:340. [Google Scholar]
- 929.Lombardi A, Nash F, Sanseverino M, Pedone C, Pavone V. Inorg Chim Acta. 1998;275–276:301. [Google Scholar]
- 930.Di Costanzo L, Geremia S, Randaccio L, Nastri F, Maglio O, Lombardi A, Pavone V. JBIC, J Biol Inorg Chem. 2004;9:1017. doi: 10.1007/s00775-004-0600-x. [DOI] [PubMed] [Google Scholar]
- 931.Ranieri A, Monari S, Sola M, Borsari M, Battistuzzi G, Ringhieri P, Nastri F, Pavone V, Lombardi A. Langmuir. 2010;26:17831. doi: 10.1021/la103744x. [DOI] [PubMed] [Google Scholar]
- 932.Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht H, Hecht MH. Science. 1993;262:1680. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 933.Rojas NR, Kamtekar S, Simons CT, McLean JE, Vogel KM, Spiro TG, Farid RS, Hecht MH. Protein Sci. 1997;6:2512. doi: 10.1002/pro.5560061204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 934.Moffet DA, Certain LK, Smith AJ, Kessel AJ, Beckwith KA, Hecht MH. J Am Chem Soc. 2000;122:7612. [Google Scholar]
- 935.Hiner ANP, Hernandez-Ruiz J, Arnao MB, Garcia-Canovas F, Acosta M. Biotechnol Bioeng. 1996;50:655. doi: 10.1002/(SICI)1097-0290(19960620)50:6<655::AID-BIT6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 936.Wei Y, Liu T, Sazinsky SL, Moffet DA, Pelczer I, Hecht MH. Protein Sci. 2003;12:92. doi: 10.1110/ps.0228003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 937.Moffet DA, Foley J, Hecht MH. Biophys Chem. 2003;105:231. doi: 10.1016/s0301-4622(03)00072-3. [DOI] [PubMed] [Google Scholar]
- 938.Das A, Hecht MH. J Inorg Biochem. 2007;101:1820. doi: 10.1016/j.jinorgbio.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 939.Wei Y, Hecht MH. Protein Eng Des Sel. 2004;17:67. doi: 10.1093/protein/gzh007. [DOI] [PubMed] [Google Scholar]
- 940.Patel SC, Bradley LH, Jinadasa SP, Hecht MH. Protein Sci. 2009;18:1388. doi: 10.1002/pro.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 941.Patel SC, Hecht MH. Protein Eng Des Sel. 2012;25:445. doi: 10.1093/protein/gzs025. [DOI] [PubMed] [Google Scholar]
- 942.Koder RL, Anderson JLR, Solomon LA, Reddy KS, Moser CC, Dutton PL. Nature. 2009;458:305. doi: 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 943.Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563. doi: 10.1021/cr950046o. [DOI] [PubMed] [Google Scholar]
- 944.Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 945.Rosenzweig AC, Sazinsky MH. Curr Opin Struct Biol. 2006;16:729. doi: 10.1016/j.sbi.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 946.Greenwood NN, Earnshaw A. Chemistry of the Elements. Elsevier Ltd; Oxford, Burlington: 1997. [Google Scholar]
- 947.Rubino JT, Franz KJ. J Inorg Biochem. 2012;107:129. doi: 10.1016/j.jinorgbio.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 948.Evans JP, Blackburn NJ, Klinman JP. Biochemistry. 2006;45:15419. doi: 10.1021/bi061734c. [DOI] [PubMed] [Google Scholar]
- 949.Pettingill TM, Strange RW, Blackburn NJ. JBIC, J Biol Inorg Chem. 1991;266:16996. [PubMed] [Google Scholar]
- 950.Chufán EE, Prigge ST, Siebert X, Eipper BA, Mains RE, Amzel LM. J Am Chem Soc. 2010;132:15565. doi: 10.1021/ja103117r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 951.Kapoor A, Shandilya M, Kundu S. PLoS One. 2011;6:e26509. doi: 10.1371/journal.pone.0026509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 952.Floris G, Mondovi B, editors. Copper Amine Oxidases: Structures, Catalytic Mechanisms, and Role in Pathophysiology. CRC Press; New York: 2009. [Google Scholar]
- 953.Bannister JV, Bannister WH, Rotilio G. CRC Crit Rev Biochem. 1987;22:111. doi: 10.3109/10409238709083738. [DOI] [PubMed] [Google Scholar]
- 954.Suzuki S, Kataoka K, Tamaguchi K. Acc Chem Res. 2000;33:728. doi: 10.1021/ar9900257. [DOI] [PubMed] [Google Scholar]
- 955.Averill BA. Chem Rev. 1996;96:2951. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 956.Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304:864. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 957.Rudzka K, Moreno DM, Eipper B, Mains R, Estrin DA, Amzel LM. JBIC, J Biol Inorg Chem. 2013;18:223. doi: 10.1007/s00775-012-0967-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 958.Jacobson F, Pistorius A, Farkas D, De Grip W, Hansson O, Sjölin L, Neutze R. J Biol Chem. 2007;282:6347. doi: 10.1074/jbc.M605746200. [DOI] [PubMed] [Google Scholar]
- 959.Suzuki S, Yamaguchi K, Kataoka K, Kobayashi K, Tagawa S, Kohzuma T, Shidara S, Iwasaki H. JBIC, J Biol Inorg Chem. 1997;2:265. [Google Scholar]
- 960.Wijma HJ, Jeuken LJC, Verbeet MP, Armstrong FA, Canters GW. J Biol Chem. 2006;281:16340. doi: 10.1074/jbc.M601610200. [DOI] [PubMed] [Google Scholar]
- 961.Tocheva EI, Rosell FI, Mauk AG, Murphy MEP. Science. 2004;304:867. doi: 10.1126/science.1095109. [DOI] [PubMed] [Google Scholar]
- 962.Tocheva EI, Eltis LD, Murphy ME. Biochemistry. 2008;47:4452. doi: 10.1021/bi7020537. [DOI] [PubMed] [Google Scholar]
- 963.Kataoka K, Furusawa H, Takagi K, Yamaguchi K, Suzuki S. J Biochem. 2000;127:345. doi: 10.1093/oxfordjournals.jbchem.a022613. [DOI] [PubMed] [Google Scholar]
- 964.Komeda N, Nagao H, Kushi Y, Adachi G, Suzuki M, Uehara A, Tanaka K. Bull Chem Soc Jpn. 1995;68:581. [Google Scholar]
- 965.Fujisawa K, Tateda A, Miyashita Y, Okamoto K, Paulat F, Praneeth VKK, Merkle A, Lehnert N. J Am Chem Soc. 2008;130:1205. doi: 10.1021/ja075071d. [DOI] [PubMed] [Google Scholar]
- 966.Kujime M, Izumi C, Tomura M, Hada M, Fujii H. J Am Chem Soc. 2008;130:6088. doi: 10.1021/ja075575b. [DOI] [PubMed] [Google Scholar]
- 967.Merkle AC, Lehnert N. Dalton Trans. 2012;41:3355. doi: 10.1039/c1dt11049g. [DOI] [PubMed] [Google Scholar]
- 968.Isoda N, Yokoyama H, Nojiri M, Suzuki S, Yamaguchi K. Bioelectrochemistry. 2010;77:82. doi: 10.1016/j.bioelechem.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 969.Kujime M, Fujii H. Angew Chem, Int Ed. 2006;45:1089. doi: 10.1002/anie.200503555. [DOI] [PubMed] [Google Scholar]
- 970.Richards RL, Durrant MC. J Chem Res. 2002;2002:95. [Google Scholar]
- 971.Woollard-Shore JG, Holland JP, Jones MW, Dilworth JR. Dalton Trans. 2010;39:1576. doi: 10.1039/b913463h. [DOI] [PubMed] [Google Scholar]
- 972.Yu F, Penner-Hahn JE, Pecoraro VL. J Am Chem Soc. 2013;135:18096. doi: 10.1021/ja406648n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 973.Maglio O, Nastri F, Martin de Rosales RT, Faiella M, Pavone V, DeGrado WF, Lombardi A. C R Chim. 2007;10:703. [Google Scholar]
- 974.Calhoun JR, Nastri F, Maglio O, Pavone V, Lombardi A, DeGrado WF. Biopolymers. 2005;80:264. doi: 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- 975.Lombardi A, Summa CM, Geremia S, Randaccio L, Pavone V, DeGrado WF. Proc Natl Acad Sci USA. 2000;97:6298. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 976.Di Costanzo L, Wade H, Geremia S, Randaccio L, Pavone V, DeGrado WF, Lombardi A. J Am Chem Soc. 2001;123:12749. doi: 10.1021/ja010506x. [DOI] [PubMed] [Google Scholar]
- 977.Torres Martin de Rosales R, Faiella M, Farquhar E, Que L, Andreozzi C, Pavone V, Maglio O, Nastri F, Lombardi A. JBIC, J Biol Inorg Chem. 2010;15:717. doi: 10.1007/s00775-010-0639-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 978.Pasternak A, Kaplan J, Lear JD, Degrado WF. Protein Sci. 2001;10:958. doi: 10.1110/ps.52101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 979.Maglio O, Nastri F, Pavone V, Lombardi A, DeGrado WF. Proc Natl Acad Sci USA. 2003;100:3772. doi: 10.1073/pnas.0730771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 980.Geremia S, Di Costanzo L, Randaccio L, Engel DE, Lombardi A, Nastri F, DeGrado WF. J Am Chem Soc. 2005;127:17266. doi: 10.1021/ja054199x. [DOI] [PubMed] [Google Scholar]
- 981.DeGrado WF, Di Costanzo L, Geremia S, Lombardi A, Pavone V, Randaccio L. Angew Chem, Int Ed. 2003;42:417. doi: 10.1002/anie.200390127. [DOI] [PubMed] [Google Scholar]
- 982.Lahr SJ, Engel DE, Stayrook SE, Maglio O, North B, Geremia S, Lombardi A, DeGrado WF. J Mol Biol. 2005;346:1441. doi: 10.1016/j.jmb.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 983.Summa CM, Rosenblatt MM, Hong J-K, Lear JD, DeGrado WF. J Mol Biol. 2002;321:923. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 984.Marsh ENG, DeGrado WF. Proc Natl Acad Sci USA. 2002;99:5150. doi: 10.1073/pnas.052023199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 985.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. J Mol Biol. 2003;334:1101. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 986.Calhoun JR, Liu W, Spiegel K, Dal Peraro M, Klein ML, Valentine KG, Wand AJ, DeGrado WF. Structure. 2008;16:210. doi: 10.1016/j.str.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 987.Calhoun JR, Bell CB, Smith TJ, Thamann TJ, DeGrado WF, Solomon EI. J Am Chem Soc. 2008;130:9188. doi: 10.1021/ja801657y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 988.Bell CB, Calhoun JR, Bobyr E, Wei P-P, Hedman B, Hodgson KO, Degrado WF, Solomon EI. Biochemistry. 2009;48:59. doi: 10.1021/bi8016087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 989.Reig AJ, Pires MM, Snyder RA, Wu Y, Jo H, Kulp DW, Butch SE, Calhoun JR, Szyperski T, Szyperski TG, Solomon EI, DeGrado WF. Nat Chem. 2012;4:900. doi: 10.1038/nchem.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 990.Zaykov AN, MacKenzie KR, Ball ZT. Chem—Eur J. 2009;15:8961. doi: 10.1002/chem.200901266. [DOI] [PubMed] [Google Scholar]
- 991.Zaykov AN, Popp BV, Ball ZT. Chem—Eur J. 2010;16:6651. doi: 10.1002/chem.200903092. [DOI] [PubMed] [Google Scholar]
- 992.Sambasivan R, Ball ZT. J Am Chem Soc. 2010;132:9289. doi: 10.1021/ja103747h. [DOI] [PubMed] [Google Scholar]
- 993.Sambasivan R, Ball ZT. Org Biomol Chem. 2012;10:8203. doi: 10.1039/c2ob26667a. [DOI] [PubMed] [Google Scholar]
- 994.Zaykov AN, Ball ZT. Tetrahedron. 2011;67:4397. [Google Scholar]
- 995.Sambasivan R, Ball ZT. Angew Chem, Int Ed. 2012;51:8568. doi: 10.1002/anie.201202512. [DOI] [PubMed] [Google Scholar]
- 996.Sambasivan R, Ball ZT. Chirality. 2013;25:493. doi: 10.1002/chir.22144. [DOI] [PubMed] [Google Scholar]
- 997.Popp BV, Ball ZT. J Am Chem Soc. 2010;132:6660. doi: 10.1021/ja101456c. [DOI] [PubMed] [Google Scholar]
- 998.Popp BV, Ball ZT. Chem Sci. 2011;2:690. [Google Scholar]
- 999.Chen Z, Vohidov F, Coughlin JM, Stagg LJ, Arold ST, Ladbury JE, Ball ZT. J Am Chem Soc. 2012;134:10138. doi: 10.1021/ja302284p. [DOI] [PubMed] [Google Scholar]
- 1000.Chen Z, Popp BV, Bovet CL, Ball ZT. ACS Chem Biol. 2011;6:920. doi: 10.1021/cb2001523. [DOI] [PubMed] [Google Scholar]
- 1001.Zaykov AN, Ball ZT. Chem Commun (Cambridge, UK) 2011;47:10927. doi: 10.1039/c1cc13169a. [DOI] [PubMed] [Google Scholar]
- 1002.Kundu R, Cushing PR, Popp BV, Zhao Y, Madden DR, Ball ZT. Angew Chem, Int Ed. 2012;51:7217. doi: 10.1002/anie.201202291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1003.Popp BV, Chen Z, Ball ZT. Chem Commun (Cambridge, UK) 2012;48:7492. doi: 10.1039/c2cc33808d. [DOI] [PubMed] [Google Scholar]
- 1004.Tegoni M. Eur J Inorg Chem. 2014 doi: 10.1002/ejic.201400057. accepted. [DOI] [Google Scholar]