

Abstract
A metabolic engineering perspective which views recombinant protein expression as a multistep pathway allows us to move beyond vector design and identify the downstream rate limiting steps in expression. In E.coli these are typically at the translational level and the supply of precursors in the form of energy, amino acids and nucleotides. Further recombinant protein production triggers a global cellular stress response which feedback inhibits both growth and product formation. Countering this requires a system level analysis followed by a rational host cell engineering to sustain expression for longer time periods. Another strategy to increase protein yields could be to divert the metabolic flux away from biomass formation and towards recombinant protein production. This would require a growth stoppage mechanism which does not affect the metabolic activity of the cell or the transcriptional or translational efficiencies. Finally cells have to be designed for efficient export to prevent buildup of proteins inside the cytoplasm and also simplify downstream processing. The rational and the high throughput strategies that can be used for the construction of such improved host cell platforms for recombinant protein expression is the focus of this review.
Keywords: Recombinant protein expression, Escherichia coli, Metabolic engineering, Genome engineering
Introduction
Host cell engineering has emerged as a powerful tool for designing microbial platforms targeted at improved metabolite production. Major successes in this area include improved production of isoprenoids, shikimic acid, isobutanol, amino acids, synthesis of artemesin, lycopene and many such metabolites [1-7]. The basic goal has been to redesign the complete pathway for the biosynthesis of these metabolites by simultaneously engineering multiple steps in the pathway. This has been achieved by a combination of many techniques such as gene knock-ins and knock-outs, promoter engineering, supplementing the expression of critical genes, enzyme engineering and modulation of the regulatory pathways. The commonly used strategies to enhance the metabolite flux through a pathway can be clubbed under the following categories a) Increase the flux through rate limiting steps in the pathway; b) Increase the supply of precursors; c) Block branched chain pathways which lead to by-product formation and d) Remove feedback controls in the pathway (Figure 1).
Figure 1.
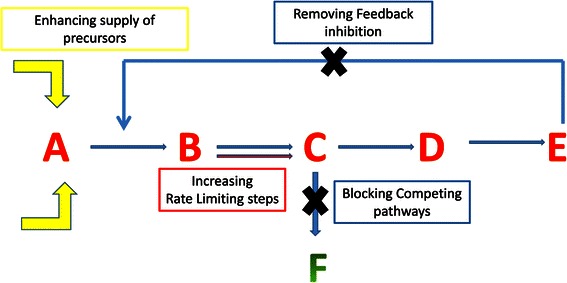
Metabolic engineering strategies to enhance the flux through a pathway. The various strategies used to improve the flux from a cellular intermediate to the desired product is shown. This includes enhancing pathways leading to the formation of intermediates (shown in yellow) and pathways which are rate limiting (B to C). Additionally branched chain pathways and feed back controls need to be blocked.
It is possible to extend the same philosophy with minor modifications to help in the design of hosts with improved recombinant protein expression capability. Just like the pathways in metabolite synthesis, recombinant protein expression also involves multiple steps viz. transcription, translation, folding and export. However unlike a typical metabolic pathway these steps are intricately linked to the cellular machinery with multiple host factors determining the flux through each step of the pathway. Hence the cellular physiology and its dynamics have a critical role in determining the overall flux through this pathway. Some important points that can be flagged by this approach are summarized as follows; traditional genetic engineering methods have mostly focused on improving the first step of this pathway i.e. transcription and hence the gains from improved vector design have tended to plateau over time. With strong promoters, the bottleneck in this pathway shifts to the translational step which needs to be up regulated to match the rates of transcription. Otherwise much of the gains of high rates of mRNA synthesis are offset by higher rates of mRNA degradation [8-11]. The supply of precursors which are critical to this pathway are the energy molecules like ATP, amino acids and nucleotides which can become the rate limiting factors in protein biosynthesis. Most importantly recombinant protein expression triggers a cellular stress response which feedback inhibits both growth and product formation, by lowering substrate uptake rates, down-regulating the ribosomal machinery and biosynthesis of ATP (Figure 2). This has a critical impact on the sustainability of the flux through this pathway and typically specific product formation rates decline sharply within a few hours post induction. Since host cell protein synthesis utilizes the same cellular machinery it can be treated as a competing pathway. Thus one way to increase recombinant protein synthesis would be to uncouple growth from product formation, thus allowing the diversion of metabolic fluxes toward product formation. Finally an efficient export mechanism needs to be in place, otherwise there is a theoretical upper limit to which the recombinant protein can accumulate inside the cells. Moreover extracellular expression would significantly simplify downstream processing steps. The challenges associated with designing such host platforms using both rational as well as high throughput strategies is the primary focus of this review.
Figure 2.
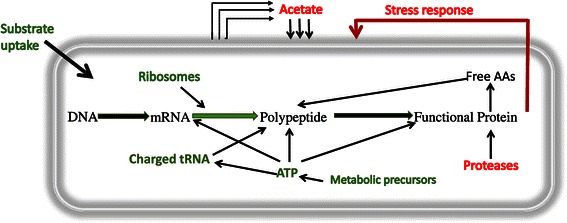
Simplified schematic of the cellular stress response on various factors affecting recombinant protein synthesis. The down-regulated pathways are shown in green (substrate uptake, ribosomes, translation rates, tRNA and ATP) and up regulated pathways (Proteases, acetate formation and stress response) are shown in red.
Improving transcriptional efficiency
The first step in the pathway for recombinant protein biosynthesis has possibly received the largest attention in terms of improved vector design. A very wide range of vectors are available both for E.coli and other microbes with specific features tailored for different applications [12-14]. The rate of mRNA synthesis is determined by both gene copy number and promoter strength, however with strong promoters like the T7 and T5, plasmid copy number has a relatively small role in expression. Rather the use of low copy number, stable vectors allows for lowered levels of ‘leaky’ expression which is important while expressing toxic proteins. Promoter design has thus focused more on titratable and tightly regulated systems rather than strength alone [15-18]. Thus a slow and controlled expression which leads to a properly folded protein can also be an important goal as is obtained with titratable promoters using low inducer concentrations [19,20]. Additionally the use of fusion tags in vector constructs can serve multiple purposes, like efficient purification, improved solubility, increased mRNA stability and more efficient translation [21-27]. To further simplify the process of protein production useful features like auto inducible systems [28-31] and self-cleavable tags have been incorporated in vector design [32-34].
Removing translational bottlenecks
Translation has been identified as the rate controlling step in recombinant protein synthesis for most high expression systems (Figure 3). Many factors have a role in controlling translational efficiency including, the first few codons of the mRNA to be translated (translation initiation) and the mRNA secondary structure. The ribosomal binding site (RBS) secondary structure is highly important for efficient initiation of translation. Recombinant protein translation in E.coli may be inhibited by presence of secondary structures in the RBS as well as 5’UTR region. Computational tools like ‘ExEnSo’ (Expression Enhancer Software) offer a platform where heterologous gene sequences can be designed on the basis of highest free energy so as to avoid translation inhibition due to mRNA secondary structures. The software also creates a 5’ primer on the basis of the ‘optimized’ sequence which can be used in PCR experiments to amplify the coding sequence of heterologous gene [35]. Similarly a predictive method for designing synthetic ribosome binding sites has been developed which enables a rational control over the protein expression level. This work combines a biophysical model of translation initiation with an optimization algorithm to predict the sequence of a synthetic RBS sequence that provides a target translation initiation rate [36]. Another work involving a random combinatorial DNA sequence library has revealed that not only the SD sequence but the entire UTR sequence, seems to play an important role in the translational process [37] implying that the rate of translation can also be rate-limiting. Translation rate calculators have been designed to estimate protein translation rates based on the sequence of the mRNA and have been shown to give good estimates of the actual level of protein expression [38].
Figure 3.
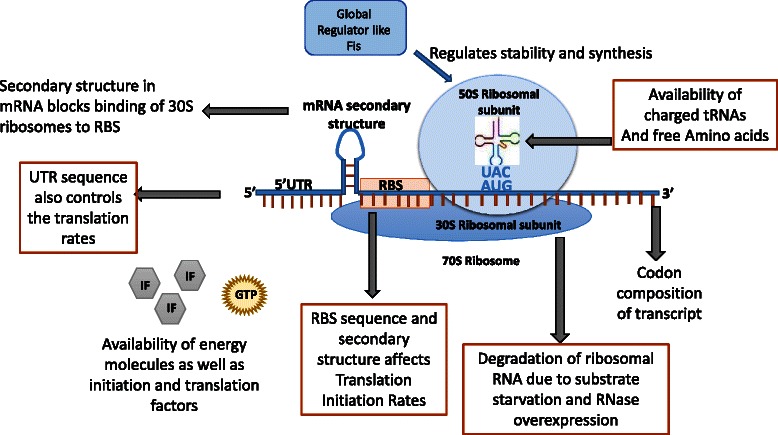
Cellular factors controlling the rate of translation. These factors which directly effect translation efficiency are a) mRNA RBS secondary structure affecting the Translation Initiation Rates (TIR) and the UTR sequences, b) Degradation of ribosomes due to substrate non-availability and RNase over expression, c) Availability of charged tRNA which depends on ATP supply and codon bias. Additionally global regulator like FIS also control translation and stability.
The increase in utilization of the protein synthetic machinery upon induction leads to a degradation of the ribosomal machinery, as a feedback stress response to over expression [39,40] that ultimately leading to a loss in the protein synthesis capacity. This decreased capacity of cells to synthesize proteins, as part of the stringent response, highlights the major challenges regarding the sustainability of recombinant protein production. It has been shown that whereas E.coli ribosomes are stable during exponential growth and in the stationary phase, degradation occurs between the transition stages and is independent from the triggering effect of the alarmone ppGpp(p) [41]. Degradation of stable RNA is also associated with conditions of starvation. Thus, depletion of any one of a number of nutrients including phosphate [42], nitrogen [43], carbon [44], or evenMg+2 [45] leads to a dramatic loss of RNA. RNase expression is also triggered during the stress response and can contribute to degradation of stable RNA [46].
Rate of translation may also slow down due to non-availability of aminoacylated transfer RNA (tRNA). The availability of charged aminoacylated transfer RNA further depends on codon composition of the transcript. The rationale behind codon usage optimization is to modify the rare codons in the target gene to mirror the codon usage of the host [47,48]. It is also known that the availability of tRNA varies significantly under different growth and stress conditions, which facilitates cellular adaptation to translational dynamics across the genome. Experimental measurements of tRNA concentrations and their charged fractions under stressful conditions have shown that tRNA availability can vary significantly between conditions and over time [49,50]. A computational workflow for estimating codon translation rates based on tRNA availability has been developed. This could be particularly important when considering the over expression of a recombinant protein, where a specific codon composition might lead to the depletion of certain charged tRNA pools [51] or under amino acid limited growth conditions that have been shown to lead to specific charging patterns [52,53]. This deficiency may lead to amino acid mis-incorporation and/or truncation of the polypeptide, thus affecting the heterologous protein expression levels and/or its activity [54]. OPTIMIZER, JCAT, Synthetic Gene Designer, DNAWorks, GeneDesign, Codon optimizer, GeMS are some of the online tools available to optimize codon usage (reviewed in [55]).
Likewise there are models like Ribosomal Flow Model (RFM) which analyses translation process on the basis of its physical and dynamical nature [56]. It considers the effect of codon order on translation rates, the stochastic nature of the translation process and the interactions between ribosomes while predicting the translation elongation step. This approach gives more accurate predictions of translation rates, protein abundance and ribosome densities in comparison to contemporary approaches. Another interesting feature that might be useful for recombinant protein expression and folding is the Translational pause at a rare codon. This provides a time delay to enable independent and sequential folding of the defined portions of the nascent polypeptide emerging from the ribosome [57].
Additionally there are regulatory genes that control the rates of ribosome biosynthesis. CsrA, is a posttranscriptional global regulator that regulates mRNA stability and translation, which in turn is regulated by two sRNAs csrB and csrC [58-60]. The E.coli DNA binding protein Fis is a transcriptional modulator involved in the regulation of many cellular processes, including the activation of rRNA synthesis. High-level expression of fis in early, mid, or late log cultures has been shown to result in growth phase and medium-specific variations in cell growth, rRNA synthesis, and ribosome content [61].
Improving energy availability
The synthesis of recombinant proteins is energy intensive and interferes with the host physiology [62]. The high energy demand during recombinant protein production leads to an enhanced need for ATP generation at the cost of biomass formation [63]. This increases maintenance energy requirements which manifests itself as an increased metabolic burden on the cells [64]. In order to sustain this energy demand, cells take up alternative pathways like substrate level phosphorylation leading to acetate formation by carbon overflow metabolism. As a consequence of reduced biomass formation, excess NADPH might be converted to NADH via the soluble transhydrogenase, filling the electron transport chain for additional ATP generation. This hypothesis is supported by a positive correlation between ATP production and productivity, while an inverse correlation exists between biomass yield and productivity. Although the ATP generation rate increases with increasing demand, the TCA cycle activity remains constant, indicating a limited capacity of the TCA cycle to overcome the postulated metabolic burden [65].
It has been shown that protein synthesis consumes approximately two-thirds of the total energy produced by a rapidly growing E.coli cell [66]. Consequently, much effort has been focused on understanding the mechanisms of ATP and GTP usage during protein synthesis. It was thus observed that phosphoenolpyruvate carboxykinase (PCK) when expressed in E.coli under glycolytic conditions helped in increasing the intracellular ATP levels, leading to enhanced protein production, of both the model proteins GFP (intracellular) and Alakaline Phosphatase (extracellular) [67]. Polymerization of amino acids as well as aminoacyl-tRNA synthetase requires large portion of ATP to mediate amino acid-charged tRNA synthesis [68]. It is known that the concentration of aminoacylated-tRNA (charged tRNA) molecules is higher in rapidly growing bacteria, and it has been postulated that the availability of the charged tRNA is one of the check points that determines the rate of protein translation [69].
Cofactor regeneration
Cofactors play an important role in generation of correctly folded, stable and functional recombinant proteins [70]. Any imbalance in cofactor consumption and regeneration can lead to a severe reduction of growth. Since these are the driving forces behind most anabolic pathways as well as oxidative phosphorylation, it is necessary to design strategies to enhance cofactor regeneration. OptSwap is a computational method which predicts strain designs by identifying optimal modifications of the cofactor binding specificities of oxidoreductase and complementary reaction knockouts [71]. Another mathematical framework, cofactor modification analysis (CMA), is a well-established constraints-based flux analysis method for the systematic identification of suitable cofactor specificity engineering (CSE) targets while exploring global metabolic effects [72]. Several genetic strategies employed for cofactor engineering have been reviewed earlier [73-76].
Facilitating protein folding and export
A major effort in recent years has focused on improved protein folding in vivo using chaperone co-expression [40,77-79]. These molecular chaperones essentially belong to Hsp70 chaperone family. Thus DnaK which is an Hsp70 homolog binds to unfolded hydrophobic stretches and helps protein folding while chaperones like GroEL encapsulates the nascent polypeptide and prevents inter molecular interactions [80]. A major issue is matching the availability of chaperones with the rates of production of the nascent polypeptide to prevent misfolding. This is a problem when high level expression systems are used. The only way to circumvent this is to have lower but sustained rates of protein expression leading to a slow buildup of the recombinant protein. Another important aspect is providing an oxidizing environment for correct disulfide bond formation in the cytoplasm or catalyzing bond formation of the oxidized protein in the periplasm [81]. This has been attempted by introducing genes for formation of disulfide bonds. Thus strains capable of producing properly folded proteins, even those with multiple disulphide bonds, are now available [82-87]. An E.coli strain has recently been designed for protein transport which oxidizes disulfide bonds in the cytoplasm and then efficiently exports these disulfide containing proteins using a signal peptide. These test proteins include alkaline phosphatase (PhoA), a phytase containing four disulfide bonds (AppA), an anti-interleukin 1bscFv and human growth hormone [88].
The more challenging task is protein export not just to the periplasm but to the extracellular medium. This would not only greatly simplify purification but also remove the upper bound on the accumulation of proteins in the culture. There are five pathways for protein secretion in E.coli Type I, II, III, IV and V. However, only the first and second secretion pathways are commonly used in recombinant protein secretion. Type I pathway directly targets proteins from cytoplasm to extracellular medium [89,90]. Studies have shown that the Type II Sec dependent pathway gets overloaded leading to an accumulation of unfolded proteins [91-95]. Plasmid based over expression of SecY, SecE and SecG proteins, which are the major interacting partners of SecA, resulted in a strong enhancement of a) translocation ATPase activity, b) preprotein translocation, c) capacity for SecA binding, and d) formation of the membrane-inserted form of SecA [96] (Figure 4). There are reports of a few proteins which get naturally secreted into the medium [97-99]. Others like GFP which do not get secreted through the Sec dependent pathway have been successfully exported via a modified TAT dependent secretion pathway [100]. In another work synthetically designed lipase ABC transporter domains (LARDs) from P.fluorescens lipase were attached to GFP and epidermal growth factor (EGF). The fused proteins were successfully secreted with the ABC transporter and showed lipase activity as an intact fused form in the supernatant [101]. These examples highlight some of the important developments in this area which has the potential of making E.coli into a truly secretory protein expression system.
Figure 4.
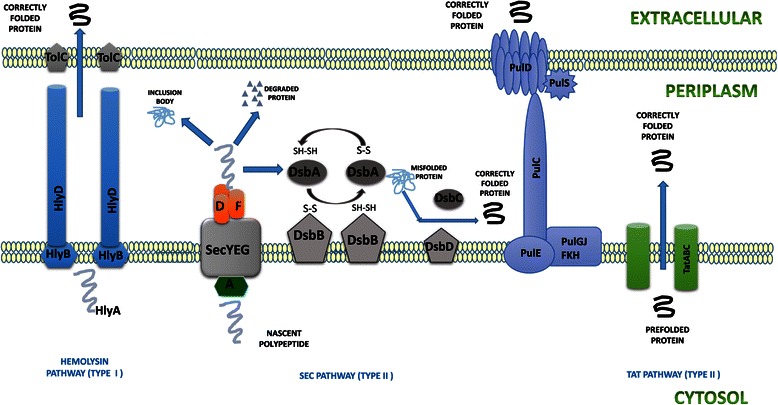
Various pathways for protein translocation (Type I and II). The type II Sec-dependent pathway is the most commonly used and gets overloaded leading to accumation of mis folded proteins in the cytoplasm. However the TAT dependent pathway is used for export of folded proteins and thus requires proper folding in the cytoplasm itself.
Feedback inhibition of product formation
It is well known that growth rates decline post induction in most cultures. It was earlier postulated that this was due to the ‘metabolic burden’ associated with the diversion of metabolic fluxes towards recombinant protein synthesis [102]. However a careful analysis of experimental data shows that this rate of decline of growth post induction, in the absence of substrate limitation is an intrinsic property of the cell and specific to the protein being expressed [103]. Thus some proteins like γ –interferon [104], α-interferon [105] even when they are expressed at high levels do not adversely affect growth while others like insulin [106], GMCSF [107,108], streptokinase [109], lead to a complete growth stoppage post–induction. Interestingly this retardation is also dependent on whether a protein is expressed as inclusion bodies or as a soluble protein like GMCSF and streptokinase [110-112]. Therefore attempts to explain growth retardation in terms of the amino acid composition of the expressed protein has limited predictive value [51,113,114] though clearly different amino acids impose different metabolic requirements on the cell. Clearly a static “diversion of fluxes” model is inadequate to explain this growth retardation rather a complex cellular dynamics controls both the growth and product formation kinetics.
This phenomenon of growth retardation is better understood by analyzing the cellular stress response to recombinant protein expression, which characteristically depends on the nature of protein, the form of expression (whether soluble or inclusion bodies) and the level of expression (whether from a strong or weak promoter) [115,116]. Moreover environmental factors such as the medium composition (presence or absence of complex nitrogen sources) [19,84,117,118] and the specific growth rate may also effect the nature of this response [119,120]. Studies have shown that this stress response mimics the features of the heat shock response, the oxidative stress response and the stringent response [39,114,121]. There are a set of common genes which get up or down-regulated due to this response which belong to the substrate uptake, amino acid and ribosomal biosynthesis genes and those involved in energy metabolism [39,121]. Others which are specific to the form of expression, like IB expression are clpBP, dnaJK, groLS, grpE, hslRUV, htpGX, ibpAB, lon, rlmE, rpoD, yrfI [122-124]. It is difficult to model this stress response using systems biology tools like Flux Balance Analysis (FBA) to predict the changes in fluxes of various pathways [125,126]. This is primarily because the commonly used metabolic model of E.coli with the largest set of pathways covers only ~30% of genes which are actually present in the organism [127,128]. Most of the differentially expressed genes are not part of this metabolic network and this unavailability prevents their expression mapping and FBA analysis using the model. Thus the stress response is better modeled as the triggering of key regulatory genes which in turn trigger a cascade of other downstream genes [121]. Efforts have been made to develop regulatory models which can analyze the complex interplay of the regulatory and metabolic networks [129-132]. These models could be applied in the analysis of the stress response due to recombinant protein over expression and provide us with leads for designing improved expression platforms. However there are as of now very few published reports on attempting to modulate this stress response by gene knock-ins or knock-outs. One would expect that knock-out of non-essential genes which get up-regulated due to the stress response or conversely supplementing gene expression of the down-regulated genes may help alleviate this stress and have a beneficial effect on recombinant protein expression [133,134].
The use of metabolic engineering strategies to remove the bottlenecks in recombinant protein production identified by analyzing this stress response has helped in improving the supply of precursors like NADPH, modification of global stress regulators and increasing the flux of the down regulated metabolic pathways including that of substrate uptake [135-140]. Thus increasing glycerol uptake by over expressing the glpK gene, lead to a 35% higher rhIFN-β expression as compared to control cultures [133]. The issue of acetate formation has been solved by knocking out genes (ackA, pta, ppc and poxB) in the acetate biosynthesis pathway leading to improve the recombinant protein production [135,138,140]. Supplementation of down regulated genes either through plasmid based expression or chromosomal integration have been successfully tried, e.g. Expression of the zwf gene coding for glucose-6-phopshate dehydrogenase in the Pentose Phosphate pathway helps to provide building blocks like nucleotides and NADPH and thus improves recombinant protein expression [139]. Knock out of ppGpp as well as the deletion of the global regulator rpoS (which is triggered by ppGpp) has been shown to enhance the recombinant protein expression [141-144]. The metabolic engineering strategies to improve the E.coli phenotype for recombinant protein production has also been reviewed earlier [145].
Uncoupling growth from product formation
The growth associated nature of recombinant protein production means that high specific growth rates need to be maintained in order to get high specific product formation rates. Since product concentration in a bioreactor is determined both by biomass concentration and specific product yield, we have the twin requirements of growing cells to high cell densities while simultaneously maintaining high specific growth rates. Together these requirements usually lead to oxygen or heat transfer limitations in the bioreactor especially during scale up. Hence the ability to produce product at high rates using slow growing or non-growing cells could greatly simplify the bioprocess strategy for high level product formation. Secondary metabolites are a very good example of how the non-growth associated nature of product formation kinetics allows the easy separation of growth and product formation phases in a bioreactor. In the case of recombinant protein synthesis, we need to ensure that the resting cells are metabolically active in terms of substrate uptake and energy metabolism as well as transcription and translation.
One interesting development in this regard was the Quiescent cell expression system where growth and product formation kinetics were decoupled [146]. Growth stoppage was achieved by over expressing a small RNA ‘Rcd’ which blocks cell division. However recombinant protein expression is unimpaired, and since the translational machinery is not required for biosynthesis, these cells have a significantly higher productivity compared to normally growing cells [147]. Further studies on the mechanism of Rcd showed that it binds to tryptophanase leading to the overproduction of indole [148]. Thus an exogenous supply of indole was also able to block cell division without affecting recombinant protein expression. However indole targets multiple sites in the cell [149-151] and may not be a preferred option for recombinant protein expression. Therefore targets downstream of indole which specifically blocks cell growth without affecting metabolic activity needs to be identified in order to achieve improved quiescence.
Tools for host cell engineering
This section deals with the vast array of techniques that have now become available, greatly simplifying the task of host modification to obtain the desirable phenotype by rational or high throughput approaches.
Single gene modification strategies
While the use of plasmid based methods for supplementing gene expression may be useful in ‘proof of principle’ studies, they have severe limitations. There is an upper limit to the number of target genes that can be supplemented; also the level of supplementation may be far higher than desired, leading to an unnecessary metabolic burden on the cells. Thus chromosomal engineering which leads to the construction of plasmid-less, marker-less strains has the advantage of extending the practical exploitation of the modified hosts in industry [152]. Also promoter engineering allows us to fine tune the expression of genes to desired levels [153,154]. One of the earliest strategies to design Single-gene knockouts was using the λ RED-ET system. Here the gene to be knocked out is replaced with an antibiotic resistance gene, usually kanamycin or chloramphenicol. If required, the selection marker can be removed by expressing the Cre or FLP recombinases that acts on the FRT or loxP site that is present in the flanking region of the selection marker or antibiotic cassette [155-161]. Another commonly used method for Single-gene knockouts is the P1-mediated transduction [162-166]. This method has gained popularity because of the availability of the Keio library of single gene knock-outs of non-essential genes in BW25113 which can be easily transferred into almost any E.coli strain [167]. Many researches prefer to use the vector plasmid pKO3 which integrates into the chromosome by homologous recombination creating tandem duplication at the non-permissive temperature. When shifted to the permissive temperature, the presence of the pSC101 replication origin in the vector ensures that it is excised from the chromosome. The presence of the sacB gene from B.subtilis in the vector allows us to screen for the loss of the vector sequence by growing the cells in the presence of sucrose [168].
The main limitation of these techniques is that they can be applied for single gene modifications and if multiple knock-ins and knock-outs have to be done, then these have to be done sequentially in a time consuming manner.
High throughput genome engineering methods
High throughput methods have been developed for genome engineering like Multiplex automated genome engineering (MAGE), Trackable multiplex recombineering (TRMR) and use of small regulatory RNAs [169-172]. These methods can create simultaneous random combinatorial modifications in the E.coli genome. Till now these approaches have mainly been used for evolutionary studies and pathway optimizations in E.coli. The same strategy can be applied for improving recombinant protein expression. As MAGE, works through oligonucleotide-mediated allelic replacement in an iterative manner it is capable of introducing multiple modifications in different locations of genome. Therefore several oligomers can be designed to perform multiple modifications iteratively, which can help in identifying the combinations which lead to the desired phenotype including that of enhanced protein capability (Figure 5). Recently, a group led by Y.S. Ryu, has modified MAGE so that it is not restricted to EcNR2 strains of E.coli [160]. This is important since it is well known that there is a wide variation in the expression levels obtained with different E.coli strains [173]. Also strains carrying different modifications and having the desired phenotype can be combined in a step wise fashion using Conjugative assembly genome engineering (CAGE) [174,175]. This approach can be used to look for synergy between various modifications. The major drawback with MAGE is that it also accumulates unwanted off-target mutations [175] and thus a method for genome engineering at multiple locations with greater precision needs to be developed [176]. Another method for rapid modification of many genes in E.coli is TRMR. This technique uses a large number of synDNAs with multiple desirable sequence features, to modulate the expression levels of genes. The synDNA contains different RBS which replaces the native RBS and a Molecular barcode is used to track the allele in mixed populations [177]. Another novel approach for high throughput metabolic engineering is the use of a transcriptional vector to express small chromosomal DNA fragments of E.coli itself. Since some fragments get inserted in the opposite orientation, they act as an anti-sense RNA and create a library of down-regulated pathways which can be screened for improved recombinant protein expression [178]. This has been extended to the use of synthetic Small regulatory RNAs (sRNAs) which helps in the modulation of gene expression [169]. This method is useful when we need to down regulate gene expression rather than completely knocking-out the gene, making it an indispensable tool for studying the effect of essential genes on cellular phenotype.
Figure 5.
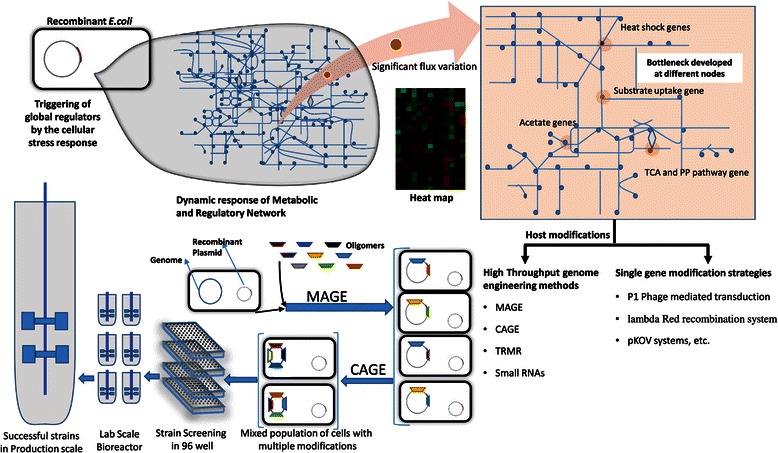
Overall Strategy for host cell engineering using rational and high throughput methods. Leads obtained from analysis of cellular physiology and Omics studies can be used to identify system level bottlenecks. These can be addressed by both single gene modifications or high throughput methods to generate improved platforms for expression. These are initially tested in microbioreactor format to select the best clones for further scale-up.
High throughput screening strategies
The screening of a large number of gene knock-in and knock-outs to select the desirable phenotype of improved expression capability is time consuming. The simplest approach to screen a very large number of clones is to use FACS based screening for cells expressing fluorescence tagged proteins like GFP [179,180]. Thus libraries with engineered genomes can be screened for the highest producers by using appropriate sorting protocols [181,182]. However such modified hosts may not necessarily over express other proteins, given the very specific nature of host-protein interactions. Another strategy would be the selection of quiescent phenotype, in order to uncouple growth and product formation. For this one can screen for a growth stoppage phenotype which typically leads to elongated cell morphologies due to stoppage of cell division [183]. Simultaneously or later these cells can be checked for recombinant protein expression capability after growth arrest. Such techniques can be coupled with automated devices where cultures can grow in 96 well plate formats. Such technologies have proven to work well in clone screening and help in quickly identifying the best performers from a large number of clones e.g. BioLector from m2p Labs [184,185], Bioscreen C from Oy Growth Curves Ab Ltd [186-190], Clone Screener from Biospectra AG and the Ambr reactor from TAPBiosystems. Apart from growth profiling, these systems can also do online monitoring of fluorescence, pH, dissolved oxygen and NADH [185] and are reviewed in [191-195].
Conclusion
The complex linkages between cellular physiology and the multiple steps in recombinant protein synthesis makes the task of removing bottlenecks in this pathway a difficult exercise. However we now have a wealth of information from transcriptomic, proteomic and metabolomic studies on the cellular factors affecting this pathway as well the changes in flux to this pathway due to the cellular stress response. The data has been useful in rational design of host cells with better expression capabilities. Also the use of high throughput screening methods have allowed us to, reverse engineer these desired phenotypes, adding vastly to the repertoire of beneficial knock-ins and knock-outs. With the development of tools for genome scale engineering to generate multiple knock-ins and knock-outs we can now study the synergistic response of these changes which were earlier limited to one or two modifications. These could lead to major improvements in the design of host platform for high level recombinant protein expression.
Acknowledgments
Financial support by Department of Biotechnology, Department of Science and Technology, Council of Scientific and Industrial Research, Government of India is deeply acknowledged.
Footnotes
Shubhashree Mahalik, and Ashish K Sharma, equal contributors.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors contributed equally in writing, design and figures of this review. All authors read and approved the final manuscript.
Contributor Information
Shubhashree Mahalik, Email: shubhashreemahalik@gmail.com.
Ashish K Sharma, Email: ashish.kumar07@gmail.com.
Krishna J Mukherjee, Email: kjmukherjee@mail.jnu.ac.in.
References
- 1.Ajikumar PK, Xiao WH, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G. Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science. 2010;330:70–74. doi: 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bongaerts J, Kramer M, Muller U, Raeven L, Wubbolts M. Metabolic engineering for microbial production of aromatic amino acids and derived compounds. Metab Eng. 2001;3:289–300. doi: 10.1006/mben.2001.0196. [DOI] [PubMed] [Google Scholar]
- 3.Cui YY, Ling C, Zhang YY, Huang J, Liu JZ. Production of shikimic acid from Escherichia coli through chemically inducible chromosomal evolution and cofactor metabolic engineering. Microb Cell Fact. 2014;13:21. doi: 10.1186/1475-2859-13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farmer WR, Liao JC. Improving lycopene production in Escherichia coli by engineering metabolic control. Nat Biotechnol. 2000;18:533–537. doi: 10.1038/75398. [DOI] [PubMed] [Google Scholar]
- 5.Flores N, Xiao J, Berry A, Bolivar F, Valle F. Pathway engineering for the production of aromatic compounds in Escherichia coli. Nat Biotechnol. 1996;14:620–623. doi: 10.1038/nbt0596-620. [DOI] [PubMed] [Google Scholar]
- 6.Paddon CJ, Keasling JD. Semi-synthetic artemisinin: a model for the use of synthetic biology in pharmaceutical development. Nat Rev Micro. 2014;12:355–367. doi: 10.1038/nrmicro3240. [DOI] [PubMed] [Google Scholar]
- 7.Pitera DJ, Paddon CJ, Newman JD, Keasling JD. Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab Eng. 2007;9:193–207. doi: 10.1016/j.ymben.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Kucharova V, Skancke J, Brautaset T, Valla S. Design and Optimization of Short DNA Sequences That Can Be Used as 5′ Fusion Partners for High-Level Expression of Heterologous Genes in Escherichia coli. Appl Environ Microbiol. 2013;79:6655–6664. doi: 10.1128/AEM.01676-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki M, Zhang J, Liu M, Woychik NA, Inouye M. Single Protein Production in Living Cells Facilitated by an mRNA Interferase. Mol Cell. 2005;18:253–261. doi: 10.1016/j.molcel.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 10.Shin J, Noireaux V. Study of messenger RNA inactivation and protein degradation in an Escherichia coli cell-free expression system. J Biol Eng. 2010;4:9. doi: 10.1186/1754-1611-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fakruddin M, Mohammad Mazumdar R, Bin Mannan KS, Chowdhury A, Hossain MN. Critical Factors Affecting the Success of Cloning, Expression, and Mass Production of Enzymes by Recombinant E. coli. ISRN Biotechnology. 2012;2013:7. doi: 10.5402/2013/590587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabrita LD, Dai W, Bottomley SP. A family of E. coli expression vectors for laboratory scale and high throughput soluble protein production. BMC Biotechnol. 2006;6:12. doi: 10.1186/1472-6750-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selleck W, Tan S. Recombinant protein complex expression in E. coli. Curr Protoc Protein Sci. 2008;5(21):21–25. doi: 10.1002/0471140864.ps0521s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schumann W, Ferreira LCS. Production of recombinant proteins in Escherichia coli. Genet Mol Biol. 2004;27:442–453. [Google Scholar]
- 15.Balzer S, Kucharova V, Megerle J, Lale R, Brautaset T, Valla S. A comparative analysis of the properties of regulated promoter systems commonly used for recombinant gene expression in Escherichia coli. Microb Cell Fact. 2013;12:26. doi: 10.1186/1475-2859-12-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dragosits M, Nicklas D, Tagkopoulos I. A synthetic biology approach to self-regulatory recombinant protein production in Escherichia coli. J Biol Eng. 2012;6:2. doi: 10.1186/1754-1611-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zwick F, Lale R, Valla S. Strong stimulation of recombinant protein production in Escherichia coli by combining stimulatory control elements in an expression cassette. Microb Cell Fact. 2012;11:133. doi: 10.1186/1475-2859-11-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giacalone MJ, Gentile AM, Lovitt BT, Berkley NL, Gunderson CW, Surber MW. Toxic protein expression in Escherichia coli using a rhamnose-based tightly regulated and tunable promoter system. Biotechniques. 2006;40:355. doi: 10.2144/000112112. [DOI] [PubMed] [Google Scholar]
- 19.Marini G, Luchese M, Argondizzo AP, de Goes ACMA, Galler R, Alves TL, Medeiros M, Larentis A. Experimental design approach in recombinant protein expression: determining medium composition and induction conditions for expression of pneumolysin from Streptococcus pneumoniae in Escherichia coli and preliminary purification process. BMC Biotechnol. 2014;14:1. doi: 10.1186/1472-6750-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi YJ, Morel L, Le François T, Bourque D, Bourget L, Groleau D, Massie B, Míguez CB. Novel, Versatile, and Tightly Regulated Expression System for Escherichia coli Strains. Appl Environ Microbiol. 2010;76:5058–5066. doi: 10.1128/AEM.00413-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nallamsetty S, Waugh DS. A generic protocol for the expression and purification of recombinant proteins in Escherichia coli using a combinatorial His6-maltose binding protein fusion tag. Nat Protocols. 2007;2:383–391. doi: 10.1038/nprot.2007.50. [DOI] [PubMed] [Google Scholar]
- 22.Chatterjee DK, Esposito D. Enhanced soluble protein expression using two new fusion tags. Protein Expr Purif. 2006;46:122–129. doi: 10.1016/j.pep.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 23.Costa SJ, Almeida A, Castro A, Domingues L, Besir H. The novel Fh8 and H fusion partners for soluble protein expression in Escherichia coli: a comparison with the traditional gene fusion technology. Appl Microbiol Biotechnol. 2013;97:6779–6791. doi: 10.1007/s00253-012-4559-1. [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Lee SB. Soluble expression of archaeal proteins in Escherichia coli by using fusion-partners. Protein Expr Purif. 2008;62:116–119. doi: 10.1016/j.pep.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 25.Ideno A, Furutani M, Iwabuchi T, Iida T, Iba Y, Kurosawa Y, Sakuraba H, Ohshima T, Kawarabayashi Y, Maruyama T. Expression of foreign proteins in Escherichia coli by fusing with an archaeal FK506 binding protein. Appl Microbiol Biotechnol. 2004;64:99–105. doi: 10.1007/s00253-003-1459-4. [DOI] [PubMed] [Google Scholar]
- 26.Esposito D, Chatterjee DK. Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol. 2006;17:353–358. doi: 10.1016/j.copbio.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Zuo X, Li S, Hall J, Mattern MR, Tran H, Shoo J, Tan R, Weiss SR, Butt TR. Enhanced expression and purification of membrane proteins by SUMO fusion in Escherichia coli. J Struct Funct Genomics. 2005;6:103–111. doi: 10.1007/s10969-005-2664-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nocadello S, Swennen E. The new pLAI (lux regulon based auto-inducible) expression system for recombinant protein production in Escherichia coli. Microb Cell Fact. 2012;11:3. doi: 10.1186/1475-2859-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Studier FW: Stable expression clones and auto-induction for protein production in E. coli. In Structural Genomics. ᅟ: Springer; 2014:17–32. [DOI] [PubMed]
- 30.da Silva AJ, Horta ACL, Velez AM, Iemma MRC, Sargo CR, Giordano R, Novo MTM, Giordano RC, Zangirolami TC. Non-conventional induction strategies for production of subunit swine erysipelas vaccine antigen in rE. coli fed-batch cultures. Springer Plus. 2013;14:322. doi: 10.1186/2193-1801-2-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 32.Sadilkova L, Osicka R, Sulc M, Linhartova I, Novak P, Sebo P. Single‐step affinity purification of recombinant proteins using a self‐excising module from Neisseria meningitidis FrpC. Protein Sci. 2008;17:1834–1843. doi: 10.1110/ps.035733.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y. Self-cleaving fusion tags for recombinant protein production. Biotechnol Lett. 2011;33:869–881. doi: 10.1007/s10529-011-0533-8. [DOI] [PubMed] [Google Scholar]
- 34.Wu W-Y, Mee C, Califano F, Banki R, Wood DW. Recombinant protein purification by self-cleaving aggregation tag. Nat Protocols. 2006;1:2257–2262. doi: 10.1038/nprot.2006.314. [DOI] [PubMed] [Google Scholar]
- 35.Care S, Bignon C, Pelissier M, Blanc E, Canard B, Coutard B. The translation of recombinant proteins in E. coli can be improved by in silico generating and screening random libraries of a− 70/+ 96 mRNA region with respect to the translation initiation codon. Nucleic Acids Res. 2008;36:e6–e6. doi: 10.1093/nar/gkm1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berg L, Lale R, Bakke I, Burroughs N, Valla S. The expression of recombinant genes in Escherichia coli can be strongly stimulated at the transcript production level by mutating the DNA‐region corresponding to the 5′‐untranslated part of mRNA. J Microbial Biotechnol. 2009;2:379–389. doi: 10.1111/j.1751-7915.2009.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reeve B, Hargest T, Gilbert C, Ellis T. Predicting translation initiation rates for designing synthetic biology. Synthetic Biol. 2014;2:1. doi: 10.3389/fbioe.2014.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffmann F, Rinas U. Stress induced by recombinant protein production in Escherichia coli. Adv Biochem Eng Biotechnol. 2004;89:73–92. doi: 10.1007/b93994. [DOI] [PubMed] [Google Scholar]
- 40.Sørensen HP, Mortensen KK. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J Biotechnol. 2005;115:113–128. doi: 10.1016/j.jbiotec.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 41.Piir K, Paier A, Liiv A, Tenson T, Maiväli Ü. Ribosome degradation in growing bacteria. EMBO Rep. 2011;12:458–462. doi: 10.1038/embor.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maruyama H, Mizuno D. Ribosome degradation and the degradation products in starved Escherichia coli. I. Comparison of the degradation rate and of the nucleotide pool between Escherichia coli B and Q-13 strains in phosphate deficiency. Biochim Biophys Acta. 1970;199:159–165. [PubMed] [Google Scholar]
- 43.Ben-Hamida F, Schlessinger D. Synthesis and breakdown of ribonucleic acid in Escherichia coli starving for nitrogen. Biochim et Biophysica Acta (BBA)-Nucleic Acids and Protein, Synthesis. 1966;119:183–191. doi: 10.1016/0005-2787(66)90049-9. [DOI] [PubMed] [Google Scholar]
- 44.Jacobson A, Gillespie D. Metabolic events occurring during recovery from prolonged glucose starvation in Escherichia coli. J Bacteriol. 1968;95:1030–1039. doi: 10.1128/jb.95.3.1030-1039.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarthy B. The effects of magnesium starvation on the ribosome content of Escherichia coli. Biochim Biophys Acta. 1962;55:880–889. [Google Scholar]
- 46.Deutscher MP. Degradation of RNA in bacteria: comparison of mRNA and stable RNA. Nucleic Acids Res. 2006;34:659–666. doi: 10.1093/nar/gkj472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgess-Brown NA, Sharma S, Sobott F, Loenarz C, Oppermann U, Gileadi O. Codon optimization can improve expression of human genes in Escherichia coli: A multi-gene study. Protein Expr Purif. 2008;59:94–102. doi: 10.1016/j.pep.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 48.Welch M, Govindarajan S, Ness JE, Villalobos A, Gurney A, Minshull J, Gustafsson C. Design parameters to control synthetic gene expression in Escherichia coli. PLoS One. 2009;4:e7002. doi: 10.1371/journal.pone.0007002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dittmar KA, Sørensen MA, Elf J, Ehrenberg M, Pan T. Selective charging of tRNA isoacceptors induced by amino‐acid starvation. EMBO Rep. 2005;6:151–157. doi: 10.1038/sj.embor.7400341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong H, Nilsson L, Kurland CG. Co-variation of tRNA Abundance and Codon Usage in Escherichia coli at Different Growth Rates. J Mol Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 51.Bonomo J, Gill RT. Amino acid content of recombinant proteins influences the metabolic burden response. Biotechnol Bioeng. 2005;90:116–126. doi: 10.1002/bit.20436. [DOI] [PubMed] [Google Scholar]
- 52.Elf J, Nilsson D, Tenson T, Ehrenberg M. Selective charging of tRNA isoacceptors explains patterns of codon usage. Science. 2003;300:1718–1722. doi: 10.1126/science.1083811. [DOI] [PubMed] [Google Scholar]
- 53.Wohlgemuth SE, Gorochowski TE, Roubos JA. Translational sensitivity of the Escherichia coli genome to fluctuating tRNA availability. Nucleic Acids Res. 2013;41:8021–8033. doi: 10.1093/nar/gkt602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22:346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 55.Puigbò P, Guzmán E, Romeu A, Garcia-Vallvé S. OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res. 2007;35:W126–W131. doi: 10.1093/nar/gkm219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reuveni S, Meilijson I, Kupiec M, Ruppin E, Tuller T. Genome-scale analysis of translation elongation with a ribosome flow model. PLoS Comput Biol. 2011;7:e1002127. doi: 10.1371/journal.pcbi.1002127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Komar AA. A pause for thought along the co-translational folding pathway. Trends Biochem Sci. 2009;34:16–24. doi: 10.1016/j.tibs.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 58.Dubey AK, Baker CS, Romeo T, Babitzke P. RNA sequence and secondary structure participate in high-affinity CsrA–RNA interaction. RNA. 2005;11:1579–1587. doi: 10.1261/rna.2990205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suzuki K, Babitzke P, Kushner SR, Romeo T. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev. 2006;20:2605–2617. doi: 10.1101/gad.1461606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weilbacher T, Suzuki K, Dubey AK, Wang X, Gudapaty S, Morozov I, Baker CS, Georgellis D, Babitzke P, Romeo T. A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol Microbiol. 2003;48:657–670. doi: 10.1046/j.1365-2958.2003.03459.x. [DOI] [PubMed] [Google Scholar]
- 61.Richins R, Chen W. Effects of FIS Overexpression on Cell Growth, rRNA Synthesis, and Ribosome Content in Escherichiacoli. Biotechnol Prog. 2001;17:252–257. doi: 10.1021/bp000170f. [DOI] [PubMed] [Google Scholar]
- 62.George H, Powell A, Dahlgren M, Herber W, Maigetter R, Burgess B, Stirdivant S, Greasham R. Physiological effects of TGFα‐PE40 expression in recombinant Escherichia coli JM109. Biotechnol Bioeng. 1992;40:437–445. doi: 10.1002/bit.260400314. [DOI] [PubMed] [Google Scholar]
- 63.Hoffmann F, Rinas U. On‐line estimation of the metabolic burden resulting from the synthesis of plasmid‐encoded and heat‐shock proteins by monitoring respiratory energy generation. Biotechnol Bioeng. 2001;76:333–340. doi: 10.1002/bit.10098. [DOI] [PubMed] [Google Scholar]
- 64.Hoffmann F, Weber J, Rinas U. Metabolic adaptation of Escherichia coli during temperature‐induced recombinant protein production: 1. Readjustment of metabolic enzyme synthesis. Biotechnol Bioeng. 2002;80:313–319. doi: 10.1002/bit.10379. [DOI] [PubMed] [Google Scholar]
- 65.Heyland J, Blank LM, Schmid A. Quantification of metabolic limitations during recombinant protein production in Escherichia coli. J Biotechnol. 2011;155:178–184. doi: 10.1016/j.jbiotec.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 66.Tempest D, Neijssel O: Growth yield and energy distribution. In Escherichia coli and Salmonella typhimurium: cellular and molecular biology American Society for Microbiology. Washington, DC: ᅟ; 1987:797–806.
- 67.Kim HJ, Kwon YD, Lee SY, Kim P. An engineered Escherichia coli having a high intracellular level of ATP and enhanced recombinant protein production. Appl Microbiol Biotechnol. 2012;94:1079–1086. doi: 10.1007/s00253-011-3779-0. [DOI] [PubMed] [Google Scholar]
- 68.Ingraham J, Maaloe O, Neidhardt FC. Growth of the Bacterial Cell. Sunderland, MA: Sinauer; 1983. pp. 381–383. [Google Scholar]
- 69.Elf J, Ehrenberg M. Near-Critical Behavior of Aminoacyl-tRNA Pools in E. coli at Rate-Limiting Supply of Amino Acids. Biophys J. 2005;88:132–146. doi: 10.1529/biophysj.104.051383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sorensen H, Mortensen K. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb Cell Fact. 2005;4:1. doi: 10.1186/1475-2859-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.King ZA, Feist AM. Optimizing Cofactor Specificity of Oxidoreductase Enzymes for the Generation of Microbial Production Strains—OptSwap. Ind Biotechnol. 2013;9:236–246. [Google Scholar]
- 72.Lakshmanan M, Chung BK, Liu C, Kim SW, Lee DY. Cofactor modification analysis: a computational framework to identify cofactor specificity engineering targets for strain improvement. J Bioinform Comput Biol. 2013;11:1343006. doi: 10.1142/S0219720013430063. [DOI] [PubMed] [Google Scholar]
- 73.Wang Y, San K-Y, Bennett GN. Cofactor engineering for advancing chemical biotechnology. Curr Opin Biotechnol. 2013;24:994–999. doi: 10.1016/j.copbio.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 74.Bastian S, Arnold FH. Reversal of NAD(P)H cofactor dependence by protein engineering. Methods Mol Biol. 2012;834:17–31. doi: 10.1007/978-1-61779-483-4_2. [DOI] [PubMed] [Google Scholar]
- 75.Berrios-Rivera SJ, Bennett GN, San KY. Metabolic engineering of Escherichia coli: increase of NADH availability by overexpressing an NAD(+)-dependent formate dehydrogenase. Metab Eng. 2002;4:217–229. doi: 10.1006/mben.2002.0227. [DOI] [PubMed] [Google Scholar]
- 76.Fan Z, Yuan L, Chatterjee R. Increased Hydrogen Production by Genetic Engineering of Escherichia coli. PLoS One. 2009;4:e4432. doi: 10.1371/journal.pone.0004432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Marco A, Deuerling E, Mogk A, Tomoyasu T, Bukau B. Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol. 2007;7:32. doi: 10.1186/1472-6750-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nannenga BL, Baneyx F. Reprogramming chaperone pathways to improve membrane protein expression in Escherichia coli. Protein Sci. 2011;20:1411–1420. doi: 10.1002/pro.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martin J, Ulrich Hartl F. Chaperone-assisted protein folding. Curr Opin Struct Biol. 1997;7:41–52. doi: 10.1016/s0959-440x(97)80006-1. [DOI] [PubMed] [Google Scholar]
- 80.Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 81.Messens J, Collet J-F. Pathways of disulfide bond formation in Escherichia coli. Int J Biochem Cell Biol. 2006;38:1050–1062. doi: 10.1016/j.biocel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 82.Masip L, Pan JL, Haldar S, Penner-Hahn JE, DeLisa MP, Georgiou G, Bardwell JCA, Collet J-F. An Engineered Pathway for the Formation of Protein Disulfide Bonds. Science. 2004;303:1185–1189. doi: 10.1126/science.1092612. [DOI] [PubMed] [Google Scholar]
- 83.Bessette PH, Åslund F, Beckwith J, Georgiou G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci. 1999;96:13703–13708. doi: 10.1073/pnas.96.24.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hortsch R, Weuster-Botz D. Growth and recombinant protein expression with Escherichia coli in different batch cultivation media. Appl Microbiol Biotechnol. 2011;90:69–76. doi: 10.1007/s00253-010-3036-y. [DOI] [PubMed] [Google Scholar]
- 85.Lobstein J, Emrich CA, Jeans C, Faulkner M, Riggs P, Berkmen M. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb Cell Fact. 2012;11:56–56. doi: 10.1186/1475-2859-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhan X, Schwaller M, Gilbert HF, Georgiou G. Facilitating the formation of disulfide bonds in the Escherichia coli periplasm via coexpression of yeast protein disulfide isomerase. Biotechnol Prog. 1999;15:1033–1038. doi: 10.1021/bp990083r. [DOI] [PubMed] [Google Scholar]
- 87.Kurokawa Y, Yanagi H, Yura T. Overexpression of Protein Disulfide Isomerase DsbC Stabilizes Multiple-Disulfide-Bonded Recombinant Protein Produced and Transported to the Periplasm in Escherichia coli. Appl Environ Microbiol. 2000;66:3960–3965. doi: 10.1128/aem.66.9.3960-3965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Matos CF, Robinson C, Alanen HI, Prus P, Uchida Y, Ruddock LW, Freedman RB, Keshavarz‐Moore E. Efficient export of prefolded, disulfide‐bonded recombinant proteins to the periplasm by the Tat pathway in Escherichia coli CyDisCo strains. Biotechnol Prog. 2014;30:281–290. doi: 10.1002/btpr.1858. [DOI] [PubMed] [Google Scholar]
- 89.Su L, Chen S, Yi L, Woodard R, Chen J, Wu J. Extracellular overexpression of recombinant Thermobifida fusca cutinase by alpha-hemolysin secretion system in E. coli BL21(DE3) Microb Cell Fact. 2012;11:8. doi: 10.1186/1475-2859-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sugamata Y, Shiba T. Improved Secretory Production of Recombinant Proteins by Random Mutagenesis of hlyB, an Alpha-Hemolysin Transporter from Escherichia coli. Appl Environ Microbiol. 2005;71:656–662. doi: 10.1128/AEM.71.2.656-662.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pugsley AP. The complete general secretory pathway in gram-negative bacteria. Microbiol Rev. 1993;57:50. doi: 10.1128/mr.57.1.50-108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mergulhao F, Summers D, Monteiro G. Recombinant protein secretion in Escherichia coli. Biotechnol Adv. 2005;23:177–202. doi: 10.1016/j.biotechadv.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 93.Choi J, Lee S. Secretory and extracellular production of recombinant proteins using Escherichia coli. Appl Microbiol Biotechnol. 2004;64:625–635. doi: 10.1007/s00253-004-1559-9. [DOI] [PubMed] [Google Scholar]
- 94.Liu CC, Schultz PG. Recombinant expression of selectively sulfated proteins in Escherichia coli. Nat Biotechnol. 2006;24:1436–1440. doi: 10.1038/nbt1254. [DOI] [PubMed] [Google Scholar]
- 95.Su L, Xu C, Woodard RW, Chen J, Wu J. A novel strategy for enhancing extracellular secretion of recombinant proteins in Escherichia coli. Appl Microbiol Biotechnol. 2013;97:6705–6713. doi: 10.1007/s00253-013-4994-7. [DOI] [PubMed] [Google Scholar]
- 96.Douville K, Price A, Eichler J, Economou A, Wickner W. SecYEG and SecA are the stoichiometric components of preprotein translocase. J Biol Chem. 1995;270:20106–20111. doi: 10.1074/jbc.270.34.20106. [DOI] [PubMed] [Google Scholar]
- 97.Khushoo A, Pal Y, Mukherjee KJ. Optimization of extracellular production of recombinant asparaginase in Escherichia coli in shake-flask and bioreactor. Appl Microbiol Biotechnol. 2005;68:189–197. doi: 10.1007/s00253-004-1867-0. [DOI] [PubMed] [Google Scholar]
- 98.Khushoo A, Pal Y, Singh BN, Mukherjee KJ. Extracellular expression and single step purification of recombinant Escherichia coli L-asparaginase II. Protein Expr Purif. 2004;38:29–36. doi: 10.1016/j.pep.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 99.Ni Y, Chen R. Extracellular recombinant protein production from Escherichia coli. Biotechnol Lett. 2009;31:1661–1670. doi: 10.1007/s10529-009-0077-3. [DOI] [PubMed] [Google Scholar]
- 100.Matos CF, Branston SD, Albiniak A, Dhanoya A, Freedman RB, Keshavarz‐Moore E, Robinson C. High‐yield export of a native heterologous protein to the periplasm by the tat translocation pathway in Escherichia coli. Biotechnol Bioeng. 2012;109:2533–2542. doi: 10.1002/bit.24535. [DOI] [PubMed] [Google Scholar]
- 101.Chung CW, You J, Kim K, Moon Y, Kim H, Ahn JH: Export of recombinant proteins inEscherichia coliusing ABC transporter with an attached lipase ABC transporter recognition domain (LARD).Microb Cell Fact 2009, 8: [DOI] [PMC free article] [PubMed]
- 102.Bentley WE, Mirjalili N, Andersen DC, Davis RH, Kompala DS. Plasmid‐encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol Bioeng. 1990;35:668–681. doi: 10.1002/bit.260350704. [DOI] [PubMed] [Google Scholar]
- 103.Panda AK, Khan RH, Rao KB, Totey SM. Kinetics of inclusion body production in batch and high cell density fed-batch culture of Escherichia coli expressing ovine growth hormone. J Biotechnol. 1999;75:161–172. doi: 10.1016/s0168-1656(99)00157-1. [DOI] [PubMed] [Google Scholar]
- 104.Vaiphei ST, Pandey G, Mukherjee KJ. Kinetic studies of recombinant human interferon-gamma expression in continuous cultures of E. coli. J Ind Microbiol Biotechnol. 2009;36:1453–1458. doi: 10.1007/s10295-009-0632-x. [DOI] [PubMed] [Google Scholar]
- 105.Srivastava P, Mukherjee KJ. Kinetic studies of recombinant human interferon-alpha (rhIFN-α) expression in transient state continuous cultures. Biochem Eng J. 2005;26:50–58. [Google Scholar]
- 106.Shin CS, Hong MS, Bae CS, Lee J. Enhanced Production of Human Mini-Proinsulin in Fed-Batch Cultures at High Cell Density of Escherichia coli BL21(DE3) [pET-3aT2M2] Biotechnol Prog. 1997;13:249–257. doi: 10.1021/bp970018m. [DOI] [PubMed] [Google Scholar]
- 107.Bhattacharya P, Pandey G, Srivastava P, Mukherjee K. Combined effect of protein fusion and signal sequence greatly enhances the production of recombinant human GM-CSF in Escherichia coli. Mol Biotechnol. 2005;30:103–115. doi: 10.1385/MB:30:2:103. [DOI] [PubMed] [Google Scholar]
- 108.Khasa YP, Khushoo A, Mukherjee KJ. Enhancing toxic protein expression in Escherichia coli fed-batch culture using kinetic parameters: Human granulocyte-macrophage colony-stimulating factor as a model system. J Biosci Bioeng. 2013;115:291–297. doi: 10.1016/j.jbiosc.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 109.Yazdani S, Mukherjee K. Continuous-culture studies on the stability and expression of recombinant streptokinase in Escherichia coli. Bioprocess Biosyst Eng. 2002;24:341–346. [Google Scholar]
- 110.Balagurunathan B, Jayaraman G. Cellular response to accumulation of recombinant proteins in the E. coli inner membrane: Implications for proteolysis and productivity of the secretory expression system. Biochem Eng J. 2008;39:74–83. [Google Scholar]
- 111.Khasa YP, Khushoo A, Tapryal S, Mukherjee K. Optimization of Human Granulocyte Macrophage-Colony Stimulating Factor (hGM-CSF) Expression Using Asparaginase and Xylanase Gene’s Signal Sequences in Escherichia coli. Appl Biochem Biotechnol. 2011;165:523–537. doi: 10.1007/s12010-011-9272-5. [DOI] [PubMed] [Google Scholar]
- 112.Ramalingam S, Gautam P, Mukherjee KJ, Jayaraman G. Effects of post-induction feed strategies on secretory production of recombinant streptokinase in Escherichia coli. Biochem Eng J. 2007;33:34–41. [Google Scholar]
- 113.Marisch K, Bayer K, Scharl T, Mairhofer J, Krempl PM, Hummel K, Razzazi-Fazeli E, Striedner G. A Comparative Analysis of Industrial Escherichia coli K–12 and B Strains in High-Glucose Batch Cultivations on Process-, Transcriptome-and Proteome Level. PloS one. 2013;8:e70516. doi: 10.1371/journal.pone.0070516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Carneiro S, Ferreira EC, Rocha I. Metabolic responses to recombinant bioprocesses in Escherichia coli. J Biotechnol. 2013;164:396–408. doi: 10.1016/j.jbiotec.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 115.Enfors SO: Physiological Stress Responses in Bioprocesses. ᅟ: Springer; 2004.
- 116.Sharma AK, Mahalik S, Ghosh C, Singh AB, Mukherjee KJ. Comparative transcriptomic profile analysis of fed-batch cultures expressing different recombinant proteins in Escherichia coli. AMB express. 2011;1:33. doi: 10.1186/2191-0855-1-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fong B, Wood D. Expression and purification of ELP-intein-tagged target proteins in high cell density E. coli fermentation. Microb Cell Fact. 2010;9:77. doi: 10.1186/1475-2859-9-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Losen M, Frölich B, Pohl M, Büchs J. Effect of Oxygen Limitation and Medium Composition on Escherichia coli Fermentation in Shake‐Flask Cultures. Biotechnol Prog. 2004;20:1062–1068. doi: 10.1021/bp034282t. [DOI] [PubMed] [Google Scholar]
- 119.Saraswat V, Kim DY, Lee J, Park Y. Effect of specific production rate of recombinant protein on multimerization of plasmid vector and gene expression level. FEMS Microbiol Lett. 1999;179:367–373. doi: 10.1111/j.1574-6968.1999.tb08751.x. [DOI] [PubMed] [Google Scholar]
- 120.Hellmuth K, Korz DJ, Sanders EA, Deckwer WD. Effect of growth rate on stability and gene expression of recombinant plasmids during continuous and high cell density cultivation of Escherichia coli TG1. J Biotechnol. 1994;32:289–298. doi: 10.1016/0168-1656(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 121.Singh AB, Sharma AK, Mukherjee KJ. Analyzing the metabolic stress response of recombinant Escherichia coli cultures expressing human interferon-beta in high cell density fed batch cultures using time course transcriptomic data. Mol Biosyst. 2012;8:615–628. doi: 10.1039/c1mb05414g. [DOI] [PubMed] [Google Scholar]
- 122.Baig F, Fernando LP, Salazar MA, Powell RR, Bruce TF, Harcum SW. Dynamic transcriptional response of Escherichia coli to inclusion body formation. Biotechnol Bioeng. 2014;111:980–999. doi: 10.1002/bit.25169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lesley SA, Graziano J, Cho CY, Knuth MW, Klock HE. Gene expression response to misfolded protein as a screen for soluble recombinant protein. Protein Eng. 2002;15:153–160. doi: 10.1093/protein/15.2.153. [DOI] [PubMed] [Google Scholar]
- 124.Smith HE. The transcriptional response of Escherichia coli to recombinant protein insolubility. J Struct Funct Genomics. 2007;8:27–35. doi: 10.1007/s10969-007-9030-7. [DOI] [PubMed] [Google Scholar]
- 125.Bordbar A, Monk JM, King ZA, Palsson BO. Constraint-based models predict metabolic and associated cellular functions. Nat Rev Genet. 2014;15:107–120. doi: 10.1038/nrg3643. [DOI] [PubMed] [Google Scholar]
- 126.Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Orth JD, Palsson B. Gap-filling analysis of the iJO1366 Escherichia coli metabolic network reconstruction for discovery of metabolic functions. BMC Syst Biol. 2012;6:30. doi: 10.1186/1752-0509-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Feist AM, Henry CS, Reed JL, Krummenacker M, Joyce AR, Karp PD, Broadbelt LJ, Hatzimanikatis V, Palsson BØ: A genome‐scale metabolic reconstruction forEscherichia coliK‐12 MG1655 that accounts for 1260 ORFs and thermodynamic information.Mol Syst Biol 2007, 3.ᅟ [DOI] [PMC free article] [PubMed]
- 129.Covert MW, Palsson BØ. Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J Biol Chem. 2002;277:28058–28064. doi: 10.1074/jbc.M201691200. [DOI] [PubMed] [Google Scholar]
- 130.Covert MW, Xiao N, Chen TJ, Karr JR. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics. 2008;24:2044–2050. doi: 10.1093/bioinformatics/btn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim J, Reed JL. OptORF: Optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst Biol. 2010;4:53. doi: 10.1186/1752-0509-4-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shlomi T, Berkman O, Ruppin E. Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc Natl Acad Sci U S A. 2005;102:7695–7700. doi: 10.1073/pnas.0406346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Singh AB, Mukherjee KJ. Supplementation of substrate uptake gene enhances the expression of rhIFN-beta in high cell density fed-batch cultures of Escherichia coli. Mol Biotechnol. 2013;54:692–702. doi: 10.1007/s12033-012-9611-y. [DOI] [PubMed] [Google Scholar]
- 134.Choi JH, Lee SJ, Lee SJ, Lee SY. Enhanced Production of Insulin-Like Growth Factor I Fusion Protein in Escherichia coli by Coexpression of the Down-Regulated Genes Identified by Transcriptome Profiling. Appl Environ Microbiol. 2003;69:4737–4742. doi: 10.1128/AEM.69.8.4737-4742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chang DE, Shin S, Rhee JS, Pan JG. Acetate metabolism in a pta mutant of Escherichia coli W3110: importance of maintaining acetyl coenzyme A flux for growth and survival. J Bacteriol. 1999;181:6656–6663. doi: 10.1128/jb.181.21.6656-6663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chou CH, Bennett GN, San KY. Effect of modified glucose uptake using genetic engineering techniques on high-level recombinant protein production in Escherichia coli dense cultures. Biotechnol Bioeng. 1994;44:952–960. doi: 10.1002/bit.260440811. [DOI] [PubMed] [Google Scholar]
- 137.De Anda R, Lara AR, Hernandez V, Hernandez-Montalvo V, Gosset G, Bolivar F, Ramirez OT. Replacement of the glucose phosphotransferase transport system by galactose permease reduces acetate accumulation and improves process performance of Escherichia coli for recombinant protein production without impairment of growth rate. Metab Eng. 2006;8:281–290. doi: 10.1016/j.ymben.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 138.Dittrich CR, Vadali RV, Bennett GN, San KY. Redistribution of metabolic fluxes in the central aerobic metabolic pathway of E. coli mutant strains with deletion of the ackA-pta and poxB pathways for the synthesis of isoamyl acetate. Biotechnol Prog. 2005;21:627–631. doi: 10.1021/bp049730r. [DOI] [PubMed] [Google Scholar]
- 139.Flores S, de Anda-Herrera R, Gosset G, Bolivar FG. Growth-rate recovery of Escherichia coli cultures carrying a multicopy plasmid, by engineering of the pentose-phosphate pathway. Biotechnol Bioeng. 2004;87:485–494. doi: 10.1002/bit.20137. [DOI] [PubMed] [Google Scholar]
- 140.Yang YT, Bennett GN, San KY. Effect of inactivation of nuo and ackA-pta on redistribution of metabolic fluxes in Escherichia coli. Biotechnol Bioeng. 1999;65:291–297. [PubMed] [Google Scholar]
- 141.Brown L, Gentry D, Elliott T, Cashel M. DksA affects ppGpp induction of RpoS at a translational level. J Bacteriol. 2002;184:4455–4465. doi: 10.1128/JB.184.16.4455-4465.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dedhia N, Richins R, Mesina A, Chen W. Improvement in recombinant protein production in ppGpp-deficient Escherichia coli. Biotechnol Bioeng. 1997;53:379–386. doi: 10.1002/(SICI)1097-0290(19970220)53:4<379::AID-BIT4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 143.Jeong KJ, Choi JH, Yoo WM, Keum KC, Yoo NC, Lee SY, Sung MH. Constitutive production of human leptin by fed-batch culture of recombinant rpoS- Escherichia coli. Protein Expr Purif. 2004;36:150–156. doi: 10.1016/j.pep.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 144.Rahman M, Hasan MR, Oba T, Shimizu K. Effect of rpoS gene knockout on the metabolism of Escherichia coli during exponential growth phase and early stationary phase based on gene expressions, enzyme activities and intracellular metabolite concentrations. Biotechnol Bioeng. 2006;94:585–595. doi: 10.1002/bit.20858. [DOI] [PubMed] [Google Scholar]
- 145.Waegeman H, Soetaert W. Increasing recombinant protein production in Escherichia coli through metabolic and genetic engineering. J Ind Microbiol Biotechnol. 2011;38:1891–1910. doi: 10.1007/s10295-011-1034-4. [DOI] [PubMed] [Google Scholar]
- 146.Rowe DC, Summers DK. The quiescent-cell expression system for protein synthesis in Escherichia coli. Appl Environ Microbiol. 1999;65:2710–2715. doi: 10.1128/aem.65.6.2710-2715.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mukherjee K, Rowe D, Watkins N, Summers D. Studies of single-chain antibody expression in quiescent Escherichia coli. Appl Environ Microbiol. 2004;70:3005–3012. doi: 10.1128/AEM.70.5.3005-3012.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chant EL, Summers DK. Indole signalling contributes to the stable maintenance of Escherichia coli multicopy plasmids. Mol Microbiol. 2007;63:35–43. doi: 10.1111/j.1365-2958.2006.05481.x. [DOI] [PubMed] [Google Scholar]
- 149.Lee JH, Lee J. Indole as an intercellular signal in microbial communities. FEMS Microbiol Rev. 2010;34:426–444. doi: 10.1111/j.1574-6976.2009.00204.x. [DOI] [PubMed] [Google Scholar]
- 150.Bury-Mone S, Nomane Y, Reymond N, Barbet R, Jacquet E, Imbeaud S, Jacq A, Bouloc P. Global analysis of extracytoplasmic stress signaling in Escherichia coli. PLoS Genet. 2009;5:e1000651. doi: 10.1371/journal.pgen.1000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Garbe TR, Kobayashi M, Yukawa H. Indole-inducible proteins in bacteria suggest membrane and oxidant toxicity. Arch Microbiol. 2000;173:78–82. doi: 10.1007/s002030050012. [DOI] [PubMed] [Google Scholar]
- 152.Huang LC, Wood EA, Cox MM. Convenient and reversible site-specific targeting of exogenous DNA into a bacterial chromosome by use of the FLP recombinase: the FLIRT system. J Bacteriol. 1997;179:6076–6083. doi: 10.1128/jb.179.19.6076-6083.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.De Mey M, Maertens J, Boogmans S, Soetaert WK, Vandamme EJ, Cunin R, Foulquie-Moreno MR. Promoter knock-in: a novel rational method for the fine tuning of genes. BMC Biotechnol. 2010;10:26. doi: 10.1186/1472-6750-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci U S A. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Marx CJ, Lidstrom ME. Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques. 2002;33:1062–1067. doi: 10.2144/02335rr01. [DOI] [PubMed] [Google Scholar]
- 157.Yang J, Sun B, Huang H, Jiang Y, Diao L, Chen B, Xu C, Wang X, Liu J, Jiang W. High-Efficiency Scarless Genetic Modification in Escherichia coli by Using Lambda Red Recombination and I-SceI Cleavage. Appl Environ Microbiol. 2014;80:3826–3834. doi: 10.1128/AEM.00313-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McCloskey D, Palsson BØ, Feist AM. Basic and applied uses of genome‐scale metabolic network reconstructions of Escherichia coli. Mol Syst Biol. 2013;9:661. doi: 10.1038/msb.2013.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chemler JA, Fowler ZL, McHugh KP, Koffas MA. Improving NADPH availability for natural product biosynthesis in Escherichia coli by metabolic engineering. Metab Eng. 2010;12:96–104. doi: 10.1016/j.ymben.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 160.Ryu YS, Biswas RK, Shin K, Parisutham V, Kim SM, Lee SK. A simple and effective method for construction of Escherichia coli strains proficient for genome engineering. PLoS One. 2014;9:e94266. doi: 10.1371/journal.pone.0094266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jeong J, Cho N, Jung D, Bang D. Genome-scale genetic engineering in Escherichia coli. Biotechnol Adv. 2013;31:804–810. doi: 10.1016/j.biotechadv.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 162.Donath MJ, Dominguez MA, Withers ST. Development of an automated platform for high-throughput P1-phage transduction of Escherichia coli. J Association for Laboratory Automation. 2011;16:141–147. doi: 10.1016/j.jala.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 163.Kim HJ, Hou BK, Lee SG, Kim JS, Lee D-W, Lee SJ. Genome-wide analysis of redox reactions reveals metabolic engineering targets for d-lactate overproduction in Escherichia coli. Metab Eng. 2013;18:44–52. doi: 10.1016/j.ymben.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 164.Chin JW, Cirino PC: Strain engineering strategies for improving whole-cell biocatalysis: engineeringEscherichia colito overproduce xylitol as an example. In Nanoscale Biocatalysis. ᅟ: Springer; 2011:185–203. [DOI] [PubMed]
- 165.Meynial-Salles I, Soucaille P: Creation of new metabolic pathways or improvement of existing metabolic enzymes by in vivo evolution inEscherichia coli. In Microbial Metabolic Engineering. ᅟ: Springer; 2012:75–86. [DOI] [PubMed]
- 166.Zhu J, Sanchez A, Bennett GN, San K-Y. Manipulating respiratory levels in Escherichia coli for aerobic formation of reduced chemical products. Metab Eng. 2011;13:704–712. doi: 10.1016/j.ymben.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006–0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Link AJ, Phillips D, Church GM. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol. 1997;179:6228–6237. doi: 10.1128/jb.179.20.6228-6237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Na D, Yoo SM, Chung H, Park H, Park JH, Lee SY. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol. 2013;31:170–174. doi: 10.1038/nbt.2461. [DOI] [PubMed] [Google Scholar]
- 170.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Warner JR, Reeder PJ, Karimpour-Fard A, Woodruff LB, Gill RT. Rapid profiling of a microbial genome using mixtures of barcoded oligonucleotides. Nat Biotechnol. 2010;28:856–862. doi: 10.1038/nbt.1653. [DOI] [PubMed] [Google Scholar]
- 172.Yoo SM, Na D, Lee SY. Design and use of synthetic regulatory small RNAs to control gene expression in Escherichia coli. Nat Protoc. 2013;8:1694–1707. doi: 10.1038/nprot.2013.105. [DOI] [PubMed] [Google Scholar]
- 173.Waegeman H, De Lausnay S, Beauprez J, Maertens J, De Mey M, Soetaert W. Increasing recombinant protein production in Escherichia coli K12 through metabolic engineering. N Biotechnol. 2013;30:255–261. doi: 10.1016/j.nbt.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 174.Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, Rohland N, Schultz PG, Jacobson JM, Rinehart J, Church GM, Isaacs FJ. Genomically recoded organisms expand biological functions. Science. 2013;342:357–360. doi: 10.1126/science.1241459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Isaacs FJ, Carr PA, Wang HH, Lajoie MJ, Sterling B, Kraal L, Tolonen AC, Gianoulis TA, Goodman DB, Reppas NB, Emig CJ, Bang D, Hwang SJ, Jewett MC, Jacobson JM, Church GM. Precise Manipulation of Chromosomes in Vivo Enables Genome-Wide Codon Replacement. Science. 2011;333:348–353. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pal C, Papp B, Posfai G. The dawn of evolutionary genome engineering. Nat Rev Genet. 2014;15:504–512. doi: 10.1038/nrg3746. [DOI] [PubMed] [Google Scholar]
- 177.Shoemaker DD, Lashkari DA, Morris D, Mittmann M, Davis RW. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat Genet. 1996;14:450–456. doi: 10.1038/ng1296-450. [DOI] [PubMed] [Google Scholar]
- 178.Ghosh C, Gupta R, Mukherjee KJ. An inverse metabolic engineering approach for the design of an improved host platform for over-expression of recombinant proteins in Escherichia coli. Microb Cell Fact. 2012;11:93. doi: 10.1186/1475-2859-11-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Crameri A, Whitehorn EA, Tate E, Stemmer WP. Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat Biotechnol. 1996;14:315–319. doi: 10.1038/nbt0396-315. [DOI] [PubMed] [Google Scholar]
- 180.Maksimow M, Hakkila K, Karp M, Virta M. Simultaneous detection of bacteria expressing GFP and DsRed genes with a flow cytometer. Cytometry. 2002;47:243–247. doi: 10.1002/cyto.10079. [DOI] [PubMed] [Google Scholar]
- 181.Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 182.Lissemore JL, Jankowski JT, Thomas CB, Mascotti DP, De Haseth PL. Green fluorescent protein as a quantitative reporter of relative promoter activity in E. coli. Biotechniques. 2000;28:82–84. doi: 10.2144/00281st02. [DOI] [PubMed] [Google Scholar]
- 183.Burke C, Liu M, Britton W, Triccas JA, Thomas T, Smith AL, Allen S, Salomon R, Harry E. Harnessing single cell sorting to identify cell division genes and regulators in bacteria. PLoS One. 2013;8:e60964. doi: 10.1371/journal.pone.0060964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kensy F, Engelbrecht C, Buchs J. Scale-up from microtiter plate to laboratory fermenter: evaluation by online monitoring techniques of growth and protein expression in Escherichia coli and Hansenula polymorpha fermentations. Microb Cell Fact. 2009;8:68. doi: 10.1186/1475-2859-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kensy F, Zang E, Faulhammer C, Tan RK, Buchs J. Validation of a high-throughput fermentation system based on online monitoring of biomass and fluorescence in continuously shaken microtiter plates. Microb Cell Fact. 2009;8:31. doi: 10.1186/1475-2859-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Fernandez-Ricaud L, Warringer J, Ericson E, Glaab K, Davidsson P, Nilsson F, Kemp GJ, Nerman O, Blomberg A. PROPHECY–a yeast phenome database, update 2006. Nucleic Acids Res. 2007;35:D463–D467. doi: 10.1093/nar/gkl1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fong SS, Marciniak JY, Palsson BO. Description and interpretation of adaptive evolution of Escherichia coli K-12 MG1655 by using a genome-scale in silico metabolic model. J Bacteriol. 2003;185:6400–6408. doi: 10.1128/JB.185.21.6400-6408.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jasnos L, Korona R. Epistatic buffering of fitness loss in yeast double deletion strains. Nat Genet. 2007;39:550–554. doi: 10.1038/ng1986. [DOI] [PubMed] [Google Scholar]
- 189.Korch SB, Henderson TA, Hill TM. Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol Microbiol. 2003;50:1199–1213. doi: 10.1046/j.1365-2958.2003.03779.x. [DOI] [PubMed] [Google Scholar]
- 190.Warringer J, Ericson E, Fernandez L, Nerman O, Blomberg A. High-resolution yeast phenomics resolves different physiological features in the saline response. Proc Natl Acad Sci U S A. 2003;100:15724–15729. doi: 10.1073/pnas.2435976100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bareither R, Pollard D. A review of advanced small-scale parallel bioreactor technology for accelerated process development: current state and future need. Biotechnol Prog. 2011;27:2–14. doi: 10.1002/btpr.522. [DOI] [PubMed] [Google Scholar]
- 192.Blomberg A. Measuring growth rate in high-throughput growth phenotyping. Curr Opin Biotechnol. 2011;22:94–102. doi: 10.1016/j.copbio.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 193.Klockner W, Buchs J. Advances in shaking technologies. Trends Biotechnol. 2012;30:307–314. doi: 10.1016/j.tibtech.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 194.Suresh S, Srivastava VC, Mishra IM. Techniques for oxygen transfer measurement in bioreactors: a review. J Chem Technol Biotechnol. 2009;84:1091–1103. [Google Scholar]
- 195.Duetz WA. Microtiter plates as mini-bioreactors: miniaturization of fermentation methods. Trends Microbiol. 2007;15:469–47. doi: 10.1016/j.tim.2007.09.004. [DOI] [PubMed] [Google Scholar]