

Abstract
RNase HII removes RNA from RNA/DNA hybrids, such as single ribonucleotides and RNA primers generated during DNA synthesis. Both, RNase HII substrates and RNase HII deficiency have been associated with genome instability in several organisms, and genome instability is a major force leading to the acquisition of drug resistance in bacteria. Understanding the mechanisms that underlie this phenomenon is one of the challenges in identifying efficient methods to combat bacterial pathogens. The aim of the present study was set to investigate the role of rnhB, presumably encoding RNase HII, in maintaining genome stability in the M. tuberculosis model organism Mycobacterium smegmatis. We performed gene replacement through homologous recombination to obtain mutant strains of Mycobacterium smegmatis lacking the rnhB gene. The mutants did not present an altered phenotype, according to the growth rate in liquid culture or susceptibility to hydroxyurea, and did not show an increase in the spontaneous mutation rate, determined using the Luria-Delbrück fluctuation test for streptomycin resistance in bacteria. The mutants also did not present an increase in the level of RNase HII substrates, measured as the level of alkaline degradation of chromosomal DNA or determined through immunodetection. We conclude that proteins other than RnhB proteins efficiently remove RNase HII substrates in M. smegmatis. These results highlight differences in the basic biology between Mycobacteria and eukaryotes and between different species of bacteria.
Introduction
RNase H proteins are ubiquitous enzymes present is eukaryotes, prokaryotes, archeons and viruses. These proteins are involved in the removal of RNA from RNA/DNA hybrids present during several cellular processes, including DNA replication, repair and transcription. Based on differences in the amino acid sequence, prokaryotic RNase H proteins have been divided into three classes and two types. RNase HI represents RNase H type 1 enzymes, whereas RNases HII and HIII represent RNase H type 2 enzymes. RNase HI and RNase HII are present in various organisms, while RNase HIII has only been detected in a limited number of bacteria, including Bacillus subtilis [1] and Chlamydophila pneumoniae [2]. The genome of Mycobacterium smegmatis encodes one gene with RNase HII fold- rnhB, similarly as the genome of Mycobacterium tuberculosis [3].
The results of in vitro experiments suggested that RNase HII recognize the RNA to DNA transition [4, 5]. Currently, two types of substrates for prokaryotic RNase HII have been identified. The first type of RNase HII substrates are RNA primers generated during DNA replication. Although the major pathway for Okazaki primer removal in eukaryotes is independent of RNase H activity [6], these structures might also be removed through RNase HII. Using an in vitro model, it was demonstrated that RNase H type 2 proteins cleave RNA primers in Okazaki fragments, leaving the last ribonucleotide of the primer attached to the DNA [4, 7, 8, 9], which is subsequently cleaved by FEN1 [10]. This mechanism was consistent with the observation that human RNase H2 localizes to replication foci [11]. The involvement of RNase H type 2 in Okazaki fragment processing has also been confirmed in archaea [12, 13]. However, in Escherichia coli, the enzyme responsible for the removal of RNA primers during Okazaki fragment maturation is polymerase I (PolI), encoded by polA [14, 15, 16]. Apart from PolI, RNase H proteins have also been implicated in RNA primer removal in bacteria [17].
The second type of substrate for RNase HII are single ribonucleotides embedded within DNA duplex. Until recently, the incorporation of ribonucleotides other than Okazaki fragments within the DNA double helix has been a neglected biological phenomenon. However, several recent studies have shown that ribonucleotides are indeed present within DNA, and ribonucleotide triphosphates incorporation is evolutionarily conserved from bacteria to eukaryotes. In fact, ribonucleotides might be the most common lesion in the DNA double helix. For example, the replicative polymerases α, δ, and ε in S. cerevisiae have been shown to insert one ribonucleotide for every 625, 5000 and 1250 deoxyribonucleotide triphosphates, respectively. This activity results in the incorporation of approximately 10 thousand ribonucleotides per round of replication [18].
In yeast, the removal of ribonucleotides embedded within the DNA double helix primarily occurs through RNase H2 [18, 19, 20] and to some extent, RNase H1 [19] and topoisomerase I [21]. Although genetic analyses in yeast have excluded the involvement of nucleotide excision repair, the minor involvement of base excision repair could not be ruled out [19]. Intriguingly, contrasting observations have been made in E. coli where, apart from RNase HII activity, the removal of ribonucleotide monophosphate primarily involved nucleotide excision repair, while the involvement of base excision repair and mismatch repair (MMR) was minimal [22].
Both, increased RNase HII substrates and RNase HII deficiency have been associated with genome instability in several organisms. Thus, the aim of the present study was to investigate whether rnhB, presumably encoding RNase HII, influences RNase HII substrate levels and the genome stability in the M. tuberculosis model organism M. smegmatis. M. smegmatis genome contains two putative RNases H- one RNase H class I enzyme encoded by rnhA gene one RNase H class II enzyme encoded by rnhB gene. Additionally, a two domain protein encoded by MSMEG4309 is homologous to Rv2228c found in M. tuberculosis, which encodes an RNase HI domain and a domain involved in cobalamin biosynthesis [23]. The protein expressed from MSMEG5849 has been shown to present RNase HII activity as well as being capable of pGpp synthesis [24]. Therefore, the genome of M. smegmatis might encode two RNases H class I and two RNases H class II.
Genome instability is a major force leading to the acquisition of drug resistance in bacteria. Understanding the mechanisms that underlie this instability is a challenge in identifying efficient methods to combat bacterial pathogens. We did not observe an increase in the levels of RNase HII substrates or mutation rates in mutant strains deficient in rnhB. Hence, we suspect that proteins other than RnhB proteins are involved in the removal of RNase HII substrates in M. smegmatis.
Materials and Methods
In silico analysis
The sequences for the genes presumably encoding RNase H proteins in M. smegmatis, E. coli and M. tuberculosis were identified and obtained from the PubMed database. The span of the domains was defined using the Simple Modular Architecture Research Tool (SMART). The homology between the domains was estimated using the National Center for Biotechnology Information (NCBI) BLAST tool. The alignments were visualized using Clustal Omega and ESPript 2.2.
Bacterial culture
M. smegmatis cultures were grown in nutrient broth (NB; Difco) during gene replacement or in 7H9 broth (Becton, Dickinson and Company) supplemented with OADC (Becton-Dickinson) and 0.05% Tween 80 (Sigma) for the determination of growth rates. Where necessary, the medium was supplemented with antibiotics and/or other supplements at the following concentrations: 25 µg/ml kanamycin (Sigma), 40 µg/ml X-gal (Sigma), 400 µg/ml hydroxyurea (HU) (Sigma), 0.4% succinate (Sigma), 12.5 µg/ml streptomycin (Serva), and 2% sucrose (Sigma). The M. smegmatis strains used in this study are listed in Table 1.
Table 1. List of the M. smegmatis strains used in this study.
| Strain | Characteristics |
|---|---|
| M. smegmatis mc2 155 | Reference strain |
| ∆rnhB | rnhB deletion mutant of mc2 155 |
| ∆rnhA/∆4305/∆rnhBattB::rnhA | Derivative of mc2 155 carrying deletions in the rnhA, MSMEG4305 and rnhB genes complemented with the full-length rnhA gene under the natural promoter at the attB site, HygR |
| ∆rnhA/∆4305/∆rnhBattB::4305 | Derivative of mc2 155 carrying deletions in the rnhA, MSMEG4305 and rnhB genes complemented with full-length MSMEG4305 gene under the acetamide promoter at the attB site, HygR |
To determine the growth rates, bacterial cells were transferred to fresh NB medium and incubated until the cultures reached an OD600 between 0.6 and 0.9. Aliquots of these seed cultures were inoculated into fresh 7H9 broth supplemented with OADC or 7H9 broth supplemented with OADC and HU at starting OD600 = 0.05. The cultures were incubated at 37°C with vigorous shaking for 24–48 hours. At desired time intervals, samples of the cultures were harvested and analyzed using a spectrophotometer (Pharmacia Biotech Ultrospec 2000). To assess the number of colony forming units, the samples were serially diluted into fresh NB broth and plated onto non-selective NB medium. The cultures were incubated at 37°C until obtaining visible colonies. Each experiment was performed at least in triplicate. To determine the cell length, samples of the cultures were harvested at 24 h and placed onto glass slides, followed by heat fixing and analysis using a Nikon Eclipse TE2000 microscope.
For susceptibility testing, cultures at starting OD600 = 0.05 were incubated at 37°C with vigorous shaking until reaching the logarithmic phase of growth (OD600 = 0.7–0.8) and the stationary phase of growth (24 hours). The logarithmic and stationary phase cultures were serially diluted, and 5 µl of each dilution was plated onto 7H10 medium supplemented OADC, succinate and 6, 10.5, 15 and 20 mM HU. As a control, 5 µl of each dilution was plated onto 7H10 medium without the addition of HU.
M. smegmatis mutants
The mutants were obtained using a gene replacement protocol as previously described [25, 26, 27]. The procedure required the construction of gene replacement and complementation plasmids. The primers used for the generation of the mutants are listed in Table 2.
Table 2. Primers used in this study.
| Primers used to generate the mutants | ||||
|---|---|---|---|---|
| Name | Sequence | Primer pair | Introduced restriction sites | Destination |
| MsGR1rnhA | TCGAGGGCAAGCTGCGCGAC | MsGR2rnhA | - | Gene replacement rnhA |
| MsGR2rnhA | CGTAGCACCGCACCCCAGCC | MsGR1rnhA | - | Gene replacement rnhA |
| MsGR3rnhA | CGGGATCCGTGCGCGCGCCACCAGGTC | MsGR4rnhA | BamHI | Gene replacement rnhA |
| MsGR4rnhA | GAAGCTTCCGCGAGGGGCCGAACACC | MsGR3rnhA | HindIII | Gene replacement rnhA |
| GR1MsRnhAII-KpnI | GGTACCCCGCCGACGATGATGCTGTC | GR2MsRnhAII-BamH | KpnI | Gene replacement MSMEG4305 |
| GR2MsRnhAII-BamH | CAAGCGGCGCAACGGGATC | GR1MsRnhAII-KpnI | - | Gene replacement MSMEG4305 |
| GR3MsRnhAII-BamH | CGGGATCCTACACAACCGCGCCGTAGCC | GR4MsRnhAII-Hind | BamHI | Gene replacement MSMEG4305 |
| GR4MsRnhAII-Hind | GCAAGCTTTCGCTGCTGGGTGCCGTGAC | GR3MsRnhAII-BamH | HindIII | Gene replacement MSMEG4305 |
| MsGR1rnhB | AACTGCAGACTACCTGCGCGAGCTCCGTG | MsGR2rnhB | PstI | Gene replacement rnhB |
| MsGR2rnhB | GAAGCTTCGCAGACCCGACGATTTCCG | MsGR1rnhB | HindIII | Gene replacement rnhB |
| RnHBgr3a | GCGAATTCCGTCGCTTCCCGTGATCGGC | RnHBgr4a | HindIII | Gene replacement rnhB |
| RnHBgr4a | GGGGTACCAAGGGTTTTGCCGCATCCGC | RnHBgr3a | KpnI | Gene replacement rnhB |
| MsA1–5562-BglIIs | CAGATCTGTGAACCACCGGCACCACGCC | MsA1–5562-XbaI | BglII | Complementation rnhA |
| MsA1–5562-XbaI | CTCTAGATGGTGGTCGGCCTGGCGGG | MsA1–5562-BglIIs | XbaI | Complementation rnhA |
| MsrnhAIIPace-sBglII | CAGATCTGTGAAGGTTCTCGTCGAGGCCGAC | MsrnhAII-rev-Xba | BglII | Complementation MSMEG4305 |
| MsrnhAII-rev-Xba | CTCTAGATGCACTCGTGAGCTACAGGTACGC | MsrnhAIIPace-sBglII | XbaI | Complementation MSMEG4305 |
Briefly, for gene replacement, the sequences flanking the desired deletion were amplified through PCR and consecutively introduced into p2NIL plasmid, followed by the introduction of the PacI suicidal cassette excised from vector pGOAL17. For complementation under an acetamide promoter, a native copy of the gene of interest was amplified through PCR and introduced into pJAM2. The gene and the acetamide promoter were subsequently excised from pJAM2 and introduced into pMV306Hyg. All cloning was performed in E. coli Top10 cells. The plasmids were introduced into mycobacterial cells, which were further subjected to a multistep selection process for the detection of the mutants.
Southern blotting
The primers used to obtain the probes and the restriction enzymes used for the digestion of the genomic DNA are listed in Table 2.
Luria-Delbrück fluctuation test
The Luria-Delbrück fluctuation test was performed as previously described [28]. Briefly, 100 ml of 7H9 medium supplemented with OADC and succinate was inoculated with approximately 100 bacteria per ml, based on the determination of colony forming units. To avoid clumping, the inoculum was passed through a syringe needle. The inoculated medium was immediately divided into 30 cultures of 3 ml each. After 72 hours at 37°C with vigorous shaking, the contents of each tube were centrifuged. A total of 26 of cultures were plated onto plates supplemented with streptomycin, while the remaining four cultures were serially diluted and used to assess the number of colony forming units. The plates were incubated for 72 h at 37°C, and subsequently the number of colony forming units on each plate was counted. The results were analyzed using the FALCOR (Fluctuation Analysis Calculator) software program.
Alkaline degradation of chromosomal DNA
The bacterial cells were transferred to fresh NB medium and cultured until reaching an OD600 between 0.6 and 0.9. Aliquots of these seed cultures were inoculated into fresh 7H9 broth supplemented with OADC at a starting OD600 = 0.05. The cultures were incubated at 37°C with vigorous shaking for 24 hours. Subsequently, the cells were harvested through centrifugation for 10 min at 8000 x g at 4°C and subjected to DNA isolation. The DNA solution was incubated in 0.3 M NaOH (Sigma) for 2 h at 55°C. As a control, the DNA solution was incubated in 0.3 M NaCl for 2 h at 55°C. Subsequently, the samples were resolved on 1% alkaline agarose gels. The level of DNA fragmentation was visually assessed through a comparison of the mobility of the DNA fragments on the gel in relative reference to the size standard (GeneRuler 1-kb DNA ladder, Thermo Scientific).
Immunodetection of RNA/DNA hybrids
The bacteria were grown in liquid culture for 24 hours in 7H9 medium supplemented with OADC and succinate. Subsequently, 3 ml of each culture was centrifuged and subjected to total nucleic acid extraction. A 50-µl sample of the bacterial cells was resuspended in 200 µl of TE buffer (10 mM Tris-HCl (Serva), pH 8.0 and 1 mM EDTA (Serva)). Approximately 50 µl of zirconia beads (BioSpec Products) was added, and the samples were homogenized using an MP Fast Prep homogenizer. Subsequently, an equal volume of phenol:chloroform (Sigma) was added, and after vigorous shaking, the samples were centrifuged at 14000 x g for 10 min. The aqueous phase was transferred to a new Eppendorf tube, and three volumes of ethanol (Sigma) were added. The samples were incubated at -20°C overnight and centrifuged for 10 min at 14000 x g. The pellet was dried at room temperature for approximately 15 min and resuspended in 100 µl of water. The samples were serially diluted, and 2 µl of each dilution was placed onto an Amersham Hybond N+ nylon membrane (GE Healthcare Life Sciences). The membrane was air-dried and blocked with 5% milk in PBST (137 mM NaCl (Sigma); 2.7 mM KCl (Sigma); 10 mM Na2HPO4 • 2 H2O (Sigma); 2 M KH2PO4 (Sigma) pH 7.4; and 0.02% Tween 20 (Sigma)) at room temperature. The membrane was washed three times in PBST for 5 min at room temperature and incubated overnight at 4°C with a primary monoclonal mouse antibody against the RNA/DNA hybrid (D5H6) (Covalab) suspended in PBST containing 1% bovine serum albumin. Subsequently, the membrane was washed three times in PBST at room temperature and incubated with a secondary anti-mouse rabbit IgG antibody (Sigma) conjugated with peroxidase suspended in PBST containing 1% milk at room temperature. The membrane was washed three times in PBST and covered with ECL detection reagent (Amersham Biosciences). The excess reagent was removed, and the membrane was tightly wrapped in Saran wrap and exposed to X-ray film (Thermo Scientific) for 1 minute. The film was developed using a Kodak Medical X-ray Processor.
Statistical analysis
To determine the growth rates of the analyzed strains, logistic or quadratic curves were fitted to the optical density measurements obtained from the liquid cultures. When the growth rates were determined after 48 h, we fitted logistic curves of the form OD = A/[1 + B*exp(-KT)], where OD is the optical density at time T, A is an asymptotic value, B is a constant of integration, and K is the growth rate constant. When the growth rates were estimated after 24 h, we fitted quadratic curves of the form OD = K*T2 + OD0, where OD is the optical density at time T, OD0 is the starting optical density, and K is the growth rate constant. Parameter K from the fitted curves was used as an indicator of the growth rate for each strain. A t-test was used to assess differences in the growth rates between the strains. The non-parametric Kruskal-Wallis test was used to compare the mutation rates between the strains. All statistical analyses were performed using Statistica 10.0 software (StatSoft, Tulsa, OK, USA).
Results
The M. smegmatis genome of encodes a protein with an RNase HII fold
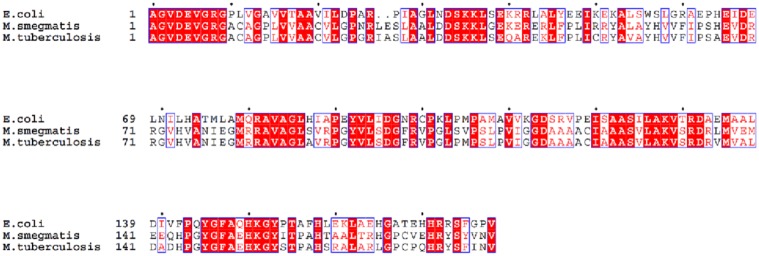
Based on the genome sequence of M. smegmatis mc2 155 deposited in the NCBI database, we identified an rnhB gene, presumably encoding RNase HII. The RNase HII domain of M. smegmatis mc2 155 RnhB shares 61% sequence homology with the same domain of E. coli K12_MG1655 RnhB with 98% of query cover of M. smegmatis RnhB (Fig. 1A). RnhB has also been identified in the genome of M. tuberculosis H37Rv. The RNase HII domains of these species share 91% amino acid sequence homology with 100% query cover of M. smegmatis RnhB (Fig. 1B).
Figure 1. Sequence comparisons of the RNase HII domains in the RnhB proteins of E. coli K12_MG1655, M. smegmatis mc2 155 and M. tuberculosis H37Rv.
Generating rnhB deficient mutants
We used gene replacement through homologous recombination to obtain genetic mutants with large deletions within the sequence of the predicted RNase HII gene, rnhB. We also obtained mutants deficient not only in the rnhB gene, but also in the genes encoding predicted RNase HI proteins (rnhA and MSMEG4305). The rnhB-deficient mutants were confirmed through Southern blot analysis (Fig. 2 and Fig. 3). We obtained a single ∆rnhB mutant. The two double mutants, ∆rnhA/∆rnhB and ∆4305/∆rnhB, were used to generate the triple conditional mutants ∆rnhA/∆4305/∆rnhBattB::rnhA and ∆rnhA/∆4305/∆rnhBattB::4305, where the expression of rnhA or MSMEG4305, controlled through the acetamide promoter, is inhibited in the presence of succinate. Because even succinate inhibition did not inhibit complemented gene expression below the native level (data not shown), the data obtained from the analysis of these strains only reflects the consequences of rnhB deletion and not consequences of the deletion of predicted RNase HI-encoding genes.
Figure 2. Southern blot analyses confirming the deletions in the single and double RNase H mutants of M. smegmatis.
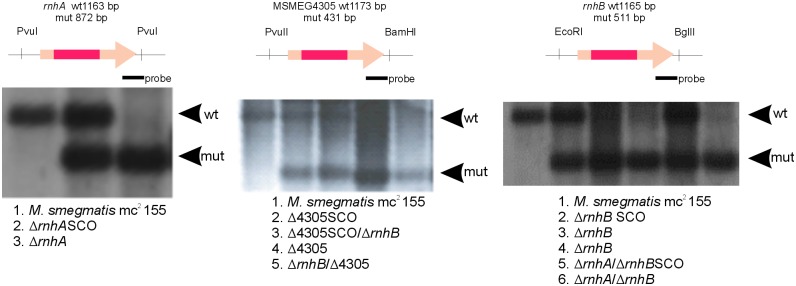
We used the gene replacement through homologous recombination technique to obtain single and double mutants deficient in rnhB and/or either rnhA or MSMEG4305. The ∆rnhA/∆rnhB mutant was obtained through the introduction of the rnhB gene replacement plasmid into the ∆rnhA-deficient strain. The mutant ∆4305/∆rnhB was obtained through the introduction of the MSMEG4305 gene replacement plasmid into the ∆rnhB strain. The intermediate steps of the gene replacement procedure are denoted SCO.
Figure 3. Southern blot analyses confirming the generation of ∆rnhA/∆4305/∆rnhBattB::rnhA and ∆rnhA/∆4305/∆rnhBattB::4305 strains.

We used the double mutant strains ∆rnhA/∆rnhB and ∆4305/∆rnhB to obtain the triple conditional mutants ∆rnhA/∆4305/∆rnhBattB::rnhA and ∆rnhA/∆4305/∆rnhBattB::4305. The intermediate steps of the gene replacement procedure are denoted SCO.
Phenotypic analysis of the M. smegmatis mutants lacking rnhB
Growth rate. We determined whether the deletion of the rnhB gene influenced the growth of mutant M. smegmatis. To this end, we measured the optical densities of the liquid cultures at determined intervals of time and fitted appropriate growth curves [29]. We did not observe any differences in growth between the ∆rnhB and wild-type M. smegmatis mc2 155 on 7H9 broth supplemented with OADC when analyzed for asymptotical values (t = 0.83, df = 5, p = 0.44) and growth rate (t = 2.18, df = 5, p = 0.08) (Fig. 4A). The morphology of the mutant cells, in terms of cell length, was not altered (t = 0.68, df = 205, p = 0.50) (Fig. 4B).
Figure 4. Growth rate and morphology of the ∆rnhB mutant and M. smegmatis mc2 155 strains.

A) Growth rates based on the optical densities of the cultures. B) The cell lengths.
Growth rate in the presence of HU. Next, we examined the growth rate of mutants lacking rnhB in the presence of HU. HU is an inhibitor of class I ribonucleotide reductases, which convert ribonucleoside 5’-diphosphates into deoxyribonucleoside 5’-diphosphates [30]. Therefore, HU is expected to change the balance of NTP pools and increase the amount of rNTPs, while limiting the amount dNTPs. These changes should increase the incorporation of rNTPs into chromosomal DNA. Notably, HU has been shown to influence the DNA content in M. smegmatis [31].
We expected that mutations that alter rNTP removal should affect HU susceptibility levels. Indeed, altered HU susceptibility was observed in RNase H-deficient mutants in yeasts [19, 32]. We did not observe any differences in HU susceptibility in ∆rnhB mutants compared with wild type (Fig. 5).
Figure 5. Susceptibility of the ∆rnhB mutant and M. smegmatis mc2 155 strains to HU.

A) Logarithmic and B) stationary phase cultures were serially diluted and plated onto medium containing different concentrations of HU. We observed that 20 mM HU inhibited the growth of the analyzed strains, but we did not observe differences in susceptibility between the strains.
We compared the growth rates of ∆rnhB mutants and the wild-type strain in rich 7H9 medium supplemented with HU. The analysis of the growth curves did not reveal any differences in the growth rates of these bacteria in the presence of HU (t = 0.90, df = 8, p = 0.39). These results were consistent with the number of the colony forming units (all p>0.10) (Fig. 6).
Figure 6. Growth rates in the presence of HU.

A) Growth rates based on the optical density of the cultures. B) Growth rates based on the number of colony forming units (white: M. smegmatis mc2 155; gray: ∆rnhB mutants).
Spontaneous mutation rate. We also assessed whether the deletion of rnhB in M. smegmatis influenced the spontaneous mutation rate. We performed the Luria-Delbrück fluctuation test and compared the results obtained from the ∆rnhA/∆4305/∆rnhBattB::rnhA, ∆rnhA/∆4305/∆rnhBattB::4305 and wild type strains grown in the presence succinate. Briefly, medium inoculated with approximately 100 bacterial cells per ml was divided into 30 parallel cultures and grown for 72 h. Subsequently, 4 cultures of each strain were used to determine the average number of viable cells in the culture, while the remaining cultures were plated onto selective media containing streptomycin. In M. tuberculosis, the resistance to streptomycin in approximately 70% of the isolates resulted from mutations in rpsL (encoding S12 ribosomal protein), rrs (16S rRNA) and gidB (ribosome methyltransferase) genes. The cause of the streptomycin resistance in the remaining 30% of the strains remains unknown [33]. While point mutations in rpsL and rrs caused streptomycin resistance [33], frameshifts and point mutations were responsible for the streptomycin resistance associated with gidB [34]. Homologs of rpsL, rss and gidB genes have been identified in M. smegmatis.
Determining the number of streptomycin-resistant colonies in each culture facilitated the calculation of the overall mutation rate of each strain. The mutation rate was calculated using FALCOR software (Table 3). We did not observe statistically significant differences in the mutation rate between the analyzed strains (Kruskal-Wallis: H2, 78 = 1.64; p = 0.48). Hence, the deletion of rnhB did not influence the spontaneous mutation rate in M. smegmatis.
Table 3. Mutation rates of M. smegmatis mc2 155, ∆rnhA/∆4305/∆rnhBattB::rnhA and ∆rnhA/∆4305/∆rnhBattB::4305 strains, estimated using the Luria-Delbrück fluctuation test.
| Strain | Mutation rate (lower bound-upper bound) |
|---|---|
| M. smegmatis mc2 155 | 4.88 (2.8–9.02) |
| ∆rnhA/∆4305/∆rnhBattB::rnhA | 9.03 (3.38–11.85) |
| ∆rnhA/∆4305/∆rnhBattB::4305 | 5.6 (3.88–6.75) |
The level of RNase HII substrates in rnhB-deficient mutants
The level of alkaline degradation of chromosomal DNA. We compared the level of ribonucleotide incorporation in the DNA isolated from ∆rnhB mutant and wild-type strains. The alkaline hydrolysis of genomic DNA has been successfully used in other studies regarding RNase H [35, 36]. As the 3’ phosphodiester bonds of ribonucleotides, but not deoxyribonucleotides, are sensitive to alkali hydrolysis, the fragmentation of genomic DNA under alkaline conditions likely indicates the presence of ribonucleotides, i.e., single ribonucleotides and unresolved RNA primers, embedded within the DNA double helix. Genomic DNA obtained from the mutant and wild-type strains were subjected to alkaline hydrolysis. The control samples were treated with an equal concentration of NaCl. The samples were separated on agarose gels. We expected that an increased level of unprocessed Okazaki primers or an increased level of unremoved single rNTP incorporated within the DNA would show an increase in the degree of genomic DNA fragmentation. We did not observe any differences in the levels of genomic DNA fragmentation under alkaline conditions between the mutant and wild-type strains based on the gel mobility of the alkaline-treated samples (Fig. 7). This observation suggests that neither the unprocessed Okazaki primers nor the single ribonucleotides in the DNA double helix were increased in ∆rnhB mutants.
Figure 7. Alkaline hydrolysis of the genomic DNA.

DNA was isolated from the ∆rnhB mutants and M. smegmatis mc2 155. The strains were grown in 7H9 medium supplemented with OADC. The DNA samples were treated with either NaOH or NaCl as a control. The fragmentation of the samples was visualized on alkaline agarose gels. Lanes 1a) GeneRuler 1-kb DNA Ladder, 2a) M. smegmatis mc2 155 control DNA, 3a) ∆rnhB mutant control DNA, 1b) GeneRuler 1-kb DNA Ladder, 2b) M. smegmatis mc2 155 DNA hydrolyzed with NaOH, and 3b) ∆rnhB mutant DNA hydrolyzed with NaOH. The level of ribonucleotide incorporated in the DNA of both strains was similar, as we did not observe differences in fragmentation of genomic DNA.
Immunodetection. To confirm that the levels of unprocessed Okazaki primers and single ribonucleotides in the DNA double helix were unaltered in rnhB-deficient M. smegmatis, we performed the immunodetection of RNA/DNA hybrids in nucleic acids isolated from ∆rnhA/∆4305/∆rnhBattB::rnhA, ∆rnhA/∆4305/∆rnhBattB::4305 and wild-type strains (Fig. 8). These antibodies have previously been used to detect the presence of RNA/DNA hybrids in eukaryotes [37, 38]. We did not observe differences in the strength of the signal generated between different strains, suggesting that the level of RNA/DNA hybrids in these strains was unchanged.
Figure 8. RNA/DNA hybrid level.

Comparison of the level of RNA/DNA hybrids in M. smegmatis mc2 155, ∆rnhA/∆4305/∆rnhBattB::rnhA and ∆rnhA/∆4305/∆rnhBattB::4305 strains grown on 7H9 medium in the presence of succinate.
Discussion
In silico analysis revealed an rnhB gene encoding a protein with a predicted RNase HII domain. We did not identify any homologs of RNase HIII, consistent with the fact the simultaneous inheritance of RNase HI and RNase HIII in the genome of M. smegmatis was avoided due to the functional redundancy of these genes [39].
There has been much confusion regarding the essentiality of RNase HII-encoding genes. Initially, RNase H type II genes were considered essential in B. subtilis [40]. However, the Yoshikawa group managed to obtain B. subtilis mutants deficient for all RNase H-encoding genes [17]. The growth rate of mutants lacking both RNase HII and RNase HIII was low, suggesting that RNase H proteins are involved in the processing of RNA primers. These suspicions were confirmed through the observation that the overexpression of YpcP exonuclease suppressed the filamentous phenotype and overexpression of the exonuclease domain of PolI in these mutants. RNase HII/RNase HIII mutants showed temperature sensitive growth at 56.5°C [17]. The O’Donell group confirmed the generation of double RNase HII/RNase HIII mutants in B. subtilis [35]. RNase HII has been demonstrated as dispensable in E. coli [41], although this enzyme was initially considered essential [42].
Using gene replacement through homologous recombination, we generated M. smegmatis mutants deficient in rnhB, suggesting that this gene is not essential for the survival of M. smegmatis. Therefore, either the function of the product of this gene is nonessential for cell survival in vitro or there are other genes in the mutant M. smegmatis genome whose products have overlapping functions with the mutated gene.
The level of RNase HII substrates and the RNase HII deficiency affect genome stability in both eukaryotes and prokaryotes. In B. subtilis, the RNase HII/RNaseHIII/YpcP-deficient mutant displayed a filamentous phenotype, and this phenotype was suppressed through the overexpression of either the deleted genes or the 5’-3’ exonuclease domain of PolI. This phenotype resulted from the induced SOS response, which, in turn, might have resulted from the accumulation of unprocessed Okazaki fragments [17]. The deletion of the 5’-3’ exonuclease domain of PolI in E. coli, which is primarily involved in the removal of Okazaki fragments in the absence of DNA damaging agents, increased the mutation rate in terms of frameshift and duplication mutations [43]. It has also been suggested that persisting Okazaki primers destabilize tetranucleotide repeats in H. influenzae. This phenotype was associated with the deletion of RNase HI or the Klenow domain of PolI [44].
The deletion of RNase H2 increased the mutation rate in budding yeast [18, 21, 45]. A recent study showed that short, 2–5 bp deletions observed in budding yeast mutants defective for RNase H2 result from topoisomerase I activity, and the deletion of topoisomerase I in RNase H2 mutants restored the mutation rate associated with these changes in the wild type [21]. Notably, the rates for mutations other than 2–5 bp deletions were not restored to the wild type in double the RNase H2/topoisomerase I mutant [21].
It has been suggested that ribonucleotide incorporated in DNA signals a newly synthesized strand for MMR. In E. coli, the MMR MutH enzyme recognizes and cleaves a newly synthesized strand at hemimethylated GATC sites [46]. E. coli mutants deficient in RNase HII grow as fast as wild type and show no increase in spontaneous mutation rates. Therefore, the authors concluded that ribonucleotides persisting in the E. coli chromosome are non mutagenic [35]. However, contrasting observations were shown in B. subtilis, which lacks a homolog of MutH. Instead MutL exhibits endonuclease activity that is not present in E. coli. It has been proposed that MutL requires a signal to direct this enzyme to the newly synthesized DNA strand. Signals from Okazaki fragments might be functional for the lagging strand, but not for the leading strand. Therefore, it has been proposed that these signals are provided from the ribonucleotides incorporated within the newly synthesized strand. To examine this hypothesis, the authors measured the spontaneous mutations rates in RNase H type II-deficient B. subtilis mutants. The deletion of RNase HII resulted in 2.4-fold increase in mutation rates, while the deletion of RNase HIII only increased mutation rates 1.3-times compared with wild type. Double mutation resulted in a five-fold increase in the mutation rates. These authors speculated that the increased mutation rate corresponded to the 10% decline in MMR efficiency [35]. The hypothesis that ribonucleotides embedded within DNA act as a strand discrimination factor during MMR has been confirmed in eukaryotes [47].
It has been shown that yeast DNA polymerase ε bypasses a single rNTP present within the DNA template [18], and the presence of ribonucleotides in the template delays bacterial replisome progression 4–30-fold [35]. Notably, mouse embryos deficient in RNase H2 show arrested development and display an increased number of ribonucleotides in the genomic DNA [48]. Thus, ribonucleotides embedded within DNA duplex might constitute a barrier for replication fork progression. While this barrier is impossible to circumvent in higher eukaryotes, based on the essentiality of RNase H, in yeast, the double deletion of RNase H1 and RNase H2 sensitizes the cells to replication stress-inducing agents, such as HU and methyl methanesulfonate [19]; however, increased HU susceptibility after single RNase H2 deletion has been observed [32]. Additionally, RNase H deletion induces the constant activation of post-replication repair, although the mechanisms of this phenomenon are poorly understood [19].
Primary phenotypic analysis of the growth rate and cell morphology showed that ∆rnhB M. smegmatis mutants exhibit growth similar to the wild-type strain, suggesting that ribonucleotides incorporated within DNA double helix after rnhB deletion do not constitute a barrier for replication fork progression. The growth rate of ∆rnhB mutants remained unaltered, even in the presence of HU, which is considered to increase ribonucleotide incorporation. These observations are in contrast to the data obtained in B. subtilis [17], yeast [19] and higher eukaryotes [36]. Moreover, Luria-Delbrück fluctuation analyses did not reveal increased mutation rates in RNase HII-deficient mutants. However, M. smegmatis does not possess homologs of the MMR system, therefore an MMR defect cannot be expected. Indeed, when we analyzed the level of RNase HII substrates in rnhB-deficient cells, based on the levels of alkaline degradation of the chromosomal DNA or the immunodetection of RNA/DNA hybrids, we did not observe differences between mutant and wild-type strains. Therefore, in contrast to B. subtilis [35], yeast [18] and higher eukaryotes [36], the RNase HII deletion did not increase the level of RNase HII substrates in M. smegmatis.
Thus, we concluded that the RNase HII activity in M. smegmatis cells after ∆rnhB deletion is sufficient to remove RNA/DNA hybrids to wild-type levels, and the genome stability in these deletion mutants is unaffected. Therefore, proteins other than RnhB proteins must be involved in the removal of RNase HII substrates in M. smegmatis. Based on the data obtained from previous studies, these proteins might include PolI [17], MSMEG5849 [24] and/or RNase HI [19]. For example in E. coli the main enzyme responsible for the removal of RNA primers during Okazaki fragment maturation is thought to be polymerase I (PolI) encoded by polA [14, 15, 16]. This enzyme possesses a number of enzymatic activities: 5’-3’ DNA dependent DNA polymerase [49], 5’-3’ RNA dependent DNA polymerase [50], 3’-5’ exonuclease activity [51] and 5’-3’ exonuclease activity [51]. There seems to be a lot of confusion regarding essentiality of PolI in bacteria. While polymerase domain of PolI can be inactivated in both E. coli [50] and M. smegmatis [52], it seemed that 5’-3’ exonuclease activity is essential for survival of E. coli [53]. In fact, it has been speculated that PolI temperature sensitive mutant strains are lethal precisely due to the failures in removal of RNA primers during DNA synthesis [54]. Other authors argued that polA mutant is in fact viable on minimal medium, but not on rich medium [55]. The same report stated that complementation of the mutant strain with either 5’-3’ exonuclease portion of PolI or polymerase 3’-5’ exonuclease portions restores the viability of the mutant on rich medium [55]. Another group was able to obtain a normally growing polA mutant on LB medium [43]. Finally, Yoshikawa group showed that polA can be deleted from the genome of B. subtilis, however only in the presence of other gene, ypcP, providing 5’-3’ exonuclease activity. They observed similar phenomenon in E. coli, when 5’-3’ activity is provided by ypcP homolog xni. The polA mutant that they obtained presented temperature sensitivity and did not grow at 56.5°C [17].
It was shown in E. coli, that RNase HI might also be involved in RNA primers removal, however its role is thought to be restricted to cleavage of longer RNA fragments [14]. Again there are contradictory reports regarding its essentiality [56, 57, 58]. The authors claiming to have obtained RNase HI deficient mutants stated that the mutants are rich broth sensitive [57, 58], which could explain the failure in obtainment of the mutant by Kanaya and Crouch.
Additional factor presumably capable of removing RNase HII substrates is a protein encoded by MSMEG5849. This protein might be sufficient to remove both single ribonucleotides embedded within DNA double helix and Okazaki primers. MSMEG5849 possesses two enzymatic activities- an RNase HII and RelA-SpoT nucleotydyl transferase domain responsible for ppGpp synthesis [24]. Until 2012 the RNase HII domain of this protein was described as a domain of unknown function 429.
Data Availability
All relevant data are within the paper and the Supporting Information file.
Funding Statement
The study was supported by POIG.01.01.02-10-107/09 project implemented under Innovative Economy Operational Programme, years 2007–2013 “Studies of the molecular mechanisms at the interface the human organism–the pathogen–environmental factors.” and by grant of Polish National Center of Science 2011/01/N/NZ6/04186 “Identification of a novel mechanism of initiation of DNA replication in Mycobacterium smegmatis”.
References
- 1. Ohtani N, Haruki M, Morikawa M, Crouch RJ, Itaya M, et al. (1999) Identification of the genes encoding Mn2+-dependent RNase HII and Mg2+-dependent RNase HIII from Bacillus subtilis: classification of RNases H into three families. Biochem 38: 605–618. 10.1021/bi982207z [DOI] [PubMed] [Google Scholar]
- 2. Liang R, Liu X, Pei D, Liu J (2007) Biochemical characterization and functional complementation of ribonuclease HII and ribonuclease HIII from Chlamydophila pneumoniae AR39. Microbiol 153: 787–793. 10.1099/mic.0.2006/003434-0 [DOI] [PubMed] [Google Scholar]
- 3. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544. 10.1038/31159 [DOI] [PubMed] [Google Scholar]
- 4. Murante RS, Henricksen LA, Bambara RA (1998) Junction ribonuclease: an activity in Okazaki fragment processing. Proc Natl Acad Sci U S A 95: 2244–2249. 10.1073/pnas.95.5.2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eder PS, Walder RY, Walder JA (1993) Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie 75: 123–126. 10.1016/0300-9084(93)90033-O [DOI] [PubMed] [Google Scholar]
- 6. Zheng L, Shen B (2011) Okazaki fragment maturation: nucleases take centre stage. J Mol Cell Biol 3: 23–30. 10.1093/jmcb/mjq048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turchi JJ, Huang L, Murante RS, Kim Y, Bambara RA (1994) Enzymatic completion of mammalian lagging-strand DNA replication. Proc Natl Acad Sci U S A 91: 9803–9807. 10.1073/pnas.91.21.9803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang L, Kim Y, Turchi JJ, Bambara RA (1994) Structure-specific cleavage of the RNA primer from Okazaki fragments by calf thymus RNase HI. J Biol Chem 269: 25922–25927. [PubMed] [Google Scholar]
- 9. Qiu J, Qian Y, Frank P, Wintersberger U, Shen B (1999) Saccharomyces cerevisiae RNase H(35) functions in RNA primer removal during lagging-strand DNA synthesis, most efficiently in cooperation with Rad27 nuclease. Mol Cell Biol 19: 8361–8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rumbaugh JA, Murante RS, Shi S, Bambara RA (1997) Creation and removal of embedded ribonucleotides in chromosomal DNA during mammalian Okazaki fragment processing. J Biol Chem 272: 22591–22599. 10.1074/jbc.272.36.22591 [DOI] [PubMed] [Google Scholar]
- 11. Bubeck D, Reijns MAM, Graham SC, Astell KR, Jones EY, et al. (2011) PCNA directs type 2 RNase H activity on DNA replication and repair substrates. Nucl Ac Res 39: 3652–3666. 10.1093/nar/gkq980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sato A, Kanai A, Itaya M, Tomita M (2003) Cooperative regulation for Okazaki fragment processing by RNase HII and FEN-1 purified from a hyperthermophilic archaeon, Pyrococcus furiosus . Biochem Biophys Res Comm 309: 247–252. 10.1016/j.bbrc.2003.08.003 [DOI] [PubMed] [Google Scholar]
- 13. Le Laz S, Le Goaziou A, Henneke G (2010) Structure-specific nuclease activities of Pyrococcus abyssi RNase HII. J Bacteriol 192: 3689–3698. 10.1128/JB.00268-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ogawa T, Okazaki T (1984) Function of RNase H in DNA replication revealed by RNase H defective mutants of Escherichia coli . Mol Gen Genet 193: 231–237. 10.1007/BF00330673 [DOI] [PubMed] [Google Scholar]
- 15. Okazaki R, Arisawa M, Sugino A (1971) Slow joining of newly replicated DNA chains in DNA polymerase I-deficient Escherichia coli mutants. Proc Nat Acad Sci USA 68: 2954–2957. 10.1073/pnas.68.12.2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olivera BM, Bonhoeffer F (1974) Replication of Escherichia coli requires DNA polymerase I. Nature 250: 513–514. 10.1038/250513a0 [DOI] [PubMed] [Google Scholar]
- 17. Fukushima S, Itaya M, Kato H, Ogasawara N, Yoshikawa H (2007) Reassessment of the in vivo functions of DNA polymerase I and RNase H in bacterial cell growth. J Bacteriol 189: 8575–8583. 10.1128/JB.00653-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McElhinny NSA, Kumar D, Clark AB, Watt DL, Watts BE, et al. (2010) Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol 6:774–781. 10.1038/nchembio.424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lazzaro F, Novarina D, Amara F, Watt DL, Stone JE, et al. (2012) RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol Cell 45: 99–110. 10.1016/j.molcel.2011.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chon H, Vassilev A, DePamphilis ML, Zhao Y, Zhang J, et al. (2009) Contributions of the two accessory subunits, RNASEH2B and RNASEH2C, to the activity and properties of the human RNase H2 complex. Nucl Ac Res 37: 96–110. 10.1093/nar/gkn913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim N, Huang SN, Williams JS, Li YC, Clark AB, et al. (2011) Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science. 332: 1561–1564. 10.1126/science.1205016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vaisman A, McDonald J, Huston D, Kuban W, Liu L, et al. (2013) Removal of misincorporated ribonucleotides from prokaryotic genomes: an unexpected role for nucleotide excision repair. PLoS Genet. 9: e1003878 Available: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3820734/. Accessed: 16 June 2014 10.1371/journal.pgen.1003878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Watkins HA, Baker EN (2010) Structural and functional characterization of an RNase HI domain from the bifunctional protein Rv2228c from Mycobacterium tuberculosis . J Bacteriol 192: 2878–2886. 10.1128/JB.01615-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murdeshwar MS, Chatterji D (2012) MS_RHII-RSD, a dual-function RNase HII-(p)ppGpp synthetase from Mycobacterium smegmatis . J Bacteriol 194: 4003–4014. 10.1128/JB.00258-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parish T, Stoker NG (2000) Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiol 146: 1969–1975. [DOI] [PubMed] [Google Scholar]
- 26. Pawelczyk J, Brzostek A, Kremer L, Dziadek B, Rumijowska-Galewicz A, et al. (2011) AccD6, a key carboxyltransferase essential for mycolic acid synthesis in Mycobacterium tuberculosis, is dispensable in a nonpathogenic strain. J Bacteriol 193: 6960–6972. 10.1128/JB.05638-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dziadek J, Rajagopalan M, Parish T, Kurepina N, Greendyke R, et al. (2002) Mutations in the CCGTTCACA DnaA box of Mycobacterium tuberculosis oriC that abolish replication of oriC plasmids are tolerated on the chromosome. J Bacteriol. 184: 3848–3855. 10.1128/JB.184.14.3848-3855.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luria SE, Delbrück M (1943) Mutations of bacteria from virus sensitivity to virus resistance. Genetics 28: 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zwietering MH, Jongenburger I, Rombouts FM, van ‘T Riet K. (1990) Modeling of the bacterial growth curve. Appl Environ Microbiol 56: 1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kolberg M, Strand KR, Graff P, Andersson KK (2004) Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta 1699: 31–34. [DOI] [PubMed] [Google Scholar]
- 31. Winder FG, Barber DS (1973) Effects of hydroxyurea, nalidixic acid and zinc limitation on DNA polymerase and ATP-dependent deoxyribonuclease activities of Mycobacterium smegmatis . J Gen Microbiol 76: 189–196. 10.1099/00221287-76-1-189 [DOI] [PubMed] [Google Scholar]
- 32. Arudchandran A, Cerritelli S, Narimatsu S, Itaya M, Shin DY, et al. (2000) The absence of ribonuclease H1 or H2 alters the sensitivity of Saccharomyces cerevisiae to hydroxyurea, caffeine and ethyl methanesulphonate: implications for roles of RNases H in DNA replication and repair. Genes to Cells 5: 789–802. 10.1046/j.1365-2443.2000.00373.x [DOI] [PubMed] [Google Scholar]
- 33. Villellas C, Aristimuño L, Vitoria MA, Prat C, Blanco S, et al. (2013) Analysis of mutations in streptomycin resistant strains reveals a simple and reliable genetic marker for identification of Mycobacterium tuberculosis Beijing genotype. J Clin Microbiol 51: 2124–2130. 10.1128/JCM.01944-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spies FS, Ribeiro AW, Ramos DF, Ribeiro MO, Martin A, et al. (2011) Streptomycin resistance and lineage-specific polymorphisms in Mycobacterium tuberculosis gidB gene. J Clin Microbiol 49: 2625–2630. 10.1128/JCM.00168-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yao NY, Schroeder JW, Yurieva O, Simmons LA, O’Donnell ME (2013) Cost of rNTP/dNTP pool imbalance at the replication fork. Proc Natl Acad Sci U S A 110: 12942–12947. 10.1073/pnas.1309506110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, et al. (2012) Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149: 1008–1022. 10.1016/j.cell.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ginno PA, Lim YW, Lott PL, Korf I, Chédin F (2013) GC skew at the 5’ and 3’ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res 23: 1590–1600. 10.1101/gr.158436.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wahba L, Gore SK, Koshland D (2013) The homologous recombination machinery modulates the formation of RNA-DNA hybrids and associated chromosome instability. eLife 2:e00505 Available: http://elifesciences.org/content/2/e00505. Accessed: 16 June 2014 10.7554/eLife.00505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kochiwa H, Tomita M, Kanai A (2007) Evolution of ribonuclease H genes in prokaryotes to avoid inheritance of redundant genes. BMC Evol Biol 7:128 Available: http://www.biomedcentral.com/1471–2148/7/128. Accessed: 16 June 2014 10.1186/1471-2148-7-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Itaya M, Omori A, Kanaya S, Crouch RJ, Tanaka T, et al. (1999) Isolation of RNase H genes that are essential for growth of Bacillus subtilis 168. J Bacteriol 181: 2118–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, et al. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. Available: http://msb.embopress.org/content/2/1/2006.0008.long. Accessed 16 June 2014 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gerdes SY, Scholle MD, Campbell JW, Balázsi G, Ravasz E, et al. (2003) Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol 185: 5673–5684. 10.1128/JB.185.19.5673-5684.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nagata Y, Mashimo K, Kawata M, Yamamoto K (2002) The roles of Klenow processing and flap processing activities of DNA polymerase I in chromosome instability in Escherichia coli K12 strains. Genetics 160: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bayliss CD, Sweetman WA, Moxon ER (2005) Destabilization of tetranucleotide repeats in Haemophilus influenzae mutants lacking RnaseHI or the Klenow domain of PolI. Nucl Ac Res 33: 400–408. 10.1093/nar/gki180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen JZ, Qiu J, Shen B, Holmquist GP (2000) Mutational spectrum analysis of RNase H(35) deficient Saccharomyces cerevisiae using fluorescence-based directed termination PCR. Nucl Ac Res 28: 3649–3656. 10.1093/nar/28.18.3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu LA (1987) Influence of GATC sequences on Escherichia coli DNA mismatch repair in vitro . J Bacteriol 169: 1254–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ghodgaonkar MM, Lazzaro F, Olivera-Pimentel M, Artola-Borán M, Cejka P, et al. (2013) Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol Cell 50: 323–332. 10.1016/j.molcel.2013.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, et al. (2012) Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med 209: 1419–1426. 10.1084/jem.20120876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De Lucia P, Cairns J (1969) Isolation of an E. coli Strain with a mutation affecting DNA polymerase. Nature 224: 1164–1166. 10.1038/2241164a0 [DOI] [PubMed] [Google Scholar]
- 50. Ricchetti M, Buc H (1993) E. coli DNA polymerase I as a reverse transcriptase. EMBO J 12: 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klenow H, Henningsen I (1970) Selective elimination of the exonuclease activity of the deoxyribonucleic acid polymerase from Escherichia coli B by limited proteolysis. Proc Natl Acad Sci USA 65: 168–175. 10.1073/pnas.65.1.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gordhan BG, Andersen SJ, De Meyer AR, Mizrahi V (1996) Construction by homologous recombination and phenotypic characterization of a DNA polymerase domain poIA mutant of Mycobacterium smegmatis . Gene 178: 125–130. 10.1016/0378-1119(96)00350-2 [DOI] [PubMed] [Google Scholar]
- 53. Kelley WS (1980) Mapping of the polA Locus of Escherichia coli K12: genetic fine structure of the cistron. Genetics 95: 15–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lehman IR, Uyemura DG (1976) DNA polymerase I: essential replication enzyme. Science 193: 963–969. 10.1126/science.781842 [DOI] [PubMed] [Google Scholar]
- 55. Joyce CM, Grindley ND (1984) Method for determining whether a gene of Escherichia coli is essential: application to the polA gene. J Bacteriol 158: 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kanaya S, Crouch RJ (1984) The rnh gene is essential for growth of Escherichia coli . Proc Natl Acad Sci U S A 81: 3447–3451. 10.1073/pnas.81.11.3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Horiuchi T, Maki H, Sekiguchi M (1984) RNase H defective mutants of Escherichia coli: a possible discriminatory role of RNase H in initiation of DNA replication. Mol Gen Genet 195: 17–22. 10.1007/BF00332717 [DOI] [PubMed] [Google Scholar]
- 58. Torrey TA, Atlung T, Kogoma T (1984) dnaA suppressor (dasF) mutants of Escherichia coli are stable DNA replication (sdrAlrnh) mutants. Mol Gen Genet 196: 350–5. 10.1007/BF00328070 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper and the Supporting Information file.