

Summary
There is currently no disease-modifying treatment for Alzheimer's disease (AD) and the need is great as the number of people diagnosed with AD is predicted to steadily increase. Inflammation is associated with AD, and is predictive of more advanced disease pathology and cognitive impairment. Moreover, preventing inflammation reduces the risk of developing AD. However, clinical trials with anti-inflammatory treatment have not been successful. One reason may be that there is diversity in the immune response and reducing immune activity with anti-inflammatories is not appropriate in all conditions. Recently, we have begun to apply categorizations, used to characterize the peripheral immune response, to the immune processes of the brain. When we do this, we are able to describe an individual's inflammatory profile within this spectrum. We have observed that patients with early AD are distributed across two broad categories of immune activation. If we recognize the diversity within this cohort of individuals with early AD and use information about immune phenotypes to guide the choice of treatment, then we may expect better clinical outcomes.
The role of inflammation in Alzheimer's disease (AD) has been questioned since the early 1900s when Alois Alzheimer identified microglia, resident brain immune cells, surrounding plaques. In the 1990s, researchers established a link between the use of NSAIDs and prevention of AD [1,2], however, later clinical trials of NSAIDs were not successful [3–10]. Numerous studies report links between AD and genes regulating immunity as well as the expression of immune factors in blood, cerebrospinal fluid (CSF) and brain tissue [11–16]. Imaging studies reveal that microglia, the primary immune cells of the brain, are activated in AD and are predictive of symptom severity [17,18]. While these studies provide evidence that inflammation is related to AD onset and progression, it is not clear how we can modulate the immune system in order to reduce AD occurrence and modulate disease outcome. Current research is focused on the dynamic range of states within the rubric of ‘immune activation’, and work from our laboratory has demonstrated that patients with early AD fall into two broad categories of immune phenotype [19]. We believe that these subsets representing heterogeneity within the early AD population reveal a therapeutic opportunity. With this information, we can make better predictions of how to modulate neuroinflammation and it may be possible to distinguish which patient population may benefit from targeted anti-inflammatory treatments.
This review will begin by describing the neuroinflammatory categories used to determine individual neuroinflammatory profiles. We will then discuss neuroinflammation in AD, how to predict an individual's inflammatory profile and, finally, discuss how this information may one day be used in a personalized treatment approach.
Neuroinflammatory categories
Microglia are the primary immune cells of the CNS, analogous to macrophages of the peripheral nervous system. Microglia activation states are often compared with macrophage activation states [20,21], although they can be qualitatively different [22,23]. These states can be described as resting (in which microglia are actively monitoring the microenvironment) [24], classical activation (defense/attack; M1), alternative activation (restoration/repair; M2a), type II macrophage (M2b) and acquired deactivation (M2c) (Figure 1) [25–33]. However, activation states are not distinct conditions, nor are they points on a continuum from resting to activated [25], but are better thought of as particular points in a nonlinear spectrum [21]. In addition, the AD brain is characterized by the presence of microglia across a range of activation states [31]. Since there is considerable overlap in microglia phenotype between activation states, variance in phenotype within a state and microglia of multiple activation states coexist, simple categorization is contrived. However, broad categories remain useful to generalize an immune profile and make predictions.
Figure 1. Inflammatory profiles in Alzheimers's disease.
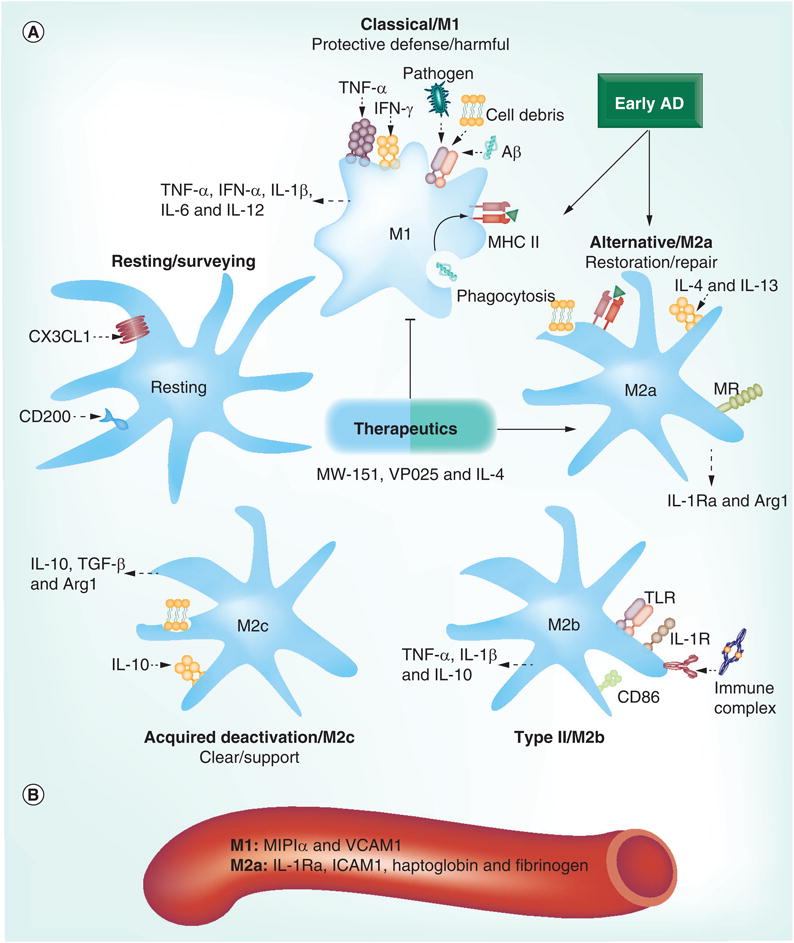
(A) Microglia activation states fall within a dynamic range that includes resting, M1, M2a, M2b and M2c. Resting microglia are kept quiescent by CD200 and CX3CL1 (fractalkine). Samples from patients with early AD indicate that their immune profile is polarized toward either an M1 or an M2a phenotype. Microglia become reactive to pathogens, neuronal debris and Aβ by activation of pattern-recognition receptors, including TLRs, which phagocytose these materials and present antigens with MHC II. Induction by and release of various pro- and anti-inflammatory cytokines differs across activation states and, taken together, can be used to identify these states. Therapeutics aimed at polarizing toward specific activation states, rather than robustly suppressing microglia activity, show promise for AD treatment. (B) Proteins from human blood (MIP1α and VCAM1 for M1; and IL-1Ra, ICAM1, haptoglobin and fibrinogen for M2a) may be useful peripheral markers to predict the central immune profile. Aβ: β-amyloid; AD: Alzheimer's disease; MR: Mannose receptor; TLR: Toll-like receptor.
Classically activated M1 microglia produce and release proinflammatory factors (e.g., TNF-α, IFN-γ, IL-1β, IL-6 and IL-12) similar to M1 macrophages, in order to attract and induce activation in other microglia and to respond to injury and pathogens, thus creating a self-amplifying cycle of activation [25–33]. Classically activated microglia also produce and release factors used for defense that can induce oxidative stress and kill pathogens, such as reactive oxygen/nitrogen species, the superoxide anion, nitric oxide and proteases. Under normal circumstances, the M1 response is transient and does not cause damage to surrounding tissue, but if the proinflammatory phase is prolonged, the inflammatory process transitions from being primarily protective to predominantly pathological [34]. For example, an M1 immune response is associated with both memory impairment and neuronal death [35–39]. A sustained neuroinflammatory environment is more likely to occur in advanced age and can be exacerbated by AD pathology [32]. Microglia from aged mice respond more robustly to INF-γ-/TNF-α-induced activation [35] and are more resistant to IL-4-induced conversion from M1 to M2a than microglia from young mice [40]. Microglia examined in vitro respond to the AD-associated protein β-amyloid (Aβ) by releasing factors characteristic of an M1 response [41], and this response is exaggerated in microglia derived from AD brains [42]. Moreover, in AD, pathological protein species, such as Aβ, as well as signals from dying and dead neurons, fuel immune activation over decades. Therefore, there is not a distinct period in AD pathogenesis in which a population of microglia is activated and later deactivated. Instead, neuroinflammation is probably sustained over time by the local activation and deactivation of individual microglia. This further suggests that the microenvironment may be influenced simultaneously by competing interests, both attack and repair, and this is consistent with our data indicating that both M1 and M2a markers are elevated in late AD [19].
The aggressive innate immune response is, under nonchronic and nondisease conditions, punctuated by a transition to an anti-inflammatory, alternative activation state, M2a, which is focused on resolution of the inflammatory response, phagocytosis of pathogens, removal of cellular debris and tissue repair [25–33]. M2a is characterized and induced by anti-inflammatory cytokines (IL-4 and IL-13), as well as chitinase-like lectins and c-type lectin receptors (e.g., the mannose receptor), and release of IL-1Ra and Arg1. The M2a activation state is generally thought to be preferred over the potentially deleterious M1 state [14,43]. Microglia may also present an M2b phenotype, also called the type II macrophage, which is characterized by markers of both M1 and M2. The M2b state is known to be triggered by immune complexes, Toll-like receptor activation and the IL-1 cytokine family. Edwards and colleagues showed that immune complexes activate Fcγ receptors and trigger the macrophage phenotype to switch to this unique state [44], which is characterized by elevations in some M1 markers (particularly IL-1β, TNF-α and IL-6, but not IL-12) and IL-10 [21,45,46]. There were also some markers that were identified to be more specific for an M2b phenotype, including CD86 [44]. We do not detect evidence of an M2b profile in AD [19], but we hypothesize that it is stimulated by the immune complexes formed by various immunotherapies that are currently in clinical trials for use in AD. Microglia can progress to an M2 state called acquired deactivation, M2c, which is characterized by immunosuppression and clearance of apoptotic cells. M2c is characterized and induced by anti-inflammatory cytokines (IL-10 and TGF-β) as well as Arg1 and absence of nitric oxide production. Finally, the immune response may transition back to a ‘resting’ state, although it probably remains primed for subsequent events.
Microglia activation & inflammatory biomarkers in AD
PET scans and immunohistological analysis in AD patients and age-matched healthy controls demonstrate that activated microglia (identified by expression of the peripheral benzodiazepine receptor or MHC II) increase in number in the brains of patients with AD; are found near primary disease pathology (Aβ plaques, neurofibrillary tangles, and dead or dying neurons) in brain regions that later degenerate; precede and are predictive of pathology and clinical symptoms; and correlate better with memory impairment than primary disease pathology, such as Aβ plaques [18,47–50]. Genome-wide association studies have identified immune factors (e.g., genes associated with IL-1, IL-18, complement receptors, human leucocyte antigen, CD33 and clusterin) with late-onset AD [11,13–15,51–53]. Measurements of cytokines and inflammatory biomarkers in blood and CSF from AD patients demonstrate elevations of proinflammatory factors, such as the proinflammatory cytokines TNF-α, I L -1β, IL-6 and IL-12 as well as complement [54,55]. Similarly, microglia collected post-mortem show a bias toward the production of proinflammatory factors when exposed to an immune challenge [22,23].
Neuroinflammation is sufficient to drive a type of dementia, known as dementia pugilistica, but it is unlikely to be the primary etiological factor in the development of AD. Neuroinflammation is present early in AD pathogenesis [12,17,18]. It is stimulated by other early features, such as Aβ fibrils and by later AD pathologies, such as Aβ plaques, neurofibrillary tangles and neuronal debris, as well as disease risk factors, such as cholesterol and adiposity [56,57]. Neuroinflammation may also be stimulated by infection, injury or comorbidity with other pathologies, such as diabetes or cardiovascular disease [58,59]. Moreover, neuroinflammation contributes to the development of AD, as reducing neuroinflammation decreases AD risk, and neuroinflammation can promote neurodegeneration through activation of the TNF-α receptor death domain, oxidative stress and excitotoxicity. Therefore, neuroinflammation is a possible therapeutic target with the potential to modify disease outcome.
How do we determine an individual's immune profile? The definition of microglia activation assumed by PET scans and immunohistological staining uses microglia surface receptor expression, most often the peripheral benzodiazepine receptor (translocator protein of 18 kDa) and MHC II; however, this, unfortunately, does not reflect the dynamic range of activation states [31,60]. It can be more clinically informative to approximate the immune profile by identifying immune factors in the CSF, such as complement [55]. Neither PET nor CSF, however, are practical as widely used forms of screening as PET scanning is expensive and CSF collection is intrusive and risky. Furthermore, immune factors from the periphery also enter and play a role in brain inflammation [23]. Therefore, it is reasonable to use more easily obtained blood serum and plasma to identify biomarkers of an individual's neuroinflammatory phenotype.
The field is currently investigating blood proteins that will serve this function. A set of 18 markers (out of 46 markers investigated) were identified by Ray et al. that include many inflammatory markers (i.e., GCSF, MCSF, ICAM, TNF-α, TNF receptor and RANTES), which, when evaluated together, are both diagnostic and predictive of AD [61]. However, two follow-up studies found that most markers did not vary between age-matched controls and AD patients, and that they did not distinguish between mild cognitive impairment and AD [62,63]. In a similar study, 11 proteins, of which many were inflammatory markers, were found to correlate with AD severity [64]. Our recent work identified six serum proteins that are reflective of the brain inflammatory state (M1: MIP1α and VCAM1; M2a: IL-1Ra, ICAM1, haptoglobin and fibrinogen) (Figure 1B) [19].
Clinical intervention may be complicated by immune phenotype
Interventional trials with NSAIDs in AD have not been promising overall, although some have indicated modest improvement. An early clinical trial of the NSAID indomethacin in mild-to-moderate AD demonstrated significantly less decline over a 6-month period; however, 50% of the nonresponders dropped out of the study [65]. Later trials of indomethacin showed small protective effects over a 12–13-month period that did not reach clinical significance owing to a small sample size [66]. A recent clinical trial of the NSAID ibuprofen with the gastroprotectant esomeprazole given to patients with mild-to-moderate AD for 1 year showed decreased cognitive decline only in those individuals who carried the APOE4 allele [67], which is in agreement with some epidemiological reports showing an interaction between NSAID use and the APOE4 genotype [68,69].
Most large-scale interventional clinical trials of anti-inflammatories, however, have shown no improvement and some serious side effects, including trials of aspirin [3], the antimalarial and anti-inflammatory hydroxychloroquine [4], the corticosteroid prednisone [5], and the NSAIDs rofecoxib, naproxen and diclofenac [6–9]. In these trials drugs were tested for 4 years or less and in patients diagnosed with mild-to-moderate AD. ADAPT compared the efficacy of the NSAID naproxen and the selective COX-2 inhibitor celecoxib to prevent onset of AD in a high-risk group and showed that treated groups tended to have worse mental scores. The trial was terminated early because of concerns about cardiovascular and cerebrovascular risk due to celecoxib, and significantly increased risk (∼60%) of these side effects in patients treated with naproxen [10,70,71].
Why was there such diversity in the outcomes of clinical trials of anti-inflammatories? It is possible that the large ADAPT study and other interventional trials showed poor outcome owing to drug choice and because the cohort, advanced in age and high risk, probably had a well-established inflammatory response years prior to the intervention with NSAIDs in these clinical trials [10,70,71]. Results from epidemiological and interventional studies are consistent with the idea that early changes occurring during a preclinical phase lead to the development of AD, and that interruption of certain processes during this period may prevent disease development, while targeting these same systems later in the disease state may be ineffective [2,67,70,72].
Although the ADAPT trials were terminated prematurely, further investigation into this cohort found that NSAIDs are protective if initiated before symptom onset, but are harmful after the development of cognitive impairment [73], and that the efficacy of NSAIDs may depend upon the rate of decline [74]. Recent work in our laboratory has found that both M1 and M2a markers are elevated in tissue from advanced AD brains. Interestingly, however, this work also identified a dichotomous distribution between M1- and M2a-baised inflammatory profiles in early AD brain tissue samples that was related to AD pathology and appeared only in diseased brain regions [19]. Heterogeneity in the immune profile may be derived from individual propensities toward certain inflammatory states, features and magnitude of AD pathology, a lasting result of previous immune challenges or injury and current general medical health, as well as common comorbidities, such as cardiovascular disorders, epilepsy, atherosclerosis and depression [58,59]. While comorbidities are often reasons for exclusion from medical trials, they must be addressed in clinical practice. We believe that heterogeneity in the immune profile may partially explain the failure of interventional anti-inflammatory trials, and addressing this heterogeneity may enable us to predict for whom a directed anti-inflammatory treatment may be successful.
Clinical intervention guided by the immune profile
Once we know a patient's immune profile, what can we do with this information? Data gathered from blood serum, blood plasma, CSF and PET imaging can be used to direct treatment, particularly in early-stage AD when the population falls into two broad categories of M1 or M2a. For example, the ADAPT trials were terminated prematurely owing to concern over vascular events and risks [10], and an M2a immune profile is associated with the same category of risk factors [19]. Therefore, performing a simple evaluation of plasma proteins that are indicative of an M2a immune profile (IL-1Ra, ICAM1, haptoglobin and fibrinogen) may allow us to make informed selections of participants for trials and immune therapy that minimizes these risks. An alternate view is that the M2a phenotype reflects the presence of cerebrovascular comorbidities with AD. Individuals with an M2a-biased phenotype also have a higher density of Aβ plaques [19]. This relationship suggests that the presence of serum proteins indicative of an M2a profile may predict a better response to therapies targeted at Aβ. There are at least 19 current clinical trials (ongoing or initiating [101]) of drugs targeting Aβ directly or indirectly by targeting enzymes that cleave APP. Similar to NSAIDs, clinical trials of drugs targeting Aβ have not been overwhelmingly successful [75], but may be improved by selecting participants who are more likely to benefit. In addition, individuals with an M1-biased profile may benefit from a therapy that selectively targets M1 immune functions, while allowing or promoting the processes of restoration and repair associated with an M2a phenotype. A blood screen for elevations in the proteins MIP1α and VCAM1, or other markers indicative of an M1 profile, will allow us to make better predictions about treatment for this population.
An individual's inflammatory profile will be most biologically informative and actionable early in the disease process. Fortunately, identifying preclinical stages of AD is becoming more realistic as we develop improved screening techniques [76]. For example, brain tau pathology can be approximated from measurements in blood and CSF. Likewise, brain plaque burden can be estimated from Aβ in the blood and CSF as well as PET scanning using Pittsburgh compound B. Other scanning methods can highlight early regional changes in metabolism or hypo-function, as well as brain atrophy. Early genetic testing can identify those who are at greater risk of developing AD, for example, carriers of the APOE4 allele. These results may be more useful when used in combination with an individual's inflammatory profile. For example, although our recent work did not identify a relationship between ApoE status and the early inflammatory profile [19], two studies of large cohorts, the Cache County Study and the Cardiovascular and Health Study found that carriers of APOE4 benefitted more from NSAID treatment [68]. Similarly, the Cardiovascular Health Study showed that NSAIDs significantly reduced the risk of AD in carriers of APOE4 [77].
If we want to manipulate the immune system, how do we do it? NSAIDs failed clinical trials, but may be more effective if an individual's inflammatory profile is known. Leoutsakos et al. found differential potential benefits from NSAIDs in the preclinical AD population of the ADAPT trials when patients were separated as slow decliners (naproxen), fast decliners (celecoxib) and those demonstrating no decline [74]. We expect differential effects, such as those between M1- and M2a- biased immune profiles, and believe that drug treatments that do not suppress immune function overall, but instead modulate these profiles, will have the most benefit (Figure 1A). One such drug, MW-151, has been recently developed in order to specifically attenuate M1-type immune activity. Early treatment with MW-151 in a transgenic mouse model of AD reduced impairment in long-term potentiation, the synaptic correlate of memory [14]. Another similar drug, VP025, reduces age- and lipopolysaccharide-induced elevations of proinflammatory cytokines, increases expression of anti-inflammatory CD200 and restores long-term potentiation in rats [78]. Another possible course of treatment may be to promote an M2a immune profile. This could potentially be accomplished by delivery of the anti-inflammatory cytokine IL-4. In mouse models of AD, IL-4 treatment induces an M2 immune profile and enhances microglial clearance of Aβ [32,79,80]. In fact, increased production of IL-4 may be partially responsible for the therapeutic effect of currently used acetocholinesterase inhibitors, although these drugs are not disease modifying [81]. We predict that directed targeting of specific immune functions will benefit individuals with preclinical or early AD.
Conclusion
The field of neuroinflammation in AD has burgeoned since the 1990s. In general, the perspective has been that neuroinflammation should be suppressed, but the interventional trials with NSAIDs in AD patients did not succeed. The field is currently applying the spectrum of inflammatory phenotypes characterized in the body periphery to the diversity of the inflammatory responses seen in the brain. It is becoming apparent that making distinctions between the different types of inflammatory states may be predictive of disease progression and may offer an opportunity to individualize the approach to treatment. Biomarkers from blood plasma and serum offer the most reasonable method for individual access to this information. There are a handful of biomarkers that correlate with disease progression and severity, and some biomarkers from blood predict the neuroinflammatory profile. The treatment approach will probably be different for patients with early AD who are M1 versus M2a biased, but the treatment approach for these situations remains to be determined.
Future perspective
We are currently beginning to understand the various immune phenotypes present in the brain of cognitively normal individuals and those with AD. As we work to determine a battery of biomarkers that reliably reflects the brain inflammatory phenotype, researchers are developing drugs that selectively target and modulate specific immune phenotypes instead of pan suppressors. It is possible that in the next 5 years both of these endeavors will have some success, allowing us to determine an individual's immune profile and adjust our therapeutic approach accordingly.
Practice Points.
Inflammatory phenotypes occur within a spectrum between resting, proinflammatory, deactivated and repair.
The immune response can be deleterious or supportive, depending upon the inflammatory state and the duration of that state.
Inflammatory profiles in early Alzheimer's disease (AD) differ from cognitively healthy individuals and those with advanced AD.
Early AD is characterized by a dichotomous distribution between M1- and M2-biased inflammatory profiles.
Inflammatory profiles may be predictive of disease progression and responsiveness to treatment.
Identifying an individual's inflammatory profile, in combination with other disease risk factors and cognitive state, will help to guide the approach to treatment.
In order to use this information to personalize treatment, it is first imperative that we better understand whether it is best to amplify or suppress immune activation under various circumstances in AD.
We also need to identify or exercise additional immune-modifying treatments that can push the immune system toward a specific profile.
Footnotes
Financial & competing interests disclosure: The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
▪ of interest
▪ ▪ of considerable interest
- 1.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47(2):425–432. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 2.Stewart WF, Kawas C, Corrada M, Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48(3):626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 3.Bentham P, Gray R, Sellwood E, Hills R, Crome P, Raftery J. Aspirin in Alzheimer's disease (AD2000): a randomised open-label trial. Lancet Neurol. 2008;7(1):41–49. doi: 10.1016/S1474-4422(07)70293-4. [DOI] [PubMed] [Google Scholar]
- 4.Van Gool WA, Weinstein HC, Scheltens P, Walstra GJ. Effect of hydroxychloroquine on progression of dementia in early Alzheimer's disease: an 18-month randomised, double-blind, placebo-controlled study. Lancet. 2001;358(9280):455–460. doi: 10.1016/s0140-6736(01)05623-9. [DOI] [PubMed] [Google Scholar]
- 5.Aisen PS, Davis KL, Berg JD, et al. A randomized controlled trial of prednisone in Alzheimer's disease. Alzheimer's Disease Cooperative Study. Neurology. 2000;54(3):588–593. doi: 10.1212/wnl.54.3.588. [DOI] [PubMed] [Google Scholar]
- 6.Scharf S, Mander A, Ugoni A, Vajda F, Christophidis N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer's disease. Neurology. 1999;53(1):197–201. doi: 10.1212/wnl.53.1.197. [DOI] [PubMed] [Google Scholar]
- 7.Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289(21):2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 8.Reines SA, Block GA, Morris JC, et al. Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62(1):66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- 9.Thal LJ, Ferris SH, Kirby L, et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005;30(6):1204–1215. doi: 10.1038/sj.npp.1300690. [DOI] [PubMed] [Google Scholar]
- 10▪.ADAPT Research Group. Cardiovascular and cerebrovascular events in the randomized, controlled Alzheimer's Disease Anti-Inflammatory Prevention Trial (ADAPT) PLoS Clin Trials. 2006;1(7):e33. doi: 10.1371/journal.pctr.0010033. Large clinical trial of anti-inflammatory treatment in Alzheimer's disease (AD) that was ended owing to adverse events, and has produced controversy over whether anti-inflammatory treatments may be useful for treatment in AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambert JC, Grenier-Boley B, Chouraki V, et al. Implication of the immune system in Alzheimer's disease: evidence from genome-wide pathway analysis. J Alzheimers Dis. 2010;20(4):1107–1118. doi: 10.3233/JAD-2010-100018. [DOI] [PubMed] [Google Scholar]
- 12.Eikelenboom P, van Exel E, Hoozemans JJ, Veerhuis R, Rozemuller AJ, van Gool WA. Neuroinflammation – an early event in both the history and pathogenesis of Alzheimer's disease. Neurodegener Dis. 2010;7(1–3):38–41. doi: 10.1159/000283480. [DOI] [PubMed] [Google Scholar]
- 13.Jones L, Holmans PA, Hamshere ML, et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS One. 2010;5(11):e13950. doi: 10.1371/journal.pone.0013950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14▪.Bachstetter AD, Norris CM, Sompol P, et al. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer's disease-related pathology. J Neurosci. 2012;32(30):10201–10210. doi: 10.1523/JNEUROSCI.1496-12.2012. Identifies one drug that preferentially attenuates the M1-biased immune response. Drugs of this type may be useful to treat AD based upon an individual's immune profile. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eikelenboom P, van Exel E, Veerhuis R, Rozemuller AJ, van Gool WA, Hoozemans JJ. Innate immunity and the etiology of late-onset Alzheimer's disease. Neurodegener Dis. 2012;10(1–4):271–273. doi: 10.1159/000334287. [DOI] [PubMed] [Google Scholar]
- 16.Pellicano M, Bulati M, Buffa S, et al. Systemic immune responses in Alzheimer's disease: in vitro mononuclear cell activation and cytokine production. J Alzheimers Dis. 2010;21(1):181–192. doi: 10.3233/JAD-2010-091714. [DOI] [PubMed] [Google Scholar]
- 17.Edison P, Archer H, Gerhard A, et al. Microglia, amyloid, and cognition in Alzheimer's disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32(3):412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Cagnin A, Brooks DJ, Kennedy AM, et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358(9280):461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 19▪ ▪.Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer's disease. Neurobiol Aging. 2013;34(4):1051–1059. doi: 10.1016/j.neurobiolaging.2012.09.012. Demonstrates that the early AD immune profile generally falls within one of two broad categories, showing diversity in the early disease state that can be capitalized on and may guide the therapeutic approach. These findings are central to the perspectives in this article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 21.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durafourt BA, Moore CS, Zammit DA, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60(5):717–727. doi: 10.1002/glia.22298. [DOI] [PubMed] [Google Scholar]
- 23.Melief J, Koning N, Schuurman KG, et al. Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia. 2012;60(10):1506–1517. doi: 10.1002/glia.22370. [DOI] [PubMed] [Google Scholar]
- 24▪.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma. in vivo Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. Demonstrates that neuroinflammation alone is sufficient to impair memory function and lead to neuronal loss. [DOI] [PubMed] [Google Scholar]
- 25.Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuro immune Pharmacol. 2009;4(4):399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64(1):110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lynch MA. The multifaceted profile of activated microglia. Mol Neurobiol. 2009;40(2):139–156. doi: 10.1007/s12035-009-8077-9. [DOI] [PubMed] [Google Scholar]
- 29.Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 30.Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer's disease. Neurobiol Dis. 2010;37(3):503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colton C, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9(2):174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 32.Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp (Warsz) 2012;60(4):251–266. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39(1):3–18. doi: 10.1111/nan.12011. [DOI] [PubMed] [Google Scholar]
- 34.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease – a double-edged sword. Neuron. 2002;35(3):419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 35.Deng XH, Bertini G, Xu YZ, Yan Z, Bentivoglio M. Cytokine-induced activation of glial cells in the mouse brain is enhanced at an advanced age. Neuroscience. 2006;141(2):645–661. doi: 10.1016/j.neuroscience.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 36.Hauss-Wegrzyniak B, Lynch MA, Vraniak PD, Wenk GL. Chronic brain inflammation results in cell loss in the entorhinal cortex and impaired LTP in perforant path-granule cell synapses. Exp Neurol. 2002;176(2):336–341. doi: 10.1006/exnr.2002.7966. [DOI] [PubMed] [Google Scholar]
- 37.Lyons A, Lynch AM, Downer EJ, et al. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J Neurochem. 2009;110(5):1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x. [DOI] [PubMed] [Google Scholar]
- 38.Griffin R, Nally R, Nolan Y, McCartney Y, Linden J, Lynch MA. The age-related attenuation in long-term potentiation is associated with microglial activation. J Neurochem. 2006;99(4):1263–1272. doi: 10.1111/j.1471-4159.2006.04165.x. [DOI] [PubMed] [Google Scholar]
- 39.Barrientos RM, Frank MG, Watkins LR, Maier SF. Memory impairments in healthy aging: role of aging-induced microglial sensitization. Aging Dis. 2010;1(3):212–231. [PMC free article] [PubMed] [Google Scholar]
- 40.Fenn AM, Henry CJ, Huang Y, Dugan A, Godbout JP. Lipopolysaccharideinduced interleukin (IL)-4 receptor-α expression and corresponding sensitivity to the M2 promoting effects of IL-4 are impaired in microglia of aged mice. Brain Behav Immun. 2012;26(5):766–777. doi: 10.1016/j.bbi.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210(1–2):3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Lue L, Rydel R, Brigham E, et al. Inflammatory repertoire of Alzheimer's disease and nondemented elderly microglia in vitro. Glia. 2001;35(1):72–79. doi: 10.1002/glia.1072. [DOI] [PubMed] [Google Scholar]
- 43.McCarty MF. Down-regulation of microglial activation may represent a practical strategy for combating neurodegenerative disorders. Med Hypotheses. 2006;67(2):251–269. doi: 10.1016/j.mehy.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 44▪.Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80(6):1298–1307. doi: 10.1189/jlb.0406249. Identifies the characteristics of macrophage polarization. These categories are applied to microglia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73(2):209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 46.Wilcock DM. A changing perspective on the role of neuroinflammation in Alzheimer's disease. Int J Alzheimers Dis. 2012;2012:495243. doi: 10.1155/2012/495243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mcgeer PL, Mcgeer EG. Glial cell reactions in neurodegenerative diseases: pathophysiology and therapeutic interventions. Alzheimer Dis Assoc Disord. 1998;12(Suppl 2):S1–S6. [PubMed] [Google Scholar]
- 48.Edison P, Archer HA, Gerhard A, et al. Microglia, amyloid, and cognition in Alzheimer's disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32(3):412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 49.Sheffield LG, Marquis JG, Berman NE. Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer's disease. Neurosci Lett. 2000;285(3):165–168. doi: 10.1016/s0304-3940(00)01037-5. [DOI] [PubMed] [Google Scholar]
- 50.Xiang Z, Haroutunian V, Ho L, Purohit D, Pasinetti GM. Microglia activation in the brain as inflammatory biomarker of Alzheimer's disease neuropathology and clinical dementia. Dis Markers. 2006;22(1–2):95–102. doi: 10.1155/2006/276239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Briones N, Dinu V. Data mining of high density genomic variant data for prediction of Alzheimer's disease risk. BMC Med Genet. 2012;13:7. doi: 10.1186/1471-2350-13-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgan K. The three new pathways leading to Alzheimer's disease. Neuropathol Appl Neurobiol. 2011;37(4):353–357. doi: 10.1111/j.1365-2990.2011.01181.x. [DOI] [PubMed] [Google Scholar]
- 53.Sleegers K, Lambert JC, Bertram L, Cruts M, Amouyel P, van Broeckhoven C. The pursuit of susceptibility genes for Alzheimer's disease: progress and prospects. Trends Genet. 2010;26(2):84–93. doi: 10.1016/j.tig.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 54.Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer's disease. Biol Psychiatry. 2010;68(10):930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 55.Fagan AM, Perrin RJ. Upcoming candidate cerebrospinal fluid biomarkers of Alzheimer's disease. Biomark Med. 2012;6(4):455–476. doi: 10.2217/bmm.12.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ringheim GE, Szczepanik AM. Brain inflammation, cholesterol, and glutamate as interconnected participants in the pathology of Alzheimer's disease. Curr Pharm Des. 2006;12(6):719–738. doi: 10.2174/138161206775474215. [DOI] [PubMed] [Google Scholar]
- 57.Misiak B, Leszek J, Kiejna A. Metabolic syndrome, mild cognitive impairment and Alzheimer's disease – the emerging role of systemic low-grade inflammation and adiposity. Brain Res Bull. 2012;89(3–4):144–149. doi: 10.1016/j.brainresbull.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 58.Bäckman L, Jones S, Small BJ, Agüero-Torres H, Fratiglioni L. Rate of cognitive decline in preclinical Alzheimer's disease: the role of comorbidity. J Gerontol B Psychol Sci Soc Sci. 2003;58(4):P228–P236. doi: 10.1093/geronb/58.4.p228. [DOI] [PubMed] [Google Scholar]
- 59.Leoutsakos JMS, Han D, Mielke MM, et al. Effects of general medical health on Alzheimer's progression: the Cache County Dementia Progression Study. Int Psychogeriatr. 2012;24(10):1561–1570. doi: 10.1017/S104161021200049X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (translocator protein 18kDa) in microglia: from pathology to imaging. Prog Neurobiol. 2006;80(6):308–322. doi: 10.1016/j.pneurobio.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med. 2007;13(11):1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 62.Björkqvist M, Ohlsson M, Minthon L, Hansson O. Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer's disease. PLoS One. 2012;7(1):e29868. doi: 10.1371/journal.pone.0029868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marksteiner J, Kemmler G, Weiss EM, et al. Five out of 16 plasma signaling proteins are enhanced in plasma of patients with mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2011;32(3):539–540. doi: 10.1016/j.neurobiolaging.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Bryant SE, Xiao G, Barber R, et al. A blood-based screening tool for Alzheimer's disease that spans serum and plasma: findings from TARC and ADNI. PLoS One. 2011;6(12):e28092. doi: 10.1371/journal.pone.0028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogers J, Kirby LC, Hempelman SR, et al. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43(8):1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 66.De Jong D, Jansen R, Hoefnagels W, et al. No effect of one-year treatment with indomethacin on Alzheimer's disease progression: a randomized controlled trial. PLoS One. 2008;3(1):e1475. doi: 10.1371/journal.pone.0001475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pasqualetti P, Bonomini C, Dal Forno G, et al. A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer's disease. Aging Clin Exp Res. 2009;21(2):102–110. doi: 10.1007/BF03325217. [DOI] [PubMed] [Google Scholar]
- 68.Fotuhi M, Zandi PP, Hayden KM, et al. Better cognitive performance in elderly taking antioxidant vitamins E and C supplements in combination with nonsteroidal anti-inflammatory drugs: the Cache County Study. Alzheimers Dement. 2008;4(3):223–227. doi: 10.1016/j.jalz.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szekely CA, Breitner JC, Fitzpatrick AL, et al. NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology. 2008;70(1):17–24. doi: 10.1212/01.wnl.0000284596.95156.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin BK, Szekely C, Brandt J, et al. Cognitive function over time in the Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65(7):896–905. doi: 10.1001/archneur.2008.65.7.nct70006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meinert CL, Mccaffrey LD, Breitner JC. Alzheimer's Disease Anti-inflammatory Prevention Trial: design, methods, and baseline results. Alzheimers Dement. 2009;5(2):93–104. doi: 10.1016/j.jalz.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zandi PP, Breitner JC, Anthony JC. Is pharmacological prevention of Alzheimer's a realistic goal? Expert Opin Pharmacother. 2002;3(4):365–380. doi: 10.1517/14656566.3.4.365. [DOI] [PubMed] [Google Scholar]
- 73.Breitner JC, Baker LD, Montine TJ, et al. Extended results of the Alzheimer's disease anti-inflammatory prevention trial. Alzheimers Dement. 2011;7(4):402–411. doi: 10.1016/j.jalz.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leoutsakos JM, Muthen BO, Breitner JC, Lyketsos CG. Effects of non-steroidal anti-inflammatory drug treatments on cognitive decline vary by phase of pre-clinical Alzheimer disease: findings from the randomized controlled Alzheimer's Disease Anti-inflammatory Prevention Trial. Int J Geriatr Psychiatry. 2012;27(4):364–374. doi: 10.1002/gps.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010;9(7):702–716. doi: 10.1016/S1474-4422(10)70119-8. [DOI] [PubMed] [Google Scholar]
- 76.Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature. 2009;461(7266):916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sweet RA, Seltman H, Emanuel JE, et al. Effect of Alzheimer's disease risk genes on trajectories of cognitive function in the Cardiovascular Health Study. Am J Psychiatry. 2012;169(9):954–962. doi: 10.1176/appi.ajp.2012.11121815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martin DSD, Walsh M, Miller AM, et al. A novel phospholipid-based drug formulation, VP025, modulates age- and LPS-induced microglial activity in the rat. Neuroimmunomodulation. 2009;16(6):400–410. doi: 10.1159/000228915. [DOI] [PubMed] [Google Scholar]
- 79.Shimizu E, Kawahara K, Kajizono M, Sawada M, Nakayama H. IL-4-induced selective clearance of oligomeric beta-amyloid peptide (1-42) by rat primary type 2 microglia. J Immunol. 2008;181(9):6503–6513. doi: 10.4049/jimmunol.181.9.6503. [DOI] [PubMed] [Google Scholar]
- 80.Lyons A, Griffin RJ, Costelloe CE, Clarke RM, Lynch MA. IL-4 attenuates the neuroinflammation induced by amyloid-beta in vivo and in vitro. J Neurochem. 2007;101(3):771–781. doi: 10.1111/j.1471-4159.2006.04370.x. [DOI] [PubMed] [Google Scholar]
- 81.Lugaresi A, Di Iorio A, Iarlori C, et al. IL-4 in vitro production is upregulated in Alzheimer's disease patients treated with acetylcholinesterase inhibitors. Exp Gerontol. 2004;39(4):653–657. doi: 10.1016/j.exger.2003.08.012. [DOI] [PubMed] [Google Scholar]
Website
- 101.ClinicalTrials.gov. www.clinicaltrials.gov