

Abstract
An enantioselective total synthesis of the polycyclic diterpene chatancin (1), a potent PAF antagonist, is reported. Proceeding in seven steps from dihydrofarnesal, this synthetic route was designed to circumvent macrocyclization-based strategies to complex, cyclized cembranoids. The described synthesis requires only six chromatographic purifications, is high yielding, and avoids protecting group chemistry. An X-ray crystal structure of this fragile marine natural product was obtained.
Keywords: terpene, biomimetic synthesis, natural products, cycloaddition reaction, asymmetric synthesis
Marine corals produce a rich array of structurally diverse natural products with intriguing biological profiles.[1] The Sarcophyton species in particular has provided chemists with an abundance of diterpenes, including many interesting architectures of cembrane biosynthetic origin.[2] In a search for naturally occurring antagonists of platelet activating factor (PAF), Sato and co-worker isolated the structurally unique diterpene Chatancin (1) from a soft coral (Sarcophyton sp.) off the coast of Okinawa, Japan (Figure 1a).[3] PAF is a small molecule phospholipid mediator of numerous biological processes including platelet aggregation, smooth muscle contraction, and hypotension, among others.[4] Altered levels of PAF have been implicated in numerous diseases, including those of the respiratory and cardiovascular systems.[5] Chatancin inhibits both PAF-induced platelet aggregation (IC50 = 2.2 μM) and PAF receptor binding (IC50 = 0.32 μM) but has no effect on platelet aggregation induced by arachidonic acid, adenosine diphosphate, or collagen.[3] Structurally, chatancin possesses a complex carbon skeleton featuring two cis-decalin motifs folded into a unique polycyclic arrangement by virtue of a hemiketal bridge. This locking element is crucial for eliciting its biological effects.[3] Chatancin bears striking structural resemblance to the diterpene sarcophytin (2), isolated from a geographically far-removed Sarcophyton species.[6]
Figure 1.
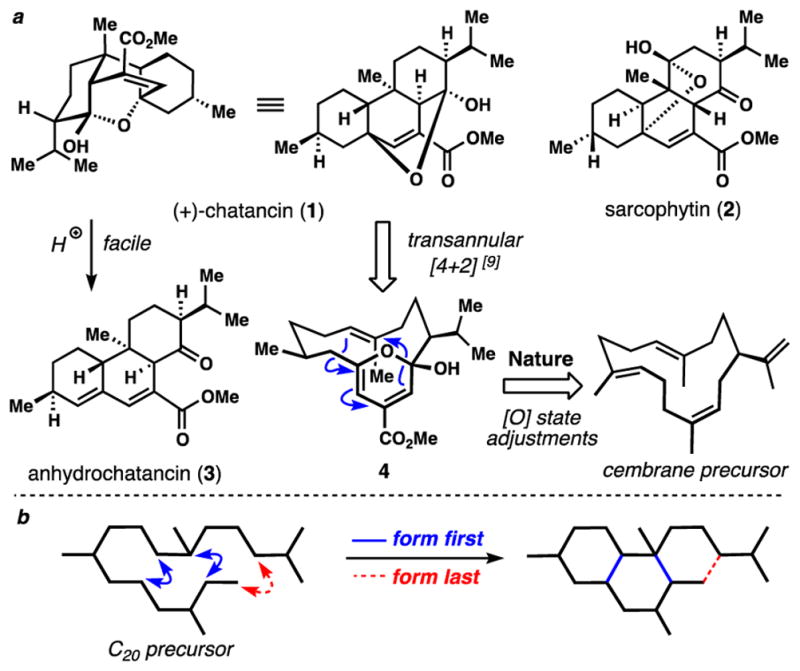
a) Chatancin (1): related diterpene 2, chemical fragility, and postulated biosynthetic origins. b) Abiotic synthetic strategy.
The compact structure of 1, containing 7 stereocenters (6 of which are contiguous), has proven to be a formidable synthetic challenge that is further exacerbated by its extreme acid sensitivity, rapidly dehydrating to anhydrochatancin (3) under even mildly acidic conditions. Gossinger and co-workers reported the first synthetic solution to (±)-1 in 33 chemical steps from thymoquinone (0.7% overall yield),[7] and in 2003, after significant chemical experimentation,[8] the group of Deslongchamps reported a fundamentally disparate synthetic strategy to (+)-1 (23 steps from cis-2-Butene-1,4-diol).[9] Guided by the biosynthetic hypothesis shown (Figure 1a), Chatancin was postulated to be of cembrane biosynthetic origins and its complex polycyclic skeleton the result of a transannular Diels-Alder cycloaddition (TADA) of pyranophane precursor 4. This hypothesis evolved from earlier attempts at eliciting a TADA reaction of a furanocembranoid-type precursor followed by ring shift; the latter of which could not be successfully executed in a flask owing to the facile conversion of 1 to 3.[8e] While the conditions employed for the conversion of 4 to 1, are not compatible with a cellular setting (temperatures >100 °C), this work did provide evidence for the possible intermediacy of pericyclic processes in the biogenesis of 1.[10] While certainly providing invaluable biosynthetic insight, many steps were required to craft the stereodefined, 14-membered ring precursor. Historically, numerous cembrane syntheses devote significant effort to linear precursor assembly and macrocycle closure.[11,12] This is at odds with the synthetic strategy employed by nature to construct such terpenes wherein large rings are made in the inaugural step and subsequently functionalized. Herein we described a non-macrocyclization based synthetic route to this complex, transannularly-cyclized cembranoid, instead relying on the alternative bond-forming orchestration shown (Figure 1b). By using this strategy, a highly concise and protecting group-free route to this fascinating bioactive substance has been realized.
Our synthetic studies commenced with the Lewis acid- mediated addition of silyl ketene acetal 7 to (S)-dihydrofarnesal 6, affording ketone 9 after in-situ oxidation of the intermediate alcohol (8) (Scheme 1). Both 6 and 7 are available in 1-step from commercial materials and this transformation could be performed reliably on a multi-gram scale.[13] Slow addition of a solution of 9 to refluxing toluene very cleanly elicited thermal acetone extrusion with concomitant cyclization to hydroxypyrone 10, conditions originally reported by Sato.[14,15] The intermediate hydroxypyrone could be triflated (Tf2O, Et3N) yielding vinyl triflate 11 after column chromatography (67% from 9). Attachment of the requisite methyl ester initially proved challenging employing standard Pd-catalyzed methoxycarbonylation conditions (Pd(OAc)2/PPh3, CO, MeOH), affording only trace amounts of product with substantial quantities of hydroxypyrone 10. Ultimately it was discovered that the catalyst system reported by Fürstner and co-workers (Pd(OAc)2/DPEPhos), utilized for similar electron-deficient substrates, was exceptionally active in this context, affording near quantitative yields of product (90–95%).[16] Notably this transformation was robust and could be performed on a gram scale with no drop in yield.
Scheme 1.

Enantioselective total synthesis of (+)-chatancin (1). Reagents and Conditions: a) 6 (1.0 equiv), 7 (1.1 equiv), BF3•OEt2 (1.5 equiv), CH2Cl2, −78 °C, 1 h, then add DMP (3.0 equiv), NaHCO3 (6.0 equiv), −78 °C → rt, 9 h, 62%; b) 9 (1.0 equiv) added slowly to PhMe, 120 °C → rt, 1.5 h, then Tf2O (1.0 equiv), Et3N (2.5 equiv), DCM, −78 °C → rt, 67% from 9; c) 11 (1.0 equiv), Pd(OAc)2 (10 mol %), DPEPhos (10 mol %), i-Pr2NEt (2.0 equiv), CO (1 atm), 4.2:1 MeCN/MeOH (v/v), rt, 8 h, 90%; d) PhMe (0.05 M), 100 °C, 4 d, 90% (12:13 = 1:1); e) 12 (1.0 equiv), SO2Cl2 (1.1 equiv), Na2CO3 (4.0 equiv), DCM, 0 °C, 30 min; Zn (40.0 equiv), THF, 65 °C, 24 h, 80% from 12; g) 17 (1.0 equiv), 5% Pd/C (10 mol %), H2 (1 atm), MeOH, 8 h, 93%; h) 13 (1.0 equiv), TMSOTf (2.0 equiv), DCM, −78 °C, 1 h, 34%; i) 14 (1.0 equiv), 5% Pd/C (10 mol %), H2 (1 atm), MeOH, 8 h, 80%. DMP = Dess Martin Periodinane, Tf = trifluoromethanesulfate, DPEPhos = Bis-[2-(diphenylphosphino)phenyl]ether.
With 4-step access to all of the requisite carbons of 1, we were in a position to test the first key C–C bond-forming reaction, a pyrone/alkene cycloaddition;[17,18] elegant synthetic work directed toward the transtaganolide and basiliolide diterpenes served as inspiration.[19] Ultimately it was discovered that heating a toluene solution of the methoxycarbonylated pyrone for 4 days at 100 °C smoothly elicited a [4+2] cycloaddition in high yield (90%) and without the need for high dilution. This process forges four stereocenters in a single operation (Scheme 1). Equimolar amounts of diastereomers 12 and 13 were formed in this process; the relative configuration of the former was confirmed by X-ray crystallography (Scheme 1). Four diastereomers are possible in this cycloaddition reaction, but only two are observed. Bicycles 12 and 13 appear to arise from favourable chair-like transition states as opposed to the alternative, boat-like structures shown (Figure 2). Owing to a lack of allylic strain, which has benefitted related intramolecular pyrone/alkene cycloadditions,[19] the pyrone group in this system does not have a biasing element favouring a given pyrone rotamer.[20] The gram- scale synthesis of 12 only became possible after significantly exploring a number of individual cycloaddition reactions, substrates, and conditions (Table 1). Notably, hydroxypyrone 10 could not be coaxed into a productive cycloaddition under either thermal or high-pressure conditions (entry 1) and pyrone triflate 11 afforded only decarboxylated diene 20 when heated (entry 2).[17] Decarboxylation was also observed for the successful ester substrate, but could be minimized by careful choice of solvent and temperature. In toluene at 80 °C, the initial [4+2] reaction did not proceed at an appreciably rate, and at 120 °C, substantial decarboxylation was observed. Polar solvents also greatly facilitated this process (entries 3–6).[17] The reaction at 100 °C in toluene, although requiring several days, was optimal for material throughput; multiple grams of 12 have been easily procured via this simple sequence.
Figure 2.
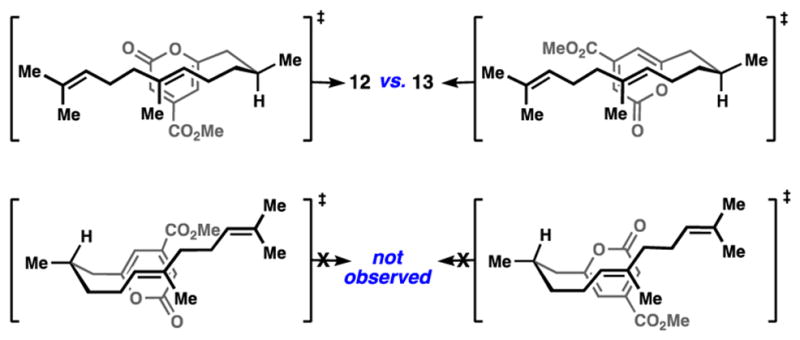
Diels-Alder transition state analysis.
Table 1.
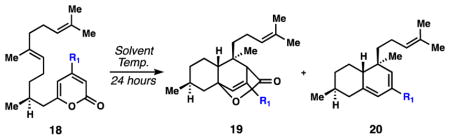
| ||||
|---|---|---|---|---|
| Entry | R1 | Solvent | Temp. (°C) | Ratio 18 : 19 : 20[b] |
| 1 | OH | toluene | 165 | 1 : 0 : 0 |
| 2 | OTf | toluene | 100 | 1 : 0 : 0.15 |
| 3 | CO2Me | heptane | 100 | 1 : 0.9 : 0.02 |
| 4 | CO2Me | PhCF3 | 100 | 1 : 1.4 : 0.07 |
| 5 | CO2Me | MeCN | 100 | 1 : 2.4 : 0.4 |
| 6 | CO2Me | DMF | 100 | 1 : 3.1 : 0.7 |
Conditions: 18 (0.03 M in solvent).
Ratios determined by 1H NMR. Cycloadducts (19) were formed as an approximate 1:1 mixture of diastereomers.
With tricyclic lactone 12 secured all that remained to construct the chatancin cembrane ring system was the forging of the C1–C11 bond (Scheme 1). This transformation turned out to be surprisingly challenging owing to the lack or reactivity at C-1 and the presence of a reactive conjugated ester. Despite their very close proximity, the lactone carbonyl in 12 (C-1) was completely inert towards Lewis-acid mediated addition of the electron-rich alkene (C-11). Interestingly, however, undesired diastereomer 13 underwent an unusual, and facile, Prins-type opening off the lactone ring affording tricyclic acid 14 when treated with TMSOTf (Scheme 1). The structure of the hydrogenation product of 14 confirmed its interesting structure (see 15, Scheme 1). While 12 could be chemo- and regioselectively hydroborated (Et2BH, 0°C) at the desired, internal position (C-11), the highly-hindered borane intermediate resisted efficient transmetallation with Zn(Et)2 or Zn(i-Pr)2.[21]
Ultimately, a practical and high-yielding solution for the conversion of 12 to chatancin was devised. Mild allylic chlorination (SO2Cl2, Na2CO3, 0°C)[22] of the electron-rich alkene produced secondary chloride 16 as a single, unassigned diastereomer, and without purification, this compound was heated with an excess of freshly activated Zn dust. These conditions forged the C1–C11 bond in excellent yield (80% from 12). Gratifyingly this transformation produced a single isomer of 17, setting both newly formed stereocenters correctly. Hydrogenation of 17 (H2, Pd/C) afforded (+)-Chatancin in high yield (93%).[23] After a number of attempts, an X-ray crystal structure of this sensitive natural product could be obtained.
In summary, a highly concise synthetic route to the complex PAF antagonist (+)-chatancin has been accomplished via a simple, 7-step sequence (13% overall yield) that avoids protecting group chemistry[24] and appears to be suitable for straightforward analog construction. Moreover, we anticipate that adaptations to the general strategy described herein will facilitate the synthesis of other complex, cembrane-derived natural products with interesting biological activity. This work will be reported in due course.
Supplementary Material
Footnotes
This work was supported by generous start-up funds from UC-Berkeley, The UC-Berkeley Hellman Fellows Fund, and The University of California Cancer Research Coordinating Committee (CRCC). We thank Dr. Chris Canlas for NMR spectroscopic assistance. Dr. Antonio DiPasquale is acknowleged for X-ray crystallographic analysis and support from NIH Shared Instrument Grant (S10-RR027172). Takasago Co. is greatfully acknowledged for providing a generous sample of l-dihydrofarnesol.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx
References
- 1.a) Radjasa OK, Vaske YM, Navarro G, Vervoort HC, Tenney K, Linington RG, Crews P. Bioorg Med Chem Lett. 2011;19:6658. doi: 10.1016/j.bmc.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Blunt JW, Copp BR, Keyzers RA, Munro MHG, Prinsep MR. Nat Prod Rep. 2014;31:160. doi: 10.1039/c3np70117d. [DOI] [PubMed] [Google Scholar]; c) Rodríguez AD. Tetrahedron. 1995;51:4571. doi: 10.1016/0040-4020(95)00216-U. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Leal MG, Puga J, Serôdio J, Gomes NCM, Calado R. PloS ONE. 2012;7:e30580. doi: 10.1371/journal.pone.0030580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Aratake S, Tomura T, Saitoh S, Yokokura R, Kawanishi Y, Shinjo R, Reimer JD, Tanaka J, Maekawa H. PloS ONE. 2012;7:e30410. doi: 10.1371/journal.pone.0030410. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Coll JC. Chem Rev. 1992;92:613. [Google Scholar]; c) Liang LF, Guo YW. Chem Biodivers. 2013;10:2161. doi: 10.1002/cbdv.201200122. [DOI] [PubMed] [Google Scholar]; d) Yang B, Zhou XF, Lin XP, Liu J, Peng Y, Yang XW, Liu Y. Curr Org Chem. 2012;16:1512. [Google Scholar]; e) Wahlberg I, Eklund A-M. Cyclized Cembranoids of Natural Occurrence. In: Herz W, Kirby GW, Moore RE, Steglich W, Tamm Ch, editors. Prog Chem Nat Prod. Springer Verlag; 1992. p. 1. [Google Scholar]
- 3.Sugano M, Shindo T, Sato A, Iijima Y, Oshima T, Kuwano H, Hata T. J Org Chem. 1990;55:5803. [Google Scholar]
- 4.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Annu Rev Biochem. 2000;69:419. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- 5.For a recent review on naturally occurring inhibitors of PAF, see: Singh P, Singh IN, Mondal SC, Singh L, Garg VK. Fitoterapia. 2013;84:180. doi: 10.1016/j.fitote.2012.11.002.
- 6.Anjaneyulu ASR, Venugopal MJRV, Sarada P, Rao GV, Clardy J, Lobkovsky E. Tetrahedron Lett. 1998;39:135. [Google Scholar]
- 7.Aigner J, Gössinger E, Kählig H, Menz G, Pflugseder K. Angew Chem Int Ed. 1998;37:2226. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2226::AID-ANIE2226>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 8.a) Toró A, Wang Y, Deslongchamps P. Tetrahedron Lett. 1999;40:2765. [Google Scholar]; b) Toró A, Wang Y, Drouin M, Deslongchamps P. Tetrahedron Lett. 1999;40:2769. [Google Scholar]; c) Toró A, L’Heureux A, Deslongchamps P. Org Lett. 2000;2:2737. doi: 10.1021/ol006220z. [DOI] [PubMed] [Google Scholar]; d) Marsault E, Toró A, Nowak P, Deslongchamps P. Tetrahedron. 2001;57:4243. [Google Scholar]; e) Toró A, Deslongchamps P. J Org Chem. 2003;68:6847. doi: 10.1021/jo034123o. [DOI] [PubMed] [Google Scholar]
- 9.Soucy P, L’Heureux A, Toró A, Deslongchamps P. J Org Chem. 2003;68:9983. doi: 10.1021/jo035193y. [DOI] [PubMed] [Google Scholar]
- 10.The biosynthesis of numerous cembrane derivatives are believed to involve pericyclic reactions, see: Roethle PA, Trauner D. Nat Prod Rep. 2008;25:298. doi: 10.1039/b705660p.Li Y, Pattenden G. Nat Prod Rep. 2011;28:1269. doi: 10.1039/c1np00023c.
- 11.Tius MA. Chem Rev. 1988;88:719. [Google Scholar]
- 12.Notable exceptions exist. For recent concise synthetic routes toward complex cembranoids, see: Roethle PA, Trauner D. Org Lett. 2006;8:345. doi: 10.1021/ol052922i.Huang Q, Rawal VH. Org Lett. 2006;8:543. doi: 10.1021/ol053054s.Tang B, Bray CD, Pattenden G. Tetrahedron Lett. 2006;47:6401.Roethle PA, Hernandez PT, Trauner D. Org Lett. 2006;8:5901. doi: 10.1021/ol062581o.
- 13.a) Fettes A, Carreira EM. J Org Chem. 2003;68:9274. doi: 10.1021/jo034964v. [DOI] [PubMed] [Google Scholar]; b) Mayer S, List B. Angew Chem Int Ed. 2006;45:4193. doi: 10.1002/anie.200600512. (S)-6 can also be prepared in 1-step from l-dihydrofarnesol which is commercially available. [DOI] [PubMed] [Google Scholar]
- 14.Sato M, Sakaki JI, Sugita Y, Yasuda S, Sakoda H, Kaneko C. Tetrahedron. 1991;47:5689. [Google Scholar]
- 15.For a recent application of this chemistry in total synthesis, see: Rentsch M Kalesse. Angew Chem Int Ed. 2012;51:11381. doi: 10.1002/anie.201206560.
- 16.Kondoh A, Arlt A, Gabor B, Fürstner A. Chem Eur J. 2013;19:7731. doi: 10.1002/chem.201300827. [DOI] [PubMed] [Google Scholar]
- 17.a) Afarinkia K, Vinader V, Nelson TD, Posner G. Tetrahedron. 1992;48:9111. [Google Scholar]; b) Afarinkia K, Bearpark MJ, Ndibwami A. J Org Chem. 2005;70:1122. doi: 10.1021/jo048213k. [DOI] [PubMed] [Google Scholar]; c) Matsumoto K, Hamana H, Iida H. Helv Chim Acta. 2005;88:2033. [Google Scholar]
- 18.For selected, recent examples of pyrone cycloaddition reactions in total synthesis, see: Baran PS, Burns NZ. J Am Chem Soc. 2006;128:3908. doi: 10.1021/ja0602997.Shin IJ, Choi ES, Cho CG. Angew Chem Int Ed. 2007;46:2303. doi: 10.1002/anie.200604612.Jung YJ, Kang HU, Cho HK, Cho CG. Org Lett. 2011;13:5890. doi: 10.1021/ol202525a.Zhao P, Beaudry CM. Org Lett. 2013;15:402. doi: 10.1021/ol303390a.Smith MW, Snyder SA. J Am Chem Soc. 2013;135:12964. doi: 10.1021/ja406546k.Zhao P, Beaudry CM. Angew Chem Int Ed. 2014;53:10500. doi: 10.1002/anie.201406621.
- 19.a) Nelson HM, Stoltz BM. Org Lett. 2008;10:25. doi: 10.1021/ol702501s. [DOI] [PubMed] [Google Scholar]; b) Kozytska MV, Dudley GB. Tetrahedron Lett. 2008;49:2899. [Google Scholar]; c) Zhou X, Wu W, Liu X, Lee CS. Org Lett. 2008;10:5525. doi: 10.1021/ol8022787. [DOI] [PubMed] [Google Scholar]; d) Larsson R, Sterner O, Johansson M. Org Lett. 2009;11:657. doi: 10.1021/ol802760z. [DOI] [PubMed] [Google Scholar]; e) Nelson HM, Stoltz BM. Tetrahedron Lett. 2009;50:1699. [Google Scholar]; f) Nelson HM, Murakami K, Virgil SC, Stoltz BM. Angew Chem Int Ed. 2011;50:3688. doi: 10.1002/anie.201008003. [DOI] [PubMed] [Google Scholar]; g) Nelson HM, Gordon JR, Virgil SC, Stoltz BM. Angew Chem Int Ed. 2013;52:6699. doi: 10.1002/anie.201301212. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Larsson R, Scheeren HW, Aben RWM, Johansson M, Sterner O. Eur J Org Chem. 2013:6955. [Google Scholar]; i) Min L, Zhang Y, Liang X, Huang J, Bao W, Lee CS. Angew Chem Int Ed. 2014;53:11294. doi: 10.1002/anie.201405770. [DOI] [PubMed] [Google Scholar]; j) Gordon JR, Nelson HM, Virgil SC, Stoltz BM. J Org Chem. 2014;79:9740. doi: 10.1021/jo501924u. [DOI] [PubMed] [Google Scholar]
- 20.Common Lewis acids examined (e.g. BF3•OEt2, Et2AlCl, AlCl3, BINOL) either exerted no influence on the diastereoselectivity of the cycloaddition reaction or resulted in decomposition of the pyrone substrate.
- 21.Hupe E, Calaza MI, Knochel P. Chem Eur J. 2003;9:2789. doi: 10.1002/chem.200204662. [DOI] [PubMed] [Google Scholar]
- 22.Li W, Li Y, Li Y. Org Prep Proceed Int. 1996;28:83. [Google Scholar]
- 23.Synthetic 1 exhibited [α]D = +10.7 (CHCl3, c = 0.01), natural 1 displays [α]D = +10.5 (CHCl3, c = 1.0)[3]
- 24.a) Young IS, Baran PS. Nature Chem. 2009;1:193. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]; b) Gaich T, Baran PS. J Org Chem. 2010;75:4657. doi: 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]; c) Saicic RN. Tetrahedron. 2014;70:8183. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.