

Abstract
Mortality and morbidity associated with oral squamous cell carcinoma (OSCC) remain unacceptably high with disfiguring treatment options and a death rate of 1 per hour in the United States. The approval of cituximab for advanced OSCC has been the only new treatment for these patients since the 1970s, although it has not significantly increased overall survival. To address the paucity of effective new therapies, we undertook a high-throughput screen to discover small molecules and natural products that could induce endoplasmic reticulum (ER) stress and enforce a terminal unfolded protein response (UPR) in OSCC. The terpenoid cantharidin (CNT), previously used to treat various malignancies in culture-specific medical practices for over 2,000 y, emerged as a hit. CNT and its analog, cantharidic acid, potently induced protein and gene expression profiles consistent with the activation of ER stress, the UPR, and apoptosis in OSCC cells. Murine embryonic fibroblasts null for the UPR-associated transcription factors Atf4 or Chop were significantly protected from CNT, implicating a key role for the UPR in the death response. These data validate that our high-throughput screen can identify novel modulators of UPR signaling and that such compounds might provide a new therapeutic approach to treating patients with OSCC.
Keywords: UPR, CHOP, ATF4, cantharide, natural products, chemotherapy
Introduction
Alcohol and tobacco are considered the major risk factors for oral squamous cell carcinoma (OSCC), and a causative role for human papillomavirus (HPV) infection has recently come to light (Smith et al. 2004). Regardless of etiology, OSCC remains the fifth most common cancer in the world, with 2013 Surveillance, Epidemiology, and End Results (SEER) data projecting 41,380 new U.S. cases of oral and pharyngeal squamous cell carcinoma and 7,890 deaths. The approval of cituximab (in 2006) for locally and regionally advanced OSCC is the only milestone on the landscape of drug discovery for these patients since the introduction of cisplatin. To address the paucity of new treatments and the myriad shortcomings associated with therapies targeting signaling intermediates, we performed a high-throughput screen (HTS) to identify small molecules and natural products that could induce endoplasmic reticulum (ER) stress and enforce a terminal unfolded protein response (UPR) in OSCC (Fribley et al. 2011).
The UPR is a cell’s coordinated effort to respond to stresses that perturb protein folding in the ER. IRE1α, ATF6, and PERK are ER transmembrane sentry proteins that monitor luminal protein folding and communicate disturbances to the cytoplasm. IRE1α and ATF6 activation leads to the upregulation of chaperones, folding enzymes, and other UPR target genes that generally attempt to restore homeostatic folding. PERK phosphorylates eIF2α, which pauses global translation and leads to nuclear accumulation of the transcription factors ATF4 and CHOP. Prolonged inhibition of translation and accumulation of ATF4 and CHOP positions the cell for apoptosis if the folding challenge cannot be remedied or if the initial stress is overwhelming. ATF4 targets proapoptotic factors such as NOXA (Fribley et al. 2006; Wang et al. 2009), and during ER stress, it collaborates with CHOP to increase protein translation through adenosine triphosphate (ATP) deletion and oxidative stress and lead to apoptosis (Han et al. 2013).
Misregulation of protein folding and aberrant UPR activation has been studied in the context of many human inflammatory and metabolic diseases, especially cancer (Ma and Hendershot 2004; Li X et al. 2011; Singleton and Harris 2012; Wang and Kaufman 2012). Angiogenesis, invasion, and metastasis are driven by the ubiquitous activation of stress responses. Stabilization of hypoxia-inducible factors, enhanced oxidative defenses, activation of AMP-activated protein kinase, and increased UPR signaling all contribute to tumor maintenance and survival. The highly secretory nature of malignant cells, coupled with their increased need for numerous protein factors that modulate survival and growth, leads to increased basal expression of UPR- and translation-related proteins (Kerekatte et al. 1995; Nathan et al. 1997; Franklin et al. 1999; Crew et al. 2000; DeFatta et al. 2000). We hypothesized that small molecules and natural products able to activate the PERK-eIF2α-ATF4-CHOP signaling axis would exacerbate the UPR and induce apoptosis in tumor cells and that healthy adjacent cells would be able to mount an effective adaptive UPR and return to homeostasis.
A cell-based HTS for UPR activators (Fribley et al. 2011) identified the PP2A inhibitor cantharidin (CNT). CNT is a defensive toxin secreted by many beetles from the family Meloidae and also known as Spanish fly. Pedanios Dioskorides, a surgeon in Nero’s army, listed cantharidin as a drug in his famous monograph Materia Medica circa 77 AD, and it has been consistently used in traditional Chinese folk medicine to treat malignancy (Wang 1989). Historically, most studies focused on liver malignancies, but in vitro esophageal carcinoma studies and even clinical trials with patients with esophageal carcinoma have been reported (Wang 1989). Cantharidin is not myelosuppressive and has been reported to increase leukocytes even at levels considered toxic (Wang 1989), two highly desirable properties of an antineoplastic. Substantial efforts by many groups have generated hundreds of analogs in attempts to overcome challenges associated with severe irritation to the urinary tract and other mucous membranes (Zhang et al. 2011; Tseng et al. 2012; Puerto Galvis et al. 2013). This is the first report we are aware of demonstrating the ability of cantharidin to induce ER stress and UPR-dependent apoptosis in cancer cell lines.
Materials and Methods
Cell Lines and Proliferation Assays
CHO-K1 cells with UPR pathway-specific luciferase reporters (CHOP or XBP1) were screened as previously described (Fribley et al. 2011). The human floor of mouth squamous cell carcinoma (SCC) lines UMSCC1, UMSCC14A, and UMSCC14B, as well as the laryngeal SCC cell lines UMSCC23, UMSCC10A, and UMSCC10B, were kindly provided by Dr. Thomas Carey at the University of Michigan. The tongue nodal metastasis cell line HN12 was provided by Dr. George Yoo at the Barbara Ann Karmanos Cancer Center at Wayne State University. The salivary epidermoid carcinoma cell line A253 was purchased from ATCC (Manassas, VA). A549 BAX–/–/–/–, BAK–/– lung adenocarcinoma cells (CLLS1015) were from Sigma-Aldrich (St. Louis, MO). All human cancer cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with penicillin-streptomycin and 10% fetal bovine serum (FBS; Invitrogen, Grand Island, NY). Murine embryonic fibroblasts (MEFs) were cultured in DMEM supplemented with penicillin-streptomycin and 10% FBS supplemented with nonessential amino acids solution (Invitrogen); Atf4 MEF medium was further supplemented (1 µL/100 mL) with 2-mercaptoethanol. Cantharidin was purchased from MicroSource Discovery Systems (Gaylordsville, CT), norcantharidin was from Sigma-Aldrich, and cantharidic acid was purchased from Enzo Life Sciences (Farmingdale, NY). For luminescent proliferation assays, 30 µL of CellTiter-Glo (Promega, Madison, WI) was added to each well, and luminescence was measured after a 10-min incubation. Alternative metabolic proliferation assays were performed by adding 0.5 mg/mL 3-[4,5-dimethylthiazol-2-yl])-2-5-diphenyltetrazolium bromide (MTT) to each well and incubating at 37 °C for 2 to 4 h. Formazan crystals were DMSO-solubilized and absorbance was measured at 570 nm. Caspase enzyme activation was measured with a luminescent Caspase-Glo 3/7 Assay (G8092; Promega). Proliferation assays were all performed at least 3 times in triplicate 96-well plates (50 µL final volume) with 7,500 cells/well. Error bars represent the standard deviation of technical replicates in a representative experiment. Significant differences between wild-type (or parental) and knockout cell lines were determined using 2-way analysis of variance (ANOVA).
Fluorescence-Activated Cell Sorting
3 × 105 cells (in 2 mL phenol red–free medium) were stained with 2.5 µg/mL JC-1 (Life Technologies, Carlsbad, CA) for 15 min at 37 °C, 5% CO2. Cells were washed with phosphate-buffered saline (PBS) and stained with DAPI to exclude dead cells prior to compensation and data acquisition. 2.5 µM valinomycin (1 h) was used as a positive control. Fluorescence-activated cell sorting (FACS) analysis was performed on a DB LSR II (BD Biosciences, San Jose, CA) and analyzed using Tree Star software (FlowJo LLC, Ashland, OR) at the Microscopy, Imaging and Cytometry Resources (MICR) core at the Karmanos Cancer Institute, Wayne State University.
Polymerase Chain Reaction Analysis
One microgram of Trizol (Invitrogen) harvested RNA was reverse transcribed for reverse transcription–polymerase chain reaction (RT-PCR) analysis of XBP1 using a single human-specific PCR primer pair producing amplicons for unspliced XBP1u and spliced XBP1s: forward, CCT TGT AGT TGA GAA CCA GG; reverse, GGG GCT TGG TAT ATA TGT GG. For quantitative RT-PCR (RT-qPCR), total RNA was harvested with the Cells-to-CT Kit (Ambion; Life Technologies) in 96-well plates, reverse transcribed, and amplified using SsoFast probes Supermix (Bio-Rad, Hercules, CA) and TaqMan (Life Technologies) primer probes, according to the manufacturer’s protocol. RT-qPCR primers were as follows: 18S (Hs99999901_s1), CHOP/DDIT3 (Hs01090850_m1), GADD34/PPP1R15 (Hs00169585_m1), ATF3 (Hs00910 173_m1), PUMA/BBC3 (Hs00248075_m1), NOXA/PMAIP1 (Hs00560402_m1), BTG2 (Hs00198887_m1), GADD45γ (Hs00198672_m1), DR5/TNFRSF1 (Hs00366 278_m1), TRIB3 (Hs00221754_m1). 18S ribosomal RNA was used as an internal control, and fold changes were determined using the ΔΔCt calculation; error bars represent standard deviation of technical replicates.
Small Interfering RNA Transfection
Five thousand OSCC cells were plated into white opaque 96-well plates in 50 µL antibiotic-free medium. Small interfering RNAs (siRNAs) (25 nM) were transfected overnight at 37 °C and 5% CO2 using DharmaFECT 1 (Thermo Scientific, Waltham, MA) in a final volume of 100 µL. The transfection medium was replaced with 50 µL complete medium, and the cells were allowed to grow for an additional 24 h prior to treatment. Gene expression analysis to validate knockdown was performed 6 h after drug treatment, and proliferation was measured after 24 h.
Immunoblotting
Whole-cell lysates were prepared using modified RIPA buffer as previously described (Fribley et al. 2004). Fifty to 80 µg protein was resolved on 7.5% or 12% sodium dodecyl sulfate (SDS)–polyacrylamide gels and transferred to PVDF membranes (Bio-Rad). Membranes were probed overnight at 4 °C with polyclonal antibodies for PARP, cleaved caspase 3, cleaved caspase 9, and phospho-eIF2α (Cell Signaling, Danvers, MA); ATF4 and CHOP (Santa Cruz Biotechnology, Santa Cruz, CA); monoclonal GAPDH or α-tubulin (Chemicon, Billerica, MA) were used to probe each membrane represented to demonstrate relative loading.
Results
Cantharides Induce ER Stress and Activate the UPR
A high-throughput screen was performed with Chinese hamster ovary (CHO) cells stably transfected with luciferase constructs that individually report on the induction of CHOP or XBP1 splicing (Fribley et al. 2011). Cantharidin, but not its demethylated analog norcantharidin, emerged as a hit that activated the UPR from a library of ~66,000 small molecules and natural products at the University of Michigan Center for Chemical Genomics. CNT, norcantharidin (NOR), and cantharidic acid (CAC) (Fig. 1A) were obtained commercially and used in culture to evaluate UPR activation. RT-qPCR analysis revealed that CNT and CAC could lead to the accumulation of transcripts for CHOP, GADD34, and ATF3 (Fig. 1B). Conventional RT-PCR analysis of the same complementary DNA (cDNA) pools revealed rapid and robust splicing of XBP1 (Fig. 1C). Immunoblot analysis of whole-cell lysates demonstrated that CNT treatment led to the phosphorylation of eIF2α (Fig. 1D) and accumulation of ATF4 and CHOP (Fig. 1E) in OSCC cell lines, confirming the ability of CNT and CAC to induce ER stress and the UPR.
Figure 1.
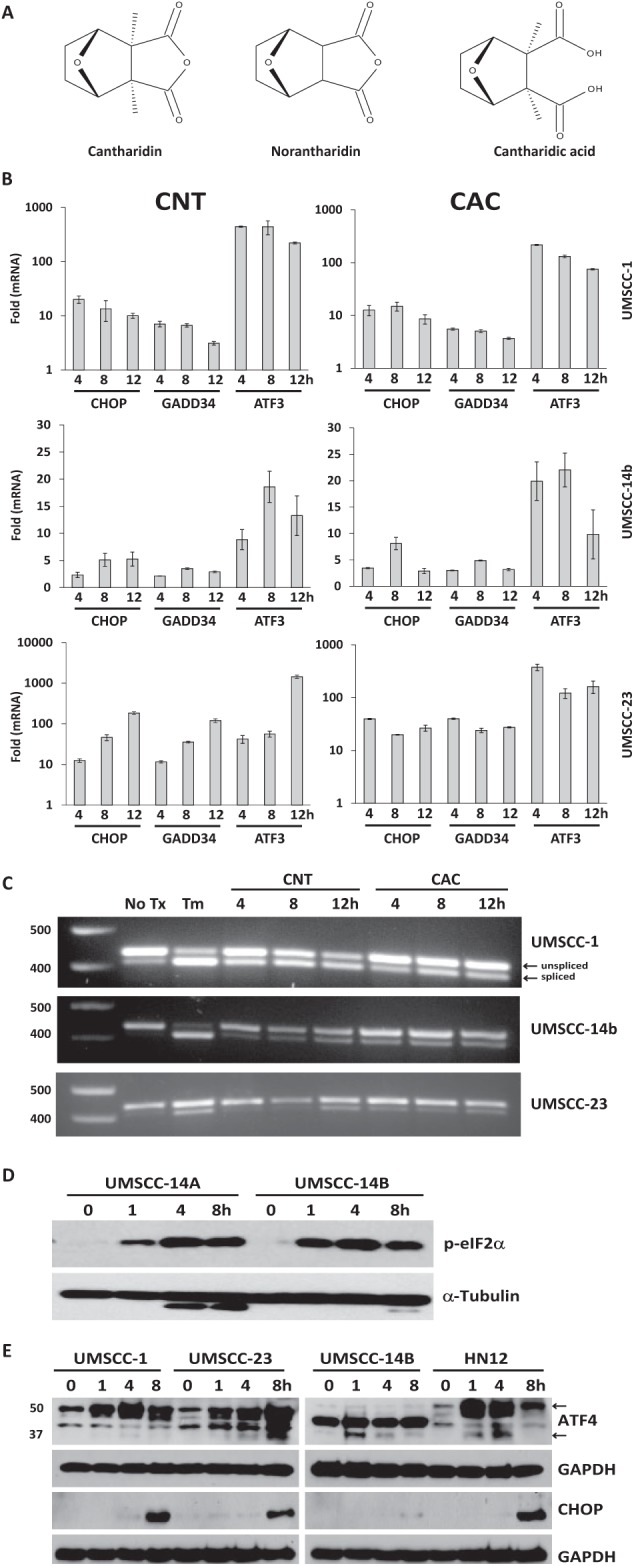
Cantharidin induces endoplasmic reticulum (ER) stress and activates the unfolded protein response (UPR). (A) Cantharidin (CNT), norcantharidin (NOR), and cantharidic acid (CAC) molecular structures. (B) Quantitative reverse transcription–polymerase chain reaction (RT-PCR) analysis of UPR-associated genes in a panel of oral squamous cell carcinoma (OSCC) treated with 10 µM CNT and CAC for 4, 8, and 12 h. (C) Complementary DNA was generated with the same RNA used in B, and conventional RT-PCR was performed with a single primer pair to appreciate relative levels of unspliced (XBP1u) and spliced (XBP1s) messenger RNA. (D) Immunoblot analysis of whole-cell lysates from cantharidin-treated OSCC cells with a monoclonal antibody for phospho-eIF2α. (E) Immunoblot analysis of ATF4 and CHOP in whole-cell lysates from OSCC cells. TM, tunicamycin. “No tx” or “0 h” indicates equimolar DMSO control.
Cantharidin Analogs Inhibit OSCC Proliferation
A panel of OSCC cell lines was used to examine the effects of cantharidin analogs on cell growth. Luminescent ATP-based proliferation assays revealed that CNT and CAC inhibited growth in 4 OSCC cell lines and that NOR did not (Fig. 2A). Metabolic MTT assays provided nearly identical results in 4 additional OSCC cell lines treated with increasing doses of CNT and NOR (Fig. 2B). RT-qPCR analysis confirmed that CNT and CAC but not NOR could induce CHOP in these cells (Appendix Fig. 1). A549 Bax–/–/–/–, Bak–/– human lung adenocarcinoma cells were significantly resistant to CNT compared with the wild-type parental cell line after 24 and 48 h (Fig. 2C). These results indicated that the cytotoxic effects of CNT are not limited to OSCC and implicated the intrinsic apoptosis cascade in the observed reductions in cancer cell proliferation.
Figure 2.

Cantharidin analogs inhibit oral squamous cell carcinoma (OSCC) proliferation. (A) Luminescent adenosine triphosphate (ATP)–based proliferation assays were performed in OSCC cell lines after a 24-h exposure to cantharidin (CNT; IC50, 8.04–28.9), norcantharidin (NOR; IC50 > 100), and cantharidic acid (CAC; IC50, 3.26–15.7). (B) Colorimetric cell viability MTT assays were performed in OSCC cell lines after 24 h of exposure to CNT (IC50, 10.6–11.6) and NOR (IC50 > 100). (C) Luminescent ATP-based proliferation assays performed with A549 parental and Bax/Bak–/– –/–, –/– cells lines after a 24- and 48-h exposure to CNT (2-way analysis of variance, P < 0.0001 for dose and for interaction).
Cantharidin and Cantharidic Acid Induce Apoptosis in OSCC
Having observed that CNT and CAC could inhibit OSCC proliferation, we sought to determine whether cantharides were cytostatic or cytotoxic. RT-qPCR analysis with 3 OSCC cell lines demonstrated that the cell death–associated transcripts NOXA, PUMA, BTG2, GADD45γ, TRB3, and DR5 were increased (Fig. 3A). RT-qPCR array analysis further demonstrated that 11 apoptosis-related genes were induced at least 2-fold after 8 h and that 16 genes were induced by 12 h (Appendix Table). Immunoblot analysis of whole-cell lysates from UMSCC1, UMSCC14A, and UMSCC14B cells exposed to CNT and CAC produced bands consistent with the active forms of PARP, caspase 9, and caspase 3 (Fig. 3B). Although proliferation assays with UMSCC14B cells demonstrated similar IC50s (as other OSCC cells) when treated with CNT or CAC (Fig. 2), only modest cleavage of PARP and caspase 9 was observed, and cleaved (active) caspase 3 could not be detected following CAC treatment. These data were confirmed with an enzymatic caspase 3/7 luciferase assay (Appendix Fig. 2). JC1 staining and FACS analysis demonstrated that CNT could significantly decrease mitochondrial membrane potential at 4 and 8 h in SCC1 and HN12 cells but not in the more resistant UMSCC23 (Fig. 3C). CNT and CAC treatment led to the fragmentation of genomic DNA into nucleosome-sized fragments, a hallmark of apoptosis (Fig. 3D). These data indicate that cantharide-induced death in OSCC cells is facilitated by the intrinsic (mitochondrial-mediated) apoptosis.
Figure 3.

Cantharidin (CNT) and cantharidic acid (CAC) induce apoptosis in oral squamous cell carcinoma (OSCC). (A) Quantitative reverse transcription–polymerase chain reaction (RT-qPCR) analysis of apoptotic messenger RNA transcripts in a panel of OSCC cell lines treated with 10 µM CNT and CAC for 4, 8, and 12 h. (B) Immunoblot analysis of whole-cell lysates from CNT- and CAC-treated OSCC cells with polyclonal antibodies for PARP and caspase 3 and a monoclonal antibody for caspase 9. (C) JC-1 analysis indicating the relative number of OSCC cells with reduced mitochondrial membrane potential following CNT (10 µM), as indicated; error bars represent standard deviation of 1.6 to 2.0 × 104 cells; see Appendix Figures 4 to 6 for original fluorescence-activated cell sorting data. (D) Electrophoretic resolution of genomic DNA harvested from OSCC cultures to appreciate DNA laddering following CNT and CAC exposure.
Intact ATF4 Signaling Is Required for Efficient Cantharidin-Induced Cell Death
MEFs null for Atf4 or Chop and wild-type littermate controls were cultured with increasing concentrations of CNT, NOR, and CAC for 16 h. ATP-based luciferase proliferation assays demonstrated that MEFs null for either Atf4 (Fig. 4A) or Chop (Fig. 4B) were protected from the apoptosis-inducing effects of CNT and CAC. Consistent with observations made with OSCC cell lines, NOR did not substantially inhibit proliferation in MEFs. PARP accumulation was delayed in Chop-null cells, but KDEL accumulation and the appearance of phospho-eIF2α were similar (Appendix Fig. 3). Murine fibroblasts are useful for mechanistic studies to evaluate the effect of drugs in the absence of specific genes but cannot be assumed to exactly recapitulate any human cancer. To address this concern, UMSCC1 and UMSCC23 were transiently transfected with pools of siRNAs for ATF4, CHOP, ATF4, and CHOP or a scrambled siRNA control. OSCC cells transfected with siATF4 grew more slowly than scrambled control or siCHOP cultures and displayed significantly higher IC50s when treated with CNT; siCHOP knockdown affected neither growth rate nor CNT IC50 in stark contrast to what was observed in MEF cultures (Fig. 4C, D, left); relative siRNA knockdown was measured by RT-qPCR (Fig. 4C, D, right). These data recapitulated our finding in MEFs that intact ATF4 signaling is required for CNT to exert its cytotoxic effect. In contrast, CHOP reduction was not sufficient to inhibit CNT-induced cell death, indicating that CHOP is dispensable or that very low mRNA levels are sufficient for CNT to induce cell death in OSCC.
Figure 4.

Intact ATF4 signaling is required for efficient cell death. (A) Luminescent adenosine triphosphate (ATP)–based proliferation assays were performed with wild-type and Atf4 –/– murine embryonic fibroblasts (MEFs) treated with cantharidin (CNT; 2-way analysis of variance [ANOVA], P < 0.0001 for dose and for interaction), norcantharidin (NOR; 2-way ANOVA, P = 0.0007 for dose and 0.0062 for interaction), and cantharidic acid (CAC; 2-way ANOVA, P < 0.0001 for dose and for interaction) at 16 h. (B) Luminescent ATP-based proliferation assays with wild-type and Chop –/– MEFs treated with CNT (2-way ANOVA, P < 0.0001 for dose and for interaction), NOR (2-way ANOVA, P = 0.0113 for dose and 0.1738 for interaction), and CAC (2-way ANOVA, P < 0.0001 for dose and for interaction). (C, D) Oral squamous cell carcinoma (OSCC) cells were transfected with small interfering RNA (siRNA) pools specific for ATF4, CHOP, or both; luminescent proliferation assays (left = performed 24 h after CNT treatment); gene expression assays were performed 6 h after treatment to demonstrate relative gene expression reduction. siRNA experiments were performed at least 3 times with 3 biological replicates, gene expression was measured in triplicate from these biological replicates.
Discussion
Cantharidin, also known as Spanish fly, is the active ingredient in the exoskeleton of the Chinese blister beetle used in traditional medicines to treat a variety of cancers, including hepatoma. Because of its apparent cytotoxicity, it is not currently approved by the Food and Drug Administration (FDA) for any internal use, and therapeutic applications are limited to topical applications for the treatment of a variety of warts (Till and Majmudar 1981; Kacar et al. 2012; Silverberg and Silverberg 2012). Cantharidin is well characterized as an inhibitor of protein phosphatases PP1 and PP2A (Li and Casida 1992; Honkanen 1993) and is a component of many commercially available protease inhibitor cocktails used to stabilize phosphorylated proteins in whole-cell lysates prior to immunoblot analysis. Cantharides have also been reported to generate oxidative stress (Li et al. 2010; Verma and Prasad 2013), activate nuclear factor (NF)–κB (Li W et al. 2011), and induce DNA damage (Efferth et al. 2005) prior to apoptosis in myriad human cancer cell lines. The multifactorial manner by which cantharides induce cell death may provide a distinct advantage over highly specific single-target small molecules to which tumors quickly become resistant (Efferth et al. 2005; Rauh et al. 2007; Kadioglu et al. 2014). There is tremendous interest in developing more potent less toxic analogs because cantharides are not myelosuppressive and can overcome multidrug resistance in several cancer cell lines (Yi et al. 1991; Efferth et al. 2002). Furthermore, the anticancer potential of phosphatase inhibitors has very recently come to light and is an active area of anticancer research (Kawada et al. 1999; Sakoff et al. 2002; Li et al. 2010; Wada et al. 2010).
We have used a variety of biochemical and genetic approaches to demonstrate for the first time that ER stress and UPR induction is a key mechanism by which CNT and CAC exert an anticancer effect. Norcantharidin, the demethylated form of cantharidin, could not induce UPR, nor was it appreciably toxic in OSCC or MEF, even at concentrations much higher than required for PP1 and PP2A inhibition (Deng et al. 2013). Using 10 µM CNT or CAC (approximate OSCC 24-h IC50), we observed phosphorylation of eIF2α and splicing of XBP1, 2 hallmarks of ER stress. Subsequently, other UPR-associated protein and gene transcripts increased confirming UPR induction. Luminescent (ATP-based) proliferation and MTT assays demonstrated that CAC was slightly more toxic than CNT in OSCC cell lines. The salivary epidermoid carcinoma cell line A253 was more resistant to CNT and CAC for reasons that are not entirely clear. However, A253 cells have a TGFβ/SMAD signaling defect that is a known clinical challenge to chemotherapy, and we have observed that relatively higher concentrations of other UPR-targeted small molecules, such as celastrol, are required to induce cell death in this cell line (Fribley et al. 2015).
During ER stress, the phosphorylation of eIF2α prevents the addition of Met to new peptide chains, thereby inhibiting general protein translation. Stressed cells are shunted along the apoptotic arm of the UPR if this pause does not occur to allow folding recovery or if eIF2α is not promptly dephosphorylated so protein synthesis can resume (Scheuner et al. 2001; Han et al. 2013). We observed rapid and robust phosphorylation of eIF2α that persisted at least 8 h in OSCC and MEF cells treated with CNT. Lysates from the same cells treated with relatively high doses of the ER stress-inducing antibiotic tunicamycin demonstrated much lower levels of phosphorylation and were able to recover from the stress. Although biochemical analysis of phosphatase status was beyond the scope of our study, the durability of eIF2α phosphorylation is a feature that would be expected of a terminal (apoptotic) UPR.
Following cantharide-induced UPR in OSCC, we observed a decrease in mitochondrial polarization, increased apoptotic gene expression, cleavage of PARP and caspases 9 and 3, and DNA fragmentation. The ability of cantharidin analogs to induce apoptosis in human cancer cells in vitro is well documented for many malignancies, including leukemia (Efferth et al. 2005), colon (Sakoff et al. 2002), pancreatic (Li et al. 2010; Li W et al. 2011), liver and lung (Shan et al. 2006), and malignant oral but not normal buccal keratinoctyes (Kok et al. 2003) or normal pancreatic duct cells (Li et al. 2010) or normal colon cells (Sakoff et al. 2002). Decreased toxicity in normal cells in vitro is an important feature of cantharides, but nonspecific gastrointestinal and urinary tract irritation and kidney damage are known side effects in CNT-treated hepatoma patients. Our observation that CHOP and eIF2α phosphorylation were increased in SV40-transformed MEFs (Appendix Fig. 3) indicates that CNT can also activate the UPR in nonmalignant fibroblasts. It is possible that some of the effects in nonmalignant cells could be mediated by the UPR. The in vivo liabilities of CNT have led to reports of scores of CNT analogs (such as NOR and CAC) in attempts to improve potency and selectivity and reduce toxicity. Our study adds to a growing body of literature that the cytotoxic effects of cantharidin are meted out via an apoptotic mechanism and, for the first time, implicates the UPR.
This is the first demonstration that intact Atf4 and Chop are required for efficient cantharide-induced cell death and that ATF4 knockdown significantly increases the IC50 of CNT in OSCC. ATF4 knockdown reduced the rate at which UMSCC1 and UMSCC23 were able to proliferate in culture, and multiple experiments demonstrated that protection from CNT increased with ATF4 knockdown efficiency. Our observation that a CHOP-targeted siRNA pool could not protect OSCC cells was unexpected. The level of CHOP knockdown achieved was significant and substantial, so this result is consistent with the idea that relatively low levels of mRNA are sufficient to facilitate cantharide-induced cell death or that the 2 OSCC cell lines we examined do not rely as heavily on CHOP for cell death as MEF. Ongoing work in our laboratory is focused toward the identification of OSCC clones that have been rendered CHOP-null using a validated CRISPR-Cas system. The fact that Atf4- and Chop-null cells are protected from cell death clearly implicates the UPR as an important mechanism by which cantharides exert their cytotoxic effect and supports our hypothesis that therapeutic targeting of the UPR might be an effective and novel approach to kill OSCC.
Author Contributions
Y. Xi, D.M. Garshott, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; A.L. Brownell, T.L. Freeburg, N.G. Yoo, contributed to design, data acquisition, analysis, and interpretation, critically revised the manuscript; G.H. Yoo, H.-S. Lin, M.U. Callaghan, contributed to conception, design, data analysis, and interpretation, critically revised the manuscript; R.J. Kaufman, contributed to conception, design, data acquisition, analysis, and interpretation, critically revised the manuscript; A.M. Fribley, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplementary Material
Footnotes
Portions of these studies were supported by National Institutes of Health (NIH) grants R00 DE019678 (AMF), R03 MH089787 (RJK and AMF), and R03 MH084182, R37 DK042394, R01 DK088227, and R01 HL052173 (RJK). The Children’s Hospital of Michigan Foundation and the Wayne State University Fund for Medical Research supported portions of these studies (AMF). The Microscopy, Imaging and Cytometry Resources Core is supported in part by an NIH Center grant P30CA22453 to the Karmanos Cancer Institute, Wayne State University, and the Perinatology Research Branch of the National Institute of Child Health and Development. Steven Buck at Children’s Hospital of Michigan provided JC1 dye and technical expertise.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Crew JP, Fuggle S, Bicknell R, Cranston DW, de Benedetti A, Harris AL. 2000. Eukaryotic initiation factor-4E in superficial and muscle invasive bladder cancer and its correlation with vascular endothelial growth factor expression and tumour progression. Br J Cancer. 82(1):161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFatta RJ, Nathan CO, De Benedetti A. 2000. Antisense RNA to eIF4E suppresses oncogenic properties of a head and neck squamous cell carcinoma cell line. Laryngoscope. 110(6):928–933. [DOI] [PubMed] [Google Scholar]
- Deng L, Dong J, Wang W. 2013. Exploiting protein phosphatase inhibitors based on cantharidin analogues for cancer drug discovery. Mini Rev Med Chem. 13(8):1166–1176. [DOI] [PubMed] [Google Scholar]
- Efferth T, Davey M, Olbrich A, Rucker G, Gebhart E, Davey R. 2002. Activity of drugs from traditional Chinese medicine toward sensitive and MDR1- or MRP1-overexpressing multidrug-resistant human CCRF-CEM leukemia cells. Blood Cells Mol Dis. 28(2):160–168. [DOI] [PubMed] [Google Scholar]
- Efferth T, Rauh R, Kahl S, Tomicic M, Böchzelt H, Tome ME, Briehl MM, Bauer R, Kaina B. 2005. Molecular modes of action of cantharidin in tumor cells. Biochem Pharmacol. 69(5):811–818. [DOI] [PubMed] [Google Scholar]
- Franklin S, Pho T, Abreo FW, Nassar R, De Benedetti A, Stucker FJ, Nathan CO. 1999. Detection of the proto-oncogene eIF4E in larynx and hypopharynx cancers. Arch Otolaryngol Head Neck Surg. 125(2):177–182. [DOI] [PubMed] [Google Scholar]
- Fribley AM, Cruz PG, Miller JR, Callaghan MU, Cai P, Narula N, Neubig RR, Showalter HD, Larsen SD, Kirchhoff PD, et al. 2011. Complementary cell-based high-throughput screens identify novel modulators of the unfolded protein response. J Biomol Screen. 16(8):825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribley AM, Evenchik B, Zeng Q, Park BK, Guan JY, Zhang H, Hale TJ, Soengas MS, Kaufman RJ, Wang CY. 2006. Proteasome inhibitor PS-341 induces apoptosis in cisplatin-resistant squamous cell carcinoma cells by induction of Noxa. J Biol Chem. 281(42):31440–31447. [DOI] [PubMed] [Google Scholar]
- Fribley AM, Miller JR, Brownell AL, Garshott DM, Zeng Q, Reist TE, Narula N, Cai P, Xi Y, Callaghan MU, et al. 2015. Celastrol induces unfolded protein response-dependent cell death in head and neck cancer. Exp Cell Res. 330(2):412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fribley A, Zeng Q, Wang CY. 2004. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol. 24(22):9695–9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. 2013. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 15(5):481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkanen RE. 1993. Cantharidin, another natural toxin that inhibits the activity of serine/threonine protein phosphatases types 1 and 2A. FEBS Lett. 330(3):283–286. [DOI] [PubMed] [Google Scholar]
- Kacar N, Tasli L, Korkmaz S, Ergin S, Erdogan BS. 2012. Cantharidin-podophylotoxin-salicylic acid versus cryotherapy in the treatment of plantar warts: a randomized prospective study. J Eur Acad Dermatol Venereol. 26(7):889–893. [DOI] [PubMed] [Google Scholar]
- Kadioglu O, Kermani NS, Kelter G, Schumacher U, Fiebig HH, Greten HJ, Efferth T. 2014. Pharmacogenomics of cantharidin in tumor cells. Biochem Pharmacol. 87(3):399–409. [DOI] [PubMed] [Google Scholar]
- Kawada M, Amemiya M, Ishizuka M, Takeuchi T. 1999. Cytostatin, an inhibitor of cell adhesion to extracellular matrix, selectively inhibits protein phosphatase 2A. Biochim Biophys Acta. 1452(2):209–217. [DOI] [PubMed] [Google Scholar]
- Kerekatte V, Smiley K, Hu B, Smith A, Gelder F, De Benedetti A. 1995. The proto-oncogene/translation factor eIF4E: a survey of its expression in breast carcinomas. Int J Cancer. 64(1):27–31. [DOI] [PubMed] [Google Scholar]
- Kok SH, Hong CY, Kuo MY, Lee CH, Lee JJ, Lou IU, Lee MS, Hsiao M, Lin SK. 2003. Comparisons of norcantharidin cytotoxic effects on oral cancer cells and normal buccal keratinocytes. Oral Oncol. 39(1):19–26. [DOI] [PubMed] [Google Scholar]
- Li W, Chen Z, Zong Y, Gong F, Zhu Y, Zhu Y, Lv J, Zhang J, Xie L, Sun Y, et al. 2011. PP2A inhibitors induce apoptosis in pancreatic cancer cell line PANC-1 through persistent phosphorylation of IKKalpha and sustained activation of the NF-kappaB pathway. Cancer Lett. 304(2):117–127. [DOI] [PubMed] [Google Scholar]
- Li W, Xie L, Chen Z, Zhu Y, Sun Y, Miao Y, Xu Z, Han X. 2010. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci. 101(5):1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang K, Li Z. 2011. Unfolded protein response in cancer: the physician’s perspective. J Hematol Oncol. 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Casida JE. 1992. Cantharidin-binding protein: identification as protein phosphatase 2A. Proc Natl Acad Sci U S A. 89(24):11867–11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. 2004. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 4(12):966–977. [DOI] [PubMed] [Google Scholar]
- Nathan CO, Liu L, Li BD, Abreo FW, Nandy I, De Benedetti A. 1997. Detection of the proto-oncogene eIF4E in surgical margins May predict recurrence in head and neck cancer. Oncogene. 15(5):579–584. [DOI] [PubMed] [Google Scholar]
- Puerto Galvis CE, Vargas Méndez LY, Kouznetsov VV. 2013. Cantharidin-based small molecules as potential therapeutic agents. Chem Biol Drug Des. 82(5):477–499. [DOI] [PubMed] [Google Scholar]
- Rauh R, Kahl S, Boechzelt H, Bauer R, Kaina B, Efferth T. 2007. Molecular biology of cantharidin in cancer cells. Chin Med. 2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakoff JA, Ackland SP, Baldwin ML, Keane MA, McCluskey A. 2002. Anticancer activity and protein phosphatase 1 and 2A inhibition of a new generation of cantharidin analogues. Invest New Drugs. 20(1):1–11. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. 2001. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 7(6):1165–1176. [DOI] [PubMed] [Google Scholar]
- Shan HB, Cai YC, Liu Y, Zeng WN, Chen HX, Fan BT, Liu XH, Xu ZL, Wang B, Xian LJ. 2006. Cytotoxicity of cantharidin analogues targeting protein phosphatase 2A. Anticancer Drugs. 17(8):905–911. [DOI] [PubMed] [Google Scholar]
- Silverberg JI, Silverberg NB. 2012. Adjunctive trichloroacetic acid therapy enhances squaric acid response to verruca vulgaris. J Drugs Dermatol. 11(10):1228–1230. [PubMed] [Google Scholar]
- Singleton DC, Harris AL. 2012. Targeting the ATF4 pathway in cancer therapy. Expert Opin Ther Targets. 16(12):1189–1202. [DOI] [PubMed] [Google Scholar]
- Smith EM, Ritchie JM, Summersgill KF, Hoffman HT, Wang DH, Haugen TH, Turek LP. 2004. Human papillomavirus in oral exfoliated cells and risk of head and neck cancer. J Natl Cancer Inst. 96(6):449–455. [DOI] [PubMed] [Google Scholar]
- Till JS, Majmudar BN. 1981. Cantharidin poisoning. South Med J. 74(4):444–447. [DOI] [PubMed] [Google Scholar]
- Tseng IJ, Sheu SY, Chen YT, Huang CY, Lin CT, Lin PY. 2012. A simple procedure for preparation of N-thiazol, thiadiazol, pyridyl and sulfanylamidocantharidinimines analogues and evaluation of their cytotoxicities against human HL-60, MCF7, Neuro-2a and A549 carcinoma cell. Chem Pharm Bull (Tokyo). 60(11):1453–1457. [DOI] [PubMed] [Google Scholar]
- Verma AK, Prasad SB. 2013. Changes in glutathione, oxidative stress and mitochondrial membrane potential in apoptosis involving the anticancer activity of cantharidin isolated from redheaded blister beetles, Epicauta hirticornis . Anticancer Agents Med Chem. 13(7):1096–1114. [DOI] [PubMed] [Google Scholar]
- Wada S, Usami I, Umezawa Y, Inoue H, Ohba S, Someno T, Kawada M, Ikeda D. 2010. Rubratoxin A specifically and potently inhibits protein phosphatase 2A and suppresses cancer metastasis. Cancer Sci. 101(3):743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS. 1989. Medical uses of mylabris in ancient China and recent studies. J Ethnopharmacol. 26(2):147–162. [DOI] [PubMed] [Google Scholar]
- Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, Ron D, Chen W, Trenkle W, Wiestner A, et al. 2009. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci U S A. 106(7):2200–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. 2012. The impact of the unfolded protein response on human disease. J Cell Biol. 197(7):857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi SN, Wass J, Vincent P, Iland H. 1991. Inhibitory effect of norcantharidin on K562 human myeloid leukemia cells in vitro. Leuk Res. 15(10):883–886. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Kelemen O, van Santen MA, Yelton SM, Wendlandt AE, Sviripa VM, Bollen M, Beullens M, Urlaub H, Lührmann R, et al. 2011. Synthesis and characterization of pseudocantharidins, novel phosphatase modulators that promote the inclusion of exon 7 into the SMN (survival of motoneuron) pre-mRNA. J Biol Chem. 286(12):10126–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.