

Abstract
Oscillations in the β-band (8–30 Hz) that emerge in the output nuclei of the basal ganglia during Parkinson's disease, along with an imbalanced activation of the direct and indirect pathways, have been linked to the hypokinetic motor output associated with the disease. Although dopamine depletion causes a change in cellular and network properties in the striatum, it is unclear whether abnormal activity measured in the globus pallidus and substantia nigra pars reticulata is caused by abnormal striatal activity. Here we use a computational network model of medium spiny neurons (MSNs)—fast-spiking interneurons (FSIs), based on data from several mammalian species, and find that robust β-band oscillations and imbalanced firing emerge from implementation of changes to cellular and circuit properties caused by dopamine depletion. These changes include a reduction in connections between MSNs, a doubling of FSI inhibition to D2 MSNs, an increase in D2 MSN dendritic excitability, and a reduction in D2 MSN somatic excitability. The model reveals that the reduced decorrelation between MSNs attributable to weakened lateral inhibition enables the strong influence of synchronous FSIs on MSN firing and oscillations. Weakened lateral inhibition also produces an increased sensitivity of MSN output to cortical correlation, a condition relevant to the parkinsonian striatum. The oscillations of FSIs, in turn, are strongly modulated by fast electrical transmission between FSIs through gap junctions. These results suggest that pharmaceuticals that desynchronize FSI activity may provide a novel treatment for the enhanced β-band oscillations, imbalanced firing, and motor dysfunction in Parkinson's disease.
Keywords: beta-band oscillation, fast-spiking interneurons, gap junctions, Parkinson's disease, striatal imbalance, striatum
Introduction
The pathophysiology of Parkinson's disease begins with the depletion of dopamine from the striatum and leads to an increase in oscillations in the globus pallidus and substantia nigra pars reticulata, especially in the 8–30 Hz range (β-band; Brown and Williams, 2005), and to an imbalance in the activation of the direct and indirect pathways (Albin et al., 1989; Bergman et al., 1990, 1994; Mallet et al., 2006). Studies report synchrony and oscillations within the cortex (Goldberg et al., 2002), the striatum (Courtemanche et al., 2003; Costa et al., 2006; Jáidar et al., 2010; López-Huerta et al., 2013), and between the cortex and striatum (Costa et al., 2006) after dopamine deletion. These observations are consistent with the hypothesis that the striatum is the source of the aberrant activity; however, the closed feedback loop of the basal ganglia could be generating striatal oscillations. Furthermore, observed striatal changes caused by dopamine depletion have not been demonstrated to produce oscillatory activity. Enhanced acetylcholinergic tone in the striatum supports generation of oscillatory activity in both cortex and striatum (McCarthy et al., 2011), but different mechanisms may be operating during dopamine depletion.
Several studies have identified changes in the cellular and circuit properties of the striatum caused by dopamine depletion, but their direct influence on the emergence of abnormal striatal oscillations and firing activity has primarily been speculative. The cellular changes include increases in Ca2+ transients in D2 medium spiny neuron (MSN) distal dendrites (Day et al., 2006, 2008) and a reduction in D2 MSN somatic excitability (Chan et al., 2012). The circuit changes include a drastic reduction in lateral inhibition (LI; MSN interconnections; Taverna et al., 2008; Tecuapetla et al., 2009) and increased feedforward inhibition (FFI) from fast-spiking interneurons (FSIs; Gittis et al., 2011) to D2 MSNs. The limited knowledge on how these changes propagate through the striatal circuit to produce oscillations and changes to firing is attributable to the technical difficulty in experimentally isolating the effects of different GABAergic inputs in vivo.
We developed a mathematical network model consisting of >1000 biophysically realistic MSN and FSI model neurons to answer the following questions. (1) Can the experimentally observed changes in connectivity and intrinsic excitability lead to emergence of β-band oscillations and imbalanced firing in the striatal network and to an increase in striatal susceptibility to cortical correlation? (2) Which cellular- or circuit-level changes are most important in modulating striatal imbalance and oscillations, and are there potential mechanisms that are suitable to target for pharmacological intervention to restore normal oscillatory activity in the striatum during dopamine depletion? The results indicate that the reduction in decorrelation by weakening of LI is critical in allowing FSI oscillatory activity to drive MSN oscillations in the dopamine depletion condition. Weakened LI makes MSNs more sensitive to cortical correlation because FSIs tend to synchronize in response to high cortical correlation. Our mathematical model further predicts that direct reduction of FSI oscillations through blocking gap junctions between FSIs is a viable mechanism for restoring normal oscillatory and firing activity in the striatum.
Materials and Methods
Striatal network.
A previously published striatal network model (Damodaran et al., 2014), consisting of 500 D1 MSNs, 500 D2 MSNs, and 49 FSIs, was modified for the present study by changing intrinsic synaptic connections (i.e., GABAergic connections) as described below. The distance between each MSN soma in the model was 25 μm both in the x-axis and the y-axis (Tunstall et al., 2002), resulting in a 775 × 775 μm2 grid. At each grid location, the assignment of either D1 or D2 MSNs was random, with p = 0.5. Each MSN received input from 55% of striatal FSIs within 100 μm (Tecuapetla et al., 2007), and between 4 and 27 converged on the same MSN (Koós and Tepper, 1999). Based on these estimates, the 49 neuron FSI network corresponded to the FSI network seen by 1000 postsynaptic MSNs. Although the percentage of FSIs is slightly larger than observed experimentally, a smaller number of FSIs would have incorrectly produced homogenous FSI input to each MSN in the network model. The 49 FSIs were evenly distributed in space.
The morphology of both MSN models consisted of 189 compartments with four primary dendrites that divide into eight secondary and then 16 tertiary dendrites. Each primary dendrite was 20 μm long, secondary dendrites were 24 μm, and tertiary dendrites comprised 11 compartments, each 36 μm long. Each MSN model had the following ionic channels: fast sodium, fast and slow transient potassium, inward-rectifying potassium, delayed-rectifier potassium, calcium-dependent potassium, L-type calcium (CaV1.2 and CaV1.3), N-type calcium, R-type calcium, and T-type calcium. In addition, each MSN had the following synaptic channels: AMPA, NMDA, and GABA. D1 and D2 MSN models were created by changing the maximal conductance of intrinsic and synaptic channels (Damodaran et al., 2014) from values used for our previous MSN model (Evans et al., 2012), based on experimental data measuring the effect of D1 and D2 receptor agonists, as summarized by Nicola et al. (2000) and Moyer et al. (2007). Each FSI in this network consisted of 127 compartments: one soma, two primary dendrites, four secondary dendrites, and eight tertiary dendrites. The channels incorporated in this model included a fast-sodium channel, Kv3.1, Kv1.3, an A-type (transient) potassium channel, and AMPA and GABA synaptic channels (Kotaleski et al., 2006). A heterogeneous network of neurons was generated by changing the A-type (transient) potassium channel conductance (both MSNs and FSIs) and NMDA channel conductance (MSN only) by ±10%. The range of activity of MSNs used in the network, in response to current injections, was within the range of experimentally observed responses (Damodaran et al., 2014).
Intrinsic synaptic inputs.
MSNs had 227 GABA synapses (one per isopotential compartment) with a distance-dependent probability of GABAergic connection between MSNs and from FSIs to MSNs. GABAergic synapses on MSNs had a rise time constant of 0.75 ms, decay time constant of 6.7 ms, and reversal potential of −60 mV (Czubayko and Plenz, 2002; Koos et al., 2004; Gittis et al., 2010). Synapses from FSIs had a maximal conductance of 3.6 nS (Gittis et al., 2010), whereas the synapses between MSNs had a maximal conductance of 0.75 nS (Koos et al., 2004). The FSI–MSN synapses also were more proximal than MSN–MSN synapses (Oorschot et al., 2013). The gap junction connections between the FSIs were modeled as resistive elements between the primary dendrites, with a conductance of 0.5 nS, coupling coefficient of 25%, and probability of gap junction connection between nearby FSIs (those within 100 μm) of 0.3 (Koós and Tepper, 1999; Tepper et al., 2004; Hjorth et al., 2009). Each FSI model had 93 GABA synaptic channels with a rise time constant of 1.33 ms, decay time constant of 4 ms, reversal potential of −60 mV, and maximal conductance of 1 nS (Gittis et al., 2010). The probability of chemical synapse connection between FSIs was 0.58 (Gittis et al., 2010) and was independent of the probability of gap junction connection. The difference in FFI and LI connectivity observed between the two MSN types in the control network was implemented for this study. Connection probability from D2 MSNs to either type of MSN was doubled, and strength of connection was doubled from D2 MSNs compared with connections from D1 MSNs (Taverna et al., 2008). Additionally, FSI connections to D1 MSNs were 15% more probable than were FSI–D2 MSN connections (Gittis et al., 2010). The transmission delays were distance-based using a conduction velocity of 0.8 m/s for both FSI and MSN synapses (Wilson, 1986; Wilson et al., 1990; Tepper and Lee, 2007).
Extrinsic synaptic input.
Both MSN classes in this model have 360 AMPA and NMDA synaptic channels, and each FSI model has 127 AMPA synaptic channels. Glutamatergic input to all neurons is simulated as Poisson distributed spike trains (generated using MATLAB, version 2007b; MathWorks) in which each Poisson train represents activity from more than one cortical neuron, and each synaptic channel represents the population of synapses in a single isopotential compartment. Each excitatory synaptic channel in the MSN model receives an input of 5 Hz (unless otherwise stated), and each excitatory synaptic channel in the FSI model also receives an input of 5 Hz (unless otherwise stated; Zheng and Wilson, 2002; Blackwell et al., 2003; Humphries et al., 2009). Because this study focuses on the relationship between striatal activity and motor deficits observed during dopamine depletion, the distribution of cortical inputs to D1 and D2 MSNs matches the values reported from motor cortices to the MSNs with the D1 MSNs receiving 20% fewer inputs than D2 MSNs (Wall et al., 2013). Extrinsic GABA input to FSIs (70% of inputs), representing input from either globus pallidus or striatal neurons not included in the network, is provided by Poisson trains of 2 Hz, producing a total of 207 GABAergic inputs per second (Kotaleski et al., 2006).
To introduce correlations within both the MSN and FSI input, each spike from the set of cortical spike trains was assigned to more than one synapse, with probability p = 1/n, where n = N − , N is the number of synapses, and c = 0.5 (Hjorth et al., 2009). To introduce between-neuron input correlation, an additional shared set of input spikes was generated. The between-neuron input correlation was then adjusted by changing the fraction of input each neuron received from this shared pool (as opposed to the spike trains that were unique to each neuron). Cortical input correlation values of 0–0.5 were referred to as low levels, whereas input correlation values of 0.6–1 were referred to as high levels. For experiments in which the contribution of different intrinsic properties to overall β-band power was investigated, cortical input correlation was fixed at 0.3 (to represent low input correlation) and at 0.6 (to represent high input correlation). This was done to evaluate the contribution of intrinsic properties with extrinsic input fixed.
Dopamine depletion: cellular changes.
The response to current injection and the dendritic excitability of MSNs during dopamine depletion were tuned to match experimental observations. The spine reduction in D2 MSNs (Arbuthnott et al., 2000; Stephens et al., 2005; Day et al., 2006) was implemented by increasing the membrane resistance and decreasing membrane capacitance to account for spine loss (Koch and Zador, 1993) because the single-neuron models did not have explicit spines. Additionally, transient K+ channels have been implicated in modulating dendritic excitability in the MSNs during dopamine depletion (Day et al., 2008), and this was implemented by decreasing KAf channel conductance in D2 MSN distal dendrites.
Dopamine depletion: circuit changes.
MSN–MSN connectivity was modulated differently based on the MSN types. All connections between D1 MSNs were removed, connection probability between D2 MSNs was reduced by 50%, and connection probability between D2 MSNs and D1 MSNs was reduced by 70% (Taverna et al., 2008). The strength of connections between MSNs was reduced by 70%. Connection probability from FSI to D2 MSNs was doubled with no change to the conductance of FSI–MSN connections, as has been reported experimentally (Gittis et al., 2011).
Analysis of spikes.
The simulation time was 2 s with no down states, and Python was used to analyze the resulting network spiking activity. To measure synchrony and oscillatory power, cross-correlograms were constructed for each directly coupled neuron pair in the FSI and MSN network and then averaged over the network (Damodaran et al., 2014). Correlation was corrected for firing frequency by subtracting the shuffled cross-correlograms (Rivlin-Etzion et al., 2006) for the same network condition. The Fourier transform was used for estimation of the power spectra for the different conditions. All simulations were repeated three times, each with a different random seed controlling intrinsic synaptic connections and extrinsic input. The firing frequency was expressed as mean ± SEM of values obtained from the three different runs of each condition. Error bars in the figures represent SEM. Statistical analyses were performed using SAS, with N = 3 networks for each condition. When only two groups were being compared, the procedure TTEST was used and p < 0.01 was considered significant. When more than two groups were compared, ANOVA was performed using the GLM procedure. Post hoc analyses used Bonferroni's correction for multiple comparisons, with p < 0.01 considered significant.
The model was implemented in GENESIS (Bower and Beeman, 2007), and simulations used a time step of 100 μs. The model was based on data from several mammalian species of both sexes. Each network simulation experiment took 3 weeks to run. The simulation and output processing software along with the files used for the simulations are available freely from the authors' website (http://krasnow.gmu.edu/CENlab/) and modelDB (http://senselab.med.yale.edu/ModelDB/).
Results
Implementation of dopamine depletion changes
To accurately model the response of the striatal network to changes in cellular and circuit properties, changes in MSN excitability and inhibitory connectivity were implemented using biophysically and anatomically realistic compartmental neuron models (Evans et al., 2012; Damodaran et al., 2014). Dopamine depletion was implemented by changing excitability through modulation of potassium channels in distal dendrites of D2 MSNs (Day et al., 2008) and reduction of spines in D2 MSNs (see Materials and Methods; Arbuthnott et al., 2000; Stephens et al., 2005; Day et al., 2006) to produce firing frequency in response to somatic current injection (Fig. 1A,C, inset) consistent with experimental findings (Day et al., 2008; Gertler et al., 2008; Chan et al., 2012). The reduction in amplitude of calcium transients with distance from the soma in D1 and D2 MSNs in normal dopamine along with the increase in calcium transient amplitude in distal D2 MSN dendrites during dopamine depletion (Fig. 1B,D) were consistent with empirical observations (Day et al., 2008). In addition, reduction in strength and connectivity of MSNs (Taverna et al., 2008) and a selective increase in FSI–D2 MSN connectivity (Gittis et al., 2011) were implemented as per experimental observations (Fig. 1E).
Figure 1.
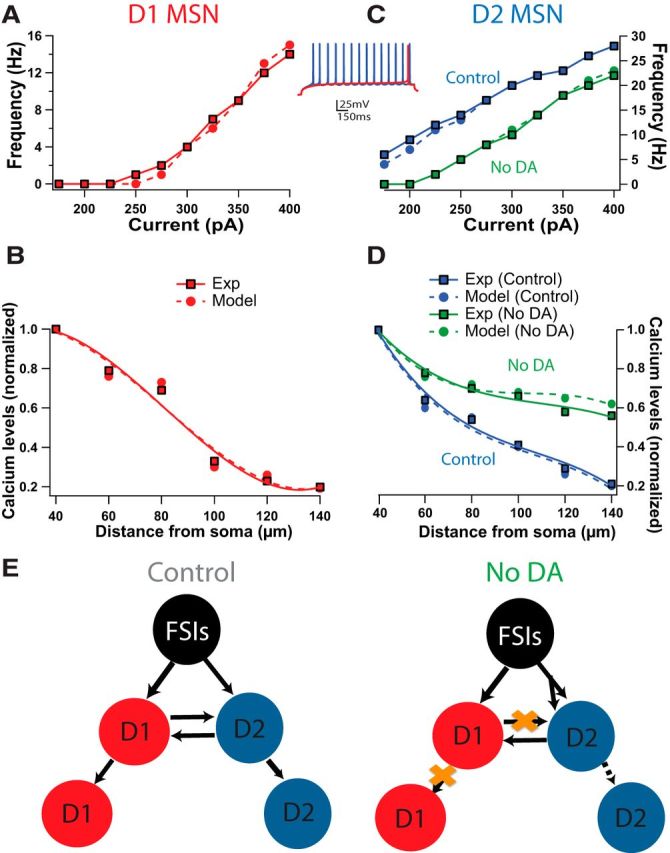
Differences in cellular and circuit properties between control and dopamine depletion conditions. A, C, Current–frequency curves for the model neuron with the median parameters (dashed lines) and experiments (solid lines). D1 MSN (A; red) and D2 MSN (C; Control, blue; No DA, green). Experimental data (solid) were replotted from published reports (Gertler et al., 2008; Chan et al., 2012) for control and dopamine depletion conditions. Inset, Models of D1 and D2 MSNs constructed with differences in intrinsic excitability reproduce electrophysiological dichotomy. The intrinsic channel conductances that differ between D1 and D2 MSNs are the L-type calcium channels, fast-sodium channels, A-type potassium channels, and inward-rectifying potassium channels. B, D, Change in amplitude of calcium transients evoked by action potentials versus distance from the soma is calculated by normalizing the distal Ca2+ transient to the most proximal transient in D1 (B; red, dashed lines) and D2 MSN (D; blue, dashed lines). Simulations are from D1 and D2 MSNs with the median parameters. Experimental data (Exp; solid lines) were replotted from a previously published report (Day et al., 2008) for Control (blue) and No-DA (green) conditions. The magnitude of the Ca2+ transients decrements more in the D1 MSNs versus the D2 MSNs. The distal dendrites of D2 MSNs have enhanced excitability during dopamine depletion versus control. E, Schematic representation of network connections during control and dopamine depletion conditions. During dopamine depletion, increased synaptic connections of D2 MSNs by FSIs have been observed. Additionally, reduced and weaker LI between MSNs, specifically an absence of presynaptic connections from D1 MSNs, has also been observed.
Emergent β-band oscillations and striatal imbalance are sensitive to level of cortical input correlation
Raster plots of our 1049-neuron striatal model (1000 heterogeneous MSNs, 49 heterogeneous FSIs; see Materials and Methods) reveal changes in MSN firing frequency and oscillatory behavior between the control (Fig. 2A) and dopamine depletion (Fig. 2B) conditions similar to that observed in experimentally induced Parkinson's disease. Each excitatory synaptic channel in the MSN and FSI models receives an input of 5 Hz (unless otherwise stated; Zheng and Wilson, 2002; Blackwell et al., 2003; Humphries et al., 2009). The average firing frequency of MSNs and the imbalance in firing between D1 and D2 MSNs are higher in the dopamine-depleted network compared with the control network (Table 1). The average rate for MSNs in the control condition in response to high cortical input correlation (0.6) is 3.6 ± 0.13 Hz, consistent with the average MSN spiking rate seen in vivo during wakefulness (Mahon et al., 2006), with D1 and D2 MSNs firing at similar rates (Fig. 2H, Table 1). MSNs fire at a higher overall rate (5.3 ± 0.2 Hz) in the dopamine-depleted network in response to high cortical input correlation, caused mostly by an increased firing of D2 MSNs (Table 1). The observed increase in MSN firing frequency after dopamine depletion is consistent with some in vivo studies (Kish et al., 1999; Tseng et al., 2001), although there is a report of overall decrease in the firing of all striatal neurons (Chang et al., 2006).
Figure 2.

Emergence of β-band oscillations in a dopamine-depleted striatal network model. A, Raster plot of 200 MSNs in the normal striatal network (Control) in response to low (0.3; A1) and high (0.6; A2) cortical input correlation. B, Raster plot of 200 MSNs in the dopamine-depleted striatal network in response to low (0.3; B1) and high (0.6; B2) cortical input correlation. Raster plots depict the first second of the 2 s simulation, with spikes from D1 MSNs indicated in red and spikes from D2 MSNs indicated in blue. C, Power spectra of the normal striatal network in response to low (0.3; light gray) and high (0.6; black) cortical input correlation. The power of α- and β-band oscillations are higher in the network in response to high cortical input correlation. D, Power spectra of the dopamine-depleted striatal network in response to low (0.3; light green) and high (0.6; dark green) cortical input correlation. Power of α- and β-band oscillations are significantly increased in the dopamine depletion condition compared with the control condition, especially in response to high cortical input correlation. E, F, Plot of β-band oscillation power versus cortical input correlation for D1 MSNs, D2 MSNs, and all MSNs for the control (E) and dopamine depletion (F) conditions. MSNs have a higher power of β-band oscillations and an increased sensitivity to changes in cortical input correlation in the dopamine depletion condition compared with control. β-Band power of D2 MSNs increases more than that of D1 MSNs in the transition from control to dopamine depletion. G, Higher β-band power of MSNs in the dopamine depletion condition compared with the control condition in response to different input frequencies (5, 7, and 10 Hz). The effect is robust to changes in input frequency. The cortical input correlation is 0.6. H, Percentage difference in firing between D1 and D2 MSNs in the control and dopamine depletion conditions in response to different input frequencies (5, 7, and 10 Hz). The percentage difference is calculated as difference divided by mean firing of D1 MSNs. The imbalance in firing between the two MSN classes is also robust to changes in input frequency. The cortical input correlation is 0.6. Error bars represent SEM. ctx corr, Cortical correlation.
Table 1.
Balance in firing for different network conditions
| Condition | D1 | D2 | D1 + D2 | % Difference |
|---|---|---|---|---|
| Control | ||||
| Low corr: 0.3 | 3.3 ± 0.1 | 3.5 ± 0.13 | 3.4 ± 0.1 | 6 |
| High corr: 0.6 | 3.5 ± 0.12 | 3.8 ± 0.14 | 3.65 ± 0.13 | 9 |
| No DA | ||||
| Low corr: 0.3 | 3.9 ± 0.16 | 5.7 ± 0.19 | 4.8 ± 0.16 | 46 |
| High corr: 0.6 | 3.9 ± 0.15 | 6.7 ± 0.2 | 5.3 ± 0.2 | 71 |
| No DA + no gaps | ||||
| Low corr: 0.3 | 3.3 ± 0.12 | 3.5 ± 0.13 | 3.4 ± 0.13 | 6 |
| High corr: 0.6 | 3.4 ± 0.14 | 3.6 ± 0.13 | 3.5 ± 0.14 | 5 |
Cortical input frequency is 5 Hz. Values shown are the mean ± SEM (in Hertz). corr, Correlation.
The raster plots reveal enhanced oscillations in addition to the altered firing rates. In response to highly correlated cortical input, the power spectra of the entire population peaks at a frequency of 21 ± 1.8 Hz in the dopamine-depleted network, whereas the power is more uniform across the different frequencies in the control network (Fig. 2C,D). The overall β-band power of MSNs increases with increasing cortical input correlation, especially after dopamine depletion (Fig. 2E,F). This was confirmed using the GLM procedure with condition and cortical input correlation as the independent variables (F(3,59) = 310.84, p < 0.001; pcondition < 0.001, pcorrelation < 0.001, pcondition × correlation < 0.001). There are also peaks at lower frequencies in the power spectra of the dopamine-depleted network, consistent with studies that report both lower and higher frequency of β-band oscillations in the basal ganglia output nuclei after dopamine depletion (Brown and Williams, 2005). The emergence of β-band oscillations (Fig. 2G) and firing imbalance between D1 and D2 MSNs (Fig. 2H) are independent of the frequency of the input trains, confirming that these effects are not unique to a particular cortical firing frequency.
In both the control and dopamine-depleted networks, β-band power is higher for D2 MSNs than for D1 MSNs (Fig. 2E,F). This was confirmed using the GLM procedure with condition and MSN class as the independent variables at a cortical input correlation of 0.6 (F(3,11) = 46.74, p < 0.001; pcondition < 0.001, pMSN class < 0.001, pcondition × MSN class < 0.001). The cause of this difference in the control network is that D2 MSNs receive less inhibitory inputs than D1 MSNs, as observed experimentally (see Materials and Methods; Taverna et al., 2008; Gittis et al., 2010). When D1 and D2 MSNs receive similar inhibitory input under control conditions, they exhibit similar β-band power (data not shown). The asymmetry between D1 MSN and D2 MSN β-band power after dopamine depletion is explained by the differences in network connections that are producing the enhanced β-band power overall, and these are explained in the next section.
Reduction of LI results in the largest change in β-band power in response to high cortical input correlation
By isolating the different classes of changes implemented in the dopamine-depleted network, we investigated whether any one specific change was critical in producing the increase in β-band oscillations. The results presented for these experiments are for two values of cortical input correlation (low = 0.3 or high = 0.6) so as to evaluate the specific roles of cellular and circuit properties of the network, independent of the changes in extrinsic input. Altering the experimentally observed changes to cellular properties but not the changes to circuit properties (only channel changes) resulted in a small increase in β-band power for both input correlation values (Fig. 3). We then investigated the distinct contribution of alterations in either FFI or LI to changes in network activity. In response to high cortical input correlation, reducing LI alone was sufficient to increase β-band power close to the levels seen in the dopamine depletion condition, whereas increasing FFI connectivity alone raised β-band power significantly less than that produced by reducing LI alone, though it was significantly higher than the power seen in control (Fig. 3A). GLM reveals that the β-band power for the control, dopamine depletion, only LI change, only FFI change, and only cellular changes groups were significantly different from each other (F(4,14) = 89.27, p < 0.001). Post hoc tests (Bonferroni's correction for paired comparisons) show that β-band power for dopamine depletion ≅ only LI change > only FFI change > only cellular changes ≅ control (p < 0.01). In response to low input correlation, reducing LI alone was not sufficient to increase β-band power to the levels seen in the dopamine depletion condition (Fig. 3B); instead, changes to both LI and FFI were required to recapitulate the dopamine-depletion condition. GLM reveals that the β-band power for the control, dopamine depletion, only LI change, only FFI change, and only cellular changes groups were significantly different from each other (F(4,14) = 105.20, p < 0.001). Post hoc tests (Bonferroni's correction for paired comparisons) show that β-band power for dopamine depletion > only LI change > only FFI change > only cellular changes ≅ control (p < 0.01). These results suggest a fundamental role for LI in the striatal network, namely to decorrelate the MSNs from synchronous input. Nonetheless, for decorrelation to play such a significant role during dopamine depletion, there must be sources of highly synchronous input to the network.
Figure 3.
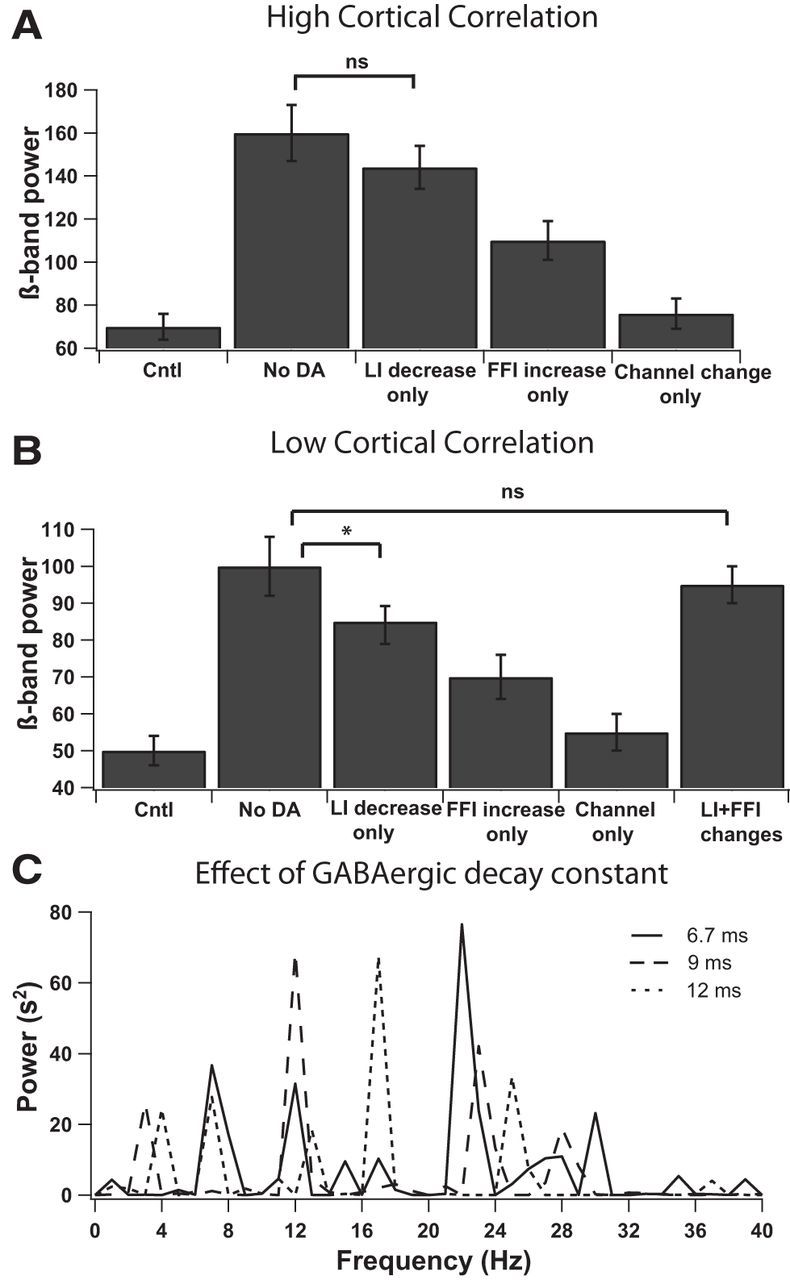
Contribution of network, cell properties and GABAergic decay constant to power of β-band oscillations. A, B, Power of β-band oscillations is significantly increased from control (Cntl) when only LI is decreased or when only FFI is increased. Decreasing LI alone is significantly higher than increasing FFI alone. Changing only cellular properties of MSNs produced small changes to the power of β-band oscillations from the levels seen in control; *p < 0.01. A, In response to high cortical input correlation, decreasing LI alone produces β-band power close to that seen in the dopamine depletion condition. B, In response to low cortical input correlation, both LI and FFI changes together were required to produce β-band power close to that seen in the dopamine depletion condition. C, When the GABAergic decay constant was changed, it shifted the peak oscillation frequency of the MSNs but the peak frequency remained with the β-band. The power spectra represented are for the dopamine depletion condition in response to high cortical input correlation (0.6). Error bars represent SEM.
We observed that the final frequency at which the MSN network oscillates is attributable to an interaction between multiple factors: level of inhibition (LI vs FFI) to MSNs, intrinsic excitability of MSNs, and GABAA receptor decay time constant of MSNs. Changing the GABAergic decay constant shifted the peak oscillation frequency of the MSNs but the peak frequency remained within the β-band, when the constants were within the range of experimentally measured values (Koos et al., 2004; Gittis et al., 2010; Fig. 3C). The power spectra shown are for the dopamine-depletion condition in response to high cortical input correlation (0.6). The oscillations at β-band in the striatal network model were thus an emergent property based on parameters constrained by experimentally observed measures.
Blocking gap junctions restores MSN oscillations and firing during dopamine depletion
Two significant feedforward inputs to MSNs are the cortical (excitatory) and FFI input, and their effects on MSN β-band power are investigated next. The power of β-band oscillations of the MSN population increases only slightly with increasing cortical input correlation under the control condition but increases dramatically during dopamine depletion (Fig. 4A), especially for higher levels of cortical input correlation (0.6–1). Previous research demonstrating that cortical input synchrony is required for gap junctions to correlate FSIs (Hjorth et al., 2009; Damodaran et al., 2014) prompted us to evaluate the oscillations of the FSI network. The power spectra of the FSI network (Fig. 4B) reveals highly oscillatory behavior among the FSIs in the control condition in response to high levels of cortical input correlation. The sharp increase in FSI β-band power at 0.6 cortical input correlation is attributable to a threshold effect, at which the influence of gap junctions between FSIs transitions from a predominantly shunting effect to a predominantly synchronizing effect (Hjorth et al., 2009; Russo et al., 2013). The threshold and the sensitivity of FSIs to cortical input correlation is robust to small changes in gap junction conductance but can be modified by large changes. Specifically, a change from 0.5 to 3 nS reduced the level of cortical input correlation required to significantly increase the power of oscillations of the FSIs to 0.5 instead of 0.6 (data not shown). Although FSIs are connected by GABAergic synapses, these are not sufficient to decorrelate the FSIs, because blocking them did not have an effect on the response of the FSIs to cortical input correlation (Fig. 4B).
Figure 4.

Blocking gap junctions compensates for weak decorrelation by LI during dopamine depletion. A, B, Plot of β-band oscillation power versus cortical input correlation and power spectra for MSNs (A) and FSIs (B). A1, B1, MSNs are more sensitive to higher levels (0.6–1) of cortical input correlation during the dopamine depletion condition (light green) compared with the control condition (black). FSIs with intact gap junctions (GJ, black) are also more sensitive to higher versus lower cortical input correlation levels, exhibiting a sigmoidal shaped input–output curve under the control condition. Removing gap junctions between FSIs (purple) reduced the FSI correlation, especially in response to high cortical input correlation. Removing gap junctions between FSIs in the dopamine-depleted network restores sensitivity of MSNs to cortical input correlation (purple) to the sensitivity seen in the control condition. Blocking chemical synapses between FSIs (light brown) or increasing their conductance to equal the strength of GABAergic synapses between MSNs (cyan) did not alter the sensitivity of FSIs to cortical input correlation. Error bars represent SEM and in some cases are smaller than the symbols. A2, B2, Power spectra of MSNs and FSIs for different conditions in response to high cortical input correlation (0.6). A2, The power of β-band oscillations increase drastically for the dopamine-depleted condition (green) compared with the control condition (black). Blocking gap junctions between FSIs (purple) during the dopamine depletion condition restores the power of the oscillations to control levels. B2, FSIs have higher power of oscillations compared with the MSNs. FSIs do not undergo any changes in the dopamine-depleted condition in the network, and thus they have the same oscillatory power in control and dopamine-depleted networks. Similar to MSNs during dopamine depletion, oscillatory power in the FSIs decreases with gap junction block. C, Blocking all LI resulted in emergent β-band oscillations higher than that seen in dopamine depletion, whereas an increase in LI connectivity from control levels lead to a decrease in β-band power. Reducing the cortical input correlation only to FSIs (0.6 to 0.3) was also sufficient to reduce β-band power of MSNs. The values shown are for the MSN network in response to high cortical input correlation (0.6). The control and dopamine depletion bars are the same as the bars depicted in Figure 3 for the respective conditions and were placed in this panel for comparison purposes. D, Percentage of reduction in β-band power by blocking gap junctions or reducing FSI input correlation for varying levels of intact LI in the MSN network. The lower the level of LI, the higher the percentage of reduction in β-band power in response to reduction in FSI correlation. *p < 0.01. Error bars represent SEM.
We separated the contribution of cortical from FSI correlation by altering FSI correlation using several methods. First, providing low cortical input correlation (0.3) to FSIs, independent of the cortical input correlation to MSNs, was sufficient to bring the MSN β-band power to that seen during the control condition (Fig. 4C; control vs reduced FSI, t(4) = −0.8, p = 0.48). Second, electrical synapses have been implicated to be responsible for oscillations of FSIs in other brain regions (Draguhn et al., 1998; Whittington and Traub, 2003). Thus, we reduced the level of FSI correlation independent of its cortical input correlation by blocking gap junctions. This manipulation drastically reduced the power of β-band oscillations of FSIs (Fig. 4B) and of MSNs in the dopamine-depleted network to that of control levels (Fig. 4A; control vs dopamine depletion + no gap junctions, t(4) = 0.5, p = 0.66). Blocking gap junctions also restored balanced firing between D1 and D2 MSNs in the dopamine-depleted network (Table 1). This effect is independent of the presence of GABAergic synapses between FSIs because blocking or increasing GABAergic conductance between FSIs did not have an effect on the response of the FSIs to cortical correlation (Fig. 4B). This is consistent with a recent study in mouse striatal slices that reported the effect of gap junctions on firing and synchronicity of FSIs being independent of GABAergic synapses (Russo et al., 2013). These results identify a specific and tangible mechanism through which balance and oscillatory power of MSNs can be modulated.
To better understand the contribution of LI toward enhanced β-band oscillations, the interaction between LI manipulation and reduced FSI correlation was evaluated in response to high cortical input correlation. Just eliminating LI in the control network produced highly oscillatory MSN activity in the β-band, whereas doubling LI (compared with control levels) attenuated the existing β-band oscillations in the control network, confirming the robust decorrelation effect of LI (Fig. 4C). Additionally, the percentage reduction in MSN β-band power was independent of the method for reducing FSI correlation (either blocking gap junctions or reducing FSI input correlation) but did depend on the level of LI between the MSNs (GLM, F(7,23) = 104.56, p < 0.001; pmethod = 0.3482; pLI < 0.001; Fig. 4D). Specifically, eliminating FSI correlation has a significantly smaller effect on the normal or high LI connection networks. Post hoc tests show that percentage reduction in β-band power in the MSNs attributable to reduction of FSI correlation for no LI > dopamine depletion > control > double LI (p < 0.01). In summary, the reduced decorrelation attributable to weaker LI connectivity allows MSNs to be controlled by increased FSI correlation and increased cortical correlation.
The importance of β-band oscillations in Parkinson's disease has been studied extensively, but the contribution of oscillations in other bands, specifically the α-band, is less clear. Pathological oscillations in the α-band (3–8 Hz) develop in the globus pallidus internal segment and in the subthalamic nucleus of some monkeys after MPTP treatment and of Parkinson's disease patients with tremor at rest (Brown, 2003; Hutchison et al., 2004). Our striatal network model is consistent with this observation and produces a doubling in the power of emergent α-band oscillations, in response to high input correlation, in dopamine depletion compared with control. Blocking gap junctions between FSIs in the dopamine-depleted network results in a reduction in the α-band power to levels slightly lower than the control condition (Table 2). Our network also produces γ-band oscillations, but they had lower power and did not change significantly across conditions. Thus, although dopamine depletion and restorative measures applied to FSIs predominantly affect β-band oscillations, the effect on α-band oscillations is consistent with existing experiments and theories on Parkinson's disease.
Table 2.
α-Band power for different conditions
| Condition | α-Band power |
|---|---|
| Control (high corr: 0.6) | 21 ± 1.8 |
| Dopamine depletion (high corr: 0.6) | 40 ± 3.3 |
| Dopamine depletion + no GJ (high corr: 0.6) | 16 ± 1.1 |
Values shown are the mean ± SEM (in s2). corr, Correlation; GJ, gap junction.
Striatal network responds quickly to increases in cortical correlation
Our simulation results suggest that synchrony and oscillations emerge from the feedback loop from the cortex to the striatum and ultimately back to the cortex. Small increases in cortical correlation, as observed experimentally (Riehle et al., 1997, 2000; Engelhard et al., 2013), are decorrelated by the normal striatum but instead are amplified by the dopamine-depleted striatum (Fig. 5A). The enhanced striatal synchrony, possibly amplified by globus pallidus and subthalamic nucleus (Plenz and Kital, 1999; Brown, 2007), are transmitted back to the cortex, enhancing its correlation even further. Our simulations show that 1 s (or longer) of cortical correlation is sufficient to produce striatal synchrony and oscillations, but what if the increases in cortical correlation are briefer than 1 s? For this positive feedback loop of correlation to produce the pathological synchrony and oscillations, the striatum should be able to respond quickly to this transient increase. We tested this hypothesis by measuring the time required for the striatum to increase its correlation after a change in cortical correlation. Results show that the increase in striatal oscillations is detectable within ∼60 ms (Fig. 5B,C), suggesting that the network does not require a long period of time to register a change in cortical correlation. Under dopamine depletion, this rapid response allows the correlation feedback loop, under dopamine depletion, to amplify the normal, transient increases in correlation observed during behavior.
Figure 5.
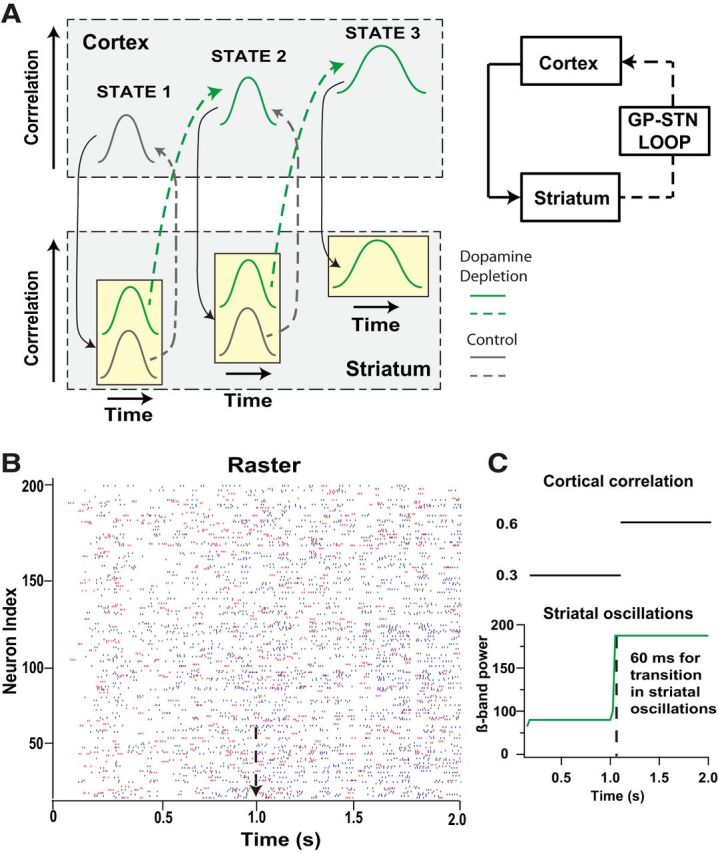
Contribution of striatum to abnormal oscillatory activity experimentally observed in Parkinson's disease. A, Feedback loop amplifies correlation during dopamine depletion. Each column represents a different state, and within each column the x-axis represents time. The tan boxes with gray and green bell-shaped traces represent choice points, in which the subsequent state of the cortex–striatum loop depends on whether dopamine is normal (gray) or depleted (green). A normal transient increase in cortical correlation (State 1, gray cortical trace) is decorrelated by the normal striatum (State 1, striatal gray trace); thus, the cortex and striatum remain in the normal state of transient increases in correlation. In the dopamine-depleted striatum (State 1, striatal green trace), the transient increase in cortical correlation leads to a higher striatal correlation, with a subsequent transition into State 2, which has a higher cortical correlation. Feedback from the striatum to the cortex through the globus pallidus–subthalamic nucleus (GP-STN) loop contributes toward increased cortical correlation (State 2, cortical green trace). The normal striatum can decorrelate even this elevated cortical correlation (State 2, striatal gray trace), but the dopamine-depleted striatum produces an additional increase in correlation (State 2, striatal green trace) with a subsequent transition into State 3 with higher and more prolonged cortical correlation. In State 3, the striatum is unable to decorrelate the cortex; thus, no gray, low correlation striatal trace is shown. B, Raster plot of 200 MSNs in the dopamine-depleted striatal network in response to a switch in cortical input correlation from 0.3 to 0.6 at t = 1 s (indicated by arrow). C, Cortical input correlation (top) and striatal oscillations, measured as power of β-band oscillations (bottom), showing that the dopamine-depleted striatal network takes only ∼60 ms to respond to the switch in cortical input correlation. The first 0.1 s was eliminated from the analysis because it represents a transient increase as the network transitions from the down state.
Discussion
Our results, using a striatal network model, demonstrate that dopamine depletion produces an increase in striatal oscillations in the β-band, as measured by multiunit activity. Because of the difficulty in recording simultaneously from multiple units in the striatum, most measurements of synchrony and oscillations in the striatum use local field recordings (Bergman et al., 1994; Mallet et al., 2006). Our measurements of correlation between MSNs is based on directly coupled pairs of MSNs, whereas measurement of correlation among the entire population reveals no correlations, suggesting that even multiunit recordings using electrode arrays in vivo may not detect such correlations. However, our result is consistent with measurements from dopamine-depleted brain slices that show increases in the number of coactive neurons (Jáidar et al., 2010). The most significant mechanism producing the increased oscillatory activity is a reduction in LI caused by dopamine depletion, making MSNs more sensitive to correlated input from the cortex and FSIs.
Firing imbalance between D1 and D2 MSNs accompanies changes to β-band oscillations
The dopamine-depleted network also exhibits emergent changes to overall MSN spiking frequency and imbalance in firing between D1 and D2 MSNs. Our network exhibits an overall increase in firing, caused by a much higher increase in D2 MSNs than in D1 MSNs, resulting in an imbalance in activity between the two classes. This result is consistent with two in vivo studies that reported increases in MSN firing in 6-OHDA animals (Kish et al., 1999; Tseng et al., 2001), although another study showed a decrease in overall striatal activity (Chang et al., 2006). The increased firing of D2 MSNs and imbalance between D2 MSNs and D1 MSNs during dopamine depletion also is consistent with in vivo studies on 6-OHDA animals with identified MSNs (Mallet et al., 2006).
Interneuron contributions to striatal β-band oscillations
In addition to FFI from FSIs and LI from MSNs, MSNs are modulated by cholinergic interneurons in the striatum (Kreitzer, 2009), whose activity is modulated by dopamine. Changes in cellular and circuit properties of the MSN network in our model are based predominantly on the 6-OHDA experimental model in which acetylcholine levels increase after dopamine depletion (Ikarashi et al., 1997). Therefore, the effects of dopamine-mediated changes on cholinergic modulation of the MSN network and consequently on MSN firing are accounted for in the model. A recent study that modeled the effects of cholinergic modulation reported that increased cholinergic modulation results in reduced potassium currents in MSNs, consequently leading to stronger effects of MSN inhibition and the emergence of β-band oscillations in dopamine depletion (McCarthy et al., 2011). In addition, bath application of acetylcholine results in a reduction of FSI–MSN conductance (Koós and Tepper, 2002); however, a recent in vivo study has shown that, during dopamine depletion, FSI–MSN conductance is similar to that recorded during healthy conditions (Gittis et al., 2011). This suggests that changes in striatal activity caused by dopamine depletion cannot be explained solely by increases in acetylcholinergic tone. Nonetheless, it remains possible that abnormalities in neuromodulators evolve during Parkinson's disease, with a phase in which dopamine depletion produces β-band oscillations in the striatal network and a phase in which increased acetylcholine levels produce β-band oscillations.
Our computational model suggests that, although FFI from FSIs strongly contributes to the high β-band oscillations in the striatum during dopamine depletion, the decrease in LI critically allows the MSNs to be synchronized by the FSIs. Interneurons in other brain regions have been shown to maintain large-scale oscillations at various frequencies (Gray, 1994; Buzsáki and Chrobak, 1995), and FSIs in our network produce strong oscillations in the 20–60 Hz range, consistent with the firing patterns observed in vivo (Sciamanna and Wilson, 2011). Although not included in our model, low-threshold spiking interneurons exhibit oscillatory activity at lower frequencies than FSIs (Beatty et al., 2012) and may contribute to MSN synchrony but are not required to produce β-band oscillations in our model. The oscillations in MSNs emerge at lower frequencies than those of FSIs, indicating that correlation between oscillations and frequency does not imply causation. Despite the strong oscillatory power of FSIs, incorporating only the increase in FSI–MSN connectivity leads to an increase in MSN β-band power smaller than that of reducing LI alone. Nonetheless, the increase in β-band power is consistent with results from a simple striatal model showing that increased FFI alone could produce synchrony (Gittis et al., 2011). In contrast to other brain regions, strong control of MSN synchrony by FSIs is not observed in the striatum because the effect of oscillatory FFI on the MSNs is opposed by strong LI under control dopamine conditions but is unmasked when LI is reduced in Parkinson's disease. The powerful role of LI seems counterintuitive given the observation of inhibition from MSNs being weaker in strength and more distal compared with FFI inhibition (Plenz, 2003). Conversely, our computational result is supported by a recent experimental study showing that LI is strong enough to decrease firing of a subset of MSNs in response to antidromic stimulation from the globus pallidus (López-Huerta et al., 2013). In summary, of the two circuits, changes to the LI circuit produce more widespread changes to MSN oscillations than do changes to the FFI circuit.
Gap junctions as a target to restore basal ganglia oscillations and balance
To restore activity to control levels in the parkinsonian striatum, mechanisms that selectively modulate FSI synchrony were targeted. FSIs are not simple “integrate and fire” devices because transient activation may activate voltage-dependent conductances and enable the cell membrane potential to oscillate (Sciamanna and Wilson, 2011). Specifically, interneurons have been demonstrated to maintain population synchrony through gap junctions in the hippocampus and cortex (Michelson and Wong, 1994; Draguhn et al., 1998; Whittington and Traub, 2003; Nimmrich et al., 2005) but not the normal striatum (Berke, 2008; Russo et al., 2013). Accordingly, removing fast electrical transmission between FSIs by blocking gap junctions was sufficient to reduce the power of β-band oscillations in the dopamine-depleted network, specifically in response to high cortical input correlation. Thus, gap junctions are critical to maintain synchrony and in the modulation of oscillations in the FSI network and consequently in the MSNs.
This action of gap junctions likely is present in vivo, although functional coupling between striatal FSIs has not yet been observed in vivo. Several reasons may explain why electrical coupling between FSIs has been demonstrated in multiple in vitro studies (Koós and Tepper, 1999; Russo et al., 2013) but not in vivo. One reason is that the coupling is quite weak and uncorrelated cortical inputs do not produce correlated FSI firing. A second reason is that, even in slices, demonstrating FSI coupling required repetitive action potential initiation in one of a pair of coupled neurons. The high cortical input correlation required to synchronize FSIs does indeed occur transiently during motor activity. Gage et al. (2010) showed that a large number of FSIs are active around the time of action selection, and motor planning has been suggested to result in increases in cortical spike synchronization (Riehle et al., 1997, 2000). Our model thus makes an important, experimentally testable prediction: selectively inhibiting gap junctions is sufficient to restore striatal firing and β-band oscillatory activity in the parkinsonian striatum to control levels.
The dopamine-depleted striatum was sensitive to changes in cortical correlation, and increased cortical correlation was critical to produce increased striatal oscillations. This suggests that a positive feedback loop, from the cortex, to the striatum, and ultimately back to the cortex, is required for the oscillations observed in vivo. If the increase in cortical correlation lasts for a long period of time, the striatum has multiple opportunities to respond to and transmit this increase in cortical correlation. Even if the increase in cortical correlation is transient, as observed during behavior (Averbeck and Lee, 2004; Harris and Thiele, 2011), the striatal network model requires <60 ms to respond to a change in cortical correlation. The striatal output reaches the cortex via the globus pallidus; thus, the feedback loop will not function if this structure decorrelates striatal output. The increased synchrony in the globus pallidus during dopamine depletion (Brown, 2007) and the oscillatory feedback loop between the globus pallidus and subthalamic nucleus (Plenz and Kital, 1999) appears to amplify, rather than reduce, striatal oscillations. Furthermore, the recently discovered feedback connections from the globus pallidus to striatum (Mallet et al., 2012) may be an additional source of synchronizing input to the striatum not yet included in our model. Collectively, our network simulations together with experimental observations suggest that the positive feedback loop of correlation from the cortex to the stratum and back to the cortex through the globus pallidus can produce the pathological synchrony and oscillations observed in Parkinson's disease. Opening up the feedback loop at any stage should normalize basal ganglia activity, and our model suggests that the striatum is a plausible stage because reducing FFI synchrony, by blocking gap junctions of the FSIs, is sufficient to restore sensitivity of the striatum to changes in cortical synchrony to control levels.
Footnotes
This work was supported by Office of Naval Research Multidisciplinary University Research Initiatives Grant N00014-10-1-0198 and through the joint National Institutes of Health/National Science Foundation Collaborative Research in Computational Neuroscience program through National Institute on Alcohol Abuse and Alcoholism Grant RO1AA-16022.
The authors declare no competing financial interests.
References
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-X. [DOI] [PubMed] [Google Scholar]
- Arbuthnott GW, Ingham CA, Wickens JR. Dopamine and synaptic plasticity in the neostriatum. J Anat. 2000;196:587–596. doi: 10.1046/j.1469-7580.2000.19640587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averbeck BB, Lee D. Coding and transmission of information by neural ensembles. Trends Neurosci. 2004;27:225–230. doi: 10.1016/j.tins.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Beatty JA, Sullivan MA, Morikawa H, Wilson CJ. Complex autonomous firing patterns of striatal low-threshold spike interneurons. J Neurophysiol. 2012;108:771–781. doi: 10.1152/jn.00283.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:507–520. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- Berke JD. Uncoordinated firing rate changes of striatal fast-spiking interneurons during behavioral task performance. J Neurosci. 2008;28:10075–10080. doi: 10.1523/JNEUROSCI.2192-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell KT, Czubayko U, Plenz D. Quantitative estimate of synaptic inputs to striatal neurons during up and down states in vitro. J Neurosci. 2003;23:9123–9132. doi: 10.1523/JNEUROSCI.23-27-09123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower JM, Beeman D. Constructing realistic neural simulations with GENESIS. Methods Mol Biol. 2007;401:103–125. doi: 10.1007/978-1-59745-520-6_7. [DOI] [PubMed] [Google Scholar]
- Brown P. Oscillatory nature of human basal ganglia activity: relationship to the pathophysiology of Parkinson's disease. Mov Disord. 2003;18:357–363. doi: 10.1002/mds.10358. [DOI] [PubMed] [Google Scholar]
- Brown P. Abnormal oscillatory synchronisation in the motor system leads to impaired movement. Curr Opin Neurobiol. 2007;17:656–664. doi: 10.1016/j.conb.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Brown P, Williams D. Basal ganglia local field potential activity: character and functional significance in the human. Clin Neurophysiol. 2005;116:2510–2519. doi: 10.1016/j.clinph.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Chrobak JJ. Temporal structure in spatially organized neuronal ensembles: a role for interneuronal networks. Curr Opin Neurobiol. 1995;5:504–510. doi: 10.1016/0959-4388(95)80012-3. [DOI] [PubMed] [Google Scholar]
- Chan CS, Peterson JD, Gertler TS, Glajch KE, Quintana RE, Cui Q, Sebel LE, Plotkin JL, Shen W, Heiman M, Heintz N, Greengard P, Surmeier DJ. Strain-specific regulation of striatal phenotype in Drd2-eGFP BAC transgenic mice. J Neurosci. 2012;32:9124–9132. doi: 10.1523/JNEUROSCI.0229-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Shi LH, Luo F, Woodward DJ. Neural responses in multiple basal ganglia regions following unilateral dopamine depletion in behaving rats performing a treadmill locomotion task. Exp Brain Res. 2006;172:193–207. doi: 10.1007/s00221-005-0312-7. [DOI] [PubMed] [Google Scholar]
- Costa RM, Lin SC, Sotnikova TD, Cyr M, Gainetdinov RR, Caron MG, Nicolelis MAL. Rapid alterations in corticostriatal ensemble coordination during acute dopamine-dependent motor dysfunction. Neuron. 2006;52:359–369. doi: 10.1016/j.neuron.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Courtemanche R, Fujii N, Graybiel AM. Synchronous, focally modulated beta-band oscillations characterize local field potential activity in the striatum of awake behaving monkeys. J Neurosci. 2003;23:11741–11752. doi: 10.1523/JNEUROSCI.23-37-11741.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czubayko U, Plenz D. Fast synaptic transmission between striatal spiny projection neurons. Proc Natl Acad Sci USA. 2002;99:15764–15769. doi: 10.1073/pnas.242428599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damodaran S, Evans RC, Blackwell KT. Synchronized firing of fast-spiking interneurons is critical to maintain balanced firing between direct and indirect pathway neurons of the striatum. J Neurophysiol. 2014;111:836–848. doi: 10.1152/jn.00382.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ. Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci. 2008;28:11603–11614. doi: 10.1523/JNEUROSCI.1840-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draguhn A, Traub RD, Schmitz D, Jefferys JG. Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro. Nature. 1998;394:189–192. doi: 10.1038/28184. [DOI] [PubMed] [Google Scholar]
- Engelhard B, Ozeri N, Israel Z, Bergman H, Vaadia E. Inducing gamma oscillations and precise spike synchrony by operant conditioning via brain-machine interface. Neuron. 2013;77:361–375. doi: 10.1016/j.neuron.2012.11.015. [DOI] [PubMed] [Google Scholar]
- Evans RC, Morera-Herreras T, Cui Y, Du K, Sheehan T, Kotaleski JH, Venance L, Blackwell KT. The effects of NMDA subunit composition on calcium influx and spike timing-dependent plasticity in striatal medium spiny neurons. PLoS Comput Biol. 2012;8:e1002493. doi: 10.1371/journal.pcbi.1002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage GJ, Stoetzner CR, Wiltschko AB, Berke JD. Selective activation of striatal fast-spiking interneurons during choice execution. Neuron. 2010;67:466–479. doi: 10.1016/j.neuron.2010.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler TS, Chan CS, Surmeier DJ. Dichotomous anatomical properties of adult striatal medium spiny neurons. J Neurosci. 2008;28:10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci. 2010;30:2223–2234. doi: 10.1523/JNEUROSCI.4870-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Hang GB, LaDow ES, Shoenfeld LR, Atallah BV, Finkbeiner S, Kreitzer AC. Rapid target-specific remodeling of fast-spiking inhibitory circuits after loss of dopamine. Neuron. 2011;71:858–868. doi: 10.1016/j.neuron.2011.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Boraud T, Maraton S, Haber SN, Vaadia E, Bergman H. Enhanced synchrony among primary motor cortex neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine primate model of Parkinson's disease. J Neurosci. 2002;22:4639–4653. doi: 10.1523/JNEUROSCI.22-11-04639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray CM. Synchronous oscillations in neuronal systems: mechanisms and functions. J Comput Neurosci. 1994;1:11–38. doi: 10.1007/BF00962716. [DOI] [PubMed] [Google Scholar]
- Harris KD, Thiele A. Cortical state and attention. Nat Rev Neurosci. 2011;12:509–523. doi: 10.1038/nrn3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjorth J, Blackwell KT, Kotaleski JH. Gap junctions between striatal fast-spiking interneurons regulate spiking activity and synchronization as a function of cortical activity. J Neurosci. 2009;29:5276–5286. doi: 10.1523/JNEUROSCI.6031-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries MD, Wood R, Gurney K. Dopamine-modulated dynamic cell assemblies generated by the GABAergic striatal microcircuit. Neural Netw. 2009;22:1174–1188. doi: 10.1016/j.neunet.2009.07.018. [DOI] [PubMed] [Google Scholar]
- Hutchison WD, Dostrovsky JO, Walters JR, Courtemanche R, Boraud T, Goldberg J, Brown P. Neuronal oscillations in the basal ganglia and movement disorders: evidence from whole animal and human recordings. J Neurosci. 2004;24:9240–9243. doi: 10.1523/JNEUROSCI.3366-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikarashi Y, Takahashi A, Ishimaru H, Arai T, Maruyama Y. Regulation of Dopamine D1 and D2 receptors on striatal acetylcholine release in rats. Brain Res Bull. 1997;43:107–115. doi: 10.1016/S0361-9230(96)00351-6. [DOI] [PubMed] [Google Scholar]
- Jáidar O, Carrillo-Reid L, Hernández A, Drucker-Colín R, Bargas J, Hernández-Cruz A. Dynamics of the parkinsonian striatal microcircuit: entrainment into a dominant network state. J Neurosci. 2010;30:11326–11336. doi: 10.1523/JNEUROSCI.1380-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish LJ, Palmer MR, Gerhardt GA. Multiple single-unit recordings in the striatum of freely moving animals: effects of apomorphine and d-amphetamine in normal and unilateral 6-hydroxydopamine-lesioned rats. Brain Res. 1999;833:58–70. doi: 10.1016/S0006-8993(99)01496-1. [DOI] [PubMed] [Google Scholar]
- Koch C, Zador A. The function of dendritic spines: devices subserving biochemical rather than electrical compartmentalization. J Neurosci. 1993;13:413–422. doi: 10.1523/JNEUROSCI.13-02-00413.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Dual cholinergic control of fast-spiking interneurons in the neostriatum. J Neurosci. 2002;22:529–535. doi: 10.1523/JNEUROSCI.22-02-00529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos T, Tepper JM, Wilson CJ. Comparison of IPSCs evoked by spiny and fast-spiking neurons in the neostriatum. J Neurosci. 2004;24:7916–7922. doi: 10.1523/JNEUROSCI.2163-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotaleski JH, Plenz D, Blackwell KT. Using potassium currents to solve signal-to-noise problems in inhibitory feedforward networks of the striatum. J Neurophysiol. 2006;95:331–341. doi: 10.1152/jn.00063.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC. Physiology and pharmacology of striatal neurons. Annu Rev Neurosci. 2009;32:127–147. doi: 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- López-Huerta VG, Carrillo-Reid L, Galarraga E, Tapia D, Fiordelisio T, Drucker-Colin R, Bargas J. The balance of striatal feedback transmission is disrupted in a model of parkinsonism. J Neurosci. 2013;33:4964–4975. doi: 10.1523/JNEUROSCI.4721-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon S, Vautrelle N, Pezard L, Slaght SJ, Deniau JM, Chouvet G, Charpier S. Distinct patterns of striatal medium spiny neuron activity during the natural sleep–wake cycle. J Neurosci. 2006;26:12587–12595. doi: 10.1523/JNEUROSCI.3987-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Ballion B, Le Moine C, Gonon F. Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J Neurosci. 2006;26:3875–3884. doi: 10.1523/JNEUROSCI.4439-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Micklem BR, Henny P, Brown MT, Williams C, Bolam JP, Nakamura KC, Magill PJ. Dichotomous organization of the external globus pallidus. Neuron. 2012;74:1075–1086. doi: 10.1016/j.neuron.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, Moore-Kochlacs C, Gu X, Boyden ES, Han X, Kopell N. Striatal origin of the pathologic beta oscillations in Parkinson's disease. Proc Natl Acad Sci USA. 2011;108:11620–11625. doi: 10.1073/pnas.1107748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson HB, Wong RK. Synchronization of inhibitory neurones in the guinea-pig hippocampus in vitro. J Physiol. 1994;477:35–45. doi: 10.1113/jphysiol.1994.sp020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JT, Wolf JA, Finkel LH. Effects of dopaminergic modulation on the integrative properties of the ventral striatal medium spiny neuron. J Neurophysiol. 2007;98:3731–3748. doi: 10.1152/jn.00335.2007. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- Nimmrich V, Maier N, Schmitz D, Draguhn A. Induced sharp wave-ripple complexes in the absence of synaptic inhibition in mouse hippocampal slices. J Physiol. 2005;563:663–670. doi: 10.1113/jphysiol.2004.079558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oorschot DE, Lin N, Cooper BH, Reynolds JNJ, Sun H, Wickens JR. Synaptic connectivity between rat striatal spiny projection neurons in vivo: Unexpected multiple somatic innervation in the context of overall sparse proximal connectivity. Basal Ganglia. 2013;3:93–108. doi: 10.1016/j.baga.2013.04.001. [DOI] [Google Scholar]
- Plenz D. When inhibition goes incognito: feedback interaction between spiny projection neurons in striatal function. Trends Neurosci. 2003;26:436–443. doi: 10.1016/S0166-2236(03)00196-6. [DOI] [PubMed] [Google Scholar]
- Plenz D, Kital ST. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus. Nature. 1999;400:677–682. doi: 10.1038/23281. [DOI] [PubMed] [Google Scholar]
- Riehle A, Grün S, Diesmann M, Aertsen A. Spike synchronization and rate modulation differentially involved in motor cortical function. Science. 1997;278:1950–1953. doi: 10.1126/science.278.5345.1950. [DOI] [PubMed] [Google Scholar]
- Riehle A, Grammont F, Diesmann M, Grün S. Dynamical changes and temporal precision of synchronized spiking activity in monkey motor cortex during movement preparation. J Physiol Paris. 2000;94:569–582. doi: 10.1016/S0928-4257(00)01100-1. [DOI] [PubMed] [Google Scholar]
- Rivlin-Etzion M, Ritov Y, Heimer G, Bergman H, Bar-Gad I. Local shuffling of spike trains boosts the accuracy of spike train spectral analysis. J Neurophysiol. 2006;95:3245–3256. doi: 10.1152/jn.00055.2005. [DOI] [PubMed] [Google Scholar]
- Russo G, Nieus TR, Maggi S, Taverna S. Dynamics of action potential firing in electrically connected striatal fast-spiking interneurons. Front Cell Neurosci. 2013;7:209. doi: 10.3389/fncel.2013.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Wilson CJ. The ionic mechanism of gamma resonance in rat striatal fast-spiking neurons. J Neurophysiol. 2011;106:2936–2949. doi: 10.1152/jn.00280.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens B, Mueller AJ, Shering AF, Hood SH, Taggart P, Arbuthnott GW, Bell JE, Kilford L, Kingsbury AE, Daniel SE, Ingham CA. Evidence of a breakdown of corticostriatal connections in Parkinson's disease. Neuroscience. 2005;132:741–754. doi: 10.1016/j.neuroscience.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Taverna S, Ilijic E, Surmeier DJ. Recurrent collateral connections of striatal medium spiny neurons are disrupted in models of Parkinson's disease. J Neurosci. 2008;28:5504–5512. doi: 10.1523/JNEUROSCI.5493-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Carrillo-Reid L, Bargas J, Galarraga E. Dopaminergic modulation of short-term synaptic plasticity at striatal inhibitory synapses. Proc Natl Acad Sci USA. 2007;104:10258–10263. doi: 10.1073/pnas.0703813104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Koós T, Tepper JM, Kabbani N, Yeckel MF. Differential dopaminergic modulation of neostriatal synaptic connections of striatopallidal axon collaterals. J Neurosci. 2009;29:8977–8990. doi: 10.1523/JNEUROSCI.6145-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, Lee CR. GABAergic control of substantia nigra dopaminergic neurons. Prog Brain Res. 2007;160:189–208. doi: 10.1016/S0079-6123(06)60011-3. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Koós T, Wilson CJ. GABAergic microcircuits in the neostriatum. Trends Neurosci. 2004;27:662–669. doi: 10.1016/j.tins.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Kasanetz F, Kargieman L, Riquelme LA, Murer MG. Cortical slow oscillatory activity is reflected in the membrane potential and spike trains of striatal neurons in rats with chronic nigrostriatal lesions. J Neurosci. 2001;21:6430–6439. doi: 10.1523/JNEUROSCI.21-16-06430.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunstall MJ, Oorschot DE, Kean A, Wickens JR. Inhibitory interactions between spiny projection neurons in the rat striatum. J Neurophysiol. 2002;88:1263–1269. doi: 10.1152/jn.2002.88.3.1263. [DOI] [PubMed] [Google Scholar]
- Wall NR, De La Parra M, Callaway EM, Kreitzer AC. Differential innervation of direct- and indirect-pathway striatal projection neurons. Neuron. 2013;79:347–360. doi: 10.1016/j.neuron.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington MA, Traub RD. Interneuron diversity series: inhibitory interneurons and network oscillations in vitro. Trends Neurosci. 2003;26:676–682. doi: 10.1016/j.tins.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Wilson CJ. Postsynaptic potentials evoked in spiny neostriatal projection neurons by stimulation of ipsilateral and contralateral neocortex. Brain Res. 1986;367:201–213. doi: 10.1016/0006-8993(86)91593-3. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Chang HT, Kitai ST. Firing patterns and synaptic potentials of identified giant aspiny interneurons in the rat neostriatum. J Neurosci. 1990;10:508–519. doi: 10.1523/JNEUROSCI.10-02-00508.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng T, Wilson CJ. Corticostriatal combinatorics: the implications of corticostriatal axonal arborizations. J Neurophysiol. 2002;87:1007–1017. doi: 10.1152/jn.00519.2001. [DOI] [PubMed] [Google Scholar]