

Abstract
Spinal cord injury (SCI) patients develop chronic pain involving poorly understood central and peripheral mechanisms. Because dysregulation of the voltage-gated Kv3.4 channel has been implicated in the hyperexcitable state of dorsal root ganglion (DRG) neurons following direct injury of sensory nerves, we asked whether such a dysregulation also plays a role in SCI. Kv3.4 channels are expressed in DRG neurons, where they help regulate action potential (AP) repolarization in a manner that depends on the modulation of inactivation by protein kinase C (PKC)-dependent phosphorylation of the channel's inactivation domain. Here, we report that, 2 weeks after cervical hemicontusion SCI, injured rats exhibit contralateral hypersensitivity to stimuli accompanied by accentuated repetitive spiking in putative DRG nociceptors. Also in these neurons at 1 week after laminectomy and SCI, Kv3.4 channel inactivation is impaired compared with naive nonsurgical controls. At 2–6 weeks after laminectomy, however, Kv3.4 channel inactivation returns to naive levels. Conversely, Kv3.4 currents at 2–6 weeks post-SCI are downregulated and remain slow-inactivating. Immunohistochemistry indicated that downregulation mainly resulted from decreased surface expression of the Kv3.4 channel, as whole-DRG-protein and single-cell mRNA transcript levels did not change. Furthermore, consistent with Kv3.4 channel dysregulation, PKC activation failed to shorten the AP duration of small-diameter DRG neurons. Finally, re-expressing synthetic Kv3.4 currents under dynamic clamp conditions dampened repetitive spiking in the neurons from SCI rats. These results suggest a novel peripheral mechanism of post-SCI pain sensitization implicating Kv3.4 channel dysregulation and potential Kv3.4-based therapeutic interventions.
Keywords: Kv3.4, pain, potassium channels, protein kinase C, spinal cord injury
Introduction
Spinal cord injury (SCI) affects ∼400,000 people in the United States (Sekhon and Fehlings, 2001). Following SCI, ∼80% of patients report chronic pain in regions below, at, and above the level of injury (below-, at-, and above-level pain; Finnerup et al., 2001; Siddall et al., 2003; Westermann et al., 2012). In rats, unilateral SCI induces bilateral pain phenotypes at and below the level of injury (Detloff et al., 2013). These chronic pain phenotypes might originate from hyperexcitability in both central and peripheral neurons (Carlton et al., 2009; Gwak and Hulsebosch, 2011; Masri and Keller, 2012; Walters, 2012; Whitt et al., 2013). Although central sensitization has received much attention, dorsal root ganglion (DRG) neurons also undergo changes after SCI (Carlton et al., 2001; Hulsebosch et al., 2009; Bedi et al., 2010, 2012; Yang et al., 2014). Peripheral nociceptors at- and below-level exhibit spontaneous activity and the incidence of this activity correlates with the severity of pain (Bedi et al., 2010). Additionally, peripheral nerves above the level of injury exhibit spontaneous activity and hyperexcitability in response to noxious stimuli (Carlton et al., 2009).
The DRG is a heterogeneous tissue, including neurons with small-diameter somas that are likely to be nociceptors (Harper and Lawson, 1985a; Gold et al., 1996a). Neuropathic chronic pain resulting from hyperexcitable of peripheral nociceptors often implicates maladaptive upregulation of voltage-gated Na+ (Nav) channels and downregulation of voltage-gated K+ (Kv) channels in small-diameter nociceptors (Dubin and Patapoutian, 2010; Gold and Gebhart, 2010; Hughes et al., 2012; Tsantoulas and McMahon, 2014). Kv3.4 is one of the Kv channels in nociceptors, which is downregulated in both nerve injury and bone cancer pain models (Chien et al., 2007; Duan et al., 2012). Furthermore, intrathecal injection of Kv3.4 antisense oligonucleotides induces pain behaviors (Chien et al., 2007). However, whether these changes also occur in DRG neurons following SCI remains unknown.
Kv3.4 channels are expressed in muscle, mossy fiber neurons, Purkinje neurons, spinal cord, and dorsal root ganglia (Rettig et al., 1992; Weiser et al., 1994; Martina et al., 2003; Brooke et al., 2004; Chien et al., 2007; Duan et al., 2012; Ritter et al., 2012); however, their role in these tissues is mostly unknown. Previously, we discovered that fast-inactivating Kv3.4 channels are expressed at high levels in small-diameter DRG neurons and confirmed that they undergo dramatic modulation of N-type inactivation by protein kinase C (PKC) in a native environment (Covarrubias et al., 1994; Ritter et al., 2012). Under normal conditions, this modulation mainly regulates action potential (AP) duration and repolarization rate (Ritter et al., 2012), and thus could drive a homeostatic response that is lost in the chronic pain state. Here, we hypothesize that SCI induces dysregulation of Kv3.4 channels, which might in part be responsible for the nociceptors' hyperexcitability and the resulting persistent pain. To test this hypothesis, we investigated three groups of rats (naive, laminectomy, and SCI cohorts) and implemented various complementary approaches to examine the biophysical properties, expression, and localization of Kv3.4 channels and the effects of SCI on spiking properties of DRG neurons. The results show dynamic changes in the inactivation kinetics and expression of Kv3.4 currents up to 6 weeks following surgery. Whereas changes in gating, mRNA and total protein could not account for Kv3.4 current downregulation, changes in surface expression emerged as the basis of Kv3.4 channel dysregulation following SCI. Excitability changes further implicate Kv3.4 channels and dynamic clamping experiments demonstrated that the Kv3.4 current can dampen the excitability of putative DRG nociceptors. This study reports novel dynamic dysregulation affecting Kv3.4 channel expression and modulation in DRG neurons and helps identify new therapeutic interventions to manage chronic pain associated with SCI.
Materials and Methods
Animals and SCI model.
Adult female Sprague-Dawley rats (200–250 g) were purchased from Taconic Farms and treated as approved by the Thomas Jefferson University Institutional Animal Care and Use Committee. SCI and laminectomy surgeries were performed as previously described (Nicaise et al., 2012). Animals were deeply anesthetized using a mixture of ketamine (95 mg/kg), xylazine (10 mg/kg), and acepromazine (0.075 mg/kg). A 1.5 inch incision was made in the skin overlying cervical levels C3–C7. The overlying musculature was incised and reflected and a laminectomy performed at C5. For SCI, the animal was then stabilized with two forceps clamping the C2 and C7 vertebral processes and placed in the Infinite Horizons Impactor (Precision Systems and Instrumentation). A 1 mm impactor tip was centered over the exposed right side of the spinal cord, raised to height of 4 mm, and a 200 kilodyne contusion delivered (range, 195–205 kilodynes). The muscle and skin were closed in layers and animals were allowed to recover for 1, 2, or 6 weeks. Laminectomy controls received all procedures in addition to receiving a contusion and time points indicate time after laminectomy. Naive controls did not experience any surgical procedure. All animals were killed by an overdose of anesthesia (ketamine at 285 mg/kg and xylazine at 30 mg/kg) followed by thoracotomy and perfusion of 400 ml 0.9% sterile saline. For immunohistochemistry saline perfusion was replaced with a 75 ml 0.9% sterile saline perfusion wash followed by perfusion of 400 ml 4% paraformaldehyde in 0.1 m PBS.
Behavioral assays.
Ugo Basile Plantar Test (Hargreaves Apparatus) was used to test for thermal hyperalgesia. The animal was acclimated to a closed clear container with a thin Plexiglas bottom. An infrared probe with an intensity of 40 Watts was applied to the footpad of each forepaw individually, and the latency to withdraw was determined. During each session, each paw was tested three times (with a 5 min break in between each trial) and the results averaged. If the animal did not remove its paw within 30 s, it was considered unresponsive. To test for mechanical hyperalgesia, von Frey hairs (North Coast Touch-Test filaments, Harvard Apparatus) were applied to the forepaw and the threshold force that induced limb withdraw measured. The “up–down” system was used to specifically determine the threshold hair diameter (Chaplan et al., 1994). Briefly, we started with 15 × g of force, corresponding to 15.18 units in the Von Frey hair scale. If the animal responded, a lower diameter was used until the animal did not respond. Conversely, if an animal was unresponsive, the next larger diameter hair was used. This pattern was continued for 20 trials with >60 s between trials. The lowest hair that produced >50% withdraws was determined as threshold. The maximum force used was 100 × g and if an animal was unresponsive to that hair, a threshold of 180 × g (next size up) was assigned. An individual blinded to the condition of the animal measured behavior; however, the majority of SCI animals had a motor phenotype on the side ipsilateral to injury.
Electrophysiology.
DRG neurons were harvested and dissociated as described previously (Ritter et al., 2012). The spinal cord was removed and C1-C6 DRG harvested. Nerve fibers coming from the DRG were trimmed and ganglia placed in Hanks buffered saline solution (HBSS) + 10 mm HEPES. DRG were then treated with collagenase (1.5 mg/ml in HBSS + 10 mm HEPES solution) for 30 min at 37°C followed by two washes with HBSS + 10 mm HEPES. This was followed by an incubation with trypsin (1.0 mg/ml in HBSS + 10 mm HEPES) for 30 min at 37°C. DRGs were then placed in L-15 Leibovitz medium supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 24 mm NaHCO3, and 2% penicillin-streptomycin and mechanically dissociated with a fire polished Pasteur pipette. Dissociated DRG neurons were plated on glass polyornithine-coated coverslips, incubated at 37°C and used within 12 h of harvesting.
Dissociated DRG neurons were used for electrophysiological experiments at room temperature (20–24°C) as previously described (Ritter et al., 2012). A PIP5 micropipette puller (HEKA Instruments) was used to pull patch electrodes made from thin-walled patch glass (Warner Instruments). Electrodes were coated with Sylgard (Dow Corning) and had tip resistances of 1–3 MΩ after fire polishing. The Multiclamp 700B amplifier (Molecular Devices) was used to amplify signals. In most experiments, signals were low-pass filtered at 2 kHz (multiclamp internal 4-pole Bessel filter) and digitized at 10 kHz (Digidata 1440, Molecular Devices). Nonstationary noise analysis experiments were filtered at 1 kHz and sampled at 10 kHz using the same equipment. A computer running Clampex 10.2 (Molecular Devices) was used to collect and store all data except for dynamic-clamp recordings, which were collected and stored using QuB v. 1.4.0.517 (www.qub.buffalo.edu). In current-clamp recordings, a +15.5 mV liquid junction potential was calculated using Clampex 10.2 and corrected offline. For dynamic-clamp experiments, the liquid junction potential of +15.5 mV was corrected online. Cell-attached macropatch recordings had a liquid junction potential of 0 mV and thus required no correction. In cell-attached macropatch experiments, the bath and pipette solution contained the following (in mm): 130 choline-Cl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, and 50 sucrose, pH 7.4. For whole-cell current-clamp and dynamic clamp recordings, the external (bath) solution contained the following (in mm): 130 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, and 10 HEPES, pH 7.4. The internal (pipette) solution in current-clamp recordings contained the following (in mm): 130 K-methanesulfonate, 1 CaCl2, 1 EGTA, 10 HEPES, 2 Mg-ATP, and 0.3 Tris-GTP, pH 7.3.
For cell-attached patch recordings, the following relation was used to determine the net command voltage V applied to the patch membrane:
 |
where VC is the command voltage given by the computer, and VR is the resting membrane potential of the cell. The VR was assumed to be −60 mV based on previous work (Ritter et al., 2012). In current-clamp experiments, rheobase was measured as the minimum 1 s current injection required to fire one AP. For repetitive firing, a stimulus of 2× rheobase was used. The AP duration was measured at 50% (APD50) of the spike height. Phase-plane plots were used to determine the threshold for firing by examining where the plot deviated from 0 mV/ms.
In dynamic-clamp experiments, synthetic currents were added to a neuron in real time while in current-clamp mode. A Kv3.4 Markov model was determined based on DRG Kv3.4 currents as previously described (Fineberg et al., 2012) and QuB dynamic clamp software was used as previously described (Milescu et al., 2008, 2010). To achieve real-time functionality, dynamic-clamp experiments were conducted using a custom built PC running Windows 7 Professional and equipped with an ASRock H61M/U3S3 Intel Motherboard, Seagate DB35 Series 80 GB internal hard drive, Intel Core i5-2320 Sandy Bridge 3.0 GHz Quad Core Processor, Corsair XMS 4 GB DDR3 SDRAM and National Instruments PCI-6251 high-speed data acquisition card (DAQ). Connections, scale factors, and dynamic clamp speed were tested and verified using a model cell with input/output speeds >76 kHz. The Kv3.4 channel Markov model was then added in real time to look at AP duration with a 0.5 ms current injection or repetitive firing in response to a 3× rheobase current injection stimulus. This was done with increasing Kv3.4 channel maximum conductance, with and without an inactivated state in the model.
Clampfit 10.2 (Molecular Devices) and Origin Pro 8.0 (OriginLab) were used to conduct data analysis. The peak chord conductance (Gp) in cell-attached patches was calculated using the following:
 |
where Ip is the peak current, Vnet is the net command voltage seen by the patch and Vrev is the observed reversal potential seen in rat pups, −72.5 mV (Ritter et al., 2012). To normalize the peak chord conductance, the maximum peak chord conductance (Gpmax) was calculated from the best-fit fourth-order Boltzmann function as follows:
 |
where k is the slope factor and Vs is the activation mid-point voltage of a single subunit. The mid-point voltage (V1/2) of peak conductance curve was calculated from the following equation:
 |
A double Boltzmann function was used to describe steady-state inactivation. Equivalent charge (z) was calculated from slope factors using the following at room temperature:
 |
where R and F are known physical constants and T is absolute temperature. Activation, deactivation, and inactivation kinetics were described assuming single exponential trajectories:
 |
where t is time, A is the amplitude, y0 is the offset and Tau is the time constant. Ip distributions were determined by fitting a sum of n Gaussian distributions as follows:
 |
where the independent variable x = Ip and the distribution is described by amplitude (A), width at 50% of A(w) and the center (xc) of the distributions. Nonstationary noise analysis was performed as previously described (Alvarez et al., 2002). Briefly, 150–300 current traces elicited from depolarizations to the test voltage (Vtest) were averaged and the variance (σ2) at each time point determined. The relation between σ2 and the mean current (I) was described by the following:
 |
where i is the unitary current and N is the number of channels in the patch. Further analysis was only pursued for recordings that were well described by the equation above. Unitary conductance (γ) and maximum open probability (PO,max) were then calculated using the following:
 |
 |
Throughout, results are expressed as mean ± SEM unless otherwise noted. Data points that were >2 SD from the mean were considered outliers. For SCI and laminectomy experiments, experimenters were blinded to the animal condition during experiments and unblinded only after analysis of individual experiments was completed. In all experiments, n > 3 animals per condition unless otherwise stated. One-way ANOVA (with post hoc Bonferroni corrections or Fisher's LSD depending on number of comparisons) and Student's independent t tests were used to evaluate differences between samples when data are normally distributed. The nonparametric Kruskal–Wallis ANOVA was used to test for differences when data were not normally distributed. Student's paired t test was used in experiments where pre- and post-data are available and the paired Wilcoxon sign rank test for nonparametric paired data.
Single-cell qPCR.
Small-diameter DRG neurons (diameter ≤25 μm) were harvested and processed as previously described (Ritter et al., 2012). In short, an individual cell was harvested in 1 μl of 1× PBS and placed into a PCR tube containing 9 μl of H2O and frozen immediately at −20°C. Reverse transcription was then initiated in a reaction containing random primers (Invitrogen), dNTPs (Roche), MMLV Buffer (Promega), RNase inhibitor (Promega), MMLV reverse transcriptase (Promega), and H2O. Quantitative real-time single-cell PCR was conducted using an Agilent Mx3005P real-time PCR machine and Brilliant SYBR Green reaction mix II (Agilent Technologies). Outer primers were used to generate cDNA fragments for standards and inner primers used for real-time qPCR detections. Tubes that showed no β-actin copy numbers were assumed not to contain a cell and were thus not included in the analysis. Significance was determined by comparing groups using Kruskal–Wallis ANOVA.
Outer primers:
Kv3.4a+b+c (forward): ACCACACCGACTTCAAGAAC
Kv3.4a (reverse): AGAGCACAGGTGTGAGACAC
Kv3.4b (reverse): CCACTGGGGGAAAAGGGATT
Kv3.4c (reverse): CCTGGGCATAGTTGGACGAG
β-actin (forward): GCTATGTTGCCCTAGACTTC
β-actin (reverse): AACGCAGCTCAGTAACAGTCC
Inner primers:
Kv3.4a+c (forward): GCAGACTCCAAGCAGAATGG
Kv3.4b (forward): TGGCCAAGCAGAAGCTTCCC
Kv3.4a (reverse): CTGCCTTCTTTCTGGACACT
Kv3.4b (reverse): CCAACCCCTAATCTCACCTG
Kv3.4c (reverse): CAGGTCTCTTTCCGGACACT
β-actin (forward): CCGATGCCCCGAGGCTCTCT
β-actin (reverse): GCCTGGGTACATGGTGGTGCC
Immunostaining.
Animals were anesthetized for euthanasia 2 weeks after surgery and were transcardially perfused with 0.9% saline followed by 4% paraformaldehyde. DRGs from C1–C6 contralateral to the hemicontusion were harvested and stored in 4% paraformaldehyde (24 h), followed by 0.1 m phosphate buffer (24 h), and finally in 30% sucrose containing phosphate buffer (3 d). The tissue was then embedded in tissue freezing medium (Tissue-Tek) and 30 μm sections were cut. Sections were collected on glass slides and stored at −20°C.
For diaminobenzidine (DAB) staining, sections were washed with TBS followed by TBS-Triton 0.2% before endogenous peroxidase blocking for 8 min with 3% H2O2 and 10% methanol in TBS. The sections were then washed in alternating buffers of TBS and TBS-Triton 0.2% (4×/5 min). Sections were then incubated for 1 h with blocking buffer consisting of 10% goat serum, 2% Blotting Grade Blocker Nonfat Dry Milk (Bio-Rad), 0.2% gelatin (Sigma-Aldrich) in TBS-Triton 0.2%. After an additional TBS-Triton 0.2% wash, primary antibody (anti-Kv3.4, Alomone Labs; 1:100) was added in a TBS-Triton 0.2% solution and incubated at 4°C overnight. Sections were then washed as previously described and incubated with secondary antibody (Sigma-Aldrich biotinylated goat anti-rabbit 1:200) at room temperature for 1 h. Sections were washed and incubated with avidin-biotin-complex (Vectastain ABC kit) for 1 h. Sections were then washed and DAB (Sigma-Aldrich) dissolved in 0.1 m Tris-HCl was added to all sections simultaneously for 6 min, and the reaction was halted by H2O addition. Slides were then exposed to ethanol and xylene before being fixed with Xylene Polymount (Sigma-Aldrich). Sections were imaged using a Zeiss Imager MZ Upright Fluorescent Microscope and optical density was quantified using ImageJ software (NIH). This experiment was performed twice and the results were subjected to a Mann–Whitney ANOVA to determine statistical significance of differences.
For immunofluorescence experiments, sections were washed with TBS (3×/10 min) and incubated with 10% goat serum and 0.2% Triton in TBS for 1 h at room temperature. Sections were then incubated with primary goat anti-Kv3.4 antibody (1:1000, Alomone Labs Catalog #APC-019 RRID:AB_2040172) overnight at 4°C. Sections were then washed with TBS as described above and then incubated with secondary antibody (1:400 AlexaFluor 488, Invitrogen) for 1.5 h at room temperature before a final wash. In some cases, sections were colabeled with mouse anti-Nav1.8 (UCDavis/NIH NeuroMab Facility Catalog #73-166 RRID:AB_10672261) and subsequently costained with AlexaFluor 546 goat anti-mouse (Invitrogen). Sections were then dried and coverslips were added along with Prolong Gold antifade reagent with DAPI (Sigma-Aldrich). Confocal microscopy was performed at the Bioimaging Facility of the Kimmel Cancer Center (Nikon C1 Plus Confocal Microscope). Images were then compiled and distributed among three blinded individuals who were asked to report the proportion of Kv3.4-positive cells that contained uninterrupted stretches of membrane staining. The data from all individuals were averaged and a Mann–Whitney ANOVA was performed to determine significant differences among the experimental groups.
For quantification of differences in membrane staining, cryosections of rat DRGs (2 weeks after laminectomy and SCI) were stained with monoclonal mouse anti-pan-cadherin antibodies (1:200, Abcam Catalog #ab22744, RRID:AB_447300) and polyclonal rabbit anti-Kv3.4 antibodies (1:1000, Alomone Labs Catalog #APC-019 RRID:AB_2040172). Primary antibodies were then conjugated to AlexaFluor 488 goat anti-rabbit (Invitrogen) and AlexaFluor 546 goat anti-mouse IgG1 (Invitrogen) secondary antibodies. Z-stacks were generated at 1 μm steps using a Nikon C1 Plus Confocal Microscope. ImageJ was used to create fluorescence intensity plots by first tracing a straight line in a random angle through a 2D axis of each cell within a field-of-view by an individual blinded to the experimental conditions. The peak pan-cadherin signals (leading and lagging edges of each cell) from each plot were averaged for all cells in view. Those Z-stack planes from which average pan-cadherin peaks were not significantly different between the SCI and laminectomy groups were compared to assess changes in Kv3.4 plasma membrane expression. Kv3.4 signals were normalized to the pan-cadherin peak signal in these planes. Graphs and corresponding statistical analyses were produced in Origin Pro 8.0 (OriginLab).
Immunoblotting.
The C1–C6 DRGs from anesthetized rats were collected, homogenized, and sonicated in lysis buffer (50 mm Tris-HCl, 1% NP40, 0.5% Na-deoxycholate, 0.1% SDS, 150 mm NaCl, 2 mm EDTA) containing a mixture of protease inhibitors (Roche). Samples were spun at 14,000× g for 10 min. The supernatant was collected and spun again at 21,000 × g for 45 min. The protein samples were separated on a 4–12% Bis-Tris gel (Life Sciences) and transferred to Nitrocellulose membrane (Millipore). The filters were blocked with Odyssey Blocking buffer for 1 h at room temperature and incubated overnight at 4°C with rabbit anti-Kv3.4 (1:100, Alomone Labs) and mouse anti-actin (1:2000, Abcam), followed by 1 h incubation with fluorescent secondary antibodies (1:20,000, Odyssey) for 1 h at 4°C. Signals were finally detected using LI-COR Odyssey CLx infrared imaging system and bands were visualized and quantified with ImageJ software (NIH). For image clarity, the background was subtracted and the brightness increased using Studio Imager (Odyssey).
Results
SCI induces persistent pain-related behaviors and DRG hyperexcitability
To investigate the peripheral mechanisms responsible for SCI-induced chronic pain, we subjected adult female rats to a spinal hemicontusion injury (see Materials and Methods). This model allows for assessment of pain-related behaviors as animals develop a bilateral pain phenotype (Detloff et al., 2013) with no motor deficits on the side contralateral to injury (Nicaise et al., 2012). Following SCI, rats developed tactile allodynia and thermal hyperalgesia on the contralateral and ipsilateral forepaws, which persisted for at least 6 weeks after the SCI compared with baseline and laminectomy controls (Fig. 1; a >50% reduction in mechanical threshold was seen in 80% and 75% of animals in the ipsilateral and contralateral forepaws, respectively). Laminectomy control rats also developed bilateral thermal hypersensitivity compared with baseline; however, it was less severe than in SCI (Fig. 1A,B). Additionally, following laminectomy there was no decrease in mechanical threshold (Fig. 1C,D; no animal displayed a >50% in mechanical threshold). To avoid the confounding effects of motor deficits affecting the ipsilateral side, we performed the study on DRG neurons from the contralateral side.
Figure 1.

Hypersensitivity phenotypes following SCI. A, Withdrawal latency of the ipsilateral paw to a thermal stimulus in SCI (cervical contusion) and laminectomy animals at 1 week intervals through 6 weeks after the injury B, Same as in A for the contralateral paw. C, Mechanical threshold stimulus on the ipsilateral paw to induce a response before (baseline) and following laminectomy or SCI. Data are normalized relative to the baseline response (withdrawal threshold after surgery/withdrawal threshold before surgery) and expressed as “fractional withdrawal threshold.” D, Same as in C for the contralateral paw. Asterisks indicate statistically significant differences between SCI and laminectomy controls (ANOVA, p < 0.05, N = 5–8 rats). In A and B, all points for laminectomy and SCI are significantly different from baseline (p < 0.05). In C and D, all SCI points are significantly different from baseline (p < 0.05).
In light of the contralateral hypersensitivities following SCI, we hypothesized that hemicontusion SCI might have caused the contralateral DRG neurons at and above the level of injury to become hyperexcitable. To test this hypothesis, we acutely dissociated DRG neurons contralateral to the surgery (2 weeks after surgery, including C1–C6) and recorded evoked spikes under current-clamping conditions (Fig. 2). Upon sustained current injection at twice the rheobase of the neuron, ∼60% of small-diameter DRG neurons from all groups fired one AP, and the rest fired two APs or more (Fig. 2A). The cumulative frequency distributions of the number of spikes in a train were not significantly different among all three groups of rats (Fig. 2B). However, the interspike intervals (ISIs) drastically changed between the groups (Fig. 2C). Compared with the control groups, the ISIs from the neurons in the SCI group was uniformly shorter throughout the repetitive spiking train (Fig. 2D). By contrast, demonstrating adaptation in naive and laminectomy controls, the ISI of these groups became progressively longer during the train (Fig. 2D). These results show that putative small-diameter DRG nociceptors increase their firing rate in response to stimuli 2 weeks after hemicontusion SCI, consistent with the idea of peripheral sensitization playing a role in SCI-induced hypersensitivity.
Figure 2.

DRG neuron hyperexcitability following SCI. A, Representative APs elicited from a 1 s current injection at 2× rheobase showing a neuron that fires once (blue trace) and another that fires multiple APs (red trace). Gray dashed line = −61 mV. B, Cumulative number of APs elicited as in A for neurons from naive, laminectomy (Lam) and SCI animals (2 weeks after surgery). C, Representative recordings from a laminectomy (blue trace) and SCI (red trace) elicited as in A and resulting in four APs. Gray dashed line = −47 mV. D, Summary of ISI versus interval number from recordings showing repetitive firing in naive (black symbols), laminectomy (blue symbols), and SCI (red symbols) animals. Number 1 on the x-axis is the interval between the first and second AP and the rest are subsequent intervals. Asterisks indicates p < 0.05 by ANOVA comparison.
Kv3.4 currents in small-diameter DRG neurons undergo dynamic changes after laminectomy and SCI
Hemicontusion SCI might induce maladaptive modulation of the Kv3.4 channel in contralateral DRG neurons, which could contribute to the excitability changes. To test this hypothesis, we investigated Kv3.4 currents expressed in acutely dissociated small-diameter neurons under minimally invasive cell-attached patch-clamping conditions (see Materials and Methods). In >80% of the patches, macroscopic Kv3.4 currents from naive rats displayed the previously reported fast inactivating profile and relatively depolarized voltage dependence characteristic of the underlying Kv channels (Fig. 3B; Table 1). Demonstrating the molecular identity of these channels, most DRG neurons exhibited anti-Kv3.4 immunostaining (Fig. 3A). Also, consistent with the expression of the Kv3.4 protein in a subset of putative nociceptors, Nav1.8-positive neurons exhibit anti-Kv3.4 immunoreactivity (Fig. 3A). The distribution of peak outward currents from the naive group alone is approximately normal (Fig. 3B). However, the aggregate results from 145 patches recorded at 1, 2, and 6 weeks after the surgery from the naive and laminectomy control groups and the SCI group is more consistent with a multimodal distribution of peak outward currents (Fig. 3C). These results suggest dynamic changes differentially affecting the functional expression of Kv3.4 channels in DRG neurons from different experimental groups. To investigate this possibility in detail, we characterized early (1 week after surgery) and late changes (2 and 6 weeks after surgery) in the current's inactivation profile and the peak current by comparing four distinct current phenotypes (Fig. 4).
Figure 3.

Kv3.4 channel expression and current phenotypes in DRG neurons. A, DRG section from a naive animal immunostained as indicated. B, Distribution of peak currents from naive neurons and best fit Gaussian distribution (solid line; inset: xc = 33.8 pA, w = 17.8 pA, and A = 1.5). Inset, Representative Kv3.4 currents from naive control neuron. Following a 1 s prepulse to −30 mV, currents were evoked by step depolarizations from −60 to +100 mV at 20 mV intervals. C, Aggregate distribution of all peak currents and the best-fit sum of three Gaussian distributions (black dashed line). Each component of the distribution is shown as a solid colored line. The solid orange distribution was fixed to that of the naive distribution (B). Parameters for the other two distributions are as follows: xc = 9.1, w = 2.2, and A = 10 for the green distribution; xc = 19, w = 3.3, and A = 14 for the blue distribution.
Table 1.
Kv3.4 biophysical properties after laminectomy and SCI
| Naïve |
1 week |
2 weeks |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Laminectomy |
SCI |
Laminectomy |
SCI |
|||||||
| N | N | N | N | N | ||||||
| Activation | ||||||||||
| V1/2 (mV) | 16.8 ± 4.8 | 18 | 11.1 ± 6.2 | 19 | 11.4 ± 4.3 | 18 | 19.1 ± 3.8 | 24 | 11.0 ± 4.5 | 24 |
| Vs (mV) | −25.5 ± 3.3 | 18 | −26.2 ± 6.2 | 19 | −29.2 ± 3.7 | 18 | −27.5 ± 4.3 | 24 | −30.2 ± 3.8 | 24 |
| zGp (e0) | 1.01 ± 0.09 | 18 | 1.14 ± 0.13 | 19 | 1.05 ± 0.13 | 18 | 0.91 ± 0.08 | 24 | 1.03 ± 0.09 | 24 |
| Inactivation | ||||||||||
| V1/2 (mV) | −30.1 ± 1.6 | 18 | −41.0 ± 2.6 | 14 | −31.6 ± 2.2 | 14 | −33.9 ± 0.7 | 11 | −29.0 ± 2.1 | 9 |
| zssi (e0) | 3.40 ± 0.58 | 18 | 2.95 ± 0.79 | 14 | 2.30 ± 0.41 | 14 | 2.32 ± 0.14 | 11 | 2.05 ± 0.31 | 9 |
| zTaui (e0) | 1.51 ± 0.07 | 17 | — | — | 1.27 ± 0.31 | 21 | — | |||
All parameters were calculated as described in Materials and Methods. For z, the subscripts indicate the parameter from which the z was calculated. Gp, Peak chord conductance relation; ssi, steady-state inactivation curve; Taui, time constant of inactivation. For some groups an exponential fit to the Taui–V relation could not be obtained and thus zTaui could not be determined.
Figure 4.

Kv3.4 current phenotypes after laminectomy and SCI. A, B, Representative Kv3.4 currents from laminectomy (Lam) and SCI neurons at 1 week and 2 weeks after surgery. Pulse protocol and scale bars are as in Figure 3A. C, Pie charts showing the percentages of four distinct Kv3.4 current phenotypes from naive (black), after laminectomy (blue), and SCI (red): Ip <16 pA; noninactivating; Ip/I500<2; Ip/I500≥2. Table 2 reports N for each group.
Early changes: laminectomy and SCI alter the inactivation properties and expression of Kv3.4 channels in DRG neurons
At 1 week after the surgery, we noticed an increase in the fraction of patches displaying slow inactivating and noninactivating currents in the laminectomy and SCI groups (Fig. 4; Table 2). To evaluate the degree of macroscopic inactivation, we compared the peak current over the current at 500 ms (Ip/I500) between the naive and the experimental groups using Ip/I500 ≥2 as the cutoff. Thus, Ip/I500 ≥2 indicates that the current exhibits more profound inactivation (Fig. 4A, right). Conversely, Ip/I500<2 indicates diminished inactivation, which might result from inactivation slowing with or without increased sustained current (Fig. 4B, top and bottom left, respectively). Whereas only 17% of the patches exhibited an Ip/I500<2 in the naive group, the percentage of patches displaying currents with an Ip/I500<2 increased ∼2.5-fold in the laminectomy and SCI groups (Fig. 4C). Relative to naive, the Ip/I500 was significantly reduced in the laminectomy and SCI groups (Fig. 5B). To evaluate the peak currents, we computed the fraction of patches exhibiting Ip<16 pA, which corresponds to 1 SD of the naive Ip distribution (Fig. 5A). This fraction was ∼6% in the naive group and increased by ∼3-fold in the laminectomy and SCI groups, indicating downregulation (Fig. 4C). These changes shifted the variability in the distributions of Ip and Ip/I500 (Fig. 5A–D). For the Ip, the coefficient of variation (COV) increased modestly in both the laminectomy and SCI groups (0.54, 0.62, and 0.66 for naive, laminectomy, and SCI, respectively; Fig. 5C); and for Ip/I500, only the SCI group exhibited a relatively large decrease in COV for the Ip/I500 (0.83, 0.78, and 0.52, for the naive, laminectomy, and SCI groups, respectively; Fig. 5D), suggesting a more uniform slowing of Kv3.4 inactivation in all neurons following SCI.
Table 2.
Kv3.4 current properties after laminectomy and SCI
| Naïve | 1 week |
2 weeks |
6 weeks |
||||
|---|---|---|---|---|---|---|---|
| Lam | SCI | Lam | SCI | Lam | SCI | ||
| Ip (pA) | 40 ± 5 | 26 ± 3 | 37 ± 6 | 54 ± 10 | 46 ± 9 | 34 ± 6 | 28 ± 5 |
| Ip/I500 | 4.7 ± 0.9 | 2.2 ± 0.4* | 2.5 ± 0.3* | 4.2 ± 0.8 | 2.4 ± 0.2* | 3.2 ± 0.9 | 1.9 ± 0.4* |
| κ (s−1) | 49 ± 6 | 33 ± 3 | 40 ± 2 | 57 ± 9 | 63 ± 13 | 51 ± 2 | 42 ± 6 |
| N | 18 | 21 | 18 | 27 | 30 | 13 | 20 |
Mean ± SEM of Kv3.4 current properties following laminectomy (Lam) and SCI. Asterisks indicate p < 0.05 compared with naïve and laminectomy controls (for direct comparisons see Fig. 5).
Figure 5.

Kv3.4 current expression and inactivation from naive, laminectomy, and SCI. A, Scatter plot of Ip for all data on a logarithmic scale. Dashed gray line is 1 SD from the center of the naive distribution (16.1 pA). B, Ip/I500 ratio for all groups examined. Asterisk indicates p < 0.05 from post hoc Fisher's LSD after ANOVA analysis. C, D, COV for Ip and Ip/I500 from all groups. E–G, 3D plots showing the Ip, Ip/I500 and ki for naive (E), 2 weeks after laminectomy (F), and 2 weeks post-SCI (G). If a current was noninactivating then ki = 0.01 (slower than can be reliably measured in these experiments). Table 2 displays mean ± SEM of all parameters.
Late changes: SCI induces persistent downregulation of Kv3.4 channels displaying slower inactivation
Further changes in both Ip/I500 and Ip occurred 2 weeks after surgery (Fig. 4; Table 2). The current phenotypes in the laminectomy group trended toward normal (15% Ip/I500<2, and 7% noninactivating; Fig. 4C), and the Ip/I500 returned to naive levels (Fig. 5B). However, the SCI group remained substantially shifted (Figs. 4C, Fig. 5B). Highlighting the variability in the current inactivation profile at 2 weeks after laminectomy and SCI (Fig. 5B), the Ip/I500 COV remained low in the SCI group (0.52) and high in laminectomy group (1.06; Fig. 5D). Meanwhile, the percentage of patches with Ip < 16 pA remained at the 1 week level for the laminectomy group, whereas that of the SCI group nearly doubled (Figs. 5A, Figs. 4C). Relative to the naive group, the Ip COV approximately doubled for both the two-week laminectomy and SCI groups (0.94 and 1.12; Fig. 5C).
At 6 weeks after surgery outward currents from the SCI group showed continued loss of inactivation and downregulation, while laminectomy currents reached the levels of naive controls (Fig. 4; Table 2). Nearly 80% of the patches from the laminectomy group were fast inactivating (Ip/I500 > 2), whereas only a third of the patches from the SCI group fell in this category (Fig. 4C). Regarding Ip/I500 variability, the COV remained reduced in the SCI group (0.67) and slightly elevated in the laminectomy group (0.93; Fig. 5D). In the SCI group, almost half of the patches had Ip<16 pA, whereas the laminectomy group had no patches in this category (Fig. 4C). Further suggesting the return to naive controls, the Ip COV for the laminectomy group returns to normal levels (0.53). By contrast, Ip COV for the SCI group remained high (0.88; Fig. 5C).
To examine the relationship between the Ip, Ip/I500 and the observed rate constant of macroscopic inactivation (ki = 1/time constant of current decay) from each recording (naive, laminectomy and SCI groups at 6 weeks), we simultaneously compared these measurements on 3D graphs (Fig. 5E–G). Whereas the data from the naive and laminectomy control groups spread out more in the 3D space, the data from the SCI group mostly cluster in a corner of the graph. This clustering indicates small peak currents exhibiting slower inactivation and increased levels of sustained current (Fig. 5G).
Overall, the analyses of Kv3.4 current profiles of inactivation and expression show that 2–6 weeks after the surgery, SCI induces more variable Kv3.4 peak currents with an excess of smaller peak currents. SCI also promotes reduced and less variable inactivation of these currents. The latter suggests uniform negative modulation of Kv3.4 current inactivation. In contrast, the properties of Kv3.4 currents in the laminectomy control group initially (1 week) resemble those in the SCI group, but become nearly normal between 2 and 6 weeks after the surgery, both in terms of current expression, degree of inactivation, and the variability of both measurements.
Kv3.4 downregulation does not result from SCI-induced dysregulation of voltage-dependent gating
Several biophysical and biochemical factors could have determined the apparent functional downregulation of the Kv3.4 current. To entertain effects on voltage-dependent activation, we examined the peak conductance-voltage relations (Gp/V curves) from all experimental groups regardless of the inactivation profile (Fig. 6). The Gp/V curves from all groups at 1 and 2 weeks after the surgery were indistinguishable (Fig. 6A,B; Table 1). To separately assess voltage-dependent gating and inactivation, we also examined the steady-state inactivation curves from all groups at the same time points. Again, there were no detectable voltage-dependent shifts among all groups. However, whereas the level of noninactivating current increased in both the laminectomy and SCI groups at 1 week after the surgery, 1 week later only the noninactivation current level of the SCI group remained high and that of the laminectomy group returned to normal levels (Fig. 6C,D). The implications of these results are threefold. First, indistinguishable Gp/V and steady-state inactivation curves from all groups at two time points strongly suggest that the same Kv channels underlie all currents. Second, the SCI-induced downregulation of the Kv3.4 current does not result from unfavorable shifts in voltage-dependent gating. Third, supporting previously described results (Figs. 4, 5), SCI persistently enhances the level of noninactivating current. Overall, this characterization rules out gating dysregulation as the mechanism responsible for SCI-induced downregulation of the Kv3.4 current.
Figure 6.

Kv3.4 channel biophysical properties from naive, laminectomy and SCI. A, B, Normalized Gp-V relation and steady-state inactivation relation for naive (black) and 1 week after laminectomy (blue) and SCI (red). Table 1 displays mean ± SEM of best-fit parameters. C, D, Same as A and B but for 2 weeks after laminectomy and SCI. Naive data are replotted from A and B (black solid lines).
Dysregulation of Kv3.4 surface expression might underlie Kv3.4 current downregulation
If gating of the remaining channels is normal, it is alternatively possible that putative nociceptors from the SCI group fail to synthesize enough Kv3.4 mRNA, produce enough Kv3.4 protein or shuttle Kv3.4 channels to the cell surface. We first examined mRNA expression at the single-neuron level from all groups at 2 weeks after the surgery. Small-diameter neurons express three distinct alternative transcripts: Kv3.4a, Kv3.4b, and Kv3.4c (Fig. 7A–C). The expression of these transcripts displayed interesting but modest changes. Relative to the naive group, Kv3.4a is not significantly altered following laminectomy and SCI (p = 0.12, Kruskal–Wallis ANOVA; Fig. 7A). Kv3.4b, the lowest expressing splice variant, is significantly reduced in the SCI group with respect to both the naive and laminectomy controls by 10–20 mRNA transcripts per cell or ∼2-fold (p = 0.007 and p = 0.01 for naive and laminectomy, respectively; Kruskal–Wallis ANOVA with post hoc Bonferroni correction; Fig. 7B). Finally, Kv3.4c copy numbers were increased in the laminectomy group compared with both naive and SCI groups (p = 0.03 and p = 0.01 for naive and SCI, respectively; Kruskal–Wallis ANOVA with post hoc Bonferroni correction, Fig. 7C).
Figure 7.

Expression of Kv3.4 mRNA and protein from naive and 2 weeks after laminectomy and SCI. A–C, Box plots of single-cell mRNA copies of Kv3.4 mRNA (black = naive; blue = laminectomy; red = SCI). Box is 25–75 percentiles with median marked by center line. Whiskers show 10–90 percentile and X marks the mean. N (Kv3.4a) = 36, 54, and 64 from naive, laminectomy, and SCI, respectively; N (Kv3.4b) = 34, 64, and 65 from naive, laminectomy, and SCI, respectively; N (Kv3.4c) = 44, 43, and 42 from naive, laminectomy, and SCI, respectively. Cells were harvested from four naive, six laminectomy, and nine SCI animals. D, Western blot analysis showing expression of Kv3.4 in C1–C6 DRGs 2 weeks after laminectomy or SCI. Actin was used as a loading control. The normalized optical density units were as follows: 2.2 and 1.9 for laminectomy and SCI, respectively. This experiment combined DRGs from four animals in each condition. E, F, Representative DAB stained sections from the laminectomy and SCI groups.
Then, we asked whether these relatively modest transcriptional and post-transcriptional changes could have affected Kv3.4 protein expression in DRG neurons. Whole DRG Western blot analysis yielded no significant intensity difference in the major Kv3.4 band at ∼65 kDa when comparing the laminectomy and SCI groups (Fig. 7D). Thus, SCI does not affect the total Kv3.4 protein in the DRG. To independently verify this conclusion, we conducted DAB staining of DRG sections using the same anti-Kv3.4 antibody and found no differences between the laminectomy and SCI groups at 2 weeks after the surgery (0.05% increase in staining intensity from SCI sections compared with laminectomy controls; N = 2 and 3 rats, respectively; Fig. 7E,F). Thus, the modest Kv3.4 transcriptional changes do not impact total Kv3.4 protein expression in DRG neurons and, consequently, cannot explain the SCI-induced downregulation of the Kv3.4 peak current.
Because voltage-dependent gating, transcription and translation of the Kv3.4 channel expressed in DRG neurons are not affected by laminectomy and SCI, we investigated possible changes in the cellular distribution of Kv3.4 channel expression at 2 weeks after the surgery. Immunostaining of DRG sections with an anti-Kv3.4 antibody revealed differences in surface expression (Fig. 8). In the naive and laminectomy groups, neurons often exhibited surface expression that delineates the perimeter of the cells (Fig. 8A,B). By contrast, surface immunoreactivity was weak and discontinuous in DRG neurons from the SCI group (Fig. 8C). The fraction of neurons exhibiting robust membrane staining in the SCI group showed a nearly twofold decrease from naive and laminectomy control groups (0.36 ± 0.04, 0.35 ± 0.02, and 0.19 ± 0.04, for the naive, laminectomy and SCI groups, respectively; p < 0.03; Fig. 8D). To quantify these membrane changes, we compared the fluorescence intensity of a pan-cadherin plasma membrane maker with that of Kv3.4 in DRG sections (Fig. 9). The ratio of Kv3.4 fluorescence to that of pan-cadherin was decreased in sections from the SCI rats compared with the laminectomy controls. This decrease is significant when either comparing many cells in sections from paired laminectomy and SCI rats (p < 0.0001, Fig. 9D) or the averages from three independent pairs (p = 0.04; Fig. 9E). Therefore, SCI dysregulates surface expression of Kv3.4 channels in putative DRG nociceptors contralateral to the side of the injury.
Figure 8.

Kv3.4 DRG immunostaining from naive and 2 weeks laminectomy and SCI. A–C, Representative immunofluorescence sections stained with anti-Kv3.4 antibody from naive, laminectomy and SCI animals. White arrows show neurons with significant staining on the cell perimeter and red arrows show neurons with no significant perimeter staining. D, Scatter plot of fraction of cells displaying anti-Kv3.4 immunostaining on the cell perimeter in the naive, laminectomy, and SCI groups. Each data point represents the mean fraction from three blinded investigators for a specific section; and each symbol represents sections from one animal (N = 3, 5, and 4 animals for naive, laminectomy, and SCI, respectively). We observed a similar change (0.36 ± 0.04, 0.16 ± 0.03, for laminectomy and SCI groups, respectively; p < 0.02) when comparing a subset of DRG neurons costained for the nociceptor marker Nav1.8 and Kv3.4 in the laminectomy and SCI groups (N = 2 and 3 animals, respectively).
Figure 9.

Quantification of Kv3.4 plasma membrane immunostaining from 2 weeks after laminectomy and SCI. A, Representative immunofluorescence sections stained with anti-Kv3.4 and pan-cadherin antibodies from laminectomy (N = 3) and SCI (N = 3) animals. B, Histogram of Kv3.4 (green) and pan-cadherin (red) fluorescence intensities corresponding to the cross-section (white line) across the cell in A from the laminectomy animal. C, Same as in B except from an SCI animal DRG neuron. D, Scatter plot of ratios of FITC/TRITC for one laminectomy and one SCI animal from the same experimental cohort. Three independent scatter plot pairs were individually compared using a Kruskall–Wallace ANOVA, which yielded the following levels of significance: p = 2 × 10−9, 7 × 10−4 and 6 × 10−14. E, Bar graph showing the mean FITC/TRITC ratio from all three independent cohorts (one-way ANOVA, p = 0.04).
SCI eliminates modulation of AP duration by PKC in small-diameter nociceptors
Previously, we reported that the Kv3.4 channel is a major regulator of AP duration in a manner that depends on the channel's direct modulation of inactivation by PKC (Ritter et al., 2012). To test whether loss of Kv3.4 channels affected the basal AP waveform of small-diameter DRG neurons, we recorded APs evoked by a brief current injection pulse (0.5 ms). At 2 weeks after surgery, there were no significant differences between all groups (Fig. 10A,B; Table 3). Then, to test whether AP duration was sensitive to PKC modulation, we recorded APs before and after exposure of the neurons to the phorbol ester PMA (Figs. 10C–F). The APs from the laminectomy group exhibited shorter durations upon PKC activation (Fig. 10C,E; Table 3). By contrast, the APs from the SCI group before and after PKC activation exhibited heterogeneous responses and on average remained indistinguishable; with 3/11 neurons exhibiting AP broadening, 3/11 exhibiting shortening and the remaining five displaying minimal shortening (Fig. 10D,F; Table 3). The altered response to PMA is reminiscent of the effect of Kv3.4 channel knockdown by siRNA on PKC-dependent AP modulation (Ritter et al., 2012). These results suggest that SCI-induced Kv3.4 channel dysregulation does not modulate the basal AP duration but alters the ability of the APs to respond to PKC activation by shortening AP duration.
Figure 10.
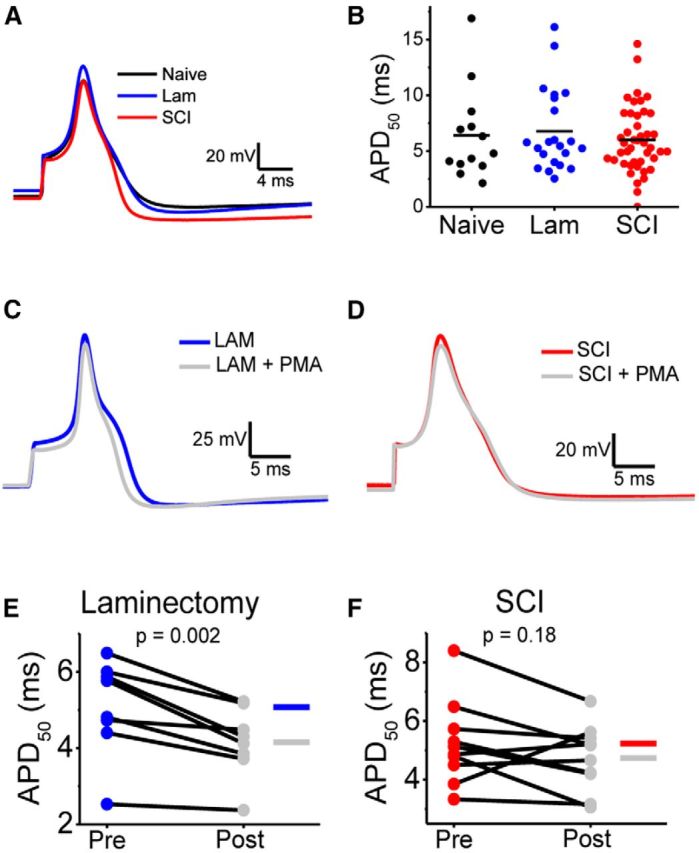
AP waveform analysis before and after PKC activation in naive and 2 weeks after laminectomy and SCI. A, Representative overlaid DRG neuron APs from the naive (black), laminectomy (blue), and SCI (red) groups. APs were elicited by a 0.5 ms current injection and aligned by their peaks. B, Summary scatter plots of APD50 in neurons from naive (black), laminectomy (blue), and SCI (red) animals. Horizontal bars indicate the mean. C, D, Representative APs elicited by a 0.5 ms current injection pulse before (blue) and after exposure to 100 nm PMA (gray) in neurons from the laminectomy and SCI groups. APs are aligned by their peak. E, F, Summary of paired scatter plots of APD50 before and after PMA. Lines join individual paired pre and postexperiments. Table 3 displays mean ± SEM of all parameters.
Table 3.
AP properties after laminectomy and SCI following PKC activation
| All neurons |
Laminectomy + PMA |
SCI + PMA |
|||||
|---|---|---|---|---|---|---|---|
| Naïve | LAM | SCI | Pre | Post | Pre | Post | |
| RMP (mV) | −56.9 ± 2.1 | −57.9 ± 2.3 | −56.0 ± 1.1 | −56.6 ± 3.3 | −68.1 ± 3.3** | −56.2 ± 2.5 | −62.3 ± 2.3** |
| Input Res. (GΩ) | 0.6 ± 0.1 | 0.7 ± 0.1 | 0.6 ± 0.1 | 0.7 ± 0.2 | 1.0 ± 0.2* | 0.6 ± 0.2 | 0.6 ± 0.2 |
| Capacitance (pF) | 15.5 ± 1.3 | 17.5 ± 1.0 | 21.8 ± 1.4* | ||||
| Diameter (μm) | 19.5 ± 0.6 | 22.2 ± 0.5 | 22.4 ± 0.4 | ||||
| Rheobase (pA) | 73 ± 23 | 108 ± 26 | 101 ± 12 | 53 ± 15 | 89 ± 29 | 83 ± 21 | 140 ± 34** |
| Threshold (mV) | −32.5 ± 1.1 | −31.1 ± 1.5 | −31.0 ± 1.0 | −31.0 ± 2.8 | −37.1 ± 2.5* | −30.7 ± 1.4 | −33.5 ± 1.5 |
| Overshoot (mV) | 30.7 ± 4.2 | 28.3 ± 3.4 | 26.9 ± 2.0 | 40.7 ± 3.0 | 31.7 ± 2.5** | 32.9 ± 4.1 | 22.2 ± 3.9** |
| AHP (mV) | −64.5 ± 1.3 | −69.6 ± 2.1 | −68.5 ± 1.0 | −71.9 ± 1.8 | −80.2 ± 2.5** | −70.6 ± 1.3 | −76.5 ± 2.4* |
| APD50 (ms) | 6.4 ± 1.1 | 6.8 ± 0.9 | 5.9 ± 0.5 | 5.1 ± 0.4 | 4.2 ± 0.3** | 5.2 ± 0.5 | 4.7 ± 0.4 |
| APD90 (ms) | 1.56 ± 0.26 | 1.66 ± 0.20 | 1.52 ± 0.09 | 1.18 ± 0.11 | 1.04 ± 0.07* | 1.40 ± 0.09 | 1.41 ± 0.13 |
| Max Dep. rate (mV/ms) | 46.9 ± 8.7 | 44.8 ± 7.4 | 40.0 ± 3.9 | 56.5 ± 11.9 | 66.0 ± 9.4 | 43.3 ± 4.9 | 46.6 ± 7.8 |
| Max Rep. rate (mV/ms) | 26.1 ± 4.1 | 25.1 ± 3.1 | 25.1 ± 2.1 | 33.4 ± 3.5 | 33.9 ± 3.5 | 26.0 ± 2.8 | 23.3 ± 3.2 |
| N | 13 | 21 | 43 | 8 | 11 | ||
AP properties were measured in naïve, laminectomy (Lam), and SCI neurons as described in Materials and Methods, and ANOVA used for comparisons. For PMA wash in experiments, paired t tests were used for comparisons;
* p ≤ 0.05;
** p ≤ 0.01. RMP, resting membrane potential; Dep., depolarization; Rep., repolarization.
The impact of Kv3.4 channels on spiking trains
Although the Kv3.4 channel underlies high voltage-activating K+ currents and is well suited to regulate the width of the AP (Ritter et al., 2012), inspection of the Gp/V curve suggested additional electrophysiological functions. The range of the Kv3.4 peak conductance between −40 and −20 mV is 7–16% of its maximum level (Fig. 6A,C). Because the AP threshold of small-diameter nociceptors is near −30 mV (Table 3), the Kv3.4 conductance might impact repetitive spiking by regulating the interspike interval. Also, Kv3.4 channels reopen during recovery from inactivation as the membrane potential hyperpolarizes, which may additionally regulate spiking (Ruppersberg et al., 1991a). To evaluate this possibility, we developed a Kv3.4 channel Markov gating model based on DRG Kv3.4 currents (Fineberg et al., 2012) and investigated the impact of the virtual Kv3.4 current on repetitive spiking of hyperexcitable small-diameter DRG neurons from the SCI group under dynamic-clamp conditions (Fig. 11; see Material and Methods).
Figure 11.

Dynamic clamping of a spiking DRG neuron in the SCI group. A, Cell-attached Kv3.4 currents evoked by 150 pulses to +100 mV (black traces) and the mean current (red trace). Pulse protocol details in Figure 3 legend. Dashed line represents 0 pA. B, Variance versus mean current plot from experiment in A. Red line presents the best-fit function to σ2 − I relation as described in Materials and Methods. C, Representative AP trains elicited by a 1 s current injection of 60 pA (3× rheobase) in a single SCI neuron with the current types indicated above the figure. The assumed Kv3.4 conductance density is indicated on the right. A-type corresponds to inactivating Kv3.4 channels and DR-type refers to noninactivating Kv3.4 channels. Duration of the recording is 1.01 s. Arrows in C mark skipped APs.
To determine a range of specific Kv3.4 conductances to be used for dynamic clamping, we estimated single channel conductance and the number of channels in a cell-attached macropatch using nonstationary noise analysis in naive small-diameter DRG neurons (Fig. 11A,B). In 13 patches, N = 88 ± 14 Kv3.4 channels/patch, γ = 11.3 ± 1.1 pS, and POmax = 0.72 ± 0.04 (see Materials and Methods). Then, to determine the conductance density, we assumed that the membrane capacitance of a cell-attached patch (Cm) is as follows: Cm = CM2πr2 were r is the radius of the patch electrode (1–2 μm) and CM is the specific membrane capacitance (1 μF/cm2). The specific Kv3.4 conductance density is 3.8–15.1 nS/pF, which agrees with previous estimates (Gold et al., 1996b). Validating the estimates, injection of synthetic Kv3.4 current reduced AP duration by 10 ± 1% (5.75 ± 1.23 to 5.07 ± 0.98 ms at a conductance density = 2.5 ± 1 nS/pF, N = 8).
In a dose-dependent manner, injection of synthetic Kv3.4 currents disrupted repetitive spiking and eventually abolished it (Fig. 11C). However, these effects did not significantly depend on whether the current was A-type (fast-inactivating) or delayed rectifier-type (noninactivating); and, indicating reversibility, repetitive spiking returned once the virtual current was removed at the end of the experiment (Fig. 11C). Providing additional support to the ability of the Kv3.4 channel to regulate repetitive spiking, the results were repeated in a second neuron from the SCI group. Thus, SCI-induced downregulation of Kv3.4 channels might be responsible for the accentuated excitability of small-diameter DRG neurons. Furthermore, they suggest that modest overexpression of Kv3.4 channels might have a beneficial effect by dampening the excitability of sensitized DRG nociceptors following SCI.
Discussion
The results from electrophysiological and immunohistochemical experiments converge to show that a major effect of hemicontusion SCI is to downregulate surface expression and to alter inactivation of Kv3.4 channels in subsets of contralateral DRG neurons. This effect might be in part responsible for the hyperexcitability of these neurons and, therefore, the persistence of pain-related behaviors in the animals. This conclusion has three significant implications. First, it supports the idea of peripheral sensitization as a major mechanism in the development of post-SCI persistent pain. Second, it implicates dysregulation of Kv3.4 channels as a common mechanism in animal models of chronic pain. Third, therapeutic interventions that enhance Kv3.4 channel functional expression might help treat SCI-induced persistent neuropathic pain.
Peripheral sensitization as a mechanism of SCI-induced persistent pain
SCI in rats induces above-, at-, and below-level pain behaviors, which may result from distinct mechanisms (Carlton et al., 2009; Hulsebosch et al., 2009; Walters, 2012). Furthermore, unilateral SCI in rats induces bilateral pain phenotypes above-, at- and below-level of injury (Detloff et al., 2013). Our results confirm the bilateral mechanical and thermal hypersensitivity following unilateral SCI. Interestingly, Detloff et al. (2013) showed mechanical allodynia in 40% of animals following SCI, whereas we found a more widespread phenotype throughout the majority of animals (Fig. 1). The discrepancy between our study and Detloff et al. (2013) most likely results from the wider impactor tip (1 vs 0.6 mm) and differences in surgical technique (e.g., strength and placement of vertebral clamps). Regardless, we show that the previously appreciated changes in thermal and mechanical thresholds is from direct SCI and not surgery as laminectomy controls only showed mild changes in thermal hyperalgesia (Fig. 1). These persistent pain phenotypes following SCI might originate from both central and peripheral neurons (Hulsebosch et al., 2009; Gwak and Hulsebosch, 2011; Masri and Keller, 2012; Walters, 2012; Whitt et al., 2013). Here, we have shown that, following SCI, the DRG undergoes a persistent increase in the excitability of contralateral small-diameter DRG neurons. This finding is consistent with a bilateral pain phenotype induced by a hemicontusion SCI. Although multiple factors could have contributed to this change (e.g., upregulation of Nav channels and downregulation of several Kv channels), we have identified dampening of Kv3.4 channel function and expression in DRG neurons as a significant factor. These results strongly support peripheral sensitization as a mechanism that contributes to neuropathic pain induced by SCI as changes were seen in DRG neurons contralateral to the side of injury. However, because unilateral injury produces bilateral pain phenotypes (Detloff et al., 2013) (Fig. 1), further work is needed to test whether similar peripheral mechanisms occur on the ipsilateral side.
Supporting a significant role of peripheral mechanisms, other studies using midline or transverse spinal cord injuries demonstrate effects on the expression Nav1.8, TRPV1, and TRPA1 channels in DRG neurons (Black et al., 2003; Andrade et al., 2011; Wu et al., 2013; Yang et al., 2014). The Nav1.8 protein is upregulated in DRG neurons following SCI and knockdown of this ion channel in the DRG reduced pain behaviors (Yang et al., 2014). Previous SCI studies also reported spontaneous firing in nociceptors (Carlton et al., 2009; Bedi et al., 2010; Yang et al., 2014), which we did not observe. This discrepancy might be attributed to the SCI model used (unilateral injury vs midline contusion), time the DRG neurons were in culture (<12 h compared with 24 h), different time points post-SCI and cervical versus lumbar DRGs. Although we did not find spontaneous activity, the excitability of the DRG neurons following SCI trended toward a higher firing frequency (Fig. 2). Altogether, based on the results reported here and previous work, we suggest that peripheral mechanisms involving DRG targets must be considered when developing new analgesics to treat SCI-induced chronic pain. Although various hypotheses have been proposed to explain how DRG neurons are alerted in SCI and become hyperexcitable (Hulsebosch et al., 2009; Walters, 2012), further studies are necessary to understand the signaling mechanisms and reveal additional therapeutic targets.
Dynamic regulation and novel SCI-induced maladaptive changes in the DRG
Neuronal populations in the DRG are characteristically heterogeneous (Harper and Lawson, 1985a,b). However, the increase in the variability of the peak Kv3.4 current and inactivation resulting from the laminectomy and SCI reflects additional heterogeneity induced by surgery and injury. The aggregate multimodal frequency distribution of all Kv3.4 peak current measurements (Fig. 3D) is consistent with this scenario. We previously proposed that Kv3.4 channels are key players in a homeostatic mechanism, which might become impaired when these channels are downregulated (Ritter et al., 2012). Here, we show evidence of two different responses: adaptive (laminectomy) and maladaptive (SCI). In both groups, the early loss of Kv3.4 channel inactivation and moderate decrease in Ip indicate an early homeostatic adaptive response triggered by the invasive surgical procedure alone. The loss of Kv3.4 inactivation in this case might be an attempt to upregulate Kv3.4 activity, which could help keep excitability in check. At 2 weeks after surgery, however, laminectomy and SCI groups diverge, as the first heals and recovers but the latter fails to restore normal function. Kv3.4 channel inactivation and expression in the laminectomy group trended toward control levels at 2 weeks and are fully restored by 6 weeks (Fig. 4). In contrast, a significant fraction of very small peak currents displaying slow-inactivating and noninactivating components remained apparent 2–6 weeks post-SCI (Fig. 4). Kv3.4 downregulation in this case is a maladaptive change, which essentially eliminates a key regulator of DRG excitability with the ability to drive a homeostatic response to oppose excitability. Recent work has suggested that long noncoding mRNAs may underlie downregulation of DRG Kv channels in chronic pain (Zhao et al., 2013). However, here we found that the downregulation was most likely mediated by a loss of Kv3.4 channel surface expression and not reduced total expression or altered biophysical properties. These changes might partly concern maladaptive modulations that could determine the persistent hyperexcitability of the neurons and the resulting persistent pain phenotype. Although our previous work suggested that Kv3.4 channel downregulation would induce AP broadening at 2 weeks post-SCI, there was no change in the AP duration. Two factors could contribute to this observation. First, two SCI-induced changes in Kv3.4 properties might have opposite consequences on AP duration. While the loss of Kv3.4 inactivation shortens the AP, Kv3.4 downregulation broadens it. Second, SCI might induce compensatory modulations of other ion channels that shape the nociceptor AP (Yang et al., 2014). These modulations could then oppose the expected broadening, which could have resulted from downregulating Kv3.4 alone. In a third possibly more realistic scenario, these two factors might be present simultaneously.
Based on the results from a previous study (Ritter et al., 2012), we hypothesize that disrupted inactivation resulted from PKC-dependent phosphorylation of the Kv3.4 N-terminal inactivation domain (Covarrubias et al., 1994; Beck et al., 1998). However, Kv3.4 channel inactivation might be modulated by other mechanisms (Ruppersberg et al., 1991b; Abbott et al., 2001). Additionally, preliminary experiments in heterologous expression systems and DRG neurons have demonstrated that persistent PKC activation induces a slow and substantial loss of the Kv3.4 current (Zemel, Ritter, and Covarrubias, unpublished observations). This response might be the main determining factor of the hyperexcitability resulting from the loss of Kv3.4 channels. Other studies have also implicated persistent PKC signaling in inflammatory pain sensitization of peripheral sensory receptors (Hucho and Levine, 2007). Although the exact mechanism of the dysregulation that ultimately causes reduced surface expression of Kv3.4 channels is not yet clear, it might involve recently discovered trafficking and localization pathways for Kv3.4 channels (Kanda et al., 2011a,b). Overall, our results suggest that Kv3.4 channels in DRG neurons namely undergo: (1) adaptive dynamic modulation of inactivation following surgical insult, and (2) maladaptive dysregulation of Kv3.4 channel surface expression following SCI.
Neuronal Kv3.4 channels under normal and pathological conditions
Due to the limited axonal and synaptic localization of Kv3.4 channels and their labile functional properties (Ruppersberg et al., 1991b; Covarrubias et al., 1994; Beck et al., 1998; Brooke et al., 2004), it has been difficult to investigate the functions and dysfunctions of these K+ channels in the nervous system. DRG nociceptors provide robust Kv3.4 channel expression in the somata, where this channel undergoes PKC-dependent modulation of inactivation and regulates AP repolarization in a manner that depends on this modulation (Ritter et al., 2012). Here, we have determined for the first time that dysfunction of Kv3.4 channels in the DRG is implicated in the SCI-induced hyperexcitability of small-diameter neurons. Others have also implicated downregulation of DRG Kv3.4 channels in chronic neuropathic pain induced by direct nerve injury and bone cancer (Chien et al., 2007; Duan et al., 2012). Furthermore, a proof-of-concept dynamic-clamp experiment demonstrated that, in addition to their previous known role in regulating AP duration, increasing densities of virtual Kv3.4 channels can also regulate the excitability of small-diameter DRG neurons by influencing the ISI and thereby dampening repetitive spiking (Fig. 11C). Although Kv3 channels are normally thought to enhance repetitive firing (Rudy and McBain, 2001), the unique combination of Kv3.4 channels with DRG specific Nav channels exhibiting slow recovery from inactivation inhibits firing. Thus, high voltage-activated Kv3.4 channels in these neurons operate with relatively low open probability near AP threshold to regulate AP firing, and above threshold with relatively high open probability to regulate AP repolarization. The potential therapeutic implications of this finding are significant. We propose that localized overexpression of Kv3.4 channels in the DRG would help normalize the excitability of DRG neurons following SCI and thereby inhibit pain sensitization, which might also be applicable in other chronic pain models.
Concluding remarks
Supporting the presence of a peripheral mechanism of persistent pain sensitization following hemicontusion SCI, we demonstrated accentuated spiking and downregulated function of Kv3.4 channels in contralateral DRG neurons. Further analysis pointed to a mechanism in which reduced surface expression is the main maladaptive change responsible for downregulation of these channels. Expression of Kv3.4 channels can dampen spiking in hyperexcitable DRG neurons and, therefore, investigating them as potentially significant targets would help develop novel interventions to treat SCI-induced chronic pain.
Footnotes
This work was supported by NINDS NS079855-01A1, Farber Family Foundation, Sigma Xi GIAR No. G20120315160396 (D.M.R.), Craig Nielsen Foundation Grant 190140 (A.C.L.), and Paralyzed Veterans of America Grant 160837 (A.C.L.). We thank Dr Cojen Ho for technical expertise, Dr Michael Gold for commenting on a previous version of the paper, Dr Lorin Milescu for help troubleshooting dynamic clamp code, Alexandra Kenny for her help with analysis and quantification of immunostaining, and members of the Covarrubias and Lepore laboratories for their feedback.
The authors declare no competing financial interests.
References
- Abbott GW, Butler MH, Bendahhou S, Dalakas MC, Ptacek LJ, Goldstein SA. MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell. 2001;104:217–231. doi: 10.1016/S0092-8674(01)00207-0. [DOI] [PubMed] [Google Scholar]
- Alvarez O, Gonzalez C, Latorre R. Counting channels: a tutorial guide on ion channel fluctuation analysis. Adv Physiol Educ. 2002;26:327–341. doi: 10.1152/advan.00006.2002. [DOI] [PubMed] [Google Scholar]
- Andrade EL, Forner S, Bento AF, Leite DF, Dias MA, Leal PC, Koepp J, Calixto JB. TRPA1 receptor modulation attenuates bladder overactivity induced by spinal cord injury. Am J Physiol Renal Physiol. 2011;300:F1223–F1234. doi: 10.1152/ajprenal.00535.2010. [DOI] [PubMed] [Google Scholar]
- Beck EJ, Sorensen RG, Slater SJ, Covarrubias M. Interactions between multiple phosphorylation sites in the inactivation particle of a K+ channel: insights into the molecular mechanism of protein kinase C action. J Gen Physiol. 1998;112:71–84. doi: 10.1085/jgp.112.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi SS, Yang Q, Crook RJ, Du J, Wu Z, Fishman HM, Grill RJ, Carlton SM, Walters ET. Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J Neurosci. 2010;30:14870–14882. doi: 10.1523/JNEUROSCI.2428-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi SS, Lago MT, Masha LI, Crook RJ, Grill RJ, Walters ET. Spinal cord injury triggers an intrinsic growth-promoting state in nociceptors. J Neurotrauma. 2012;29:925–935. doi: 10.1089/neu.2011.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA, Cummins TR, Yoshimura N, de Groat WC, Waxman SG. Tetrodotoxin-resistant sodium channels Na(v)1.8/SNS and Na(v)1.9/NaN in afferent neurons innervating urinary bladder in control and spinal cord injured rats. Brain Res. 2003;963:132–138. doi: 10.1016/S0006-8993(02)03957-4. [DOI] [PubMed] [Google Scholar]
- Brooke RE, Atkinson L, Batten TF, Deuchars SA, Deuchars J. Association of potassium channel Kv3.4 subunits with pre- and post-synaptic structures in brainstem and spinal cord. Neuroscience. 2004;126:1001–1010. doi: 10.1016/j.neuroscience.2004.03.051. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Du J, Davidson E, Zhou S, Coggeshall RE. Somatostatin receptors on peripheral primary afferent terminals: inhibition of sensitized nociceptors. Pain. 2001;90:233–244. doi: 10.1016/S0304-3959(00)00407-3. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, Yamani A, Lin Q, Willis WD, Hulsebosch CE. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain. 2009;147:265–276. doi: 10.1016/j.pain.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chien LY, Cheng JK, Chu D, Cheng CF, Tsaur ML. Reduced expression of A-type potassium channels in primary sensory neurons induces mechanical hypersensitivity. J Neurosci. 2007;27:9855–9865. doi: 10.1523/JNEUROSCI.0604-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M, Wei A, Salkoff L, Vyas TB. Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron. 1994;13:1403–1412. doi: 10.1016/0896-6273(94)90425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detloff MR, Wade RE, Jr, Houlé JD. Chronic at- and below-level pain after moderate unilateral cervical spinal cord contusion in rats. J Neurotrauma. 2013;30:884–890. doi: 10.1089/neu.2012.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan KZ, Xu Q, Zhang XM, Zhao ZQ, Mei YA, Zhang YQ. Targeting A-type K(+) channels in primary sensory neurons for bone cancer pain in a rat model. Pain. 2012;153:562–574. doi: 10.1016/j.pain.2011.11.020. [DOI] [PubMed] [Google Scholar]
- Dubin AE, Patapoutian A. Nociceptors: the sensors of the pain pathway. J Clin Invest. 2010;120:3760–3772. doi: 10.1172/JCI42843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineberg JD, Ritter DM, Covarrubias M. Modeling-independent elucidation of inactivation pathways in recombinant and native A-type Kv channels. J Gen Physiol. 2012;140:513–527. doi: 10.1085/jgp.201210869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerup NB, Johannesen IL, Sindrup SH, Bach FW, Jensen TS. Pain and dysesthesia in patients with spinal cord injury: a postal survey. Spinal Cord. 2001;39:256–262. doi: 10.1038/sj.sc.3101161. [DOI] [PubMed] [Google Scholar]
- Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16:1248–1257. doi: 10.1038/nm.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Dastmalchi S, Levine JD. Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience. 1996a;71:265–275. doi: 10.1016/0306-4522(95)00433-5. [DOI] [PubMed] [Google Scholar]
- Gold MS, Shuster MJ, Levine JD. Characterization of six voltage-gated K+ currents in adult rat sensory neurons. J Neurophysiol. 1996b;75:2629–2646. doi: 10.1152/jn.1996.75.6.2629. [DOI] [PubMed] [Google Scholar]
- Gwak YS, Hulsebosch CE. Neuronal hyperexcitability: a substrate for central neuropathic pain after spinal cord injury. Curr Pain Headache Rep. 2011;15:215–222. doi: 10.1007/s11916-011-0186-2. [DOI] [PubMed] [Google Scholar]
- Harper AA, Lawson SN. Conduction velocity is related to morphological cell type in rat dorsal root ganglion neurones. J Physiol. 1985a;359:31–46. doi: 10.1113/jphysiol.1985.sp015573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper AA, Lawson SN. Electrical properties of rat dorsal root ganglion neurones with different peripheral nerve conduction velocities. J Physiol. 1985b;359:47–63. doi: 10.1113/jphysiol.1985.sp015574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Hughes JP, Chessell I, Malamut R, Perkins M, Bačkonja M, Baron R, Farrar JT, Field MJ, Gereau RW, Gilron I, McMahon SB, Porreca F, Rappaport BA, Rice F, Richman LK, Segerdahl M, Seminowicz DA, Watkins LR, Waxman SG, Wiech K, Woolf C. Understanding chronic inflammatory and neuropathic pain. Ann N Y Acad Sci. 2012;1255:30–44. doi: 10.1111/j.1749-6632.2012.06561.x. [DOI] [PubMed] [Google Scholar]
- Hulsebosch CE, Hains BC, Crown ED, Carlton SM. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60:202–213. doi: 10.1016/j.brainresrev.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda VA, Lewis A, Xu X, Abbott GW. KCNE1 and KCNE2 inhibit forward trafficking of homomeric N-type voltage-gated potassium channels. Biophys J. 2011a;101:1354–1363. doi: 10.1016/j.bpj.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda VA, Lewis A, Xu X, Abbott GW. KCNE1 and KCNE2 provide a checkpoint governing voltage-gated potassium channel α-subunit composition. Biophys J. 2011b;101:1364–1375. doi: 10.1016/j.bpj.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Yao GL, Bean BP. Properties and functional role of voltage-dependent potassium channels in dendrites of rat cerebellar Purkinje neurons. J Neurosci. 2003;23:5698–5707. doi: 10.1523/JNEUROSCI.23-13-05698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri R, Keller A. Chronic pain following spinal cord injury. Adv Exp Med Biol. 2012;760:74–88. doi: 10.1007/978-1-4614-4090-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milescu LS, Yamanishi T, Ptak K, Mogri MZ, Smith JC. Real-time kinetic modeling of voltage-gated ion channels using dynamic clamp. Biophys J. 2008;95:66–87. doi: 10.1529/biophysj.107.118190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milescu LS, Yamanishi T, Ptak K, Smith JC. Kinetic properties and functional dynamics of sodium channels during repetitive spiking in a slow pacemaker neuron. J Neurosci. 2010;30:12113–12127. doi: 10.1523/JNEUROSCI.0445-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicaise C, Hala TJ, Frank DM, Parker JL, Authelet M, Leroy K, Brion JP, Wright MC, Lepore AC. Phrenic motor neuron degeneration compromises phrenic axonal circuitry and diaphragm activity in a unilateral cervical contusion model of spinal cord injury. Exp Neurol. 2012;235:539–552. doi: 10.1016/j.expneurol.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Rettig J, Wunder F, Stocker M, Lichtinghagen R, Mastiaux F, Beckh S, Kues W, Pedarzani P, Schröter KH, Ruppersberg JP. Characterization of a Shaw-related potassium channel family in rat brain. EMBO J. 1992;11:2473–2486. doi: 10.1002/j.1460-2075.1992.tb05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter DM, Ho C, O'Leary ME, Covarrubias M. Modulation of Kv3.4 channel N-type inactivation by protein kinase C shapes the action potential in dorsal root ganglion neurons. J Physiol. 2012;590:145–161. doi: 10.1113/jphysiol.2011.218560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/S0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Frank R, Pongs O, Stocker M. Cloned neuronal IK(A) channels reopen during recovery from inactivation. Nature. 1991a;353:657–660. doi: 10.1038/353657a0. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature. 1991b;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- Sekhon LH, Fehlings MG. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine (Phila Pa 1976) 2001;26:S2–12. doi: 10.1097/00007632-200112151-00002. [DOI] [PubMed] [Google Scholar]
- Siddall PJ, McClelland JM, Rutkowski SB, Cousins MJ. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain. 2003;103:249–257. doi: 10.1016/S0304-3959(02)00452-9. [DOI] [PubMed] [Google Scholar]
- Tsantoulas C, McMahon SB. Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. 2014;37:146–158. doi: 10.1016/j.tins.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters ET. Nociceptors as chronic drivers of pain and hyperreflexia after spinal cord injury: an adaptive-maladaptive hyperfunctional state hypothesis. Front Physiol. 2012;3:309. doi: 10.3389/fphys.2012.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann A, Krumova EK, Pennekamp W, Horch C, Baron R, Maier C. Different underlying pain mechanisms despite identical pain characteristics: a case report of a patient with spinal cord injury. Pain. 2012;153:1537–1540. doi: 10.1016/j.pain.2012.02.031. [DOI] [PubMed] [Google Scholar]
- Whitt JL, Masri R, Pulimood NS, Keller A. Pathological activity in mediodorsal thalamus of rats with spinal cord injury pain. J Neurosci. 2013;33:3915–3926. doi: 10.1523/JNEUROSCI.2639-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Yang Q, Crook RJ, O'Neil RG, Walters ET. TRPV1 channels make major contributions to behavioral hypersensitivity and spontaneous activity in nociceptors after spinal cord injury. Pain. 2013;154:2130–2141. doi: 10.1016/j.pain.2013.06.040. [DOI] [PubMed] [Google Scholar]
- Yang Q, Wu Z, Hadden JK, Odem MA, Zuo Y, Crook RJ, Frost JA, Walters ET. Persistent pain after spinal cord injury is maintained by primary afferent activity. J Neurosci. 2014;34:10765–10769. doi: 10.1523/JNEUROSCI.5316-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Tang Z, Zhang H, Atianjoh FE, Zhao JY, Liang L, Wang W, Guan X, Kao SC, Tiwari V, Gao YJ, Hoffman PN, Cui H, Li M, Dong X, Tao YX. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat Neurosci. 2013;16:1024–1031. doi: 10.1038/nn.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]