

Abstract
Adult progenitor cells proliferate in the acutely injured spinal cord and their progeny differentiate into new oligodendrocytes (OLs) that remyelinate spared axons. Whether this endogenous repair continues beyond the first week postinjury (wpi), however, is unknown. Identifying the duration of this response is essential for guiding therapies targeting improved recovery from spinal cord injury (SCI) by enhancing OL survival and/or remyelination. Here, we used two PDGFRα-reporter mouse lines and rats injected with a GFP-retrovirus to assess progenitor fate through 80 d after injury. Surprisingly, new OLs were generated as late as 3 months after injury and their processes ensheathed axons near and distal to the lesion, colocalized with MBP, and abutted Caspr+ profiles, suggesting newly formed myelin. Semithin sections confirmed stereotypical thin OL remyelination and few bare axons at 10 wpi, indicating that demyelination is relatively rare. Astrocytes in chronic tissue expressed the pro-OL differentiation and survival factors CNTF and FGF-2. In addition, pSTAT3+ NG2 cells were present through at least 5 wpi, revealing active signaling of the Jak/STAT pathway in these cells. The progenitor cell fate genes Sox11, Hes5, Id2, Id4, BMP2, and BMP4 were dynamically regulated for at least 4 wpi. Collectively, these data verify that the chronically injured spinal cord is highly dynamic. Endogenous repair, including oligodendrogenesis and remyelination, continues for several months after SCI, potentially in response to growth factors and/or transcription factor changes. Identifying and understanding spontaneous repair processes such as these is important so that beneficial plasticity is not inadvertently interrupted and effort is not exerted to needlessly duplicate ongoing spontaneous repair.
Keywords: demyelination, glial scar, growth factor, NG2 cell, stem cell, transplant
Introduction
Progenitor cells present throughout the CNS give rise to myelinating oligodendrocytes (OLs) during development and in adulthood (Nishiyama et al., 2009). These cells are traditionally identified by NG2 and platelet-derived growth factor receptor α (PDGFRα) expression and are the most proliferative cells of the adult CNS (Nishiyama et al., 1996; Horner et al., 2002; Dawson et al., 2003 Richardson et al., 2011).
NG2/PDGFRα cells retain their role as progenitors after injury or demyelination (Watanabe et al., 2002; Keirstead et al., 2005; Lytle et al., 2009; Tripathi et al., 2010). This is well demonstrated in the context of spinal cord injury (SCI), which causes extensive cell death and significant cell proliferation (McTigue et al., 2001; Zai and Wrathall, 2005; Horky et al., 2006; Sellers et al., 2009; Barnabé-Heider et al., 2010). OLs are lost in the first hours after SCI (Zai and Wrathall, 2005), and protracted OL apoptos is continues for 2–3 weeks postinjury (wpi), especially distal to the lesion (Crowe et al., 1997; Grossman et al., 2001). Surviving OLs are postmitotic and cannot remyelinate axons (Keirstead and Blakemore, 1997). Therefore, surviving progenitor cells are the major source for OL replacement and remyelination after SCI. Previous work demonstrated that SCI induces robust proliferation of NG2 cells (McTigue et al., 2001; Horky et al., 2006; Lytle et al., 2006; Rabchevsky et al., 2007). It also has been shown that, over the first 2 wpi, the progeny of the dividing NG2 cells differentiates into OLs, particularly along lesion borders (Zai and Wrathall, 2005; Horky et al., 2006; Lytle and Wrathall, 2007; Tripathi and McTigue, 2007). These new OLs likely contribute to remyelination, as recent and classic studies have shown evidence of OL remyelination beginning 2–3 wpi (Gledhill et al., 1973; Harrison et al., 1975; Sellers et al., 2009).
Although acute progenitor cell responses after SCI are well documented, there is a substantial gap in the literature regarding progenitor cell behavior chronically. It is unknown whether new OLs are generated beyond 2 wpi and, if so, if they can engage and remyelinate axons. Therefore, we tested the hypothesis that protracted progenitor cell proliferation occurs and results in chronic oligodendrogenesis after SCI. As lesions evolve differently between rats and mice, two different PDGFRα-reporter mice lines and adult rats injected with a GFP-expressing retrovirus were used to track progenitor cell fate for 12 wpi. Results reveal that oligodendrocyte replacement occurs continuously through 3 months after injury, along with signs of remyelination by late-born OLs. NG2 cells upregulate Jak/STAT signaling up to 5 wpi, alongside chronic increases in astrocytic expression of prosurvival growth factors for OLs and their progenitors. Finally, progenitor cell fate genes display dynamic changes for several wpi. Collectively, these data highlight that endogenous cell replacement continues for at least 3 months after SCI and that the chronic lesion milieu remains in a highly dynamic state. This long-term endogenous repair should be considered when designing drug treatments or cell transplantation strategies that may either take advantage of or inadvertently disrupt this chronic spontaneous repair.
Materials and Methods
Spinal cord injury.
All surgical and postoperative care procedures were performed in accordance with The Ohio State University Institutional Animal Care and Use Committee. Adult male and female mice (∼12 weeks old) and adult female Sprague Dawley rats (∼250 g) were anesthetized with a ketamine/xylazine mixture (10 and 80 mg/kg, respectively) and the spinal cord exposed at the T9 (mouse) or T8 (rat) vertebral level via a single-level laminectomy. The animals then received either a moderate-severe (75 kDyne force; mouse) or moderate (150 kDyne force; rat) spinal contusion injury using the Infinite Horizons device (Precision Instruments). A subset of rats received a comparable moderate injury with the Ohio State University electromechanic spinal cord injury device (0.9 mm displacement). The muscles overlying the spinal cord were then sutured and the skin was closed using surgical clips. Animals were given 2 cc (mouse) or 5 cc (rat) of saline and placed in warm recovery cages. Postsurgical care included 5 d treatment with antibiotics (gentomicin, 5 mg/kg) and saline to maintain hydration and twice-a-day manual bladder expression until spontaneous voiding returned. Sham animals received a laminectomy as described above but did not receive spinal cord injuries. All animals were assessed before injury for normal walking (score of 9 on BMS scale; 21 on BBB scale).
Retrovirus production and injection.
A gammaretrovirus based on the murine leukemia virus (MLV) was used to transfect dividing cells. Retroviral encoding the green fluorescence protein (RV-GFP, catalog #16664; Addgene) was produced by calcium chloride transient transfection into HEK293 cells, followed by supernatant viral purification by ultracentrifugation (Tashiro et al., 2006). Determination of viral titer was performed by serial dilution and visualization of colonies expressing GFP. The viral titer obtained ranged from 0.5 to 1 × 109 infectious units per milliliter. The virus was aliquoted and stored at −80°C until used.
For intraspinal virus injections, rats were anesthetized as above on day 1, 2, 7, 14, 21, or 28 after injury and the laminectomy site was reexposed (see Table 2 for group details). A volume of 1 μl of GFP-retrovirus solution was carefully drawn up into a sterile Hamilton syringe, which was gently inserted into the center of the lesion site. The solution was slowly injected into the spinal cord and the syringe maintained in place for an additional 2 min to prevent back-flux from the injection site. The surgery site was closed as above and animals returned to their home cages. Sham animals (n = 4) that had received a laminectomy but no injury were similarly injected with the 1 μl of GFP-retrovirus solution and killed 3 weeks later.
Table 2.
Animal number and time course information
| Rat | ||
|---|---|---|
| GFP-Virus | Kill date | n |
| 1 or 2 d | 1 wk | 6 |
| 1 d | 4 wk | 8 |
| 7 d | 4 wk | 4 |
| 14 d | 5 wk | 4 |
| 21 d | 6 wk | 4 |
| 28 d | 7 wk | 5 |
| N/A | 9 wk | 4 |
| PDGFRa-ROSA mice | ||
|---|---|---|
| Tamoxifen | Kill date | n |
| 28–31 d | 7 wk | 4 |
| 56–59 d | 11 wk | 6 |
| mTmG mice | ||
|---|---|---|
| Tamoxifen | Kill date | n |
| 1–4 d | 4 wk | 4 |
| 14–17 d | 5 wk | 5 |
| 21–24 d | 6 wk | 2 |
| 28–31 d | 7 wk | 5 |
| 42–45 d | 9 wk | 3 |
Generation of PDGFRα-CreER:mT/mG transgenic mice.
Because NG2 is expressed by pericytes and some macrophages after SCI, PDGFRα reporter mice were used for this study. PDGFRα-CreERT2 transgenic mice have been described previously (for detailed information, see supplementary information in Rivers et al., 2008). Heterozygous PDGFRα-CreER mice (generous gift from Dr. William Richardson) that were backcrossed with C57BL/6J mice for four generations were bred with mT/mG mice (Jackson Laboratories, strain B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) in which all cells express membrane-targeted tandem dimer Tomato (mT) before Cre-mediated recombination (Muzumdar et al., 2007) (see Fig. 4A). All mice used for experiments were PDGFRα+ and heterozygous for mT/mG. After recombination, cells transcribing the PDGFRα promoter express membrane-targeted enhanced GFP (mG). Recombination was induced by oral administration of tamoxifen (300 mg/kg) for 4 consecutive days starting at 1, 14, 21, 28, or 42 d postinjury (dpi) and animals were killed 3 weeks later (see Table 2 for group details).
Figure 4.

Reporter mice successfully label NG2+ cells that can be followed for at least 3 weeks after recombination. A, Schematic of breeding paradigm to generate PDGFRα-CreER:mT/mG mice. PDGFRα-CreER mice were crossed with ROSA26-mGFP (mT/mG) mice to generate mice that ubiquitously express mT. Upon recombination by oral administration of tamoxifen, most cells expressing the PDGFRα promoter begin to express mG (∼90%) and downregulate mT. pCA, Promoter with CMV enhancer; pA, polyadenylation sequences; arrows indicate the direction of transcription. Single-channel and merged confocal images of GFP+ NG2 cells in naive spinal cord white (B) and gray (C) matter. Single-channel and merged confocal images of GFP+ NG2 cells from spared white matter labeled at 1 wpi and examined at 4 wpi (D), labeled at 2 wpi and examined at 5 wpi (E), or labeled at 4 wpi and examined at 7 wpi. F, Low-power visual comparison of naive (G) and injured spinal cord (7 wpi, 250 μm rostral to the epicenter; H) labeled with GFP and NG2. Nuclei are labeled with DAPI in blue. Scale bars: B, D, E, 50 μm; C, G, H, 100 μm; F, 10 μm.
Generation of PDGFRα-ROSA transgenic mice.
Heterozygous PDGFRα-CreER transgenic mice were crossed with ROSA26-EYFP mice (Jackson Laboratories, strain B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J) to generate PDGFRα-ROSA mice that express cytoplasmic YFP under the control of the PDGFRα promoter after recombination. Recombination was induced by oral administration of tamoxifen (200 mg/kg) for 4 consecutive days starting at 28 or 52 dpi, and animals were killed at 52 and 80 dpi (∼7 and 12 wpi), respectively (see Table 2 for group details).
Tissue processing: immunohistochemistry.
For tissue collection, rats were deeply anesthetized with ketamine and xylazine (1.5× surgery dose above) and then perfused transcardially with PBS, followed by 250 ml of 4% paraformaldehyde in PBS. Spinal cords were removed, postfixed for 2 h at 4°C, and placed in 0.2 m PB overnight. Tissue was cryoprotected in 30% sucrose at 4°C for 48 h. For tissue embedding, spinal cords were frozen on dry ice and cut into 1 cm blocks centered on the injection site. After submersion in OCT compound (Electron Microscopy Sciences), blocks were frozen and cross-sections were cut at 10 μm on a cryostat and mounted onto slides. Tissue was stored at −20°C until use.
Tissue processing: epon embedding.
Rats received moderate spinal contusions as above and were perfused with 0.1 m PBS followed by 2% paraformaldehyde/2% glutaraldehyde solution at 28 d or 70 dpi (n = 3–4/group). Spinal cords were removed and a 1 cm segment of tissue centered on the lesion site was divided into 1 mm pieces and processed for epon embedding as described previously (Tripathi and McTigue, 2007). Semithin sections were cut at 1 μm in a transverse orientation on an ultramicrotome (Ultracut MZ6; Leica Microsystems). Sections were stained with 2% toluidine blue/2% borax and coverslipped.
Immunohistochemistry.
Sections were rinsed in 0.1 m PBS and blocked for nonspecific antigen binding using 4% BSA/0.1% Triton-100/PBS (BP+) for 1 h. Next, sections were incubated in primary antibody overnight at 4°C. Sections were rinsed and treated with biotinylated antiserum (1:800 in BP+; Vector Laboratories) for 1 h at room temperature. After rinsing, endogenous peroxidase activity was quenched using a 4:1 solution of methanol/30% hydrogen peroxide for 15 min in the dark. Sections were then treated with Elite avidin–biotin enzyme complex (ABC; Vector Laboratories) for 1 h. Visualization of labeling was achieved using DAB or SG substrates (Vector Laboratories). Sections were rinsed, dehydrated, and coverslipped with Permount (Fisher Scientific). For a complete list of antibodies used, see Table 1.
Table 1.
List of all primary antibodies used in this study
| Primary antibody: specificity | Concentration | Host species | Vendor |
|---|---|---|---|
| AldH1L1: astrocytes | 1/200 | Rabbit | Abcam |
| Caspr: functional paranodal junctions | 1/3000 | Rabbit | Abcam |
| CC1: oligodendrocytes | 1/100 | Mouse | Abcam |
| CNTF: ciliary neurotrophic factor | 1/100 | Goat | R&D Systems |
| FGF-2: fibroblast growth factor-2 | 1/500 | Mouse | Upstate |
| GFAP: astrocytes | 1/10,000 | Rabbit | Dako |
| GFP: green fluorescent protein | 1/800 | Chicken | Aves |
| Ki-67: proliferating cells | 1/100 | Mouse | Dako |
| MBP: myelin | 1/500 | Mouse | Abcam |
| 1/500 | Chicken | Aves | |
| NF: axons | 1/1000 | Chicken | Aves |
| 1/500 | Mouse | DSHB | |
| NG2: progenitor cells | 1/500 | Rabbit | US Biological |
| 1/200 | Mouse | US Biological | |
| pSTAT3: phosphorylated STAT3 | 1/200 | Rabbit | Cell Signaling Technology |
Immunofluorescence.
Sections were rinsed in 0.1 m PBS and blocked for nonspecific antigen binding using BP+ or 4% BSA/0.3% Triton-100/PBS (BP3+) for 1 h. Next, sections were incubated in primary antibody overnight at 4°C. Sections were rinsed and incubated with an Alexa Fluor secondary antibody (1:500; Invitrogen) for 1 h. For double- and triple-label immunofluorescence, sections were blocked and treated with primary and secondary antibodies as above. For mTmG tissue, Alexa Fluor 633 secondary antibodies were used and the signal was pseudocolored red. After rinses, slides were coverslipped with Immu-Mount (Thermo Scientific). For cases in which the primary antibody was raised in the same species as the tissue being labeled (mouse anti-CC1), the primary and secondary antibodies were first complexed together in BP3+ with 3% normal goat serum at 37°C for 1 h, followed by the addition of normal mouse serum (1:500 dilution) for another 1 h, and then left on ice for 2 more hours.
Microscopy and quantitative analysis.
Immunofluorescence labeling was analyzed by confocal microscopy (Olympus FV1000 laser scanning confocal microscope) and Fluoview software. Due to the high recombination efficiency of PDGFRα-CreER:mT/mG mice (>90% PDGFRα+ cells expressed GFP) and the variability of retroviral uptake in rats, all GFP+ cell quantifications were performed in reporter mice. For quantification of fluorescent GFP+/CC1+ cells, nine nonoverlapping images per spinal cord section (eight in the perimeter of spared tissue and one at the central canal region) were collected at the epicenter, 0.15 mm, and 0.45 mm rostral and caudal to the injury site. The total number of GFP+/CC1+ cells from all nine images per section were summed and expressed as a fraction over the total area (in cubic millimeters) of spinal cord represented in the same images. A cell was counted if the CC1 label colocalized with DAPI and was completely surrounded by GFP labeling. A two-way non-repeated-measures ANOVA was used to analyze CC1/GFP/DAPI+ cell counts with a Bonferroni post hoc test and significance was reported when p < 0.05.
A Zeiss Axioskop 2 Plus microscope with a Sony 970 three-chip color camera was used to analyze non-fluorescent sections. Cells double labeled for NG2 and Ki-67 or NG2 and pSTAT3 were manually counted at high power (40×) throughout spinal cord cross-sections. Cell counting was conducted in a conservative manner and cell counts are expressed as cells per cubic millimeter. Criteria for a proliferating NG2 cell to be counted included a clearly defined border immunoreactive for NG2 surrounding an entire nucleus immunoreactive for Ki-67 or pSTAT3. These criteria had to be met in the same plane of focus. Care was taken to avoid counting NG2+ pericytes or macrophages, which are both easily distinguished by cellular morphology. A one-way ANOVA was used to analyze cells counts where significance was reported when p < 0.05. Total counts of NG2/pSTAT3+ cell reflect the summation of nine evenly spaced 40× reticule boxes placed around the lesion border, while Ki-67/NG2+ counts represent the entire cross-section.
Epon-embedded tissue was analyzed for axon integrity/myelination and g-ratio measurements. Using a Zeiss 100× oil objective, nine evenly spaced images were systematically placed around the pial border of each section (epicenter and 2 mm rostral and caudal). A rectangle (covering ∼1/3 of each image) was centered over the image and all axons within this rectangle were categorized as “bare,” “pathological,” or “myelinated.” g-ratios were determined by dividing the axon diameter by the axon + myelin diameter. Noncircular (oblong-shaped) axons were excluded from counts. g-ratios were analyzed using a repeated-measures 2-way ANOVA and a Bonferroni post hoc test, where significance was reported when p < 0.05.
Quantitative real-time PCR.
cDNA was prepared from RNA isolated from mouse SCI tissue centered around the injury site as previously described using the TRIzol method (Kigerl et al., 2007). Samples were collected at 1 dpi, 3 dpi, 1 wpi, 2 wpi, and 4 wpi, as well as from naive tissue (n = 4–5 per group). Levels of PCR product were measured using SYBR green fluorescence. The primer sequences used were as follows: 18S(F) TTCGGAACTGAGGCCATGAT, (R) TTTCGCTCTGGTCCGTCTTG; Id2(F) GCTCTACAACATGAACGACTGCTACT, (R) TGCAGGTCCAAGATGTAATCGA; Id4(F) GAGACTCACCCTGCTTTGCT, (R) AGAATGCTGTCACCCTGCTT; Hes5(F) GCTGAGTGCTTTCCTATGAGGAA, (R) GCCCTGGGCACATTTGC; BMP2(F) CGTGCGCAGCTTCCATCACG, (R) GAAGAAGCGCCGGGCCGTTT; BMP4(F) GCATCCGAGCTGAGAGACCCCA, (R) ATCCCATCAGGGACGGAGACCA; Sox11(F) ATGGTGCAGCAGGCCG, (R) TCAATACGTGAACACCAGGTCG. Data were analyzed using the Ct method (Livak and Schmittgen, 2001) and a one-way ANOVA with a Tukey post hoc test. Significance was reported when p < 0.05.
Results
OL progenitors proliferate for at least 4 weeks after SCI in rats
Studies have shown that dividing NG2+ progenitors differentiate into new OLs acutely after SCI. The long-term response and distribution of progenitors, however, is not known. To track the fate of these cells after rat SCI, a GFP-expressing retrovirus was injected into lesioned rat spinal cord, which induced GFP expression in cells dividing within 2 h of viral injection. Spinal cords were examined 1–4 weeks later (Table 2) to determine the fate of cells dividing at the time of virus injection.
We first confirmed that retroviral-labeled cells could be detected 4 weeks later. In spinal cords injected at 1 or 2 dpi, robust GFP cell labeling was present at 4 wpi throughout ∼1 cm of tissue centered on the injury site (Fig. 1A). GFP+ cells were present in the lesion cavity and in spared white and gray matter surrounding the injury site. To ensure that the large number of GFP+ cells was not an artifact of viral injection, GFP-retrovirus was injected into laminectomy-control naive rat spinal cords (n = 4) and the tissue examined 3 weeks later. In contrast to SCI tissue (Fig. 1A), GFP labeling in uninjured spinal cords was minimal and confined to ± 0.5 mm around the injection site (Fig. 1B), indicating that the injection procedure stimulated only minor local proliferation.
Figure 1.
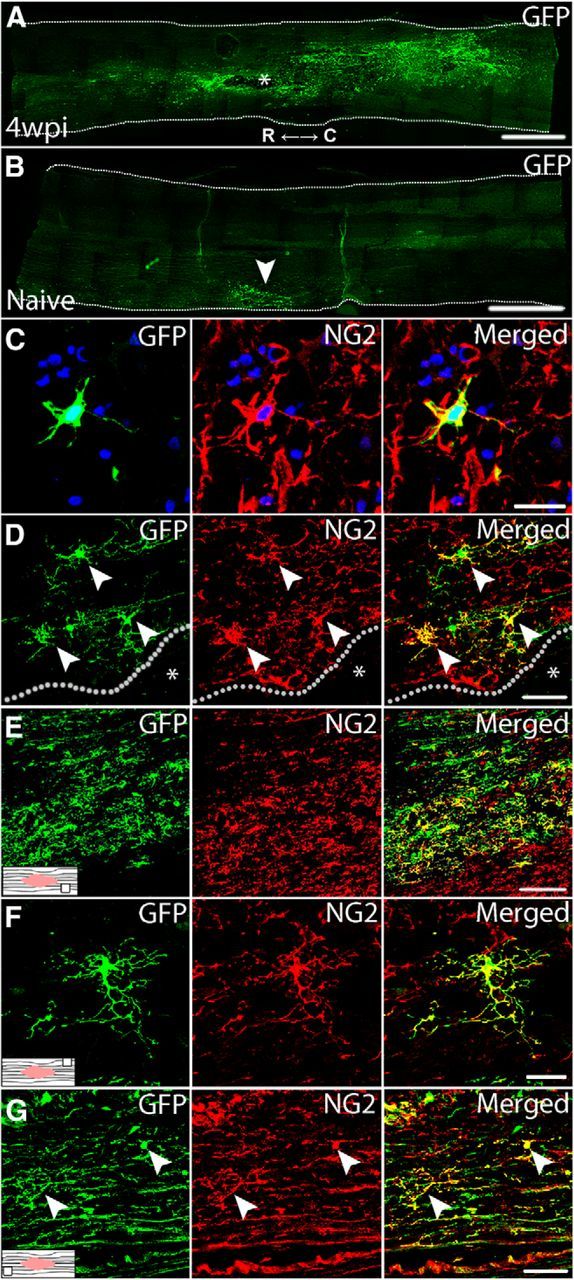
Intraspinal injection of GFP-retrovirus successfully labels proliferating NG2 cells after SCI in rat. A, Widespread GFP-expression in the spinal cord at 4 wpi (virus at 1 dpi). Injury cavity is denoted by *. R, Rostral; C, caudal. B, In naive spinal cord, intraspinal virus injection stimulates only a local cell proliferation (GFP+ cells) around the injection site. Single-channel and merged confocal images of GFP+/NG2+ cells at 1 wpi (virus at 1 dpi; C), 4 wpi (virus at 1 dpi; D, E), and 7 wpi (virus at 4 wpi; F, G) at various locations throughout the spinal cord. D, High-power view of the lesion border from A. Location within the spinal cord where images were taken is indicated by associated schematics. Nuclei are labeled with DAPI in blue. Lesion cavity denoted by *; lesion border denoted by dotted line. Scale bars: A, B, 1250 μm; C, F, 20 μm; D, G, 50 μm; E, 150 μm.
Next, we tested whether the virus labeled proliferating NG2+ cells. For this, virus was injected at 1 or 2 dpi (a time of robust NG2 cell proliferation) and spinal cords examined 1 or 4 weeks later. At both times, numerous GFP+ NG2 cells were detected in all spinal cords examined (Fig. 1C–E). GFP+ NG2 cells were prevalent along lesion borders and in spared gray and white matter up to 5 mm distal to the injury epicenter (Fig. 1D,E).
Our prior work showed that NG2 cells divide for at least 4 weeks after SCI in rats (McTigue et al., 2001). To determine the fate of cells dividing throughout the first month after injury, a bolus of GFP-retrovirus was injected into SCI lesions at 1, 2, 3, or 4 wpi and spinal cords examined 3 weeks later (i.e., 4–7 wpi) for the presence and distribution of GFP-expressing cells (Table 2). At every time examined, GFP+ NG2 cells had distributed throughout 1 cm of tissue centered on the injury site (Fig. 1F,G), revealing that the NG2 cell population proliferates continuously for at least 1 month after SCI and their progeny are maintained throughout the tissue for several weeks thereafter.
The distribution and number of GFP+ NG2 cells varied based on time at which they were labeled. Most NG2 cells dividing at 1 dpi accumulated in a dense band within the lesion border/glial scar. They were also present in the distal dorsal column lesion extensions, and scattered throughout the spared tissue. At later times, the number of NG2 cell progeny decreased progressively and had a more outwardly radial distribution. For example, in tissue examined at 7 wpi after 4 wpi virus injection, the overall number of GFP+ cells was lower than the earlier groups and few new cells accumulated along the lesion border. Rather, new NG2 cells born at 4 wpi were primarily found in spared tissue outside the glial scar, followed by distal spared white matter (Fig. 1G).
Because cell labeling by viral injections can be nonuniform between animals, NG2 cell division was quantified at 1–10 wpi using NG2/Ki-67 double-label immunohistochemistry. NG2/Ki-67+ cells were significantly increased compared with naive controls between 1 and 4 wpi (Fig. 2A–C). Interestingly, the most robust NG2 cell proliferation occurred rostral to the epicenter (Fig. 2). The number of Ki-67+ NG2 cells declined after 4 wpi but remained 3- to 9-fold above controls as late as 10 wpi, revealing protracted proliferation of this cell population after SCI. These data also verify that NG2 cells were dividing at each time of virus injection. In summary, NG2 cells divide for at least 4 wpi, the progeny of which survive and incorporate throughout injured and spared tissue after SCI. In addition, cells dividing acutely appear to contribute to the glial scar.
Figure 2.

NG2 cells proliferate for at least 28 dpi. Number of Ki-67/NG2+ cells/mm3 in the rat spinal cord 0.9 mm rostral to the epicenter (A), at the epicenter (B), and 0.9 mm caudal to the epicenter (C) from 7 to 70 dpi. *p < 0.05; **p < 0.01; ***p < 0.001 versus naive.
Dividing progenitors differentiate into new OLs for at least 4 weeks after SCI in rats
Although it is established that new OLs are produced for the first 2 wpi (Tripathi and McTigue, 2007), it is not known whether more chronic lesion environments support new OL formation. Therefore, sections from rats injected with virus at 1 dpi through 4 wpi were examined 3–4 weeks later for GFP+ OLs. Surprisingly, new OLs (GFP+/CC1+ cells) were generated in spared tissue at every time examined, including as late as between 4 and 7 wpi (Fig. 3A–C). The distribution of new OLs was similar to that of GFP/NG2+ cells; that is, at 4 wpi, OLs derived from progenitors dividing at 1–2 dpi were mainly located in the glial scar/lesion border and in spared tissue directly around the lesion, which matches the distribution of NG2 cells proliferating at 1–2 dpi. In contrast, new OLs derived from progenitors dividing at 4 wpi were more diffusely distributed and were mainly located in distal spared white matter close to the pial border. These results reveal that oligodendrogenesis occurs as late as the second month after SCI in rats and that new OLs distribute throughout the tissue surrounding the lesion cavity. Because the virus only survives ∼2 h after injection, these results provide a snapshot of the fate of cells dividing at each injection time and likely are conservative estimates of cell turnover and differentiation.
Figure 3.

Chronic oligodendrogenesis from proliferating progenitors occurs in the injured rat spinal cord. Single-channel and merged confocal images of GFP+ oligodendrocytes (red, CC1+ cells) formed around the lesion at different times after injury. A, Rats received intraspinal virus injection at 1 dpi; tissue was examined at 4 wpi. B, Rats received intraspinal virus injection at 3 wpi; tissue was examined at 6 wpi. C, Rats received intraspinal virus injection at 4 wpi; tissue was examined at 7 wpi. Nuclei are labeled with DAPI in blue. Scale bars: A, 50 μm; B, C, 30 μm.
Oligodendrogenesis continues for at least 80 d after SCI in mice
Because SCI lesion pathology differs between rats and mice, two different reporter mouse lines with inducible GFP/YFP expression under the PDGFRα promoter were used to examine post-SCI oligodendrogenesis in mice. PDGFRα-CreER:mT/mG mice express mG (Muzumdar et al., 2007) in PDGFRα+ cells after tamoxifen treatment, which allows visualization of fine cellular processes and myelin generated after GFP induction (Kang et al., 2010; Powers et al., 2013; Fig. 4A). The second line, PDGFRα-CreER-ROSA mice, express cytoplasmic YFP in PDGFRα+ cells upon Cre-recombination by tamoxifen. In both lines, GFP/YFP expression was induced in PDGFRα+ progenitors at different times after injury and the fate of the labeled cells was examined 3 weeks later (Table 2).
To confirm efficient labeling of OL progenitors in tamoxifen-treated PDGFRα-CreER:mT/mG mice, cross-sections of naive spinal cords were immunolabeled for NG2 or PDGFRα. In these sections ∼95% of GFP+ cells expressed NG2 and were present throughout white and gray matter (Fig. 4B,C). Likewise, all GFP+ cells coexpressed PDGFRα and ∼90% of all PDGFRα+ cells were GFP+, indicating a high level of recombination (data not shown). However, because recombination was not 100%, our results may underestimate changes in post-SCI OL genesis. Although there are NG2+ pericytes in the CNS, the only PDGFRα+ cells in the brain are OL progenitors, which avoids the potential confound of examining GFP+ pericytes (Daneman et al., 2010). Likewise, all GFP/NG2+ and GFP/PDGFRα+ cells morphologically resembled multiprocessed OL progenitors and not pericytes.
To examine progenitor cell distribution and fate at different times after SCI, cohorts of PDGFRα-CreER:mT/mG mice received a 4 d regimen of tamoxifen starting at 1 d or 2, 3, 4, or 6 weeks after SCI and were killed 3 weeks later (Table 2). Compared with naive tissue, GFP+/NG2+ cells were markedly increased at every time examined (4, 5, 6, 7, and 9 wpi) throughout the gray and white matter (Fig. 4D–F). Low-magnification comparison between naive and 7 wpi tissue illustrated that robust GFP and NG2 expression in the injured tissue was maintained ∼2 months after injury (Fig. 4G,H). This demonstrates that, similar to rats, an expanded progenitor population is maintained long-term, as noted previously (McTigue et al., 2001; Rosenberg et al., 2005; Rabchevsky et al., 2007).
Acute generation of new OLs after SCI has been reported in mice (Lytle and Wrathall, 2007). To determine whether oligodendrogenesis continues after 1 week post-SCI, sections from PDGFRα-CreER:mT/mG mice were examined for GFP+ OLs after tamoxifen administration as described above (Table 2). Because GFP is induced in progenitor cells at the time of tamoxifen treatment, any new OLs derived over the subsequent 3 weeks from progenitors can be identified by GFP expression. Tissue from different mouse cohorts revealed that new OLs were generated throughout spared tissue during every time interval examined, including 1–4, 2–5, 3–6, 4–7, and 6–9 wpi (Fig. 5A–D). In longitudinal sections, it was clear that OLs born as late as 6–9 wpi integrated into spared white matter, with their GFP+ processes running parallel to axons, resembling myelin profiles (Fig. 5D).
Figure 5.

Oligodendrogenesis occurs for at least 12 wpi in PDGFRα-CreER:mT/mG mice. Single-channel and merged confocal images of GFP+ oligodendrocytes (CC1; red) from spared white matter labeled at 1 wpi and examined at 4 wpi (A), from spared white matter labeled at 3 wpi and examined at 6 wpi (B), from spared white matter labeled at 4 wpi and examined at 7 wpi (C), and from spared white matter labeled at 6 wpi and examined at 9 wpi (D). Single-channel and merged confocal images of sections triple-labeled for CC1, NG2 and GFP from PDGFRα-ROSA mice labeled at 4 wpi and examined at 8 wpi (E) or labeled at 8 wpi and examined at 12 wpi (F). Nuclei are labeled with DAPI in blue. Scale bars: A, E, F, 30 μm; B, C, 20 μm; D, 50 μm.
To confirm chronic oligodendrogenesis in the second reporter mouse line, PDGFRα-CreER-ROSA mice were given tamoxifen at 4 or 8 wpi and killed at 8 and 12 wpi, respectively (Table 2). In both groups, YFP+ NG2 cells (labeled with GFP in images) and OLs were prevalent throughout spared white and gray matter (Fig. 5E,F). Therefore, OL progenitors divide and differentiate into new OLs that integrate into spared tissue for at least 3 months after SCI.
The distribution of new OLs in mice after SCI was similar to that in rats. The majority of new OLs generated from progenitors labeled during the first or second week after injury were mainly present around the lesion border with some new OLs located more distally (see Fig. 12). However, this pattern began to shift when progenitors were labeled during the third week after injury, with fewer new OLs around the lesion border and more located distally, in particular along the pial border. This trend was even more pronounced when progenitors were labeled at 6 wpi.
Figure 12.

Schematic illustrating change in pattern of new oligogenesis occurring at different times after SCI. Between 1 dpi and 4 wpi, most new OLs were generated around the lesion border with scattered new cells distally as well. Between 2 and 5 wpi, oligogenesis was more robust but still primarily concentrated around the lesion border. Between 3 and 6 wpi, fewer new OLs were generated around the lesion border and instead were mostly located along distal pial borders and throughout the dorsal column. Between 6 and 9 wpi, most new OLs were found in distal spared white matter along the pial border with few new OLs generated along lesion borders.
Quantification of new OLs in mouse SCI tissue
New OLs were quantified in naive and 4, 5, and 7 wpi tissue from PDGFRα-CreER:mT/mG mice. In naive tissue, 1–2 GFP/CC1+ cells at most per section were present, which is consistent with a low rate of oligodendrogenesis in adults (Horner et al., 2000; Psachoulia et al., 2009). After SCI, new OLs increased 5- to 26-fold, with the greatest accumulation of new OLs at 5 wpi rostral and caudal to epicenter (Fig. 6A). The number of new OLs in this tissue (labeled at 2 wpi) was significantly greater than in naive and tissue labeled at 1 dpi or 4 wpi. When collapsed over distance, new OLs increased 14-fold in 2–5 wpi tissue compared with naive (p < 0.01) and 5- to 6-fold in 1 dpi to 4 wpi and 4–7 wpi tissue (data not shown). Collectively, this reveals robust oligodendrogenesis occurs throughout the first 7 wpi.
Figure 6.
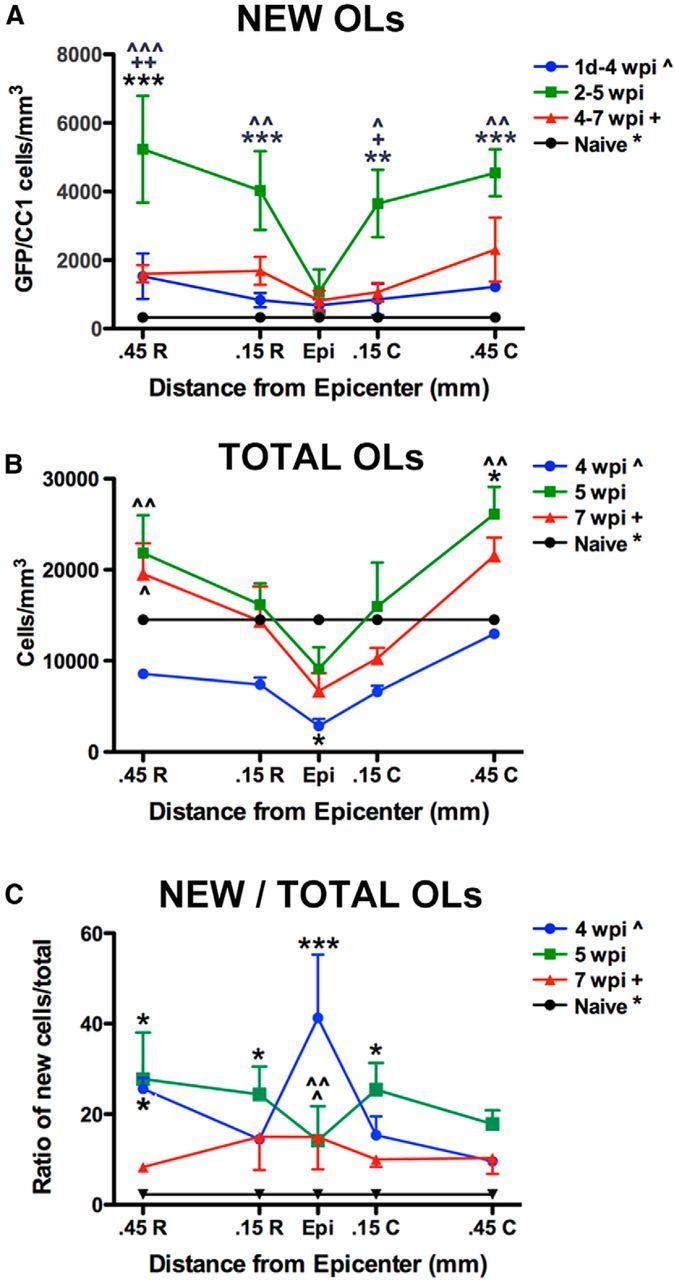
Quantification of new OLs after SCI in PDGFRα-CreER:mT/mG mice labeled and examined (respectively) at 1 dpi to 4 wpi, 2–5 wpi, and 4–7 wpi. A, Quantification of new OLs (GFP/CC1/DAPI+ cells) at each time point and distance examined (epicenter, Epi) and 0.45 and 0.15 mm rostral (R) and caudal (C). The greatest accumulation of new OLs occurred between 2 and 5 wpi rostral and caudal to epicenter. B, The overall number of OLs in each sampled region was determined. At 4 wpi, the total number of OLs was significantly reduced in epicenter compared with naive values. By 5 wpi, OL numbers had increased such that no regions were significantly lower than naive; 0.45 mm caudal to epicenter OLs significantly outnumbered those in naive. At 5 and 7 wpi, OL numbers 0.45 mm rostral to epicenter were significantly greater than the same area at 4 wpi. C, Percentage of new OLs of total OLs counted. At 4 wpi, the percentage of OLs that were new was significantly greater than naive at the epicenter and 0.45 mm rostral, and significantly greater than 5 and 7 wpi at the epicenter. Percentages remained higher than naive at 5 wpi in tissue 0.15 and 0.45 mm rostral to epicenter and 0.15 mm caudal. *p < 0.05; **p < 0.01; ***p < 0.001 versus naive; +p < 0.05, ++p < 0.01 versus 4–7 wpi group; ∧p < 0.05, ∧∧p < 0.01, ∧∧∧p < 0.001 versus 1 dpi to 4 wpi group.
To determine how overall OL numbers changed between 4 and 7 wpi, the number of OLs in each sampled region was counted. In spared tissue at 4 wpi, OLs were reduced 80% in the epicenter (p < 0.05; Fig 6B) and 10–55% distally compared with naive. Between 4 and 5 wpi, OL numbers tripled in the epicenter and rose 2- to 2.5-fold in distal sections, resulting in significantly greater OLs than 4 wpi (rostral and caudal) and naive (caudal; Fig. 6B). These data reflect the dynamic changes in OL numbers after SCI and again suggest that peak OL accumulation occurred between 4 and 5 wpi. At 7 wpi, OLs were slightly lower but still elevated rostrally compared with 4 wpi. This reveals that oligodendrogenesis over the first 5 wpi restores OLs and results in distal OL numbers greater than before injury, as seen previously in rat SCI tissue (Tripathi and McTigue, 2007).
Finally, the percentage of OLs composed of new cells was determined by calculating the ratio of new OLs to total OLs sampled (Fig 6C). At 4 wpi, when OL numbers were lowest, almost half (41%) of OLs in the epicenter were new (GFP+; p < 0.001 vs naive). In distal sections, 10–26% of OLs were new (p < 0.05 vs naive) at 4 wpi, revealing that many of the OLs lost during the first month after injury are replaced by new cells. By 5 wpi, the percentage of new OLs ranged from 14% to 28% versus 2% in naive (p < 0.05). Although oligodendrogenesis declined thereafter, ∼10–15% of OLs present at 7 wpi were generated during the third month after injury (4–7 wpi; Fig 6C). Therefore, progenitor differentiation into new OLs after SCI restores OL density to naive levels or greater by 5 wpi.
New OL lineage cells ensheathe and remyelinate axons for at least 2 months after injury in rats and mice
To assess whether new OLs initiate remyelination, sections from rat spinal cords injected with the GFP-retrovirus at 1 dpi or 4 wpi were examined at 4 and 7 wpi, respectively, for GFP, neurofilament (NF), and/or myelin basic protein (MBP). At 4 wpi, GFP+ processes encircled and wrapped NF+ axons along lesion borders and new cells integrated into spared white matter and engaged axons therein (Fig. 7A–C). GFP processes colocalized with MBP around axons, which was confirmed with Z-stack analysis (Fig. 7B), revealing that OLs generated during the first month after injury formed myelin around spinal cord axons along the lesion border and in spared tissue (Fig. 7B,C).
Figure 7.
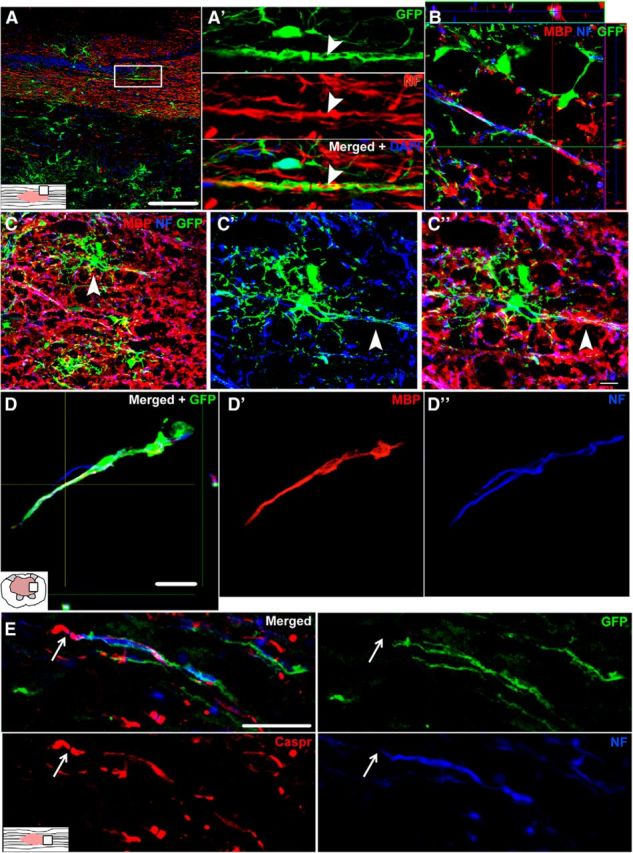
Single-channel and merged confocal images showing new OL lineage cells wrap axons and express markers of mature myelin after SCI in rat. A, Longitudinal spinal cord section at 4 wpi (virus injected at 1 dpi) labeled with GFP (green) and NF (red). A′, Single-channel and merged high-power view of rectangle from A. A GFP+ process ensheathes an NF+ axon (red) in spared white matter adjacent to the lesion (arrowhead). B, Z-stack and orthogonal view of an axon (blue) wrapped with a process double-labeled for GFP and MBP (red). GFP is cytoplasmic and therefore does not label the entire myelin membrane. C, Image of 4 wpi spared white matter labeled with GFP-virus at 1 dpi. Lesion cavity is near upper left corner. GFP+ cells (and therefore new) integrate into the spared white matter. C′, C′′, High-power images of cell indicated by arrowhead in C. The GFP+ cell has an OL morphology and extends processes double-labeled with MBP (red) along an axon (blue). D, D′, D′′, 7 wpi lesion site from spinal cord labeled with virus at 4 wpi. An axon (blue) extending into the lesion cavity is double-labeled with GFP+ MPB (red). E, A GFP+ process from an OL lineage cell in 7 wpi rat tissue wraps an NF+ axon (blue) and ends adjacent to a Caspr+ paranodal junction (red). Scale bars: A, 100 μm; C–C′′, 10 μm; D, E, 20 μm.
New OLs produced during the second month after injury in rats also wrapped axons. GFP+ processes again colocalized with NF and MBP, indicating that progenitor cells labeled at 4 wpi subsequently gave rise to new OLs that formed myelin around axons (Fig. 7D). Evidence of chronic remyelination in rats was bolstered by triple labeling for GFP, NF, and the paranodal protein Caspr. At 7 wpi, GFP+ processes that wrapped NF+ axons were adjacent to Caspr+ paranodal junctions, which are indicative of the formation of nodes of Ranvier between two myelinated segments (Fig. 7E).
PDGFRα-CreER:mT/mG mice revealed similar findings of ongoing axon ensheathment by OLs born between 1 dpi and 9 wpi. In these mice, GFP+ processes of new OL lineage cells engaged and wrapped NF+ axons at all time intervals examined, specifically between 0 and 4 wpi (Fig. 8A), 3–6 wpi (Fig. 8B), 4–7 wpi (Fig. 8C,D), and 6–9 wpi (Fig. 8E,F). At more chronic times, most newly GFP-wrapped axons were located along the distal pial border, which is where the majority of new OLs were generated chronically. Sections from 9 wpi mouse tissue triple-labeled for GFP, NF, and Caspr revealed that, similar to rat tissue, Caspr+ paranodal junctions were present along NF+ axons adjacent to GFP+ processes wrapping the axon (Fig 8F). In some axons, the myelin sheath between two paranodal Caspr+ profiles was abnormally short, which is a hallmark of early remyelination (Fig 8F). The presence of GFP+ myelin on only one side of this node suggests that the other internode was myelinated either by spared myelin or myelin generated before 6–9 wpi.
Figure 8.

Single-channel and merged confocal images show OL lineage cells continually wrap axons from 0 to 9 wpi in mouse SCI tissue. GFP+ processes colocalize with NF+ axons (red) in spared tissue during the time periods of 0–4 wpi (A), 3–6 wpi (B), 4–7 wpi (C, D), and 6–9 wpi (E). Examples in B and D clearly demonstrate the sheaths that GFP+ processes form around axons. The image in B also shows the adjacent OL lineage cell that gives rise to the GFP+ processes wrapping an NF+ axon. Examples of GFP/NF colocalization could be found in both spared white matter (C–E) as well as along lesion borders (A, B). F, GFP+ processes in 9 wpi tissue wrap a NF+ axon (blue; Alexa Fluor 405) that express Caspr+ paranodal junctions (red). Nuclei are labeled with DAPI in blue except in F. Scale bars: A, C, E, F, 10 μm; D, 20 μm; B, E, 30 μm.
Semithin sections reveal evidence of chronic OL remyelination with limited demyelination
To better examine the ultrastructure of new myelin, rats were injured as above and spinal cords were collected at 4 or 10 wpi. This tissue was embedded in epon and 1 μm sections were stained with toluidine blue. At both 4 and 10 wpi, axons with abnormally thin myelin sheaths, indicative of OL remyelination, were prevalent along lesion borders (Fig 9A–C) and in spared white matter near the pia (Fig 9D). At 10 wpi, there was still ongoing tissue pathology, including myelin degeneration (Fig 9B–D) and macrophages with engulfed myelin profiles throughout the white matter. Although fairly rare, a small number of axons completely lacking myelin were present in tissue bordering the lesion (Fig 9C) and in spared tissue along the pial border (Fig 9D).
Figure 9.
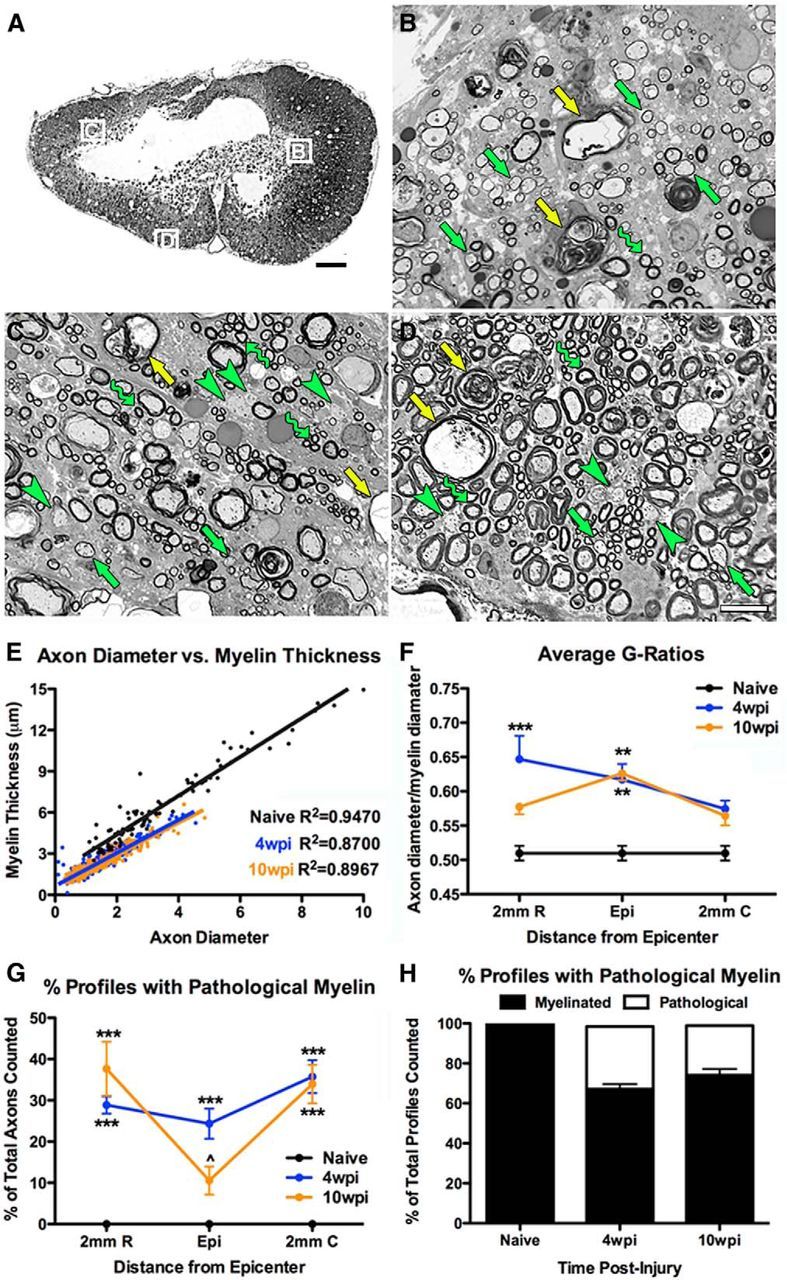
Semithin sections from 10 wpi rat spinal cords reveal evidence of ongoing OL remyelination and spared demyelinated axons. A, Low-power image of a typical 1 μm epon-embedded rat spinal cord cross-section from the lesion epicenter at 10 wpi. Boxes along lesion borders are shown at higher power in B–D. B–D, Images showing examples of spared myelin profiles (wavy arrow) and myelin rings thinner than would be expected for spared myelin (green arrows). These are typically classified as newly formed myelin or remyelination and are in the area of prominent new OL generation. Axons with apparent healthy morphology but lacking myelin are also present (green arrowheads). Ongoing pathology including myelin debris and axon pathology is still prominent (yellow arrows), indicating the dynamic nature of this 10 wpi lesion. E, Comparison of axon diameter versus myelin thickness shows that myelinated axons after injury are smaller than in naive tissue. F, Comparison of g-ratios after SCI demonstrates that, at least in rostral areas of the injured cord, the average g-ratio of axons decreased between 4 and 10 wpi, meaning increased myelin thickness. G–H, Between 4 wpi and 10 wpi, the number of axons displaying pathological myelin decreased, especially in the epicenter, whereas the number of myelinated axon profiles increased. **p < 0.01 versus naive; ***p < 0.001 versus naive; ∧p < 0.05 versus 4 wpi. Scale bars: A, 200 μm; B–D, 20 μm.
To better characterize changes in myelin thickness in chronic tissue, g-ratios were calculated on a subset of randomly sampled axon profiles from naive, 4 wpi, and 10 wpi sections (Fig 9E,F). This revealed a significant increase in g-ratios in the epicenter at both times, indicating an increase in axons with thinner myelin than normal. Rostral to the epicenter, g-ratios were significantly greater than naive at 4 wpi but not 10 wpi, suggesting either myelin thickening over time, as previously reported by Powers et al. (2013), or loss of the more thinly myelinated axons. Pathological myelin profiles indicate that tissue degeneration continues as late at 10 wpi. To determine whether this changed between 4 and 10 wpi, the number of pathological myelin profiles of the total number of myelin profiles counted was quantified. This showed a significant increase throughout the tissue at 4 wpi compared with naive, with a decline in number at the epicenter by 10 wpi (Fig 9G,H). Collectively, these data support the hypothesis that oligodendrocyte myelination continues chronically and that the lesioned tissue remains in a dynamic state, including remyelination and axon/myelin breakdown, for over 2 months after injury.
CNTF, FGF-2, and pSTAT3 expression remain elevated chronically after SCI in mice and rats
Our previous work showed that CNTF and FGF-2, two factors that promote OL formation and survival, are elevated for at least 4 wpi along the gliogenic lesion border after SCI in rats (Tripathi and McTigue, 2008). To determine whether these factors remain elevated beyond 4 wpi, FGF-2 and CNTF immunolabeling was performed. In naive rat spinal cords, FGF-2+ cells were present in the gray matter, as expected (Fig. 10A). After SCI, FGF-2 increased and was maintained throughout the spared tissue proximal to the lesion and in particular along the lesion border for at least 9 wpi in rats (Fig. 10B,C). FGF-2 did not colocalize with NG2 or CC1 (data not shown) and was primarily present in astrocytes (Fig. 10C).
Figure 10.
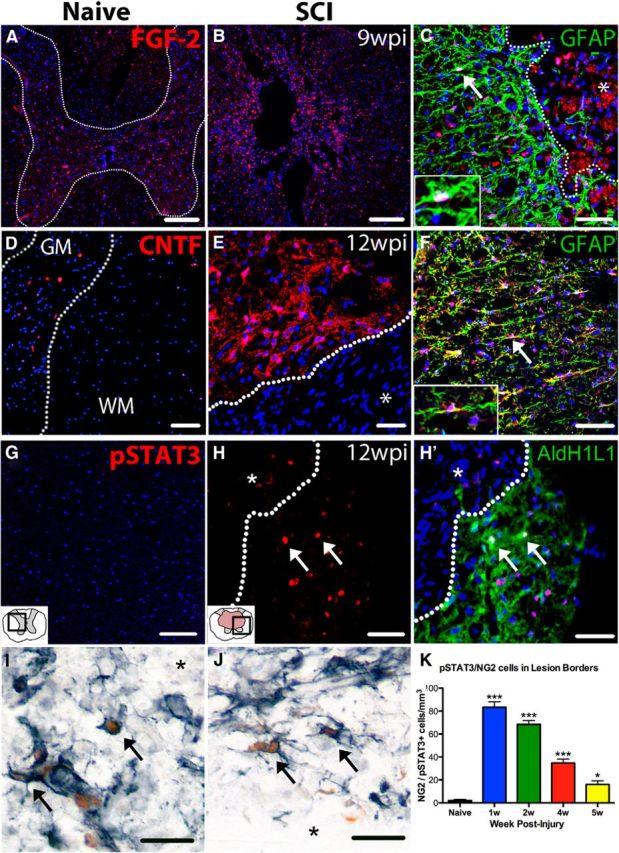
Confocal images reveal that FGF-2, CNTF, and pSTAT3 expression is increased chronically after SCI in mice and rats. A, Expression of FGF-2 (red) in naive rat spinal cord is mainly restricted to the gray matter. B, At 9 wpi in rat spinal cords, expression of FGF-2 is upregulated and spread more diffusely throughout spared gray and white matter as well as within the lesion cavity (center of image). C, At 9 wpi, FGF-2 is primarily expressed by GFAP+ astrocytes (green). Cell indicated with arrow shown at higher power in the inset. D, Naive mouse spinal cord tissue shows very little CNTF immunoreactivity (pseudocolored red). E, At 12 wpi, robust CNTF is present in cells bordering the lesion cavity (*). F, CNTF is primarily expressed by GFAP+ astrocytes (green), especially around the lesion border. G, In naive mouse spinal cord, there is virtually no expression of pSTAT3 (red). H, H′, Single and merged confocal images from 12 wpi mouse spinal cord show that pSTAT3 (red) is upregulated and found in many AldH1L1+ astrocytes (green). I–J, Acutely after injury (1 wpi), many lesion border NG2 (black) cells contain pSTAT3+ nuclei (brown). K, NG2/pSTAT3 double-labeled cells were quantified along the lesion border in naive and 1–5 wpi cross-sections from rats. The number of pSTAT3+ NG2 cells was significantly increased through 5 wpi. Nuclei are labeled by DAPI in blue. *Lesion cavity (C–J). In K: *p < 0.05 versus naive; ***p < 0.001 versus naive. Scale bars: A, B, 200 μm; C, F, H, H′, I, J, 50 μm; D, E, G, 100 μm.
Naive and 12 wpi mouse spinal cord sections were immunolabeled for CNTF. Minimal CNTF immunoreactivity was present in naive tissue, as expected (Fig. 10D). At 12 wpi, CNTF immunoreactivity was robust both along the glial scar/lesion border (Fig. 10E) and in spared tissue further from the lesion (Fig 10F). The majority of CTNF was expressed by astrocytes (Fig. 10F).
CNTF and related factors signal through the Jak/STAT pathway. To determine whether this pathway was active chronically, 12 wpi sections were immunolabeled for pSTAT3. Naive mouse tissue contained virtually no pSTAT3+ cells (Fig. 10G). In contrast, at 12 wpi, pSTAT3+ cells were abundant throughout spared white and gray matter (Fig. 10H), some of which were astrocytes (Fig. 10H′).
To determine whether CNTF (or related factors) initiate Jak/STAT signaling in OL lineage cells and potentially contribute to chronic oligodendrogenesis, sections were labeled for NG2 and pSTAT3 and the number of double-labeled cells counted. NG2 cells expressing pSTAT3 were prevalent after SCI, especially along the lesion borders (Fig 10I,J). The number of pSTAT3/NG2+ cells significantly increased in the epicenter and distal sections from 1 wpi through at least 5 wpi (Fig 10K). These data suggest that post-SCI factors such as CNTF stimulate the Jak/STAT pathway in NG2 cells, which may promote NG2 cell differentiation.
Expression of OL lineage cell-fate-associated genes is dynamically regulated after SCI
To determine whether endogenous signaling pathways that negatively regulate OL differentiation are altered by SCI, gene expression of Id2, Id4, Hes5, and Sox11 was analyzed by real time qPCR in epicenter SCI mouse tissue from 1 dpi to 4 wpi. Expression of Id2 increased significantly at 4 wpi (Fig. 11A), whereas Id4 increased significantly at 2 and 4 wpi compared with naive (Fig. 11B). This chronic increase in Id proteins may dampen oligodendrogenesis over time. In contrast, there were significant reductions in Hes5 mRNA at 1 dpi to 4 wpi (Fig. 11C) and Sox11 mRNA at 3 dpi to 4 wpi (Fig. 11D) compared with naive. Because these factors are part of pathways that prevent OL differentiation, these factors likely do not inhibit OL generation after SCI and, in fact, their reduction may help promote oligodendrogenesis (Swiss et al., 2011).
Figure 11.

Genes with possible roles in OL lineage cell fate are chronically modulated in tissue from the SCI lesion site in rats. Relative gene expression of Id2 (A), Id4 (B), Hes5 (C), Sox11 (D), BMP2 (E), and BMP4 (F) was examined in naive and SCI tissue using real-time PCR. *p < 0.05; **p < 0.01; ***p < 0.001 versus naive.
Another set of signaling molecules known to regulate oligodendrogenesis is BMP2 and BMP4 (See et al., 2004; Cheng et al., 2007; Porlan et al., 2013). BMP2 expression was dynamically regulated over the first week after injury; it increased significantly at 1 dpi, and then significantly declined by 7 dpi (Fig. 11E). BMP4, in contrast, rose over time to a significant increase at 2 wpi and 4 wpi (Fig. 11F). Because BMP4 can upregulate Id2 and Id4 (Samanta and Kessler, 2004), it may contribute to their upregulation at 2–4 wpi and thereby contribute to the deceleration in chronic oligodendrogenesis and myelination.
Discussion
Numerous studies have examined the time course and causes of OL death after SCI. Similarly, the extent of post-SCI demyelination and its clinical importance have been discussed and debated (for reviews, see Mekhail et al., 2012; Plemel et al., 2014). The belief that OL loss and demyelination are important therapeutic targets after SCI has led to multiple preclinical transplantation studies with the goal of replacing OLs and promoting remyelination; these studies demonstrated varying degrees of improved remyelination and/or functional recovery (Karimi-Abdolrezaee et al., 2006; Cao et al., 2010; Plemel et al., 2011; Sun et al., 2013), with work by Keirstead et al. (2005) forming the basis for a phase 1 clinical trial in acute SCI patients. Therefore, OL replacement is clearly viewed as a valuable clinical target.
Given that, it is somewhat surprising that the duration of spontaneous oligodendrogenesis has not received more attention. Adult progenitors are highly responsive to injury and can proliferate, differentiate, and remyelinate axons (McTigue and Tripathi, 2008), and studies have shown all these responses occur during the first 2 weeks post-SCI (McTigue et al., 2001; Zai and Wrathall, 2005; Horky et al., 2006; Lytle and Wrathall, 2007; Rabchevsky et al., 2007; Tripathi and McTigue, 2007; Sellers et al., 2009). How long this reparative response continues, however, is unknown.
Using Cre-ER technology and a GFP-expressing retrovirus, we tracked the long-term post-SCI fate of progenitors in mice and rats, respectively, to determine the duration of oligodendrogenesis and remyelination. Because retroviruses only incorporate into dividing cells, this approach allowed us to examine whether cells dividing after injury survive and/or differentiate into OLs. The results verified that NG2 cells proliferate for at least 4 wpi in rats and that new cells accumulate around the lesion border/glial scar and incorporate into the surrounding spared tissue. In mice, progenitor fate was tracked by inducing GFP/YFP in PDGFRα-expressing cells at different times after injury. This also revealed a chronic accumulation of progenitors at a higher than normal density, which is consistent with data showing NG2 cell density in spared tissue is 2–3× higher at 6 wpi compared with naive (Rosenberg et al., 2005).
The present data show that new OLs were produced as late as the 2 months after injury in rats (the latest time examined) and 3 months after injury in mice. New GFP+ OLs were generated throughout spared white and gray matter adjacent to the lesion and 4–5 mm distal to epicenter, showing widespread and protracted gliogenesis. GFP+ OL processes wrapped axons, expressed MBP, and abutted Caspr+ profiles, indicative of functional myelination and formation of paranodal junctions. Therefore, endogenous progenitors undergo oligodendrogenesis for at least 3 months after injury, during which new OLs myelinate/remyelinate axons. Although central canal ependymal cells also divide after SCI and produce a small number of OLs after dorsal spinal hemisection (Meletis et al., 2008), retrovirus injections in the present study were directed into the contusion epicenter in which all ependymal cells were killed, suggesting that their contribution, if anything, was minimal.
A point of controversy may be that long-term demyelination after SCI has not been widely reported, raising the question of exactly which axons did new OLs wrap? However, it may be exactly because of the unappreciated chronic production of new OLs that widespread demyelination is not typically noted. If axons are continually demyelinated over time, they may quickly become remyelinated by newly born OLs, as suggested in the epon tissue and reporter mice/rats. Further, work by Powers et al. (2013) using the mT/mG reporter mouse line used here suggested that new OL-derived myelin segments generated after SCI in the rubrospinal tract become progressively thicker and longer, resulting in a return to “baseline” conditions. Our g-ratio data agree with this because g-ratios in rostral tissue decreased between 4 and 10 wpi (meaning that myelin was thicker). If remyelinated segments indeed continuously thicken over time after SCI, the detection herein of thin myelin with short internodes at 2 and 3 months after injury further supports the notion that OL generation and remyelination occur continuously and chronically after SCI.
The present work also shows that the primary distribution of new OLs and remyelination shifts from proximal to the lesion core acutely (first month after injury) to tissue closer to the pial border more chronically (second to third months; Fig. 12). This could be explained by several mechanisms. First, an acute gradient of oligogenic signals may be highest in areas of greatest demyelination and damage (lesion epicenter), which induces local progenitor migration, division, and differentiation. The high expression of growth factors (CNTF, FGF-2) around the lesion border chronically, however, suggests that if such a gradient exists, it likely remains for several months. A second possibility is that distal axons undergo demyelination more chronically than axons along the lesion border. Because demyelinated axons are thought to be vulnerable and do not survive long-term after SCI (Lasiene et al., 2008), remyelination in chronic SCI likely occurs on axons that had only recently become demyelinated. Work by Crowe et al. (1997) supports chronic, distal myelin loss because they showed demyelination and OL apoptosis in distal white matter tracts (but not the lesion border) at 3 wpi. Our present work showing pathological degeneration of myelin at 10 wpi supports the idea that demyelination may occur chronically after SCI. A third possible explanation for chronic distal remyelination is that these areas may contain protracted axon sprouting, which in turn could stimulate oligodendrogenesis. This is not incompatible with the above mechanisms and, indeed, all three may develop concurrently after SCI.
The present work also illustrates that the chronic lesion environment remains dynamic, with markedly altered growth factors and cell fate gene expression changes. Previous work using immunohistochemistry and Western blot showed that CNTF and FGF-2 are elevated for 4 wpi around the lesion border (Tripathi and McTigue, 2008); the present work extends this increased growth factor expression to 12 wpi. CNTF is an astrocyte-derived factor that promotes OL lineage cell survival, differentiation, and myelination (Barres et al., 1993, 1996; Mayer et al., 1994; Salehi et al., 2013; Vernerey et al., 2013). It initiates phosphorylation of STAT3 and enhances the effects of FGF-2, which is also important for oligodendrogenesis (Dell'Albani et al., 1998; Baron et al., 2000; Yokogami et al., 2000). NG2 cells, especially around the lesion, expressed pSTAT3 for at least 5 wpi, indicating that this pathway remains active in NG2 cells. Interestingly, the time frame of peak pSTAT3 expression in NG2 cells (1–2 wpi) occurs concomitantly with the greatest NG2 cell proliferation (McTigue et al., 2001). Therefore, the chronic SCI environment contains robust expression of oligogenic factors that may promote ongoing gliogenesis/differentiation.
Another favorable factor for chronic post-SCI oligodendrogenesis was the significant reduction in Hes5 and Sox11, factors that inhibit OL differentiation. However, at least one factor that prevents oligodendrogenesis, BMP4, progressively increased after SCI. BMP4 can push NG2 cells toward an astrocytic fate (See et al., 2004; Cheng et al., 2007; Porlan et al., 2013), although BMP4 injection into the injured spinal cord actually reduced astrogliosis (Sellers et al., 2009). BMP4 can induce Id2 and Id4 expression, transcription factors that inhibit OL differentiation, and, indeed, RNA for both factors increased at 2–4 wpi. Therefore, the chronic injury milieu contains signaling molecules that can both promote and inhibit OL differentiation and myelination. The balance must favor long-term oligodendrogenesis, although the progressively increasing anti-oligogenic factors likely contribute to its reduction over time.
Cell quantification in this study revealed that greatest accumulation of new OLs after SCI was derived from cells differentiating between 2 and 5 wpi. Present data also show that there was a significant jump in total OL numbers between 4 and 5 wpi, which would suggest that peak new OL formation occurred during the fourth week after injury. However, it is possible (and even likely) that OL genesis was at least as robust before 4 wpi. Indeed, despite a significant reduction in OLs at 4 wpi, almost half of the OLs in epicenter sections were already GFP+ (and therefore new) at that time, indicating that they had differentiated from progenitors before 4 wpi. Therefore, robust OL genesis must occur throughout the first month after injury, which partially compensates for OL apoptosis and prevents OL numbers from falling drastically further.
In summary, this work demonstrates that the window of spontaneous oligodendrogenesis and remyelination after SCI is much longer than previously appreciated. New OLs incorporate throughout spared white matter tracks, where they initiate myelination for at least 3 months after injury. The knowledge of ongoing oligodendrogenesis and fluctuating progliogenic and antigliogenic factors provides insight into the chronic injury milieu, which may be crucial for predicting how strategies such as stem cell transplantation will fare long-term or whether therapeutically induced regenerated axons will become myelinated. Further, this may provide opportunities for leveraging endogenous progenitors, thereby obviating the need for exogenous cell transplantation. It is also important to understand the dynamics of this response so that pharmaceutical-based strategies targeting other post-SCI pathologies do not interfere with this reparative process. Last, knowing the potential negative regulators of oligodendrogenesis may provide new targets for treatments aimed at enhancing myelination of either spared or newly regenerated axons.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke (Grants NS059776 and P30-NS045758). Images presented in this report were generated using the instruments and services at the Campus Microscopy and Imaging Facility, The Ohio State University. We thank Ping Wei and Andrea Houchin for excellent technical assistance and William Richardson for the PDGFRα-CreER transgenic mice.
The authors declare no competing financial interests.
References
- Barnabé-Heider F, Göritz C, Sabelström H, Takebayashi H, Pfrieger FW, Meletis K, Frisén J. Origin of new glial cells in intact and injured adult spinal cord. Cell Stem Cell. 2010;7:470–482. doi: 10.1016/j.stem.2010.07.014. [DOI] [PubMed] [Google Scholar]
- Baron W, Metz B, Bansal R, Hoekstra D, de Vries H. PDGF and FGF-2 signaling in oligodendrocyte progenitor cells: regulation of proliferation and differentiation by multiple intracellular signaling pathways. Mol Cell Neurosci. 2000;15:314–329. doi: 10.1006/mcne.1999.0827. [DOI] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118:283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- Barres BA, Burne JF, Holtmann B, Thoenen H, Sendtner M, Raff MC. Ciliary neurotrophic factor enhances the rate of oligodendrocyte generation. Mol Cell Neurosci. 1996;8:146–156. doi: 10.1006/mcne.1996.0053. [DOI] [PubMed] [Google Scholar]
- Cao Q, He Q, Wang Y, Cheng X, Howard RM, Zhang Y, DeVries WH, Shields CB, Magnuson DS, Xu XM, Kim DH, Whittemore SR. Transplantation of ciliary neurotrophic factor-expressing adult oligodendrocyte precursor cells promotes remyelination and functional recovery after spinal cord injury. J Neurosci. 2010;30:2989–3001. doi: 10.1523/JNEUROSCI.3174-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Wang Y, He Q, Qiu M, Whittemore SR, Cao Q. Bone morphogenetic protein signaling and olig1/2 interact to regulate the differentiation and maturation of adult oligodendrocyte precursor cells. Stem Cells. 2007;25:3204–3214. doi: 10.1634/stemcells.2007-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–788. doi: 10.1016/S1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- Dell'Albani P, Kahn MA, Cole R, Condorelli DF, Giuffrida-Stella Am, de Vellis J. Oligodendroglial survival factors, PDGF-AA and CNTF, activate similar JAK/STAT signaling pathways. J Neurosci Res. 1998;54:191–205. doi: 10.1002/(SICI)1097-4547(19981015)54:2<191::AID-JNR7>3.0.CO%3B2-9. [DOI] [PubMed] [Google Scholar]
- Gledhill RF, Harrison BM, McDonald WI. Demyelination and remyelination after acute spinal cord compression. Exp Neurol. 1973;38:472–487. doi: 10.1016/0014-4886(73)90169-6. [DOI] [PubMed] [Google Scholar]
- Grossman SD, Rosenberg LJ, Wrathall JR. Temporal-spatial pattern of acute neuronal and glial loss after spinal cord contusion. Exp Neurol. 2001;168:273–282. doi: 10.1006/exnr.2001.7628. [DOI] [PubMed] [Google Scholar]
- Harrison BM, Gledhill RF, McDonald WJ. Remyelination after transient compression of the spinal cord. Proc Aust Assoc Neurol. 1975;12:117–122. [PubMed] [Google Scholar]
- Horky LL, Galimi F, Gage FH, Horner PJ. Fate of endogenous stem/progenitor cells following spinal cord injury. J Comp Neurol. 2006;498:525–538. doi: 10.1002/cne.21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner PJ, Power AE, Kempermann G, Kuhn HG, Palmer TD, Winkler J, Thal LJ, Gage FH. Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J Neurosci. 2000;20:2218–2228. doi: 10.1523/JNEUROSCI.20-06-02218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner PJ, Thallmair M, Gage FH. Defining the NG2-expressing cell of the adult CNS. J Neurocytol. 2002;31:469–480. doi: 10.1023/A:1025739630398. [DOI] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi-Abdolrezaee S, Eftekharpour E, Wang J, Morshead CM, Fehlings MG. Delayed transplantation of adult neural precursor cells promotes remyelination and functional neurological recovery after spinal cord injury. J Neurosci. 2006;26:3377–3389. doi: 10.1523/JNEUROSCI.4184-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keirstead HS, Blakemore WF. Identification of post-mitotic oligodendrocytes incapable of remyelination within the demyelinated adult spinal cord. J Neuropathol Exp Neurol. 1997;56:1191–1201. doi: 10.1097/00005072-199711000-00003. [DOI] [PubMed] [Google Scholar]
- Keirstead HS, Nistor G, Bernal G, Totoiu M, Cloutier F, Sharp K, Steward O. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci. 2005;25:4694–4705. doi: 10.1523/JNEUROSCI.0311-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Lai W, Rivest S, Hart RP, Satoskar AR, Popovich PG. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem. 2007;102:37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- Lasiene J, Shupe L, Perlmutter S, Horner P. No evidence for chronic demyelination in spared axons after spinal cord injury in a mouse. J Neurosci. 2008;28:3887–3896. doi: 10.1523/JNEUROSCI.4756-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lytle JM, Wrathall JR. Glial cell loss, proliferation and replacement in the contused murine spinal cord. Eur J Neurosci. 2007;25:1711–1724. doi: 10.1111/j.1460-9568.2007.05390.x. [DOI] [PubMed] [Google Scholar]
- Lytle JM, Vicini S, Wrathall JR. Phenotypic changes in NG2+ cells after spinal cord injury. J Neurotrauma. 2006;23:1726–1738. doi: 10.1089/neu.2006.23.1726. [DOI] [PubMed] [Google Scholar]
- Lytle JM, Chittajallu R, Wrathall JR, Gallo V. NG2 cell response in the CNP-EGFP mouse after contusive spinal cord injury. Glia. 2009;57:270–285. doi: 10.1002/glia.20755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M, Bhakoo K, Noble M. Ciliary neurotrophic factor and leukemia inhibitory factor promote the generation, maturation and survival of oligodendrocytes in vitro. Development. 1994;120:143–153. doi: 10.1242/dev.120.1.143. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem. 2008;107:1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Wei P, Stokes BT. Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J Neurosci. 2001;21:3392–3400. doi: 10.1523/JNEUROSCI.21-10-03392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekhail M, Almazan G, Tabrizian M. Oligodendrocyte-protection and remyelination post-spinal cord injuries: a review. Prog Neurobiol. 2012;96:322–339. doi: 10.1016/j.pneurobio.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Meletis K, Barnabé-Heider F, Carlén M, Evergren E, Tomilin N, Shupliakov O, Frisén J. Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol. 2008;6:e182. doi: 10.1371/journal.pbio.0060182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluroescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Co-localization of NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells in the developing rat brain. J Neurosci Res. 1996;43:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<299::AID-JNR5>3.0.CO%3B2-E. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, Zhu X. Polydendrocytes (NG2 cells): multifunctional cells with lineage plasticity. Nat Rev Neurosci. 2009;10:9–22. doi: 10.1038/nrn2495. [DOI] [PubMed] [Google Scholar]
- Plemel JR, Chojnacki A, Sparling JS, Liu J, Plunet W, Duncan GJ, Park SE, Weiss S, Tetzlaff W. Platelet-derived growth factor-responsive neural precursors give rise to myelinating oligodendrocytes after transplantation into the spinal cords of contused rats and dysmyelinated mice. Glia. 2011;59:1891–1910. doi: 10.1002/glia.21232. [DOI] [PubMed] [Google Scholar]
- Plemel JR, Keough MB, Duncan GJ, Sparling JS, Yong VW, Stys PK, Tetzlaff W. Remyelination after spinal cord injury: is it a target for repair? Prog Neurobiol. 2014;117:54–72. doi: 10.1016/j.pneurobio.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Porlan E, Morante-Redolat JM, Marqués-Torrejón MÁ, Andreu-Agulló C, Carneiro C, Gómez-Ibarlucea E, Soto A, Vidal A, Ferrón SR, Fariñas I. Transcriptional repression of Bmp2 by p21Waf1/Cip1 links quiescence to neural stem cell maintenance. Nat Neurosci. 2013;16:1567–1575. doi: 10.1038/nn.3545. [DOI] [PubMed] [Google Scholar]
- Powers BE, Sellers DL, Lovelett EA, Cheung W, Aalami SP, Zapertov N, Maris DO, Horner PJ. Remyelination reporter reveals prolonged refinement of spontaneously regenerated myelin. Proc Natl Acad Sci U S A. 2013;110:4075–4080. doi: 10.1073/pnas.1210293110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psachoulia K, Jamen F, Young KM, Richardson WD. Cell cycle dynamics of NG2 cells in the postnatal and ageing brain. Neuron Glia Biology. 2009;5:57–67. doi: 10.1017/S1740925X09990354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Sullivan PG, Scheff SW. Temporal-spatial dynamics of oligodendrocyte and glial progenitor cell numbers throughout ventrolateral white matter following contusion spinal cord injury. Glia. 2007;55:831–843. doi: 10.1002/glia.20508. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Young KM, Tripathi RB, McKenzie I. NG2-glia as multipotent neural stem cells–fact or fantasy? Neuron. 2011;70:661–673. doi: 10.1016/j.neuron.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg LJ, Zai LJ, Wrathall JR. Chronic alternations in the cellular composition of spinal cord white matter following contusion injury. Glia. 2005;49:107–120. doi: 10.1002/glia.20096. [DOI] [PubMed] [Google Scholar]
- Salehi Z, Hadiyan SP, Navidi R. Ciliary neurotrophic factor role in myelin oligodendrocyte glycoprotein expression in Cuprizone-induced multiple sclerosis mice. Cell Mol Neurobiol. 2013;33:531–535. doi: 10.1007/s10571-013-9918-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J, Kessler JA. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development. 2004;131:4131–4142. doi: 10.1242/dev.01273. [DOI] [PubMed] [Google Scholar]
- See J, Zhang X, Eraydin N, Mun SB, Mamontov P, Golden JA, Grinspan JB. Oligodendrocyte maturation is inhibited by bone morphogenetic protein. Mol Cell Neurosci. 2004;26:481–492. doi: 10.1016/j.mcn.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Sellers DL, Maris DO, Horner PJ. Postinjury niches induce temporal shifts in progenitor fates to direct lesion repair after spinal cord injury. J Neurosci. 2009:6722–6733. doi: 10.1523/JNEUROSCI.4538-08.2009. 29(20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Xu CC, Li J, Guan XY, Gao L, Ma LX, Li RX, Peng YW, Zhu GP. Transplantation of oligodendrocyte precursor cells improves locomotor deficits in rats with spinal cord irradiation injury. PLoS ONE. 2013;8:e57534. doi: 10.1371/journal.pone.0057534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiss VA, Nguyen T, Dugas J, Ibrahim A, Barres B, Androulakis IP, Casaccia P. Identification of a gene regulatory network necessary for the initiation of oligodendrocyte differentiation. PLoS ONE. 2011;6:e18088. doi: 10.1371/journal.pone.0018088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Zhao C, Gage FH. Retrovirus-mediated single-cell gene knockout technique in adult newborn neurons in vivo. Nat Protoc. 2006;1:3049–3055. doi: 10.1038/nprot.2006.473. [DOI] [PubMed] [Google Scholar]
- Tripathi R, McTigue DM. Prominent oligodendrocyte genesis along the border of spinal contusion lesions. Glia. 2007;55:698–711. doi: 10.1002/glia.20491. [DOI] [PubMed] [Google Scholar]
- Tripathi RB, McTigue DM. Chronically increase ciliary neurotrophic factor and fibroblast growth factor-2 expression after spinal contusion in rats. J Comp Neurol. 2008;510:129–144. doi: 10.1002/cne.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi RB, Rivers LE, Young KM, Jamen F, Richardson WD. NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J Neurosci. 2010;30:16383–16390. doi: 10.1523/JNEUROSCI.3411-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernerey J, Macchi M, Magalon K, Cayre M, Durbec P. Ciliary neurotrophic factor controls progenitor migration during remyelination in the adult rodent brain. J Neurosci. 2013;33:3240–3250. doi: 10.1523/JNEUROSCI.2579-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Toyama Y, Nishiyama A. Differentiation of proliferated NG2-positive glial progenitor cells in a remyelinating lesion. J Neurosci Res. 2002;69:826–836. doi: 10.1002/jnr.10338. [DOI] [PubMed] [Google Scholar]
- Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol. 2000;10:47–50. doi: 10.1016/S0960-9822(99)00268-7. [DOI] [PubMed] [Google Scholar]
- Zai LJ, Wrathall JR. Cell proliferation and replacement following contusive spinal cord injury. Glia. 2005;50:247–257. doi: 10.1002/glia.20176. [DOI] [PubMed] [Google Scholar]