

Abstract
The plasma membrane Ca2+-ATPase (PMCA) is found near postsynaptic NMDARs. This transporter is a Ca2+-H+ exchanger that raises cell surface pH. We tested whether the PMCA acts in an autocrine fashion to boost pH-sensitive, postsynaptic NMDAR currents. In mouse hippocampal slices, NMDAR EPSCs in a singly activated CA1 pyramidal neuron were reduced when buffering was augmented by exogenous carbonic anhydrase (XCAR). This effect was blocked by the enzyme inhibitor benzolamide and mimicked by the addition of HEPES buffer. Similar EPSC reduction occurred when PMCA activation was prevented by dialysis of BAPTA or the PMCA inhibitor carboxyeosin. Using HEPES, BAPTA, or carboxyeosin, the effect of XCAR was completely occluded. XCAR similarly curtailed NMDAR EPSCs of minimal amplitude, but had no effect on small AMPAR responses. These results indicate that a significant fraction of the postsynaptic NMDAR current is reliant on a perisynaptic extracellular alkaline shift generated by the PMCA.
Keywords: benzolamide, CA1, Ca2+ ATPase, carbonic anhydrase, carboxyeosin, hippocampus
Introduction
NMDA receptors are a major pathway of postsynaptic Ca2+ entry in pyramidal neurons (Kovalchuk et al., 2000) and have important roles in synaptic plasticity (Malenka and Bear, 2004). After its influx, Ca2+ is pumped from dendritic spines by the plasma membrane Ca2+-ATPase (PMCA) (Scheuss et al., 2006) via an electroneutral, 1:2 exchange for extracellular H+ (Carafoli and Stauffer, 1994; Thomas, 2009). Therefore, Ca2+ extrusion causes a fall in cytosolic pH and a rise in cell surface pH (Schwiening et al., 1993; Kreitzer et al., 2007; Makani and Chesler, 2010). A widespread rise in extracellular pH (pHe) results when many neurons are coactivated (Chesler, 2003). These pHe shifts can be detected within milliseconds (Gottfried and Chesler, 1996; Tong et al., 2006), indicating rapid activation of the PMCA by submembrane Ca2+.
NMDARs are highly sensitive to pHe (Traynelis et al., 2010). Factors governing this alkalosis have therefore been the subject of study, especially in hippocampal area CA1 (Chen and Chesler, 1992; Shah et al., 2005; Tong et al., 2006). The size and speed of this pHe shift is dependent on extracellular carbonic anhydrase (ECAR). This enzyme catalyzes the hydration of CO2 to HCO3− and H+, and thereby controls the rate of buffering. Although the CO2/HCO3− system has a high buffering capacity at equilibrium, this requires seconds because the overall ECAR activity is low. During the rise of an alkaline transient over milliseconds, the system is far from equilibrium. Therefore, the extracellular fluid is poorly buffered in the time frame of synaptic events (Tong et al., 2006).
In brain slices, rapid buffering efficacy was doubled by addition of exogenous CAR (XCAR) to the saline (Tong et al., 2006; Makani and Chesler, 2007). This curtailed the alkaline pHe transients and also reduced the NMDAR component of the EPSC in CA1 pyramidal cells, indicating that a fraction of the NMDAR current arose from the population alkalosis. AMPAR currents, by contrast, were unaffected (Makani and Chesler, 2007).
The relevance of these observations would seem limited to the occasions of synchronous activation sufficient to cause an effective population alkalosis. However, the description of PMCA isoforms adjacent to NMDARs suggests that this modulation may be far more discrete. Splice variants of PMCA2 were localized to dendritic spines (Burette et al., 2010) and coimmunoprecipitated with postsynaptic density proteins (DeMarco and Strehler, 2001). Given this proximity to postsynaptic NMDARs, an alkaline boost of the current might occur in a local, autocrine fashion. This modulation could be independent of population activity, requiring afferent input to just one or a few dendritic spines. We tested this hypothesis by studying the effect of XCAR on NMDAR-mediated EPSCs evoked solely in the patched cell. Our results indicate that postsynaptic NMDAR currents of a pyramidal neuron are boosted by approximately one third through activity of the same cell's PMCA. Part of this work has appeared previously in abstract form (Chen and Chesler, 2013).
Materials and Methods
Experimental preparation and solutions.
Procedures were performed with approval of the New York University School of Medicine Institutional Animal Care and Use Committee. Transverse hippocampal slices (250–300 μm) were prepared from P7–P14 Swiss Webster mice of either sex. A cut was made between the CA3 and CA1 regions to prevent reverberating synaptic activation of CA1 neurons. Slices were made and kept in low Ca2+ artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 3.0 KCl, 0.5 CaCl2, 3.0 MgCl2, 26 NaHCO3, 1.0 NaH2PO4, and 10 d-glucose plus 1 mm kynurenic acid. Slices recovered in the cutting ACSF at room temperature for at least 30 minutes and were then transferred to the recording chamber and superfused with standard ACSF for another 20 minutes. Standard ACSF contained the following (in mm): 124 NaCl, 3.0 KCl, 2.0 CaCl2, 1.5 MgCl2, 26 NaHCO3, 1.0 NaH2PO4, and 10 d-glucose, pH 7.4, with 95% O2 and 5% CO2. In some cases, a modified ACSF was used with 20 mm HEPES acid in addition to 26 mm NaHCO3. The HEPES ACSF was made using initial addition of 35 mm NaHCO3 with a compensatory reduction of NaCl to 109 mm. In 95% O2 and 5% CO2, titration of the HCO3− by HEPES acid produced a final HCO3− level of 26 mm, as judged by the final pH of 7.4. Experiments were conducted in a submersion-style chamber at 32°C.
Drugs were added to the external ACSF or to the pipette solution in the following concentrations: 6,7-dinitroquinoxaline-2,3-dione (DNQX) 10 μm, picrotoxin 100 μm, benzolamide (10 μm), ethylisopropylamiloride (EIPA) 10 μm, cariporide 100 μm, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) 100 μm, S0859 30 μm, niflumic acid 100 μm, lidocaine N-ethyl bromide (QX-314) 4 mm, 1,2-Bis (2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) 20 mm, carboxyeosin (CE) 1 or 5 μm (see below), and N-methyl verapamil bromide (D-890) 500 μm. In most cases, XCAR consisted of 1 μm bovine type II enzyme in ACSF. In three experiments, we used 2 μm human, recombinant type IV carbonic anhydrase. d-890, QX-314, and cariporide were obtained from Abcam, Tocris Bioscience (now R&D Systems), and Santa Cruz Biotechnology, respectively. Benzolamide was a gift from Dr. E. Swenson (University of Washington). Recombinant type IV carbonic anhydrase was a gift from Dr. W. Sly (St. Louis University). All other agents were obtained from Sigma-Aldrich.
Whole-cell voltage clamp.
CA1 pyramidal neurons were viewed under infrared differential interference contrast microscopy using a Zeiss Axioscop 2 Plus fixed-stage microscope with a 40×, water-immersion objective (0.75 numerical aperture), and a Dage-MTI video camera. Patch pipettes were pulled from 1.5 mm OD × 1.12 mm ID borosilicate tubing (World Precision Instruments). The pipette filling solution contained the following (in mm): 120 Cs-gluconate, 20 KCl, 20 HEPES, 2 Mg2+-ATP, 1.0 EGTA, and 4 QX-314 and was titrated to pH 7.3 with CsOH (osmolarity, 280–290 mOsm). Pipettes had resistances of 3–5 MΩ. With addition of 20 mm BAPTA to the filling solution, Cs-gluconate was reduced to maintain osmolarity. With BAPTA, CE, or d-890 in the patch pipette, 10 minutes of dialysis was allowed before analysis of drug effects. In CE occlusion experiments, pipettes contained 5 μm CE and initial effects were apparent within 5 minutes. Therefore, to study EPSCs before and after dialysis, CE concentration was reduced to 1 μm, allowing time for acquisition of control traces before its effects occurred.
After breakthrough, recordings stabilized for 3–5 minutes before data acquisition. Cells were accepted if the series resistance was <20 MΩ and did not change by >20%. Data were acquired using an Axopatch 1D amplifier and Digidata board 1320A controlled by Clampex 10.2 and analyzed with ClampFit (Molecular Devices). Traces were sampled at 5 kHz and filtered at 2 kHz. Records in figures are averages of 10 raw traces, except for d-890 experiments, in which 5 traces were averaged. Electrical artifacts were truncated for clarity. EPSC reversal potential was obtained from the zero current intercept of the EPSC I–V curve obtained in 20 mV steps between holding potentials of −70 and +50 mV.
Stimulation.
The Schaffer collateral fibers in stratum radiatum were activated with a 100 μs constant current stimulus via a 2–4 μm tip diameter pipette filled with ACSF. Stimulus intensity and duration were set to elicit EPSCs of either 100–400 pA or 30–55 pA and were delivered at a 30 s interval in all experiments.
pH microelectrodes.
Concentric pH microelectrodes (tip diameter of 2–4 μm) were fabricated and calibrated as described previously (Makani and Chesler, 2007). pHe readings were obtained in mid-CA1 stratum radiatum at a depth of ∼150 μm and referenced to the overlying ACSF, pH 7.4.
Data analysis.
EPSC peak, charge transfer, and half-time of decay were obtained in the 5 minute period before the addition of XCAR. Although effects of XCAR appeared rapidly, data were not analyzed during a 7 minute wash-in period to insure full effect of the enzyme. A similar 7 minute period was allowed for delay experiments and for wash-in of HEPES-bicarbonate ACSF. After the 7 minute delay, data were taken over the next 5 minute period. Statistics were presented as means with SE with significance at p < 0.05. Comparisons were made with a two-tailed, Student's paired or unpaired t test or one-way ANOVA, as appropriate. Values of n refer to the number of recorded cells. In the Discussion, we calculated the reversal potential (Er) for a putative anion conductance with a given HCO3− to Cl− permeability ratio using the zero current condition of the constant field equation (Hodgkin and Katz, 1949), assuming a [HCO3−]i of 45 mm or lower.
Results
Slices were bathed in standard ACSF with DNQX and picrotoxin to block AMPAR and GABA-A receptor-mediated responses in all cells. A holding potential (VH) of +50 mV removed the Mg2+ block of NMDARs, allowing a large NMDAR-mediated EPSC solely in a patched neuron. Addition of XCAR consistently reduced the EPSC peak (−23 ± 3.0%, n = 9, p < 0.001, Fig. 1a,b). This effect was virtually instantaneous as the enzyme reached the slice and was comparable using bovine CAR type II (n = 6) or human recombinant CAR type IV (n = 3, dashed lines Fig. 1b). Charge transfer was likewise decreased (−20 ± 4.2%, n = 9, p < 0.01, Fig. 1c), but the decay half-time was unaffected (89 ± 3.5 ms in control vs 91 ± 5.2 ms in XCAR, n = 9, p = 0.54, Fig. 1d), nor was the reversal potential changed (+2.7 ± 2.9 mV in control vs +2.3 ± 2.6 mV in XCA, n = 5, p = 0.90, Fig. 1e). EPSC reduction was not due to run down, because a 7 minute delay had no effect on the peak (−1.9 ± 1.4%, n = 5, p = 0.29, Fig. 1f,g). Moreover, in saline containing benzolamide, an ECAR inhibitor (Travis et al., 1964), XCAR had no effect on the peak EPSC (−1.1 ± 2.3%, n = 6, p = 0.89, Fig. 1h,i), indicating that the enzyme active site was required to reduce the NMDAR current.
Figure 1.

XCAR reduced NMDAR-mediated EPSCs. a, EPSC in XCAR (red) was reduced compared with control (CTL) at a VH of +50 mV. Scaled trace (green) shows no effect on time course. b, Effect of XCAR on peak EPSC in 9 cells. Dashed lines indicate use of human, recombinant type IV carbonic anhydrase in 3 cells. c, XCAR reduced charge transfer. d, Half-time of decay was unaffected by XCAR. e, XCAR had no effect on the EPSC reversal potential. f, Averaged EPSCs at 5–10 minutes (black) versus 17–22 minutes (delay, red) after breakthrough. g, Peak EPSC of 5 cells in delay experiments. h, XCAR had no effect on peak EPSC in presence of benzolamide. I, Peak EPSC in 6 cells exposed to XCAR in the presence of benzolamide. Calibrations: 100 pA/50 ms. VH = +50 mV for a–d and f–i.
XCAR did not lower pHe (ΔpHe = 0.027 ± 0.01, n = 4 slices, p = 0.55; see also Makani and Chesler, 2007), so acidosis could not explain the EPSC reduction. Because a more localized, transport-mediated surface acidosis might occur, we tested XCAR in the presence of the Na+-H+ exchange blockers EIPA (n = 4) or cariporide (n = 4). To cover HCO3−-dependent acid extruders, we used DIDS (n = 4), which blocks the Slc4 transporters of brain, with the exception of Slc4a7 (Ruffin et al., 2014), for which we used the drug S0859 (n = 4) (Ch'en et al., 2008). With these drugs present, respective reductions of the EPSC by XCAR were −29 ± 4.0%, −24 ± 2.2%, −23 ± 3.2%, and −21 ± 1.8%, which did not differ from controls (9 cells of Fig. 1b) or from one another.
Alternatively, apparent EPSC reduction could occur if XCAR diminished the Ca2+ activated Cl− conductance of these cells (TMEM16B; Huang et al., 2012; but see Discussion). In the presence of its blocker niflumic acid (n = 4), EPSC curtailment by XCAR was −28 ± 5.0%, which did not differ from controls or from the reduction seen with the four drugs above.
XCAR curtailed the EPSC similarly at a VH of −30 mV (−26 ± 4.5%, n = 5, p < 0.01, Fig. 2a,b), which did not differ from its effect at +50 mV in paired trials (p = 0.91). Therefore, the EPSC reduction was not voltage dependent. This also suggested that the effect of XCAR was not reliant on a Ca2+ influx triggered by the step to +50 mV, for example, via l-type Ca2+ channels in postsynaptic spines (Leitch et al., 2009). This was tested directly with the l-type blocker d-890 in the pipette. If these channels were required, then a smaller effect of XCAR would be expected at +50 mV. With d-890, however, reductions of the EPSC by XCAR were −34 ± 5.0% at +50 mV and −26 ± 3.7% at −30 mV (n = 5, p = 0.13, Fig. 2c–f).
Figure 2.
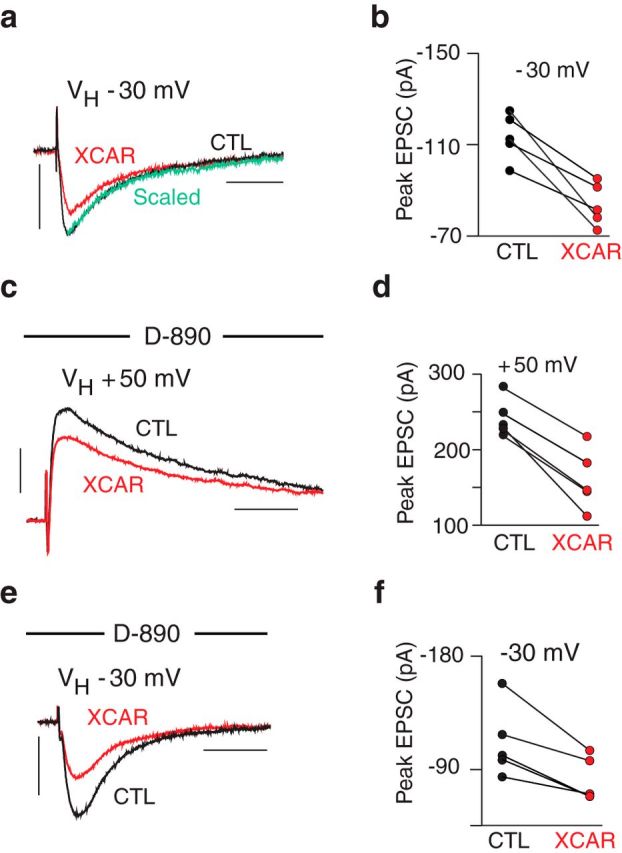
Effect of XCAR was not voltage dependent. a, EPSC curtailed by XCAR (VH = −30 mV.) b, XCAR reduced EPSC amplitude in 5 cells (VH = −30 mV). c, XCAR reduced the EPSC after dialysis of d-890 (VH = +50 mV). d, Reduction of EPSC by XCAR in 5 cells after dialysis of d-890 (VH = +50 mV) e, XCAR reduction of EPSC after dialysis of d-890 (VH = −30 mV). f, Reduction of EPSC by XCAR in 5 cells after dialysis of d-890 (VH = −30 mV). Calibration: 100 pA/50 ms.
If XCAR curtailed the EPSC by buffering a Ca2+-triggered, PMCA-mediated alkalosis, then the EPSC should be reduced and the effect of XCA occluded by the addition of a fast extracellular buffer or by agents that prevent PMCA activation. Adding 20 mm HEPES to normal HCO3−-buffered ACSF diminished the EPSC (−24 ± 2.9%, n = 5, p < 0.05, Fig. 3a,b) and occluded the effect of XCAR (−0.5 ± 3.4%, n = 5, p = 0.99, Fig. 3c,d). Inclusion of BAPTA in the pipette similarly reduced the EPSC (−23 ± 4.2%, n = 7, p < 0.001, Fig. 3e,f) and occluded the effect of XCAR (−1.8 ± 1.6%, n = 5, p = 0.31, Fig. 3g,h). Inhibiting the PMCA with 1 μm CE in the pipette (Makani and Chesler, 2010) also curtailed the EPSC (−19 ± 4.0%, n = 7, p < 0.01, Fig. 3i,j) and occluded further reduction by XCAR (+1.8 ± 2.6%, n = 5, p = 0.6, Fig. 3k,l). EPSC curtailment with both 20 mm BAPTA and 5 μm CE in the patch pipette (−24 ± 3.1%, n = 6, p < 0.001) was no different from that caused by BAPTA or CE alone, which is consistent with a common mechanism. These data demonstrate that the recorded cell was the sole source of the EPSC modulation; tie the EPSC reduction to pHe buffering, Ca2+ influx, and the PMCA; and show by occlusion that EPSC reduction was not due to NMDAR antagonism by XCAR.
Figure 3.

XCAR effect was due to buffering and required the PMCA. a, EPSC reduced by 20 mm HEPES. b, Effect of HEPES in 5 cells. c, In 20 mm HEPES, XCAR had no effect on the EPSC. d, EPSC peak from 5 cells exposed to XCAR in 20 mm HEPES. e, Peak EPSC was reduced by dialysis of BAPTA. f, Effect of BAPTA on peak EPSC in 7 cells. g, After dialysis of BAPTA for 10 minutes, XCAR had no effect on the peak EPSC. h, Peak EPSC before and after XCAR in 5 cells dialyzed with BAPTA. i, Peak EPSC was reduced by dialysis of CE. j, Effect of CE on peak EPSC in 7 cells. k, After dialysis of CE, exposure to XCAR had no effect on peak EPSC. l, Peak EPSC before and after XCAR in 5 cells dialyzed with CE. VH = +50 mV for a–l. Calibration: 100 pA/50 ms. For e, f and I, j, the data were compared at 3–8 minutes versus 12–17 minutes after breakthrough.
If a PMCA-mediated alkalosis boosts adjacent NMDARs, then the effect of XCAR should occur with just a few afferents activated. This was tested using a minimal stimulus to elicit EPSCs averaging 43 ± 3.3 pA (range 30–55 pA). XCAR reduced these responses by −25 ± 3.2% (n = 9, p < 0.001, Fig. 4a,b). Small EPSCs were unchanged in delay experiments without XCAR (−2.0 ± 3.8%, n = 5, p = 0.72, Fig. 4c,d). The reduction of small EPSCs did not differ from that of the larger EPSCs of Figure 1b (p = 0.65). Pooling all XCAR trials at +50 mV, the mean EPSC reduction was −26 ± 1.2% (n = 52).
Figure 4.
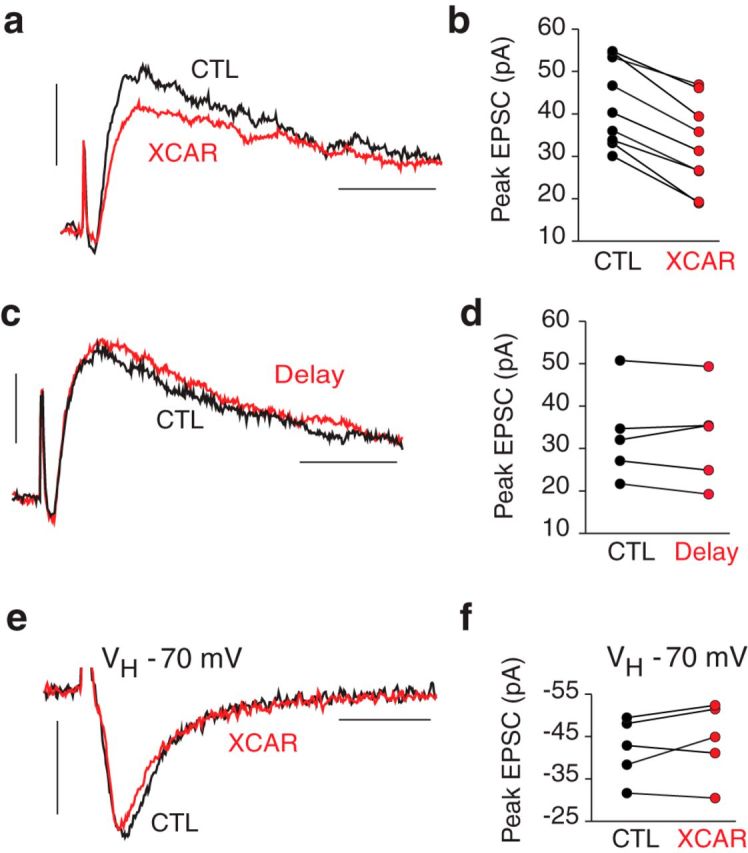
Small NMDAR EPSCs reduced by XCAR. a, Small EPSC curtailed by XCAR. b, Effect of XCAR on small EPSCs in 9 cells. c, Small EPSC recorded 5–10 minutes (CTL, black) versus 17–22 minutes after breakthrough (Delay, red). d, Peak EPSC in 5 delay experiments. e, XCAR had no effect on AMPAR peak EPSC (no DNQX, VH = −70 mV). f, Effect of XCAR on peak of small, AMPAR EPSCs in 5 cells. VH = +50 mV for a–d. Calibration: 20 pA/20 ms (a,c), 20 pA/10 ms (e).
To test whether XCAR had reduced small EPSCs by an increase in afferent threshold and dropout of axons, we studied its effect on small AMPAR-mediated EPSCs (without DNQX; VH = −70 mV). AMPAR EPSCs (−42 ± 3.3 pA, n = 5, range −32 to −50 pA) were not changed by XCAR (+4.5 ± 3.9%, n = 5, p = 0.26, Fig. 4e,f). Therefore, the enzyme did not reduce the number of activated afferents nor was its effect due to diminished presynaptic release (Makani and Chesler, 2007).
Discussion
Our data argue that the postsynaptic NMDAR current is boosted in an autocrine manner by a PMCA-mediated alkalosis. This interpretation is based on reduction of the EPSC by XCAR, which augments buffering in a rapid time frame (Tong et al., 2006). In support of this view, HEPES both curtailed the EPSC and occluded the effect of XCAR, as did dialyzed BAPTA or CE. Therefore, XCAR's effect required Ca2+-mediated activation of the PMCA in just the patched cell, most likely via an NMDAR-mediated Ca2+ influx.
Several alternative hypotheses can be excluded. XCAR did not cause a fall in pHe that could reduce the EPSC and a more localized acidosis due to transport was unlikely because a host of transport blockers did not alter XCAR's effect. XCAR did not change EPSC time course, consistent with an action upon NMDAR channels alone. An alternate notion is that Ca2+ entry activated a Cl− conductance of similar time course that was diminished by XCAR (see below). This is unlikely because neither niflumic acid nor DIDS (which inhibits the ClC-3 Cl− channel; Wang et al., 2006) altered the effect of XCAR.
A role for a pure Cl− conductance or mixed Cl−-HCO3− conductance is doubtful in principle because XCAR had the same effect on the peak EPSC at +50 vs −30 mV. At +50 mV, an apparent reduction of the outward EPSC could occur if a coactivated, outward current (ECl = −50 mV) were reduced by XCAR. At −30 mV, however, the EPSC was inward, but a Cl− current would still be outward. Therefore, its reduction would have been expected to increase, rather than decrease, the EPSC. The same argument would hold for typical Cl− channels with some permeability to HCO3− because the reversal potential of the anion current would remain negative to −30 mV if the HCO3− to Cl− permeability ratio were as high as 0.6 (Kaila, 1994) and the intracellular HCO3− was elevated to as much as 45 mm.
A similar argument applies if a surface pH shift linked to an HCO3− conductance were postulated to account for our results. At +50 mV, an HCO3− influx could elicit a surface acidosis, given a typical HCO3− reversal potential of 0 to −20 mV. If this acid shift were augmented by XCAR (by speeding CO2 hydration), it could inhibit NMDARs and reduce the EPSC. At −30 mV, however, an HCO3− efflux and consequent alkalosis would occur. This alkaline shift would be boosted by XCAR (by speeding HCO3− dehydration; Kaila, 1994), so an increase in the EPSC would have been expected instead of the observed decrease.
In summary, our data imply that a perisynaptic alkalosis normally increases the postsynaptic NMDAR current by approximately one-third. This is likely to be a minimal estimate because increased buffering should reduce but not fully abolish a surface alkalosis. The modulation is highly localized because XCAR caused similar reduction of large and small EPSCs. Given expression of PMCA2 variants in association with the postsynaptic density (DeMarco and Strehler, 2001), a current boost at the level of single postsynaptic boutons appears plausible.
Whereas prior studies using XCAR relied upon simultaneous activation of the pyramidal neuron population, the present results indicate that alkaline modulation of NMDARs occurs with modest synaptic input to a single neuron and is therefore a normal, significant attribute of this form of synaptic transmission. Functionally, this modulation may provide a means of regulating NMDAR responses via control of the alkalosis. Because the size of this pH shift is affected greatly by ECAR activity (Chesler, 2003), changes in the expression of one or more of its isoforms could provide a form of plasticity. In fact, increased excitability was reported in mice with a knock-out of extracellular CAR14 (Makani et al., 2012). Another means of altering the magnitude of the alkalosis is via electrogenic Na+-HCO3− cotransport by glia. These mechanisms of pHe control may differ regionally, as evidenced by the variability in alkaline transients in brain and spinal cord (Chesler, 2003). Evolution may have favored a large alkalosis in the CA1 stratum radiatum, where long-term potentiation depends on the activation of postsynaptic NMDARs (Collingridge et al., 1983).
Footnotes
This work was supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke (Grant NS032123) and the Attilio and Olympia Ricciardi Fund. We thank N. Galifianakis, A. Paulson, S. Makani, W. Sly, and A. Waheed for critical feedback.
The authors declare no competing financial interests.
References
- Burette AC, Strehler EE, Weinberg RJ. A plasma membrane Ca2+ ATPase isoform at the postsynaptic density. Neuroscience. 2010;169:987–993. doi: 10.1016/j.neuroscience.2010.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E, Stauffer T. The plasma membrane calcium pump: functional domains, regulation of the activity, and tissue specificity of isoform expression. J Neurobiol. 1994;25:312–324. doi: 10.1002/neu.480250311. [DOI] [PubMed] [Google Scholar]
- Ch'en FF, Villafuerte FC, Swietach P, Cobden PM, Vaughan-Jones RD. S0859, an N-cyanosulphonamide inhibitor of sodium-bicarbonate cotransport in the heart. Br J Pharmacol. 2008;153:972–982. doi: 10.1038/sj.bjp.0707667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HY, Chesler M. NMDAR-mediated EPSCs in single pyramidal neurons are highly dependent on a concomitant rise in peri-synaptic pH. Soc Neurosci Abstr. 2013;39:514.10. [Google Scholar]
- Chen JC, Chesler M. pH transients evoked by excitatory synaptic transmission are increased by inhibition of extracellular carbonic anhydrase. Proc Natl Acad Sci U S A. 1992;89:7786–7790. doi: 10.1073/pnas.89.16.7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMarco SJ, Strehler EE. Plasma membrane Ca2+-atpase isoforms 2b and 4b interact promiscuously and selectively with members of the membrane-associated guanylate kinase family of PDZ (PSD95/Dlg/ZO-1) domain-containing proteins. J Biol Chem. 2001;276:21594–21600. doi: 10.1074/jbc.M101448200. [DOI] [PubMed] [Google Scholar]
- Gottfried JA, Chesler M. Temporal resolution of activity-dependent pH shifts in rat hippocampal slices. J Neurophysiol. 1996;76:2804–2807. doi: 10.1152/jn.1996.76.4.2804. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of the giant axon of the squid. J Physiol. 1949;108:37–77. doi: 10.1113/jphysiol.1949.sp004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC, Xiao S, Huang F, Harfe BD, Jan YN, Jan LY. Calcium-activated chloride channels (CaCCs) regulate action potential and synaptic response in hippocampal neurons. Neuron. 2012;74:179–192. doi: 10.1016/j.neuron.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman J, Konnerth A. NMDA receptor-mediated subthreshold Ca(2+) signals in spines of hippocampal neurons. J Neurosci. 2000;20:1791–1799. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer MA, Collis LP, Molina AJ, Smith PJ, Malchow RP. Modulation of extracellular proton fluxes from retinal horizontal cells of the catfish by depolarization and glutamate. J Gen Physiol. 2007;130:169–182. doi: 10.1085/jgp.200709737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitch B, Szostek A, Lin R, Shevtsova O. Subcellular distribution of L-type calcium channel subtypes in rat hippocampal neurons. Neuroscience. 2009;164:641–657. doi: 10.1016/j.neuroscience.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Makani S, Chesler M. Endogenous alkaline transients boost postsynaptic NMDA receptor responses in hippocampal CA1 pyramidal neurons. J Neurosci. 2007;27:7438–7446. doi: 10.1523/JNEUROSCI.2304-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makani S, Chesler M. Rapid rise of extracellular pH evoked by neural activity is generated by the plasma membrane calcium ATPase. J Neurophysiol. 2010;103:667–676. doi: 10.1152/jn.00948.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makani S, Chen HY, Esquenazi S, Shah GN, Waheed A, Sly WS, Chesler M. NMDA receptor-dependent afterdepolarizations are curtailed by carbonic anhydrase 14: Regulation of a short-term potentiation. J Neurosci. 2012;32:16754–16762. doi: 10.1523/JNEUROSCI.1467-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Ruffin VA, Salameh AI, Boron WF, Parker MD. Intracellular pH regulation by acid-base transporters in mammalian neurons. Front Physiol. 2014;5:43. doi: 10.3389/fphys.2014.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuss V, Yasuda R, Sobczyk A, Svoboda K. Nonlinear [Ca2+] signaling in dendrites and spines caused by activity-dependent depression of Ca2+ extrusion. J Neurosci. 2006;26:8183–8194. doi: 10.1523/JNEUROSCI.1962-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening CJ, Kennedy HJ, Thomas RC. Calcium-hydrogen exchange by the plasma membrane Ca-ATPase of voltage-clamped snail neurons. Proc Biol Sci. 1993;253:285–289. doi: 10.1098/rspb.1993.0115. [DOI] [PubMed] [Google Scholar]
- Shah GN, Ulmasov B, Waheed A, Becker T, Makani S, Svichar N, Chesler M, Sly WS. Carbonic anhydrase IV and XIV knockout mice: roles of the respective carbonic anhydrases in buffering the extracellular space in brain. Proc Natl Acad Sci U S A. 2005;102:16771–16776. doi: 10.1073/pnas.0508449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RC. The plasma membrane calcium ATPase (PMCA) of neurones is electroneutral and exchanges 2 H+ for each Ca2+ or Ba2+ ion extruded. J Physiol. 2009;587:315–327. doi: 10.1113/jphysiol.2008.162453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong CK, Chen K, Chesler M. Kinetics of activity-evoked pH transients and extracellular pH buffering in rat hippocampal slices. J Neurophysiol. 2006;95:3686–3697. doi: 10.1152/jn.01312.2005. [DOI] [PubMed] [Google Scholar]
- Travis DM, Wiley C, Bohdan RN, Maren TH. Selective renal carbonic anhydrase inhibition without respiratory effect: pharmacology of 2-benzenesulfonamido-1,3,4-thiadiazole-5-sulfonamide (CL 11,366) J Pharmacol Exp Ther. 1964;143:383–394. [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Deriy LV, Foss S, Huang P, Lamb FS, Kaetzel MA, Bindokas V, Marks JD, Nelson DJ. CLC-3 channels modulate excitatory synaptic transmission in hippocampal neurons. Neuron. 2006;52:321–333. doi: 10.1016/j.neuron.2006.08.035. [DOI] [PubMed] [Google Scholar]