

Abstract
Background and Objective
Data from two similarly designed studies of tapentadol extended release (ER) for managing neuropathic pain associated with diabetic peripheral neuropathy (DPN; NCT00455520, NCT01041859) in adults were pooled for this analysis, allowing a detailed evaluation of efficacy in patient subgroups and secondary endpoints.
Methods
In each study, patients were titrated to their optimal dose of open-label tapentadol ER [100–250 mg twice daily (bid)] over 3 weeks. Patients with ≥1-point improvement in average pain intensity [11-point numerical rating scale (NRS)] were randomized (1:1) to receive placebo or tapentadol ER during a 12-week, double-blind maintenance period.
Results
Mean (standard deviation [SD]) changes in pain intensity from baseline to week 12 of maintenance in the placebo (n = 343) and tapentadol ER (n = 360) groups, respectively, were 1.28 (2.41) and 0.08 (1.87) [least squares mean difference (LSMD): −1.14 (95 % confidence interval [CI]: −1.435, −0.838); P < 0.001, in favour of tapentadol ER]. Significant between-group differences were also observed in changes from the start of the double-blind treatment period to the double-blind endpoint for the Short Form-36 physical functioning, role-physical, bodily pain, social functioning and role-emotional subscale and physical component summary scores, and the EuroQol 5-Dimension health status index (all P < 0.05, in favour of tapentadol ER). No clinically relevant differences were observed in the efficacy of tapentadol ER across patient subgroups divided by age, sex, race, opioid experience and pain intensity. Incidences of treatment-emergent adverse events were 56.0 % (192/343) with placebo and 74.7 % (269/360) with tapentadol ER during maintenance.
Conclusion
Results of this pooled analysis indicate that tapentadol ER was effective for managing DPN-related pain, and provided consistent analgesic efficacy across different patient subgroups.
Key Points
| Results of this pooled analysis indicate that tapentadol extended release (ER) is effective and well tolerated for the management of neuropathic pain associated with diabetic peripheral neuropathy (DPN) in adults. |
| Tapentadol ER is associated with improvements in measures of health-related quality of life in patients with painful DPN. |
| Tapentadol ER provides consistent efficacy across different patient subgroups divided by age, sex, race, opioid experience and pain intensity. |
Introduction
Painful diabetic peripheral neuropathy (DPN) is a common complication of diabetes [1, 2], affecting an estimated 10–26 % of diabetic patients [2, 3]. Symptoms of painful DPN may include burning pain, pain radiating down the legs, stabbing pains, paraesthesia and allodynia [2]. Painful DPN is often very distressing for patients, and may be associated with a reduction in physical functioning, poor sleep quality and an increased risk of anxiety or depression [2–5].
Although the pathophysiology of painful DPN is not fully understood [1], both central and peripheral mechanisms are likely involved in the genesis of pain [2, 3, 6], which complicates the management of patients with painful DPN [1, 2]. Duloxetine and pregabalin, which are approved by the US Food and Drug Administration (FDA) for the management of painful DPN, have generally been considered first-line treatment options for pain related to DPN [7].
Although opioid analgesics (e.g. morphine, oxycodone) have been shown to be efficacious for the management of neuropathic pain, they have generally been considered second-line treatments because of the potential for tolerability and safety issues, the development of physical dependency and the risk of abuse or misuse with long-term therapy [2, 7, 8]. Long-term opioid therapy may be associated with hypogonadism, suppressed immune functioning or various side effects, such as constipation, nausea and sedation [8]. Nausea and sedation generally resolve with time and can often be avoided by the use of low starting doses and slow up-titration [8]. Opioid-induced constipation is often chronic [8] and may require reduction or discontinuation of opioids or switching to an alternative therapy for pain [9, 10]. Because single pharmacological agents may provide insufficient relief for painful DPN, combinations of two or more drugs with different mechanisms of action are considered as an option for patients who fail to achieve adequate analgesia with a single treatment [2, 8].
Tapentadol represents a proposed new class of centrally acting analgesic combining two mechanisms of action, µ-opioid receptor agonism and norepinephrine reuptake inhibition [11]. The norepinephrine reuptake inhibitor activity of tapentadol may contribute to an opioid-sparing effect [11], resulting in the improved gastrointestinal tolerability compared with traditional opioid analgesics (controlled-release morphine and oxycodone) that has been demonstrated in phase 3 studies of an extended-release formulation of tapentadol [12–17]. Tapentadol extended release [ER; prolonged release (PR) in Europe] has been shown to be effective and well tolerated for the management of moderate to severe chronic pain in randomized, double-blind, placebo- and/or active-controlled phase 3 studies [12–14, 18, 19]. Results of two of these studies [randomized-withdrawal, multicentre, placebo-controlled, phase 3 trials (ClinicalTrials.gov Identifiers: NCT00455520 [18] and NCT01041859 [19]), which had a similar design] demonstrated that tapentadol ER [100–250 mg twice daily (bid)] is efficacious and well tolerated in patients with moderate to severe, chronic, painful DPN. In August of 2012, tapentadol ER received FDA approval for the management of neuropathic pain associated with DPN in adults when a continuous, around-the-clock opioid analgesic is needed for an extended period of time. Both studies [18, 19], upon which FDA approval was based, were powered with sample sizes to evaluate the primary endpoint. Neither study [18, 19] was powered to allow for subgroup analyses or the analyses of secondary endpoints. The objective of the present post hoc analysis was to evaluate data pooled from those two studies [18, 19] to allow for a more detailed evaluation of efficacy in different patient subgroups and of secondary efficacy and quality-of-life endpoints.
Patients and Methods
Patient Population and Disallowed Concomitant Medications
In the two studies included in this pooled analysis, eligible patients were adults at least 18 years of age with a diagnosis of type 1 or 2 diabetes mellitus and painful DPN, with signs and symptoms present for at least 6 months prior to screening and pain present at the time of screening. Patients were required to have their diabetes well controlled by diet, oral hypoglycaemic agents or insulin for a minimum of 3 months prior to study enrolment. In the first study [18], adequate blood glucose control must have been demonstrated by a glycated haemoglobin (HbA1c) level of no more than 11 % for at least 3 months prior to enrolment. In the second study [19], adequate blood glucose control was demonstrated by a stable, optimized diabetic regimen (consisting of diet, oral hypoglycaemic therapy or insulin therapy) for at least 3 months prior to enrolment. Patients must have been taking an analgesic medication for their painful DPN for at least 3 months prior to screening; for patients taking an opioid, the dose equivalent of oral morphine must have been no more than 160 mg/day. Patients taking opioid analgesics must have been dissatisfied with their treatment (in terms of pain relief or tolerability), while patients taking non-opioid analgesics must have been dissatisfied with their current level of pain relief. Patients were required to have a mean pain intensity score of at least 5 on an 11-point numerical rating scale (NRS; 0 = “no pain” to 10 = “pain as bad as you can imagine”); the mean pain intensity score for this inclusion criterion was based on twice-daily pain intensity assessments (patient-rated average pain levels over the past 12 h) that were recorded during a 3-day evaluation period prior to the start of open-label treatment.
In both studies, patients were excluded if they had participated in another trial within 30 days of enrolment or if they had participated in another tapentadol trial. Patients were also excluded if they had a history of alcohol or drug abuse or were known or suspected to be unable to comply with the study protocol and use of the study drug. Additional exclusion criteria common to both studies included significant pulmonary, gastrointestinal, metabolic (except diabetes mellitus), neurological, psychiatric or other disorders that could affect efficacy or safety assessments or compromise patient safety; significant cardiac or vascular disease; a history of seizure disorder or epilepsy; a history of mild or moderate traumatic brain injury, stroke, transient ischaemic attack or brain neoplasm within 1 year; a history of severe traumatic brain injury within 15 years; a history of malignancy within the past 2 years (except for successfully treated basal cell carcinoma); severe renal impairment; moderate or severe hepatic impairment; chronic hepatitis B or C; active hepatitis B or C within the past 3 months; severe or extensive diabetic foot ulcers, limb amputation, or Charcot neuroarthropathy; a clinically relevant history of sensitivity to acetaminophen or opioid analgesics (or their ingredients); any scheduled surgery or painful procedure during the study that could affect efficacy or safety assessments; and any condition (other than painful DPN) that could confound the assessment or self-evaluation of pain (e.g. fibromyalgia, rheumatoid arthritis, ankylosing spondylitis [6]). Women who were pregnant or breastfeeding were also excluded from both studies.
The use of other analgesics, besides permitted doses of acetaminophen and tapentadol ER (as described in the ‘Study Design’ section), was not permitted during either study. In both studies, the use of neuroleptics, monoamine oxidase inhibitors, serotonin norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants, anticonvulsants and antiparkinsonian drugs was prohibited within 14 days of screening and throughout the study, and the use of corticosteroids (other than topical or inhaled steroids) was prohibited within 4 weeks to 6 months of screening (depending on the method of administration) and throughout the study. Neuroleptics, SNRIs, anticonvulsants and antiparkinsonian drugs were not permitted because of the potential that their use could confound analgesic efficacy assessments. Selective serotonin reuptake inhibitors were permitted if patients were taking a stable dose for at least 3 months prior to screening.
Study Design
Both studies had similar randomized-withdrawal, double-blind, placebo-controlled designs. The first study [18] used the conventional hypromellose-based formulation employed in other phase 3 clinical trials [12, 13, 20], while the second study [19] used a new formulation of tapentadol ER that has a high mechanical strength resulting from a polyethylene oxide matrix and melt extrusion manufacturing process, is less susceptible to crushing or extraction than the conventional hypromellose-based formulation and is intended to reduce the potential for tampering with the tablet that might overcome the extended-release characteristics. The new formulation of tapentadol ER, which has a similar release profile to the hypromellose-based formulation [21], has been approved for the management of chronic pain in the United States.
The initial open-label phase of both studies included a screening period, a washout period, a pre-titration pain intensity evaluation period and an open-label titration period. During the washout period (typically 3–14 days in the first study [18] and 5 days in the second study [19]), patients discontinued all prior medications that were being taken to manage painful DPN. In both studies [18, 19], any medication that was being taken to manage pain related to DPN was to be washed out for at least five times the elimination half-life of that medication prior to starting open-label treatment. In the first study [18], eligible patients taking SNRIs, anticonvulsants or tricyclic antidepressants, solely for pain related to DPN and not for other disorders, could proceed to washout immediately after signing the informed consent document, such that washout could extend through the combined screening and washout period (14 days). If patients in that study [18] were taking a drug with an extended elimination half-life (e.g. methadone) for their painful DPN, the washout period could be extended beyond 14 days. For the second study [19], the screening and washout periods could be combined (total, 18 days) if more than 5 days of washout was required to allow for a washout period of at least five half-lives of a medication. The washout period was followed by a 3-day, pre-titration pain intensity evaluation period to determine eligibility to enter the study; patients with a pre-titration mean pain intensity score of at least 5 then entered a 3-week, open-label titration period [pre-titration mean pain intensity score was calculated from the average daily pain intensity scores during that 3-day period, with average daily pain intensity based on twice-daily pain intensity assessments (morning and evening)]. During the open-label titration period, all patients initially received tapentadol ER 50 mg bid for 3 days, then the dose was increased to 100 mg bid. Doses could be titrated upwards in increments of 50 mg bid every 3 days (to a maximum dose of 250 mg bid) or downward in decrements of 50 mg bid without a time restriction (to a minimum dose of 100 mg bid) for the remainder of this period. The purpose of titration was to allow each patient to reach a dose of tapentadol ER that provided an optimal balance of pain reduction and tolerability. Throughout the titration period (except during the last 4 days to allow for evaluation of baseline pain intensity, as described in Sect. 2.3), acetaminophen (≤2,000 mg/day) was permitted as additional analgesia.
Patients who tolerated tapentadol ER and had at least a 1-point improvement in average pain intensity from the pre-titration evaluation period to the last 3 days of the titration period were randomized (1:1) to receive placebo or tapentadol ER during a 12-week, double-blind maintenance period. To minimize the possibility of withdrawal symptoms, patients who were randomized to placebo received blinded tapentadol ER 100 mg bid for 3 days before switching to placebo twice daily from day 4 of the maintenance period onwards. Patients who were randomized to tapentadol ER continued taking the same optimal dose of tapentadol ER that was determined during the open-label titration period. Patients in both treatment groups were allowed to take supplemental tapentadol ER 25 mg bid as additional analgesia during the first 4 days of the maintenance period; from day 5 of the maintenance period onwards, patients were permitted to take a supplemental dose of tapentadol ER 25 mg once daily. At approximately 4 days after the last intake of study drug, a follow-up clinic visit was scheduled and, at approximately 10–14 days after the last intake of study drug, a follow-up phone call was scheduled.
Efficacy Evaluations
Patients recorded their average pain intensity (11-point NRS) over the past 12 hours twice daily during the 3-day, pre-titration pain intensity evaluation period and throughout the titration and maintenance periods. Daily pain intensity was defined as the mean of the average pain intensity scores in a 24-h period. Baseline pain intensity was defined as the mean of the average daily pain intensity scores during the last 3 days prior to randomization (i.e. the last 3 days of the open-label titration period). The baseline score was based on the mean of average daily pain intensity scores for the last 3 days of the titration period to reflect pain intensity on the final optimal dose of tapentadol ER that was determined during the titration period. Weekly average pain intensities during the double-blind treatment period were defined as the means of the average daily pain intensities in each 7-day period starting from the first dose of double-blind study drug. The primary efficacy endpoint was the mean change in average pain intensity (11-point NRS) from baseline to week 12 (mean of seven daily average pain intensity scores during week 12) of the double-blind maintenance period. Secondary efficacy endpoints included the following: responder rates (based on the percentage improvement in pain intensity from pre-titration) at week 12 of the maintenance period; the Patient Global Impression of Change (PGIC) [22, 23] at the double-blind endpoint; and changes in the Brief Pain Inventory—Short Form (BPI-SF) [24] pain interference and pain intensity subscale scores, Short Form-36 (SF-36) Health Survey [25] subscales and summary scales and EuroQol 5-Dimension (EQ-5D) health status index [26] from the start of the double-blind maintenance period to the double-blind endpoint.
For the responder rate analysis, the number and percentage of patients achieving at least a 30 % improvement in pain intensity and at least a 50 % improvement in pain intensity from pre-titration to week 12 of the double-blind maintenance period were evaluated by treatment group. The percentage change in pain intensity from pre-titration to week 12 of the double-blind maintenance period was calculated as follows: 100 × (average pain intensity during week 12 − average pre-titration pain intensity)/(average pre-titration pain intensity). Patients who discontinued during treatment, had pain intensity that worsened or had no change in pain intensity were considered to be non-responders. For the PGIC [22, 23], patients rated the change in their overall status since beginning study treatment on a scale from 1 (“very much improved”) to 7 (“very much worse”). The BPI-SF [24] was used to assess the pain severity and interference with daily activity. The BPI-SF includes one item evaluating the level of pain relief over the past 24 hours (scored from 0 % = “no relief” to 100 % = “complete relief”); four items evaluating the pain intensity (11-point NRS) at the time the questionnaire was completed, at its worst, at its least and on average over the previous week; and seven items evaluating pain interference with mood, walking, other physical activity, work, social activity, relations with others and sleep (each rated on an 11-point scale; 0 = “does not interfere” to 10 = “completely interferes”). Scores for the individual items of the BPI-SF were combined to yield pain severity and pain interference subscales. The SF-36 [25] health status survey includes physical functioning, role-physical, bodily pain, general health, vitality, social functioning, role-emotional and mental health subscales; each subscale was scored from 0 (“poor health”) to 100 (“good health”). Weighted combinations of the eight subscale scores were used to derive two summary scores, the mental and physical component summary scores. The EQ-5D [26] measures five dimensions of health status (mobility, self-care, usual activities, pain or discomfort, and anxiety or depression); each dimension was scored using one of three possible responses (“no problems,” “some problems” or “extreme problems”). An overall EQ-5D health status index score (possible score, 0 = “death” to 1 = “full health”) was obtained from a combination of responses to the individual dimensions, which were scored using a utility-weighted algorithm.
For the primary efficacy outcome (average pain intensity on an 11-point NRS), subgroup analyses were performed on the basis of sex, age group (≥65 vs <65 years), race (white, black and other), prior opioid experience (opioid naive vs opioid experienced) and pain intensity category at the start of the double-blind maintenance period (11-point NRS; none, 0; mild, >0 to <4; moderate, ≥4 to <6; severe, ≥6). Opioid-experienced patients were defined as those patients who had previously received an opioid analgesic for the treatment of painful DPN for at least 3 weeks continuously or intermittently, regardless of their response to treatment.
Safety and Tolerability Evaluations
Treatment discontinuations and treatment-emergent adverse events (TEAEs) were monitored and recorded throughout both studies. For each treatment period (the open-label titration period and the double-blind treatment period), a TEAE was defined as any adverse event that newly occurred or worsened in intensity after the first intake of study drug during that period. All TEAEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA), version 13.1. HbA1c levels, which served as a marker of glycaemic control, were measured at screening and at the end of treatment or at early withdrawal in both studies. Other safety evaluations performed in both studies included additional laboratory tests, physical examinations (including a standard neurological examination), vital sign measurements and electrocardiograms; the results of these evaluations were not pooled and will not be presented here.
Statistical Analyses
The intent-to-treat (ITT) populations were used for efficacy evaluations. The open-label ITT and safety populations of each study included all patients who received at least one dose of study drug during the open-label titration period. The double-blind ITT and safety populations of each study included all randomized patients who received at least one dose of study drug during the double-blind maintenance period.
The last observation carried forward (LOCF) was used for imputing missing pain intensity assessments for the analysis of the primary efficacy endpoint. The primary efficacy endpoint was analysed using an analysis of covariance (ANCOVA) model similar to that used in the individual studies [18, 19], which included treatment, study, country and dose category at the end of the open-label titration period (low dose, 100–150 mg bid; high dose, 200–250 mg bid) as factors and mean pain intensity at the start of the double-blind maintenance period as a covariate. Treatment effects were estimated on the basis of the least squares mean difference (LSMD) in the change from baseline to week 12 of the double-blind maintenance period. The test for efficacy analysis was two-sided at the 0.05 level of significance.
Exploratory sensitivity analyses of the primary efficacy endpoint were performed using alternative imputation methods, including baseline observation carried forward (BOCF), worst observation carried forward (WOCF), placebo mean imputation (PMI) and modified BOCF (as described previously for the first DPN study [18]). In addition, an observed-case analysis was performed for the primary efficacy endpoint. As a post hoc sensitivity analysis, a longitudinal evaluation of the change in average pain intensity from baseline to week 12 of the double-blind maintenance period, including all observed data, was performed using a mixed-effects model for repeated measures (MMRM), including treatment, week, treatment-by-week interaction, country and dose level at baseline as factors and baseline average pain intensity score as a covariate.
The distribution of responder rates at week 12 of the maintenance period was evaluated using a Kaplan–Meier estimate, and between-group comparisons were performed using a log-rank test. The numbers and percentages of patients with at least a 30 % and at least a 50 % improvement in pain intensity from pre-titration to week 12 of the maintenance period were evaluated for each treatment group. The numbers and percentages of patients were also summarized by treatment group for PGIC assessments at the double-blind endpoint. Between-group comparisons for the percentages of patients with at least a 30 % and at least a 50 % improvement in pain intensity and PGIC evaluations were performed using a Cochran–Mantel–Haenszel test, controlling for treatment, study, country and dose category at the end of the open-label titration period.
The absolute values and changes from the start of the double-blind maintenance period to the double-blind endpoint in the BPI-SF pain intensity and pain interference subscale scores, SF-36 subscale and summary scores, and EQ-5D health status index were summarized using descriptive statistics. Between-group comparisons of the changes from the start of the double-blind maintenance period to the double-blind endpoint in these measures were analysed using the same ANCOVA model as defined for the primary efficacy endpoint.
For subgroup analyses of the change in average pain intensity (11-point NRS) from baseline to week 12 of the double-blind maintenance period, results for the subgroups were summarized if at least 10 % of the total number of patients were in at least two of the subgroups. An ANCOVA model similar to that used for the primary efficacy analysis was used to evaluate differences between the tapentadol ER and placebo groups in the change in average pain intensity from baseline to week 12 of the double-blind maintenance period for each of the subgroups (divided according to sex, age, race, prior opioid experience and pain intensity category at the start of the double-blind maintenance period, as described previously).
Incidences of all reported TEAEs during the double-blind maintenance period were summarized descriptively. Changes in HbA1c levels were evaluated from the start of the open-label titration period to endpoint of the open-label titration period for patients who discontinued treatment during the open-label titration period, and from the start of the open-label titration period to endpoint of the double-blind maintenance period for patients who entered the double-blind maintenance period. The endpoint values for efficacy and safety measures during the open-label titration period and double-blind maintenance period were defined as the last available measures during the respective period.
Results
Patients
Across the two studies included in this analysis, 1,040 patients received treatment during the open-label titration period and 703 patients received treatment during the double-blind maintenance period. Of the patients who were treated in the open-label titration period, the percentage who failed to meet the randomization criterion (≥1-point improvement in pain intensity) was low (5.0 % [52/1,040]). For the double-blind safety population, demographic and baseline characteristics were similar across the placebo and tapentadol ER treatment groups (Table 1). In the double-blind ITT population (n = 703), 59.7 % of patients were male, 74.8 % of patients were white, 69.3 % of patients were under 65 years of age and 66.9 % of patients were opioid naive. The majority of patients (83.9 % [587/700]) in the double-blind ITT population with available pain intensity scores had severe pain intensity (≥6 on an 11-point NRS) at the start of the open-label titration period, while more than half of the patients (57.3 % [400/698]) had no pain or mild pain intensity (<4 on an 11-point NRS) at the start of the double-blind maintenance period.
Table 1.
Demographic and baseline characteristics [double-blind (DB) intent-to-treat (ITT) population]
| Characteristic | Placebo (n = 343) | Tapentadol ER (n = 360) |
|---|---|---|
| Sex, n (%) | ||
| Male | 202 (58.9) | 218 (60.6) |
| Female | 141 (41.1) | 142 (39.4) |
| Race, n (%)a | ||
| White | 253 (73.8) | 273 (75.8) |
| Black | 41 (12.0) | 49 (13.6) |
| Asian | 3 (0.9) | 4 (1.1) |
| American Indian or Alaskan Native | 3 (0.9) | 3 (0.8) |
| Other | 43 (12.5) | 31 (8.6) |
| Mean (SD) age, years | 59.9 (9.94) | 59.1 (10.62) |
| Age category (years) | ||
| <65 | 229 (66.8) | 258 (71.7) |
| ≥65 | 114 (33.2) | 102 (28.3) |
| Prior opioid experience, n (%)b | ||
| Opioid naive | 229 (66.8) | 241 (66.9) |
| Opioid experienced | 114 (33.2) | 119 (33.1) |
| Mean (SD) duration of DPN, yearsc | 6.1 (5.41) | 5.3 (4.81) |
| Mean (SD) start OL pain intensity scored,e | 7.4 (1.29) | 7.3 (1.41) |
| Start OL pain intensity category, n (%)f | ||
| Mild | 0 | 2 (0.6) |
| Moderate | 44 (12.9) | 67 (18.7) |
| Severe | 298 (87.1) | 289 (80.7) |
| Mean (SD) start DB pain intensity scoreg,h | 3.5 (2.02) | 3.7 (1.84) |
| Start DB pain intensity category, n (%)a,f | ||
| None | 10 (2.9) | 5 (1.4) |
| Mild | 196 (57.3) | 189 (53.1) |
| Moderate | 88 (25.7) | 117 (32.9) |
| Severe | 48 (14.0) | 45 (12.6) |
DPN diabetic peripheral neuropathy, ER extended release, NRS numerical rating scale, OL open-label, SD standard deviation
aPercentages may not total 100 % because of rounding
bOpioid experience was defined as having previously received an opioid analgesic for the treatment of painful DPN for ≥3 weeks continuously or intermittently, regardless of the response to treatment
cPlacebo, n = 212; tapentadol ER, n = 225
dStart OL pain intensity score is the average of pain scores over the 3 days prior to start of titration
ePlacebo, n = 342; tapentadol ER, n = 358
fPain intensity categories: none, 0; mild, >0 to <4; moderate, ≥4 to <6; severe, ≥6 on an 11-point NRS (0 = “no pain” to 10 = “pain as bad as you can imagine”)
gStart DB pain intensity score is the average of pain scores over the 72 h prior to randomization
hPlacebo, n = 342; tapentadol ER, n = 356
During the open-label titration period, 28.5 % of patients (296/1,040) discontinued treatment; the most common reason for treatment discontinuation during titration was adverse events (16.3 % [169/1,040]; Table 2). During the double-blind maintenance period, 30.0 % of patients (103/343) in the placebo group and 28.9 % of patients (104/360) in the tapentadol ER group discontinued treatment. In the placebo and tapentadol ER groups, the most common reasons for treatment discontinuation were a lack of efficacy (placebo, 11.1 % [38/343]; tapentadol ER, 3.1 % [11/360]) and adverse events (placebo, 8.2 % [28/343]; tapentadol ER, 14.2 % [51/360]; Table 2).
Table 2.
Reasons for treatment discontinuation [intent-to-treat (ITT) population]
| Reason for discontinuation, n (%) | OL titration | DB maintenance | |
|---|---|---|---|
| Tapentadol ER (n = 1,040) | Placebo (n = 343) | Tapentadol ER (n = 360) | |
| Total discontinuations for any reason | 296 (28.5) | 103 (30.0) | 104 (28.9) |
| Adverse event | 169 (16.3) | 28 (8.2) | 51 (14.2) |
| Withdrawal of consent | 32 (3.1) | 16 (4.7) | 21 (5.8) |
| Ineligible for DB maintenance | 26 (2.5) | – | – |
| Lack of efficacy | 22 (2.1) | 38 (11.1) | 11 (3.1) |
| Study drug noncompliance | 22 (2.1) | 8 (2.3) | 6 (1.7) |
| Lost to follow-up | 5 (0.5) | 2 (0.6) | 2 (0.6) |
| Physician decision | 1 (0.1) | 1 (0.3) | 2 (0.6) |
| Protocol violation | 1 (0.1) | 2 (0.6) | 1 (0.3) |
| Other | 18 (1.7) | 8 (2.3) | 10 (2.8) |
DB double-blind, ER extended release, OL open-label
Treatment Exposure
The median duration of treatment exposure during the double-blind maintenance period was 84 days in both the placebo and tapentadol ER groups. The majority of patients in both treatment groups (placebo, 71.4 % [245/343]; tapentadol ER, 71.7 % [258/360]) received double-blind treatment for 71 days or more.
At the end of the open-label titration period, almost half of patients (48.4 % [340/703]) were taking tapentadol ER 250 mg bid. The median modal total daily dose of tapentadol ER was 300 mg during the open-label titration period and 400 mg during the double-blind maintenance period. There was no notable increase in the mean total daily dose of tapentadol ER during the double-blind maintenance period (start of double-blind maintenance, 391.3 mg; week 12 of double-blind maintenance, 393.6 mg). During the double-blind maintenance period, 57.4 % of patients (197/343) in the placebo group and 50.8 % of patients (183/360) in the tapentadol ER group took supplemental tapentadol ER. The mean percentage of days that patients in the placebo group took supplemental tapentadol ER was higher than in the tapentadol ER group (47.8 % and 31.8 %, respectively).
Efficacy
Mean (standard error) pain intensity scores over the course of the study are shown in Fig. 1. For the open-label ITT population, mean [standard deviation (SD)] pain intensity (observed-case analysis) was 7.29 (1.38) at the start of the open-label titration period and 4.15 (2.10) at week 3 of the open-label titration period; the mean change from the start to week 3 of the open-label titration period was −3.19 (2.00). For patients who tolerated tapentadol ER, had at least a 1-point improvement in pain intensity during the open-label titration period and were randomized to treatment in the double-blind maintenance period, mean (SD) pain intensity scores (LOCF) were 3.48 (2.02) in the placebo group and 3.67 (1.84) in the tapentadol ER group at baseline (average pain over the 3 days prior to the start of the double-blind maintenance period), and 4.76 (2.52) in the placebo group and 3.77 (2.19) in the tapentadol ER group at week 12 of the double-blind maintenance period. The mean (SD) change from baseline to week 12 of the maintenance period was 1.28 (2.41) in the placebo group and 0.08 (1.87) in the tapentadol ER group [LSMD for tapentadol ER vs placebo (95 % confidence interval [CI]) −1.14 (−1.435, −0.838); P < 0.001]. These results indicate that pain intensity worsened upon switching from open-label tapentadol ER treatment to placebo during the double-blind maintenance period but was relatively unchanged with continued treatment with tapentadol ER.
Fig. 1.
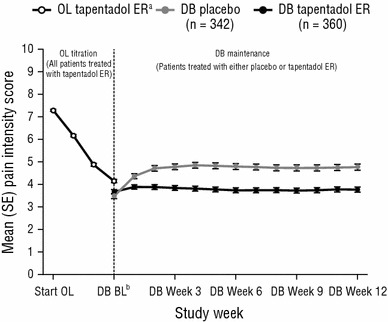
Mean [standard error (SE)] pain intensity over time [intent-to-treat (ITT) population]. Values during the OL titration period are based on observed-case analysis, and values during the DB maintenance period are based on the last observation carried forward (LOCF). Patients were required to have ≥1-point improvement in pain intensity during the OL titration period to be eligible for randomization to treatment during the DB maintenance period. BL baseline, DB double-blind, ER extended release, OL open-label. aOL tapentadol ER population: start OL, n = 1,034; week 1, n = 1,038; week 2, n = 951; week 3, n = 869. bValue for week 3 of the OL titration period is also shown at this timepoint
Sensitivity analyses using alternative imputation methods supported the results of the primary efficacy analysis. Statistically significant differences were observed between placebo and tapentadol ER for the change in average pain intensity from baseline to week 12 of the double-blind maintenance period, using all alternative imputation methods (P < 0.001, in favour of tapentadol ER for all comparisons; Fig. 2). The results of the post hoc MMRM analysis were consistent with those of the primary efficacy analysis using LOCF and the other imputation methods summarized in Fig. 2; using the MMRM, the estimated difference between the tapentadol ER and placebo groups for the change from baseline to week 12 of the double-blind maintenance period was −1.14 (95 % CI −1.47, −0.82; P < 0.001).
Fig. 2.
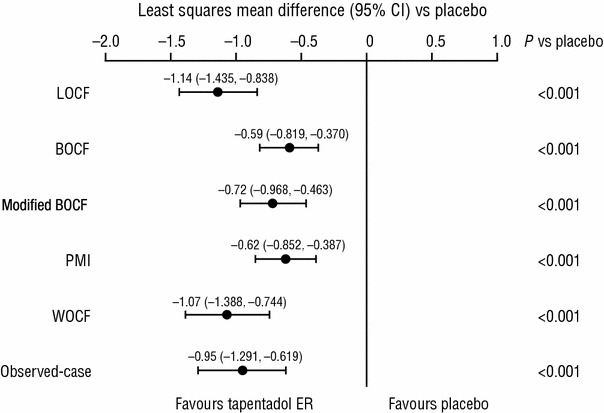
Sensitivity analyses of the primary efficacy analysis [double-blind (DB) intent-to-treat (ITT) population]. BOCF baseline observation carried forward, CI confidence interval, ER extended release, LOCF last observation carried forward, PMI placebo mean imputation, WOCF worst observation carried forward
A significant difference was observed in the overall distribution of responder rates between the placebo and tapentadol ER groups (P = 0.0167, in favour of tapentadol ER). As shown in Fig. 3, the percentages of patients who achieved at least a 30 % improvement and at least a 50 % improvement in pain intensity from the start of the open-label titration period to week 12 of the double-blind maintenance period were significantly lower in the placebo group than in the tapentadol ER group (both P ≤ 0.005).
Fig. 3.
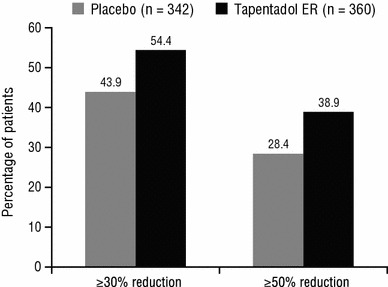
Responder rates for a ≥30 % and a ≥50 % reduction in pain intensity from the start of the open-label (OL) titration period to week 12 of the double-blind (DB) maintenance period [DB intent-to-treat (ITT) population]. P ≤ 0.005 for tapentadol extended release (ER) versus placebo for both responder rates
For the PGIC, a significant difference in the distribution of PGIC scores was observed between the placebo and tapentadol ER groups (P < 0.001, in favour of tapentadol ER). The percentage of patients who reported that their overall status was “very much improved” or “much improved” on the PGIC at the double-blind endpoint was 41.7 % (131/314) in the placebo group and 65.5 % (215/328) in the tapentadol ER group. For the BPI-SF, the mean (SD) changes in the pain intensity subscale score from the start of the double-blind treatment period to the double-blind endpoint were significantly different between the placebo (1.0 [2.22]) and tapentadol ER groups (−0.0 [1.87]; P < 0.001, in favour of tapentadol ER). A significant difference was also observed in the mean (SD) changes from the start of the double-blind treatment period to the double-blind endpoint in the BPI-SF pain interference subscale score between treatment groups (placebo, 0.7 [2.27]; tapentadol ER, −0.2 [1.79]; P < 0.001, in favour of tapentadol ER). For both BPI-SF subscales, mean scores increased in the placebo group over the course of the double-blind maintenance period, indicating that pain intensity and pain interference worsened after the switch from open-label tapentadol ER treatment to placebo, while mean pain intensity and interference subscale scores were unchanged or improved slightly with continued tapentadol ER treatment.
For the SF-36 physical functioning, role-physical, bodily pain, social functioning and role-emotional subscale scores and the physical component summary score, significant differences were observed in the changes from the start of the double-blind treatment period to the double-blind endpoint between the placebo and tapentadol ER groups (P < 0.05, in favour of tapentadol ER; Fig. 4). A significant difference was also observed in the change from the start of the double-blind treatment period to the double-blind endpoint for the EQ-5D health status index between the placebo and tapentadol ER groups (LSMD for tapentadol ER vs placebo [95 % CI], 0.09 [0.056, 0.122]; P < 0.001), also in favour of tapentadol ER.
Fig. 4.
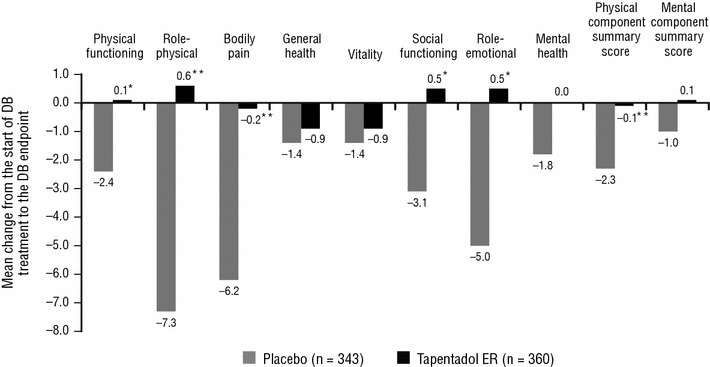
Mean change in Short Form-36 (SF-36) subscale and summary scale scores from the start of the double-blind (DB) treatment period to the DB endpoint. Negative values indicate deterioration. ER extended release. *P < 0.05 versus placebo. **P ≤ 0.001 versus placebo
Subgroup Analyses
Slight differences were observed between patients grouped according to age or race in the mean change in pain intensity from baseline to week 12 of the maintenance period between the placebo and tapentadol ER groups (LSMD for tapentadol ER vs placebo: <65 years of age, −1.18; ≥65 years of age, −1.01; black, −1.12; white, −1.15; other, −1.05). Differences between the placebo and tapentadol ER groups in the mean change in pain intensity from baseline to week 12 of the maintenance period were numerically greater for patients with mild pain intensity (LSMD for tapentadol ER vs placebo: −1.25) than for those with moderate pain intensity (−0.97) or severe pain intensity (−0.82) at the start of the double-blind maintenance period. A numerically greater difference was observed in the mean change in pain intensity from baseline to week 12 of the maintenance period between the placebo and tapentadol ER groups for female patients (LSMD for tapentadol ER vs placebo: −1.51) than for male patients (−0.89), and for opioid-naive patients (−1.21) than for opioid-experienced patients (−1.00). Although numerical differences in the LSMD between the placebo and tapentadol ER groups in the mean change in pain intensity from baseline to week 12 of the maintenance period were observed between subgroups of patients divided according to pain intensity at the start of the double-blind maintenance period, sex and opioid experience, these differences were not considered to be clinically relevant.
Safety and Tolerability
At least one TEAE was reported by 73.1 % of patients (760/1,040) during the open-label titration period. Gastrointestinal TEAEs (40.7 % [423/1,040]), including nausea and constipation, and nervous system TEAEs (38.7 % [402/1,040]), including dizziness and somnolence, were among the most commonly reported TEAEs during titration (Table 3).
Table 3.
Treatment-emergent adverse events (TEAEs) reported by ≥5 % of patients during the open-label (OL) titration period (OL safety population)a
| TEAE, n (%) | Tapentadol ER (n = 1,040) |
|---|---|
| Gastrointestinal disorders | 423 (40.7) |
| Nausea | 236 (22.7) |
| Constipation | 116 (11.2) |
| Vomiting | 93 (8.9) |
| Dry mouth | 67 (6.4) |
| Diarrhoea | 47 (4.5) |
| Nervous system disorders | 402 (38.7) |
| Dizziness | 168 (16.2) |
| Somnolence | 137 (13.2) |
| Headache | 89 (8.6) |
| General disorders and administration site conditions | 156 (15.0) |
| Fatigue | 84 (8.1) |
| Skin and subcutaneous tissue disorders | 124 (11.9) |
| Pruritus | 73 (7.0) |
| Metabolism and nutrition disorders | 77 (7.4) |
| Decreased appetite | 49 (4.7) |
ER extended release
aIncidence is based on the number of patients experiencing ≥1 TEAE, not the number of TEAEs
During the double-blind maintenance period, 56.0 % of patients (192/343) in the placebo group and 74.7 % of patients (269/360) in the tapentadol ER group reported at least one TEAE. Among the most common TEAEs in the placebo and tapentadol ER groups were gastrointestinal TEAEs (placebo, 17.8 % [61/343]; tapentadol ER, 34.7 % [125/360]), such as nausea and vomiting, and nervous system TEAEs (placebo, 12.8 % [44/343]; tapentadol ER, 20.0 % [72/360]), such as dizziness and somnolence (Table 4).
Table 4.
Treatment-emergent adverse events (TEAEs) reported by ≥5 % of patients in either treatment group during the double-blind (DB) maintenance period (DB safety population)a
| TEAE, n (%) | Placebo (n = 343) | Tapentadol ER (n = 360) |
|---|---|---|
| Gastrointestinal disorders | 61 (17.8) | 125 (34.7) |
| Nausea | 27 (7.9) | 61 (16.9) |
| Vomiting | 9 (2.6) | 34 (9.4) |
| Diarrhoea | 18 (5.2) | 27 (7.5) |
| Constipation | 2 (0.6) | 21 (5.8) |
| Nervous system disorders | 44 (12.8) | 72 (20.0) |
| Dizziness | 6 (1.7) | 27 (7.5) |
| Somnolence | 1 (0.3) | 17 (4.7) |
| Headache | 18 (5.2) | 14 (3.9) |
| Psychiatric disorders | 31 (9.0) | 55 (15.3) |
| Anxiety | 15 (4.4) | 26 (7.2) |
| Insomnia | 11 (3.2) | 19 (5.3) |
| Musculoskeletal and connective tissue disorders | 44 (12.8) | 46 (12.8) |
| Myalgia | 15 (4.4) | 18 (5.0) |
ER extended release
aIncidence is based on the number of patients experiencing ≥1 TEAE, not the number of TEAEs
Serious TEAEs were reported for 1.9 % of patients (20/1,040) during the open-label titration period, and for 3.5 % of patients (12/343) in the placebo group and 5.0 % of patients (18/360) in the tapentadol ER group during the double-blind maintenance period. The majority of serious TEAEs were reported for no more than one patient during the open-label titration period or double-blind maintenance period. The only serious TEAEs reported for more than one patient during the open-label titration period were chest pain (0.5 % [5/1,040]) and dehydration (0.2 % [2/1,040]). During the double-blind maintenance period, the only serious TEAEs reported for more than one patient in either treatment group were dehydration (placebo, 0 %; tapentadol ER, 0.6 % [2/360]) and coronary artery disease (placebo, 0.6 % [2/343]; tapentadol ER, 0 %).
The mean (SD) change in HbA1c levels was −0.12 (0.467) from the start of the open-label titration period to endpoint of the open-label titration period. Mean (SD) changes in HbA1c levels from the start of the open-label titration period to endpoint of the double-blind maintenance period were 0.11 (1.040) for patients in the placebo group and −0.13 (1.009) for patients in the tapentadol ER group.
Discussion
For this analysis, efficacy and selected safety data were pooled from two randomized-withdrawal, placebo-controlled, phase 3 studies of similar design in patients with moderate to severe, chronic, painful DPN [18, 19]. Results of this pooled analysis support those of the two individual studies [18, 19] and indicate that treatment with tapentadol ER (100–250 mg bid) is associated with clinically meaningful reductions in pain intensity during the titration period. Improvements in pain intensity achieved during the open-label titration period for patients who achieved at least a 1-point reduction in pain intensity and tolerated tapentadol ER treatment were maintained for patients who continued taking tapentadol ER, while pain intensity increased for patients who switched to placebo. A significant difference, in favour of tapentadol ER, was observed between the tapentadol ER and placebo groups in the mean change from baseline to week 12 of the double-blind maintenance period. Sensitivity analyses using alternative imputation methods supported the results of the primary analysis, showing significant differences between the tapentadol ER and placebo groups in the mean change from baseline to week 12 of the double-blind maintenance period. Changes in HbA1c levels over the course of the two pooled studies were minimal, suggesting that tapentadol ER treatment had no relevant impact on glycaemic control, and that the improvements in pain intensity observed with tapentadol ER treatment observed in the current analysis were not likely related to changes in glycaemic control. The therapeutic dose range of tapentadol ER (100–250 mg bid) was well tolerated and effective when patients were individually titrated to their optimal dose (median modal total daily dose during titration, 300 mg; median modal total daily dose during maintenance, 400 mg).
In addition to the positive results observed for the primary efficacy endpoint, patients who received tapentadol ER achieved clinically meaningful improvements in pain intensity and health status, based on responder rate analyses and the PGIC, respectively. More than 50 % of patients who received tapentadol ER throughout the study achieved a clinically meaningful 30 % improvement in pain intensity [23] from the start of titration to the end of the maintenance period, and approximately 39 % achieved a 50 % improvement in pain intensity. On the PGIC, more than 60 % of patients reported clinically meaningful improvements (“very much improved” or “much improved” [23]) in their health status over 15 weeks of continuous tapentadol ER treatment.
Painful DPN is often associated with a substantial negative impact on patients’ physical function, mental and emotional well-being, social interactions and overall quality of life [27–29]. For that reason, improvements in quality of life and physical function, as well as reductions in pain, have been established as a goal of therapy for painful DPN [7, 30]. In their evidence-based guidelines, the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine noted that evidence for improvements in quality of life and physical function is lacking for a number of treatments for painful DPN [7]. Results of the current pooled analysis showed that tapentadol ER treatment was associated with significant improvements in health-related quality of life and physical functioning compared with placebo, based on differences in the mean change from the start to the end of the double-blind treatment period in the SF-36 physical functioning, role-physical, bodily pain, social functioning and role-emotional subscale scores; the SF-36 physical component summary score; and the EQ-5D health status index score. One of the available treatment options for painful DPN, pregabalin [7], has been shown to provide improvements in measures of sleep quality, as well as quality of life [31]. Results of separate phase 3b studies in patients with severe, chronic low back pain with or without a neuropathic pain component, as diagnosed using the painDETECT questionnaire, showed that tapentadol ER (50–250 mg bid) was associated with improvements from baseline in measures of anxiety [32, 33], depression [32, 33] and sleep quality [32, 33]. Further research is needed to support the efficacy of tapentadol ER for improving these outcomes in patients with painful DPN.
Pooling of efficacy data from the individual phase 3 studies [18, 19] of tapentadol ER for the management of painful DPN allowed for the evaluation of the effects of patient-specific factors on the efficacy of tapentadol ER. Slight numerical differences were observed in the difference in the mean change in pain intensity from baseline to week 12 of the maintenance period between the placebo and tapentadol ER groups for all subgroup analyses, but these differences were not considered to be clinically relevant. Tapentadol ER was effective for the management of moderate to severe, chronic, painful DPN across all analysed subgroups.
Opioid analgesics provide relief for neuropathic pain, including painful DPN, but poor tolerability, particularly gastrointestinal tolerability, may hinder their use [2, 7, 8]. In the current pooled analysis, tapentadol ER (100–250 mg bid) was shown to be well tolerated on the basis of the incidences of TEAEs and low incidence of discontinuations due to adverse events; the side effect profile in the current analysis was consistent with that observed in previous studies of tapentadol ER for the management of chronic pain [12–14, 20]. The two studies [18, 19] in this pooled analysis included no active comparator; however, results from previous phase 3 studies comparing tapentadol ER and oxycodone controlled release (CR) in patients with moderate to severe chronic osteoarthritis or low back pain showed that tapentadol ER treatment was associated with better gastrointestinal tolerability than oxycodone CR [12, 13, 20]. A pooled analysis [14] of results from three phase 3 studies in patients with osteoarthritis knee pain or low back pain showed that tapentadol ER (100–250 mg bid) provided non-inferior analgesic efficacy to oxycodone HCl CR (20–50 mg bid) but had superior gastrointestinal tolerability, based on the incidences of common gastrointestinal TEAEs (i.e. constipation, nausea and vomiting). The improved tolerability of tapentadol ER compared with the pure μ-opioid agonist oxycodone CR may result from the opioid-sparing effect engendered by the contribution of a second mechanism of action, norepinephrine reuptake inhibition, to its analgesic activity [11].
As a post hoc analysis of pooled data from two separate phase 3 clinical studies [18, 19], this analysis was associated with certain inherent limitations. Although both 15-week studies [18, 19] had similar randomized-withdrawal, double-blind, placebo-controlled designs and included similar patient populations, there were minor differences between the two studies that could possibly have affected the results. The formulations of tapentadol ER used in the two studies differed; a conventional hypromellose-based formulation was used in the first study [18], while a new formulation of tapentadol ER, with a high mechanical strength resulting from a polyethylene oxide matrix and melt extrusion manufacturing process, was used in the second study [19]. Bioequivalence has generally been demonstrated for these two formulations [21]. In addition, the enrolment criteria related to glycaemic control differed in the two studies. In the first study [18], patients were required to have HbA1c levels of no more than 11 % for at least 3 months prior to enrolment; in the second study [19], patients were required to have a stable, optimized diabetic regimen for at least 3 months prior to enrolment. The pre-specified washout time periods also differed between the two studies (3–14 days in the first study [18] and 5 days in the second study [19]); however, the duration of the washout period was flexible to accommodate the washout of drugs with longer elimination half-lives. Despite these differences in study design, efficacy and tolerability results were generally comparable across the two individual phase 3 studies [18, 19] and the current pooled analysis. Furthermore, these trials formed the basis for the approval in the United States of tapentadol ER for the management of neuropathic pain associated with DPN in adults severe enough to require daily, around-the-clock, long-term opioid treatment and for whom alternative treatment options are inadequate [34].
The degree of enrichment, based on the requirement that patients responded to tapentadol ER with at least a 1-point improvement in pain intensity during the open-label titration period, was relatively small in the current study. Therefore, these results should at least be generalizable to the population of patients who can tolerate treatment with tapentadol ER.
Conclusion
Results of this pooled analysis support the efficacy of tapentadol ER (100–250 mg bid) for the management of neuropathic pain associated with DPN in adults, which was previously demonstrated in two individual phase 3 studies [18, 19]. In addition, tapentadol ER was shown to provide consistent efficacy, regardless of race, sex, age, prior opioid experience or pain intensity at the start of the double-blind maintenance period. This analysis also provided evidence for the positive impact of tapentadol ER on health-related quality of life and physical functioning in patients with painful DPN, which may be of particular relevance because patients with DPN often experience a decline in physical functioning and quality of life [2–5]. In addition to pain intensity and quality of life, the effects of treatment on other comorbidities of painful DPN (e.g. anxiety, depression and insomnia) should be considered [6].
Acknowledgments
Janssen Research & Development, LLC and Grünenthal GmbH supported both studies included in this analysis. Editorial support for the writing of this manuscript was provided by Megan Knagge, PhD, of MedErgy, and was funded by Janssen Research & Development, LLC and Grünenthal GmbH. The authors retained full editorial control over the content of the manuscript.
Conflict of interest
M.S.E., C.R., K.C. and J.H. are employees of Janssen Research & Development, LLC, and are Johnson & Johnson stockholders. D.Y.S. was an employee of Janssen Research & Development, LLC, and a Johnson & Johnson stockholder at the time of the writing of this manuscript. I.V.H. is an employee of Janssen Research & Development, Beerse, Belgium. B.L. is an employee of Grünenthal GmbH. S.S. and A.I.V. are not employed by Janssen Research & Development or Grünenthal GmbH, and were not compensated for this manuscript.
Footnotes
ClinicalTrials.gov Identifiers: NCT00455520 and NCT01041859.
References
- 1.Smith HS, Argoff CE. Pharmacological treatment of diabetic neuropathic pain. Drugs. 2011;71:557–589. doi: 10.2165/11588940-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 2.Tesfaye S, Vileikyte L, Rayman G, et al. Painful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assessment and management. Diabetes Metab Res Rev. 2011;27:629–638. doi: 10.1002/dmrr.1225. [DOI] [PubMed] [Google Scholar]
- 3.Veves A, Backonja M, Malik RA. Painful diabetic neuropathy: epidemiology, natural history, early diagnosis, and treatment options. Pain Med. 2008;9:660–674. doi: 10.1111/j.1526-4637.2007.00347.x. [DOI] [PubMed] [Google Scholar]
- 4.Jensen TS, Backonja MM, Hernandez JS, et al. New perspectives on the management of diabetic peripheral neuropathic pain. Diab Vasc Dis Res. 2006;3:108–119. doi: 10.3132/dvdr.2006.013. [DOI] [PubMed] [Google Scholar]
- 5.Zelman DC, Brandenburg NA, Gore M. Sleep impairment in patients with painful diabetic peripheral neuropathy. Clin J Pain. 2006;22:681–685. doi: 10.1097/01.ajp.0000210910.49923.09. [DOI] [PubMed] [Google Scholar]
- 6.Vinik A. The approach to the management of the patient with neuropathic pain. J Clin Endocrinol Metab. 2010;95:4802–4811. doi: 10.1210/jc.2010-0892. [DOI] [PubMed] [Google Scholar]
- 7.Bril V, England J, Franklin GM, et al. Evidence-based guideline: treatment of painful diabetic neuropathy. Report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76:1758–1765. doi: 10.1212/WNL.0b013e3182166ebe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dworkin RH, O’Connor AB, Audette J, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc. 2010;85:S3–S14. doi: 10.4065/mcp.2009.0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panchal SJ, Muller-Schwefe P, Wurzelmann JI. Opioid-induced bowel dysfunction: prevalence, pathophysiology and burden. Int J Clin Pract. 2007;61:1181–1187. doi: 10.1111/j.1742-1241.2007.01415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Candrilli SD, Davis KL, Iyer S. Impact of constipation on opioid use patterns, health care resource utilization, and costs in cancer patients on opioid therapy. J Pain Palliat Care Pharmacother. 2009;23:231–241. doi: 10.1080/15360280903098440. [DOI] [PubMed] [Google Scholar]
- 11.Tzschentke TM, Jahnel U, Kogel B, et al. Tapentadol hydrochloride: a next-generation, centrally acting analgesic with two mechanisms of action in a single molecule. Drugs Today (Barc). 2009;45:483–496. doi: 10.1358/dot.2009.45.7.1395291. [DOI] [PubMed] [Google Scholar]
- 12.Afilalo M, Etropolski MS, Kuperwasser B, et al. Efficacy and safety of tapentadol extended release compared with oxycodone controlled release for the management of moderate to severe chronic pain related to osteoarthritis of the knee: a randomized, double-blind, placebo- and active-controlled phase III study. Clin Drug Investig. 2010;30:489–505. doi: 10.2165/11533440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 13.Buynak R, Shapiro DY, Okamoto A, et al. Efficacy and safety of tapentadol extended release for the management of chronic low back pain: results of a prospective, randomized, double-blind, placebo- and active-controlled phase III study. Expert Opin Pharmacother. 2010;11:1787–1804. doi: 10.1517/14656566.2010.497720. [DOI] [PubMed] [Google Scholar]
- 14.Lange B, Kuperwasser B, Okamoto A, et al. Efficacy and safety of tapentadol prolonged release for chronic osteoarthritis pain and low back pain. Adv Ther. 2010;27:381–399. doi: 10.1007/s12325-010-0036-3. [DOI] [PubMed] [Google Scholar]
- 15.Imanaka K, Tominaga Y, Etropolski M, et al. Efficacy and safety of oral tapentadol extended release in Japanese and Korean patients with moderate to severe, chronic malignant tumor-related pain. Curr Med Res Opin. 2013;29:1399–1409. doi: 10.1185/03007995.2013.831816. [DOI] [PubMed] [Google Scholar]
- 16.Imanaka K, Tominaga Y, Etropolski M, et al. Ready conversion of patients with well-controlled, moderate to severe, chronic malignant tumor-related pain on other opioids to tapentadol extended release. Clin Drug Investig. 2014;34:501–511. doi: 10.1007/s40261-014-0204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kress HG, Koch ED, Kosturski H, et al. Tapentadol prolonged release for managing moderate to severe, chronic malignant tumor-related pain. Pain Physician. 2014;17:329–343. [PubMed] [Google Scholar]
- 18.Schwartz S, Etropolski M, Shapiro DY, et al. Safety and efficacy of tapentadol ER in patients with painful diabetic peripheral neuropathy: results of a randomized-withdrawal, placebo-controlled trial. Curr Med Res Opin. 2011;27:151–162. doi: 10.1185/03007995.2010.537589. [DOI] [PubMed] [Google Scholar]
- 19.Vinik AI, Shapiro DY, Rauschkolb C, et al. A randomized withdrawal, placebo-controlled study evaluating the efficacy and tolerability of tapentadol extended release in patients with chronic painful diabetic peripheral neuropathy. Diabetes Care. 2014;37:2302–2309. doi: 10.2337/dc13-2291. [DOI] [PubMed] [Google Scholar]
- 20.Wild JE, Grond S, Kuperwasser B, et al. Long-term safety and tolerability of tapentadol extended release for the management of chronic low back pain or osteoarthritis pain. Pain Pract. 2010;10:416–427. doi: 10.1111/j.1533-2500.2010.00397.x. [DOI] [PubMed] [Google Scholar]
- 21.Zannikos PN, Smit JW, Stahlberg H-J, et al. Pharmacokinetic evaluation of tapentadol extended-release tablets in healthy subjects. J Opioid Manag. 2013;9:291–300. doi: 10.5055/jom.2013.0171. [DOI] [PubMed] [Google Scholar]
- 22.Dworkin RH, Turk DC, Farrar JT, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain. 2005;113:9–19. doi: 10.1016/j.pain.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 23.Dworkin RH, Turk DC, Wyrwich KW, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain. 2008;9:105–121. doi: 10.1016/j.jpain.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Cleeland CS, Ryan KM. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Singapore. 1994;23:129–138. [PubMed] [Google Scholar]
- 25.Ware JE, Jr, Snow KK, Kosinski M, et al. SF-36 health survey manual and interpretation guide. Boston: The Health Institute, New England Medical Center; 1993. [Google Scholar]
- 26.Brooks R. EuroQol: the current state of play. Health Policy. 1996;37:53–72. doi: 10.1016/0168-8510(96)00822-6. [DOI] [PubMed] [Google Scholar]
- 27.Davies M, Brophy S, Williams R, et al. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care. 2006;29:1518–1522. doi: 10.2337/dc05-2228. [DOI] [PubMed] [Google Scholar]
- 28.Galer BS, Gianas A, Jensen MP. Painful diabetic polyneuropathy: epidemiology, pain description, and quality of life. Diabetes Res Clin Pract. 2000;47:123–128. doi: 10.1016/S0168-8227(99)00112-6. [DOI] [PubMed] [Google Scholar]
- 29.Schmader KE. Epidemiology and impact on quality of life of postherpetic neuralgia and painful diabetic neuropathy. Clin J Pain. 2002;18:350–354. doi: 10.1097/00002508-200211000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Lindsay TJ, Rodgers BC, Savath V, et al. Treating diabetic peripheral neuropathic pain. Am Fam Physician. 2010;82:151–158. [PubMed] [Google Scholar]
- 31.Vinik A, Emir B, Cheung R, et al. Relationship between pain relief and improvements in patient function/quality of life in patients with painful diabetic peripheral neuropathy or postherpetic neuralgia treated with pregabalin. Clin Ther. 2013;35:612–623. doi: 10.1016/j.clinthera.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Gálvez R, Schäfer M, Hans G, et al. Tapentadol prolonged release versus strong opioids for severe, chronic low back pain: results of an open-label, phase IIIb study. Adv Ther. 2013;30:229–259. doi: 10.1007/s12325-013-0015-6. [DOI] [PubMed] [Google Scholar]
- 33.Steigerwald I, Muller M, Davies A, et al. Effectiveness and safety of tapentadol prolonged release for severe, chronic low back pain with or without a neuropathic pain component: results of an open-label, phase 3b study. Curr Med Res Opin. 2012;28:911–936. doi: 10.1185/03007995.2012.679254. [DOI] [PubMed] [Google Scholar]
- 34.Nucynta. Nucynta® ER (tapentadol) extended-release oral tablets C-II [package insert]. Raritan: Janssen Pharmaceuticals, Inc. 2014.