

Abstract
Background
The development of novel anesthetics has historically been a process of combined serendipity and empiricism, with most recent new anesthetics developed via modification of existing anesthetic structures.
Methods
Using a novel high-throughput screen employing the fluorescent anesthetic 1-aminoanthracene (1-AMA) and apoferritin as a surrogate for on-pathway anesthetic protein target(s), we screened a 350,000 compound library for competition with 1-AMA-apoferritin binding. Hit compounds meeting structural criteria had their binding affinities for apoferritin quantified with isothermal titration calorimetry and were tested for γ-aminobutyric acid type A-receptor binding using a flunitrazepam binding assay. Chemotypes with a strong presence in the top 700 and exhibiting activity via isothermal titration calorimetry were selected for medicinal chemistry optimization including testing for anesthetic potency and toxicity in an in vivo Xenopus laevis tadpole assay. Compounds with low toxicity and high potency were tested for anesthetic potency in mice.
Results
From an initial chemical library of over 350,000 compounds, we identified 2,600 compounds that potently inhibited 1-AMA binding to apoferritin. A subset of compounds chosen by structural criteria (700) was successfully reconfirmed using the initial assay. Based upon a strong presence in both the initial and secondary screens the 6-phenylpyridazin-3(2H)-one chemotype was assessed for anesthetic activity in tadpoles. Medicinal chemistry efforts identified four compounds with high potency and low toxicity in tadpoles, two were found to be effective novel anesthetics in mice.
Conclusions
We demonstrate the first use of a high-throughput screen to successfully identify a novel anesthetic chemotype and show mammalian anesthetic activity for members of that chemotype.
Introduction
Despite more than 150 yr of study and hundreds of millions of administrations annually,1 the precise molecular mechanisms by which general anesthetics achieve the hallmarks of the anesthetized state- hypnosis, amnesia, and analgesia- remain shrouded in uncertainty. Despite their widespread use, these drugs are among the more dangerous employed in medicine, with therapeutic indices ranging from 2–4 and troubling nonfatal off-target effects including neuroapoptosis and potentiation of neurocognitive decline.2–5 Historically, the search for newer, safer general anesthetics was a mixture of empiricism and serendipity; modification of structures of existing anesthetics has been the primary means of recent drug development, while it was the rare occasion where an entirely new anesthetic class was discovered (the last major advancement being the alkylphenols in the 1970s.) In part, the explanation for this state-of-affairs is uncertainty with regard to the major target(s) underlying the desirable actions of these important drugs.
Fortunately, some progress has been made. Investigations over the past twenty years suggest certain proteins as functional targets of general anesthetics.6,7 Studies demonstrating key functional interactions between general anesthetic agents and specific ion channels, including two-pore K+ channels and ligand-gated ion channels such as N-methyl-D-aspartate and γ-aminobutyric acid type A receptors (GABAAR), lend credence to the theory that altering the activity of one or more of these channels is important to producing general anesthesia.8–10 While one of these putative ion-channel targets, the GABAAR, is activated by many known anesthetic agents, the lack of a high resolution structure for this heteromeric ion channel has impeded novel drug design targeted at modifying this channel’s activity. This is further complicated by the recent findings that ligand gated ion channels like GABAAR have multiple allosterically active sites;11–13 attaching functional significance to each will be challenging.
We have previously reported that apoferritin, a soluble protein that readily submits to structural studies using x-ray crystallography, binds anesthetics proportionally to their anesthetic potency in a well-defined interhelical site14. Moreover, this site bears secondary, tertiary and quaternary structural similarity to the transmembrane region of ligand gated channels.15 We further have described a screening assay based on competitive binding, employing apoferritin as a surrogate for the unknown anesthetic protein target and the fluorescent anesthetic 1-aminoanthracene (1-AMA) as a reporter molecule.16 Having miniaturized the assay, it became possible to perform high-throughput screens on large numbers of small molecules, to better explore chemical-space. Here, we describe this high-throughput screening assay to determine anesthetic activity in a library of over 350,000 compounds (National Institutes of Health Chemical Genomics Center, Bethesda, MD).17 Compounds found to be “active” were confirmed as interacting with apoferritin using isothermal titration calorimetry (ITC), followed by testing of anesthetic activity first in a tadpole model and subsequently in an in vivo mammalian (mouse) model. This process has yielded a novel chemotype with general anesthetic activity and potencies approaching those of the alkylphenols.
Materials and Methods
Probe Report
As a requirement of funding through the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research (NIH U54MH084681) we issued a nonpeer-reviewed technical report detailing portions of this project; the report is indexed on PubMed.18
Quantitative high-throughput screening assay
All quantitative high-throughput screening (qHTS) assays employed black 1,536-well plates (Greiner Bio-One, Frickenhausen, Germany) and used Phosphate Buffered Saline, pH 7.2, with 0.1% polyethylene glycol 400 as the assay buffer. An integrated robotic system (Kalypsys, San Diego, CA) with one RX-130 and two RX-90 robotic arms (Staubli, Duncan, SC) executed all screening procedures. Two (no. 3,4) of the 48 columns per plate served as negative controls, containing 3 μL 50% saturated solution of 1-AMA in assay buffer. The other 46 columns contained 3 μL 10 μM apoferritin and 50% 1-AMA in assay buffer. 23 μL of 160 mM propofol dissolved in dimethyl sulfoxide (DMSO), the positive control, was added to the top two rows of one column (no. 2). The remainder of the positive control column consisted of twofold dilutions of propofol in duplicate, for a total of 16 dilutions of propofol. The robotic system delivered all propofol by pin transfer from separate control plates to the experimental plates. The Kalypsys system similarly delivered 23 μL of experimental compounds to the remaining wells via the 1,536 pin Kalypsys pintool array (10 nL slotted pins, V&P Scientific, San Diego, CA.) The plates were incubated at room temperature for 10 min prior to reading on an Envision HTS multilabel reader (Perkin-Elmer, Waltham, MA) equipped with a Fura2 Blue Fluorescent Protein excitation filter (380 nm, 10 nm bandwidth) and fluorescein emission filter (535 nm, 25 bandwidth) combined with a LANCE/DELFIA dichroic mirror (wavelength cutoff 400 nm.) In order to speed the overall screen, we used two robotic dispensers and two Envision scanners. Throughout the screen, all liquid lines and reagent containers were made light-tight to prevent reagent degradation.
Compound library plates were screened with concentrations ranging from 6.9 to 153 μM, with a total of 5 concentrations compared to the baseline 1-AMA/apoferritin. Vehicle-only plates (containing DMSO only in the “compound” region of the plates) were inserted uniformly at the beginning and the end of each library to evaluate any shifts in the background signal. Activity was computed as the fluorescence response normalized to free 1-AMA and 1-AMA/apoferritin complex values. Concentration-effect relationships were derived by using publicly available curve fitting algorithms developed in-house.19,20 A four parameter Hill equation was used to fit the concentration-response data by minimizing the residual error between the modeled and observed responses. Those compounds that both displayed full or partial inhibitory curves and which had a maximum response six standard deviations above the mean in either normalized or uncorrected data sets were classified as “top actives.”
General methods for chemistry
All compounds used for ITC, the [3H] flunitrazepam assay, and all biologic assays were either purchased via commercial sources and purified, or synthesized at the National Institutes of Health. Specific chemical synthesis procedures are detailed in Supplemental Digital Content 1. All air or moisture sensitive reactions were performed under positive pressure in nitrogen using oven-dried glassware. We obtained the anhydrous solvents, including dichloromethane, N,N-dimethylforamide, acetonitrile, methanol and triethylamine, from Sigma-Aldrich (St. Louis, MO). We employed a Waters semi-preparative high-performance liquid chromotography system for all preparative purification. This included a Phenomenex Luna C18 (5 micron, 30 × 75 mm) column (Phenomenex, Torrence, CA); the mobile phase was acetonitrile and water (both with 0.1% trifluoroacetic acid) using a 10% to 50% acetonitrile gradient over 8 min at 45 milliliters per minute (ml/min). Analytical analyses were carried out on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA) (see supplemental digital content 1 for details on chemical synthesis.)
Isothermal titration calorimetry
Compounds meeting our criteria as top actives and possessing high ligand efficiency were reevaluated using ITC to determine direct binding signatures between the compounds of interest and apoferritin. All ITC experimentation took place on a MicroCal VP-ITC system (MicroCal, Northampton, MA.) The compounds were dissolved to produce ~1 mM solution in qHTS buffer without DMSO (described under quantitative high-throughput screening assay section) via vigorous shaking and repeated sonication, followed by filtration through a 0.2 μm polytetrafluoroethylene syringe filter. Absorption spectroscopy compared to standards prepared in organic solvents determined the precise concentration of the prepared solutions (since some were not soluble at 1 mM). For each compound, the 1.43 mL ITC sample cell was loaded with apoferritin in qHTS buffer, the reference cell was loaded with water and the syringe loaded with 286 μL of the investigative-compound solution. The ligand-containing syringe was then titrated into the sample cell at room temperature. Titrations were performed in duplicate. In order to correct for heat of dilution, parallel titrations of buffer into buffer, buffer into apoferritin solution, and compound solution into buffer were performed. The corrected data were fitted to single-class binding models using Origin software (version 5.0, OriginLab, Northampton, MA.)
GABAAR [3H] flunitrazepam binding assay
[3H] flunitrazepam (specific activity 84.5Ci/mmol) was purchased from Perkin-Elmer (Life Sciences Inc., Boston, MA) and dissolved in 50 mM Tris-HCl buffer (pH 7.4.) Piglet brain homogenate was used for the assay. 0.5 mg membrane protein was incubated in Tris–HCl buffer (50 mM, pH 7.4) with 1 nM of [3H]flunitrazepam with or without 10 μM of the experimental compound (final volume of 500 μL) at 4 °C for 60 min. Nonspecific binding was determined by incubating membranes with [3H]flunitrazepam in the presence of 10 μM lorazepam. Binding studies were terminated by separating the membrane material from the incubation solution using vacuum filtration. The samples were washed three times with ice-cold Tris–HCl buffer (50 mM, pH 7.4) and the filters were transferred to scintillation vials with 10 mL scintillation fluid for scintillation counting with a Tri-Carb 2800 TR instrument (Perkin Elmer Inc., Shelto, CT). Scintillation counts in samples with experimental compounds were compared with those of [3H]flunitrazepam alone.
Xenopus tadpole anesthetic activity and toxicity assay
All animal studies were performed with approval from the Institutional Animal Care and Use Committee at the University of Pennsylvania (Philadelphia, Pennsylvania) and in accordance with National Institutes of Health guidelines. Prelimb Xenopus tadpoles were obtained from Nasco (Fort Atkinson, WI.) All tadpole experiments were carried out in a ~20-fold dilution of Ringers saline solution (artificial pond water). All compounds used in animal studies were obtained from commercial sources. For each experiment, 10 tadpoles were placed in 20 mm Petri dishes containing approximately 20 mL pond water. Experimental compounds in DMSO stock were added to the pond water to create final concentrations of 100, 10, and 1 μM. After incubation in these solutions for one hour, tadpoles were scored for the anesthetic phenotype, defined here as a lack of startle response (no movement in response to a sharp tap on the Petri dish lid.) The lack of startle response corresponds well to loss-of-righting reflex in mice and loss of consciousness in humans.21 Following exposure, the pond water was changed, and return of startle response monitored for 24 h. If tadpoles remained immobile after 24 h, toxicity was assumed, and death was typically confirmed by observing cessation of heartbeat.
Tadpoles were exposed to compounds from the 6-phenylpyridazin-3(2H)-one chemotype at 100 μM. Those compounds producing 100% immobility at 1 hour were retested at 10 μM and 1 μM. Compounds that produced delayed death without immobility as well as those that produced death at 1 and 10 μM concentrations were eliminated from further testing. Importantly, compounds that produced death at 100 μM (a group that included propofol) were tested at the lower concentrations.
Loss-of-righting reflex anesthetic potency assay in mice
All experimental mice were wild-type female C57BL/6J aged 2–4 months (Jackson Laboratories, Bar Harbor, ME). Compounds that demonstrated anesthetic activity in tadpoles at 1 μM, and were devoid of apparent toxicity, were additionally tested in mice. Whereas DMSO was used as a solvent for the tadpole exposures, the volumes needed for reliable tail-vein injection in mice (greater than 50 μL) approached the LD50 for DMSO in mice. As an alternative vehicle, a solution of 60% 2-hydroxypropyl-β-cyclodextrin (HP- β-CD) in 0.9% sterile sodium chloride (pH 7.4) was used. The final concentration for the ML306 (5) solution was 28.5 mg/mL (54.2 mM) while the concentration of the administered 14, 13, and 12, solutions were between 30.0 mg/mL and 45.0 mg/mL (74.1–116 mM.) Injections were given at 40, 80, and 100 mg/kg. Injection volumes were calculated to the nearest 2.5 μl. Investigators were blinded to compound identity though not dose. The mice were restrained and injections made into their tail veins with a 30 ga needle. At the onset of hypnosis, mice were placed on their backs, kept warm (>36.0 °C) via a heating pad, and the time until return of their righting reflex (consistently able to maintain three of four paws on the ground) recorded. Group sizes for ML306 were n = 4, n = 6, n = 3 for 40, 80, and 100 mg/kg, respectively. Group sizes for 14 were n = 5, n = 5, n = 6 for 40, 80, and 100 mg/kg respectively. Group sizes for 12 and 13 were both n = 5 for 100 mg/kg. The number of mice per exposure group was based upon prior experience with agents of unknown potency and compound availability.
Statistical Analysis
High-throughput screening (HTS) data were corrected and normalized using GeneData Screener Software as previously described.19 Concentration-effect relationships for the HTS were derived using the publicly-available curve fitting algorithm developed in-house, Grid, as previously described.20 All non-HTS statistical analyses conducted using Prism 6.0 (Graphpad Software, Inc., San Diego, CA) Flunitrazepam assay curve assessed with best-fit linear regression. Dose-response results were computed using four-parameter Hill equation. Loss-of-righting-reflex (LoRR) assays were compared with one-way ANOVA using Tukey’s multiple comparisons post-hoc test. We used an alpha value of 0.05 for significance (all significant findings had p < 0.05) and all analyses were two-tailed.
Results
High-throughput screening
The screen of the compound library was conducted (as detailed in Methods) at six concentrations between 6.9 and 153 μM, a significant advantage afforded by the qHTS approach. The positive control, propofol, showed a consistent concentration-dependent pattern of fluorescence inhibition, with an average IC50 of 23 μM (fig. 1). The compounds that both displayed full or partial inhibitory curves and had a maximum response six standard deviations above the mean in either normalized or uncorrected data sets were classified as “top actives” (dark blue, fig. 1), a total of 2,593 compounds. Other compounds that caused signal decrease but did not reach the standard for top active were categorized as weak active/indeterminate (light blue, fig. 1). To further characterize the top actives, they were grouped by structural similarity. There were a total of 261 structural clusters among the top actives, defined using a lower limit of 0.7 for the Tanimoto coefficient to define structural similarity. There were an additional 176 compounds among the top actives not structurally similar to any other in the group.
Figure 1.

Three-dimensional plot of quantitative high-throughput screen showing inhibition of the apoferritin/1-aminoanthracene interaction by propofol control (green,) top active compounds (dark blue,) and weak actives/indeterminate compounds (blue.) Top actives and weak actives/indeterminates represent 7.3% of the total compound library screened. This figure is publically available at http://www.ncbi.nlm.nih.gov/books/NBK153223/ and permission to reproduce is not required.
Compound selection and secondary screens
From among the top active compounds, 700 were chosen for additional follow-up based upon structure-activity relationships, maintaining the broad diversity of structures, molecular weight, partition coefficient, and ligand efficiency (Gibbs free energy per nonhydrogen atom of the compound.).22 Those 700 compounds were retested in the original qHTS assay to confirm activity, which was reproducible for all compounds (data not shown). A subset of the 70 compounds with the highest ligand efficiency from among the 700 retested compounds were selected for confirmation of apoferritin interaction in a secondary assay using ITC (fig. 2). In addition to ITC, we measured the ability of a sampling of those compounds to allosterically modulate [3H] flunitrazepam binding in rat cortical membranes. Increased [3H] flunitrazepam binding indicates positive modulation of these receptors, interpreted here as specific binding of the compounds under investigation to GABAA R.14,23,24 Interestingly, [3H] flunitrazepam binding did not appear to correlate with either apoferritin binding (as all compounds tested showed significant apoferritin interaction in the qHTS) or anesthetic potency (fig. 3, also see section on anesthetic potency), suggesting that efficacious compounds might act through GABAAR-independent mechanisms.
Figure 2.

Degree of interaction as measured by isothermal titration calorimetry between screened library compounds and apoferritin. Top interactions shown, along with propofol as reference.
Figure 3.

There is no correlation between anesthetic potency and tritiated flunitrazepam activity (an indirect measure of for γ-aminobutyric acid receptor interaction) [p = 0.42 (not significantly non-zero), best-fit linear regression] suggesting that it is unlikely that this high-throughput screening method is discovering strictly γ-aminobutyric acid receptor interacting novel anesthetics.
Anesthetic potency and medicinal chemistry
Based on the initial screens demonstrating high ligand efficiency, confirmed strong interactions with apoferritin as measured by ITC, and the largest number of members in the top active list, the 6-phenylpyridazin-3(2H)-one chemotype (fig. 4) was chosen for resynthesis, medicinal chemistry and further investigation of anesthetic activity via tadpole assays. Among members of the 6-phenylpyridazin-3(2H)-one chemotype, efficacious compounds with high potency, inducing tadpole immobility after 60 minutes exposure, and low toxicity tended to fall within the 2-(6-oxopyridazin-1-yl)-N-(2-phenylethyl)acetamide family (fig. 4.) As shown in figure 4, our initial exploration at 6-position of the pyridazin-3-one showed that smaller groups or rings are not well tolerated. For example, entries 1–3 showed reduced activity or no effect on immobilizing the tadpoles. Similarly, replacing with more hydrophilic groups such as pyridine analog 15 and N, N-dimethylphenyl analog 11 resulted in substantial loss of anesthetic activity in tadpoles. This phenomenon was consistently observed with similar analogs that are not included here. However, para-substitution of phenyl ring at 6-position with lipophilic groups improved the potency that is exemplified by the percentage of immobile tadpoles at 10 μM for compounds 5 and 11–14. Fortunately, these compounds showed minimal or no toxicity against tadpoles at 10 μM concentrations. However, similarly substituted analogs at ortho and meta positions showed decreased potency with increased toxicity as seen with compounds 7–6 in figure 4 as representative examples. The 6-substitution pattern of the pyridazin-3-one core appears to be essential for the modulation of anesthetic activity as evidenced by complete loss of potency for 5 and 6 substituted analogs 32, 37, and 38 (fig. 5). Our attempts to replace the pyridazin-3-one core with smaller or similar heterocycles resulted in inactive compounds (representative analogs shown in fig. 5).
Figure 4.
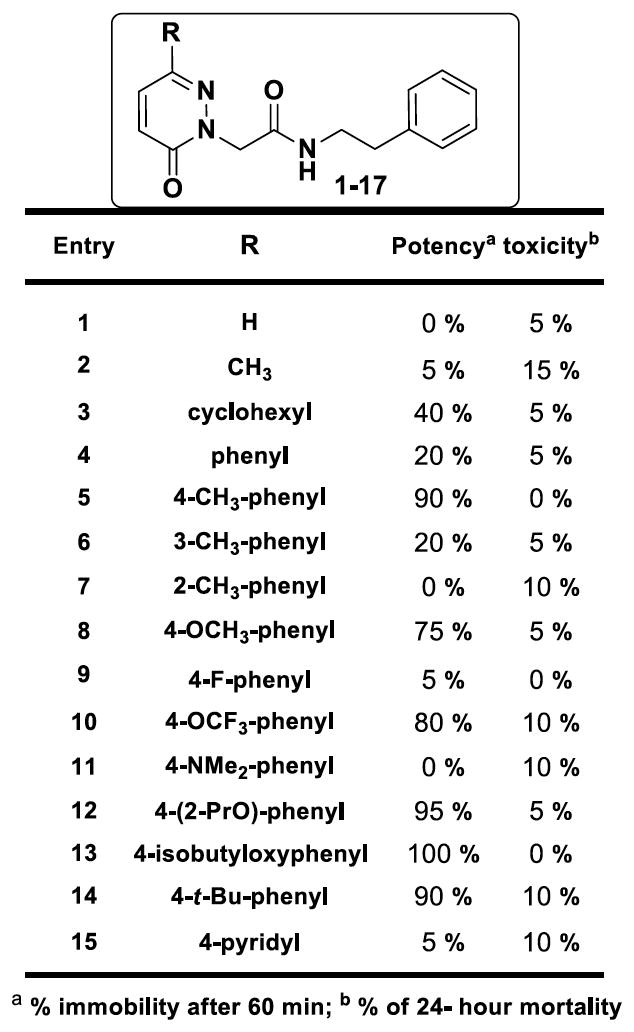
Structure-activity relationship studies with modifications p-tolyl region. The data represents tadpole anesthetic activity (as measured by immobility) and toxicity (mortality at 24 h).
Figure 5.

Structure-activity relationship studies with modifications at the core. The data represents tadpole anesthetic activity (as measured by immobility) and toxicity (mortality at 24 h).
Our next focus of structure-activity relationship (SAR) exploration was modification of the side chain at the 2-position as shown in figure 6 represented by selected analogs. The amide linkage is critical for the anesthetic activity as evidenced by complete loss of activity for analogs 27–29. Increased potency was observed upon introducing more lipophilic side chains containing phenyl, furan, thiophene and cycloalkyl groups as noticed with analogs 16–23 and 26. Analogs 24–25 showed no anesthetic profile against tadpoles suggesting that hydrophilic groups are less tolerated in this region. It is also important to note that a two carbon linker (analog 5) is needed to manifest optimal anesthetic activity whereas a one carbon or ≥3 carbon linkers decreased the potency as shown for entries 21 and 22 respectively. Our SAR exploration resulted in para-substituted phenyl groups at the 6-position of the pyrazinone core bearing a phenylethyl amide moiety side chain provided best anesthetic profile (analogs 5, 12, 13, and 14) and minimal toxicity with favorable in vitro absorption, distribution, metabolism, and excretion and in vivo pharmacokinetic properties for further profiling. Figure 7 provides a summary of the SAR assessed during the medicinal chemistry optimization. We found four compounds that produced greater than 90% immobility after a 1-h exposure at 10 μM as well as less than 10% lethality at 24-h postexposure (fig. 4). These four compounds were advanced for testing of anesthetic activity in mice.
Figure 6.
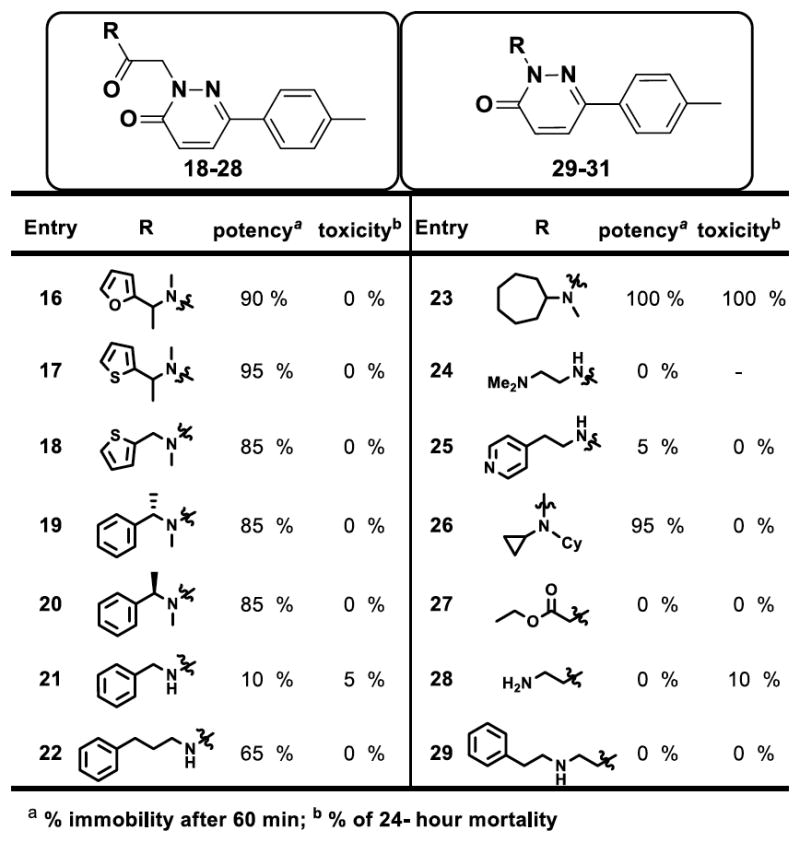
Structure-activity relationship studies with modifications at the side chain. The data represents tadpole anesthetic activity (as measured by immobility) and toxicity (mortality at 24 h).
Figure 7.
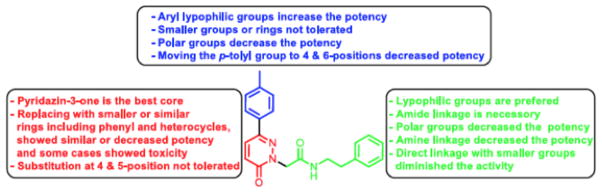
Structure-activity relationship summary. This figure is publically available at http://www.ncbi.nlm.nih.gov/books/NBK153223/ and permission to reproduce is not required.
Mammalian anesthetic activity
The cyclodextrin vehicle alone produced no behavioral effects whatsoever. Similarly, two of the compounds, 12 and 13, failed to produce LoRR in mice at the highest doses tested (100 mg/kg, data not shown.),. At intravenous moles-per-kilogram doses roughly twice that of propofol, both ML306 (5) and 14 produced rapid (<20-s onset) LoRR in mice (at the highest doses, three mice were used for ML306 (5) and six mice for 14), an endpoint shown to correlate with loss of consciousness in humans (fig. 8A). Mean duration of LoRR with ML306 (5) at 100 mg/kg (0.29 mmol/kg) was 245 ± 65 s, while 14 at the same dose (0.26 mmol/kg) produced a mean LoRR of 860 ± 242 s (fig. 8B and C). This compared favorably to propofol where 20 mg/kg (0.11 mmol/kg) produced 445 ± 27 of LoRR.
Figure 8.

Effects of murine intravenous injection of ML306 and 14. A) Dose response curves: LogED50 of ML306 (5) was −0.761 and the logED50 of 14 was −0.667. The logED50 of propofol (mmol/kg) in mice has previously been reported as −1.03.25 Loss-of-righting reflex after iv administration of ML306 (B) and 14 (C.) Both ML306 and 14 produced increased duration of loss of righting reflex in a dose-dependent fashion (p < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test), though duration was considerably longer with administration of 14.
Anesthetic potency for the members of the chemotype did not follow the Meyer-Overton rule, with the two inactive compounds having a lipid solubility in between the anesthetically-active compounds (CLogP was 1.62, 1.97, 2.59, 2.95 for compounds 5, 12, 13, and 14, respectively.) In terms of bioavailability, pharmacokinetic profiling of an index member of the chemotype (compound 5) did not show any remarkable patterns of uptake, distribution, or elimination (data not shown.)
With both compounds, immediately after return of righting reflex the mice continued to appear sedated, demonstrating slow exploration of their environment and little aversion to handling, effects that dissipated 10 to 20 min after return of righting reflex. Mice injected with either compound did not suffer any gross residual behavioral effects or toxicities for up to 2 weeks after injection.
Discussion
This initial investigation used an objective, protein-based approach to ultimately focus on the 6-phenylpyridazin-3(2H)-one chemotype, and the 2-(6-oxopyridazin-1-yl)-N-(2-phenylethyl)acetamide subtype in particular The two final novel anesthetics described here should still only be considered as “probes”, that will require further medicinal chemistry optimization. Even within the 6-phenylpyridazin-3(2H)-one chemotype, there were multiple compounds demonstrating high anesthetic potency and low toxicity via the tadpole assay that have yet to be fully characterized18 in a mammalian model and could very well yield useful alternative probe compounds. Moreover, beyond the 6-phenylpyridazin-3(2H)-one chemotype are the many top active compounds that have not yet been characterized; many may have anesthetic activity as predicted by the surrogate assay.
Our ability to deploy a highly miniaturized surrogate assay based on the apoferritin-AMA system has enabled the testing of several hundred thousand small molecules in a concentration-response mode within the span of several days, thus providing a facile entry to potential general anesthetics. However, despite the success in identifying a novel chemotype of anesthetic (albeit one that requires further development) there were a number of limitations inherent in the screening process that require consideration. The qHTS relies upon a readout of fluorescence inhibition as the marker for successful compound competition with 1-AMA, or binding to the apoferritin general anesthetic site. This reliance on fluorescence yield does not take into account the possible confounding effect of optically-active compounds (inner-filter or flourescent resonance energy transfer) rather than actually interacting with apoferritin. This results in false positives, which are always expected in a large screen and which would ideally be corrected using a secondary confirmatory screen. A full confirmation of the apoferritin-compound interaction for all active compounds via ITC would have served this purpose, except the secondary assays has limitations as well. For example, the limited solubility of an average ‘hit’ molecule in some cases prevented effective evaluation with ITC, and ITC is low to moderate through-put. Another approach could involve reversing the common paradigm of starting with a virtual screen (“docking”) followed by confirmatory tests,25 to start with the physical screen and confirm hits with docking to the target in question (in this case apoferritin) It is important to note that the success of any virtual “docking” approach depends on the quality of target three-dimensional structures.
While a strong correlation exists between apoferritin binding and anesthetic potency,15 it was striking that a correlation between apoferritin binding and flunitrazepam binding to cortical membranes was absent. Select top active compounds, including the 6-phenylpyridazin-3(2H)-one chemotype representative, appear to not only not enhance, but to inhibit flunitrazepam binding. This probably represents allosteric inhibition, as is seen with bicuculine, but it could also reflect direct competition at the benzodiazepine binding site. Future studies of these candidate probes will examine this possible direct interaction at the benzodiazepine site. The lack of a correlation between allosteric potentiation of flunitrazepam binding and anesthetic potency suggests that although our initial hypothesis was that apoferritin binding would select for GABAAR specific anesthetics, apoferritin binding appears to select for general anesthetics targeting other functional substrates. For example, the apoferritin deep hydrophobic site mimics the critically important pharmacophoric features of hydrophobicity and cavity volume. Important electrostatic contacts such as hydrogen bonding, are not represented in this binding site. Given our recent finding that the appoferritin “anesthetic site” is a fatty-acid binding site,26 the apoferritin qHTS may prove to be a useful screening tool for finding fatty acid receptor or enzyme ligands. Additionally, the lack of GABAAR specificity could be considered an asset in finding anesthetic chemotypes that engage entirely novel anesthetic target, as may have occurred here.
Much of the difficulty in developing novel anesthetics stems from the fact that the exact mechanism(s) or target(s) for the anesthetic state remains unknown. Even relying on pharmacophoric information has provided only vague assistance. In the present study, for example, four very similar members of the same chemotype have significantly different anesthetic phenotypes in mammals that could not have been predicted structurally or physicochemically. While the two inactive compounds, 12 and 13, both contain a similar ether functionality, and the anesthetically active compounds do not, we cannot say if and how this produces the change in anesthetic potency without knowing the structure of sites within their functional target(s).
Thus, the definition of anesthesia relies ultimately on an in vivo phenotype. Since anesthesia is currently defined behaviorally, the only reliable type of test to evaluate anesthetic potency is necessarily in vivo, performed here for throughput purposes in tadpoles. While less time- and compound-consuming than, for instance, a mammalian assay, it is still ill-suited to screening the hundreds of compounds classified as top actives from the qHTS. Using the stereotypic photoresponse of zebrafish embryos, a method pioneered by the Peterson Lab at Harvard,27,28 holds promise as a secondary in vivo phenotypic screen that might more efficiently analyze the top actives from this and other qHTS assays in the future.
Ultimately, the development of a truly effective screen for novel anesthetics will require identification of the specific molecular targets responsible for both the on- and off-target effects of anesthetics. Knowledge of those specific targets would reduce the need for a secondary phenotypic screen, and also permit a more focused optimization of probe molecules. However, the surrogate screen described herein provides another benefit, the discovery of novel chemotypes with unknown targets, the search for which may yield new insight into the state of anesthesia.
Supplementary Material
Acknowledgments
Funding: Andrew McKinstry-Wu, Weiming By, Brian Weiser and Roderic Eckenhoff were supported by the National Institutes of Health (Bethesda, Maryland) through the Small Grant Program (R03 MH 084836) and a Program Project Grant (P01 GM 55876). Ganesha Rai, Wendy Lea, Anton Simenov, Ajit Jadhav, and David Maloney were supported by the intramural research program of the National Center for Advancing Translational Sciences and the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research (U54MH084681, National Institutes of Health Bethesda, Maryland). The authors wish to thank Sam Michael, B.S.E., and Richard Jones, B.S.E., for automation support (National Center for Advancing Translational Sciences, National Institutes of Health Bethesda, Maryland); Paul Shinn, B.A. and Danielle van Leer, B.S. for the assistance with compound management (National Institutes of Health Chemical Genomics Center, National Center for Advancing Translational Sciences, National Institutes of Health, Bethesda, Maryland); William Leister, M.S., Heather Baker, B.S., Elizabeth Fernandez, B.S., and Christopher Leclair, Ph.D. for analytical chemistry and purification support (National Institutes of Health Chemical Genomics Center, National Center for Advancing Translational Sciences, National Institutes of Health, Bethesda, Maryland.)
Footnotes
The authors declare no competing interests. The authors or their institutions have no intellectual property rights regarding the compounds generated by this reported research.
References
- 1.Weiser TG, Regenbogen SE, Thompson KD, Haynes AB, Lipsitz SR, Berry WR, Gawande AA. An estimation of the global volume of surgery: A modelling strategy based on available data. Lancet. 2008;372:139–44. doi: 10.1016/S0140-6736(08)60878-8. [DOI] [PubMed] [Google Scholar]
- 2.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–4. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 3.Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X, Dissen GA, Creeley CE, Olney JW. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:834–41. doi: 10.1097/ALN.0b013e3181d049cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckenhoff RG, Johansson JS, Wei H, Carnini A, Kang B, Wei W, Pidikiti R, Keller JM, Eckenhoff MF. Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology. 2004;101:703–9. doi: 10.1097/00000542-200409000-00019. [DOI] [PubMed] [Google Scholar]
- 5.Xie Z, Dong Y, Maeda U, Moir RD, Xia W, Culley DJ, Crosby G, Tanzi RE. The inhalation anesthetic isoflurane induces a vicious cycle of apoptosis and amyloid beta-protein accumulation. J Neurosci. 2007;27:1247–54. doi: 10.1523/JNEUROSCI.5320-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–9. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- 7.Eckenhoff RG, Johansson JS. Molecular interactions between inhaled anesthetics and proteins. Pharmacol Rev. 1997;49:343–67. [PubMed] [Google Scholar]
- 8.Reynolds DS, Rosahl TW, Cirone J, O’Meara GF, Haythornthwaite A, Newman RJ, Myers J, Sur C, Howell O, Rutter aR, Atack J, Macaulay AJ, Hadingham KL, Hutson PH, Belelli D, Lambert JJ, Dawson GR, McKernan R, Whiting PJ, Wafford KA. Sedation and anesthesia mediated by distinct GABA(A) receptor isoforms. J Neurosci. 2003;23:8608–17. doi: 10.1523/JNEUROSCI.23-24-08608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, Andreasen A, Viner A, Trudell JR, Harrison NL. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21:RC136. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linden A, Aller MI, Leppä E, Vekovischeva O, Aitta-Aho T, Veale EL, Mathie A, Rosenberg P, Wisden W, Korpi ER. The in vivo contributions of TASK-1-containing channels to the actions of inhalation anesthetics, the alpha(2) adrenergic sedative dexmedetomidine, and cannabinoid agonists. J Pharmacol Exp Ther. 2006;317:615–26. doi: 10.1124/jpet.105.098525. [DOI] [PubMed] [Google Scholar]
- 11.Brannigan G, LeBard DN, Hénin J, Eckenhoff RG, Klein ML. Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc Natl Acad Sci U S A. 2010;107:14122–7. doi: 10.1073/pnas.1008534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li G-D, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci. 2006;26:11599–605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW, Cohen JB. Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human α1β3γ2 γ-aminobutyric acid type A (GABAA) receptor. J Biol Chem. 2013;288:19343–57. doi: 10.1074/jbc.M113.479725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butts CA, Xi J, Brannigan G, Saad AA, Venkatachalan SP, Pearce RA, Klein ML, Eckenhoff RG, Dmochowski IJ. Identification of a fluorescent general anesthetic, 1-aminoanthracene. Proc Natl Acad Sci U S A. 2009;106:6501–6. doi: 10.1073/pnas.0810590106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ebalunode JO, Dong X, Ouyang Z, Liang J, Eckenhoff RG, Zheng W. Structure-based shape pharmacophore modeling for the discovery of novel anesthetic compounds. Bioorg Med Chem. 2009;17:5133–8. doi: 10.1016/j.bmc.2009.05.060. [DOI] [PubMed] [Google Scholar]
- 16.Lea WA, Xi J, Jadhav A, Lu L, Austin CP, Simeonov A, Eckenhoff RG. A high-throughput approach for identification of novel general anesthetics. PLoS One. 2009;4:e7150. doi: 10.1371/journal.pone.0007150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasgar A, Shinn P, Jadhav A, Auld D, Michael S, Zheng W, Austin CP, Inglese J, Simeonov A. Compound management for quantitative high-throughput screening. JALA Charlottesv Va. 2008;13:79–89. doi: 10.1016/j.jala.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rai G, Bu W, Lea WA, Liang D, Weiser B, Setola V, Austin CP, Simeonov A, Jadhav A, Eckenhoff R, Maloney DJ. Discovery of novel general anesthetics using apoferritin as a surrogate system. Bethesda, MD: National Center for Biotechnology Information (US); 2012. [PubMed] [Google Scholar]
- 19.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006;103:11473–8. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Jadhav A, Southal N, Huang R, Nguyen D-T. A grid algorithm for high throughput fitting of dose-response curve data. Curr Chem Genomics. 2010;4:57–66. doi: 10.2174/1875397301004010057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility. J Pharmacol Exp Ther. 2001;297:338–51. [PubMed] [Google Scholar]
- 22.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–1. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 23.Vedula LS, Brannigan G, Economou NJ, Xi J, Hall Ma, Liu R, Rossi MJ, Dailey WP, Grasty KC, Klein ML, Eckenhoff RG, Loll PJ. A unitary anesthetic binding site at high resolution. J Biol Chem. 2009;284:24176–84. doi: 10.1074/jbc.M109.017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong EH, Iversen LL. Modulation of [3H]diazepam binding in rat cortical membranes by GABAA agonists. J Neurochem. 1985;44:1162–7. doi: 10.1111/j.1471-4159.1985.tb08739.x. [DOI] [PubMed] [Google Scholar]
- 25.Heusser Sa, Howard RJ, Borghese CM, Cullins Ma, Broemstrup T, Lee US, Lindahl E, Carlsson J, Harris RA. Functional validation of virtual screening for novel agents with general anesthetic action at ligand-gated ion channels. Mol Pharmacol. 2013;84:670–8. doi: 10.1124/mol.113.087692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bu W, Liu R, Cheung-Lau JC, Dmochowski IJ, Loll PJ, Eckenhoff RG. Ferritin couples iron and fatty acid metabolism. FASEB J. 2012;26:2394–400. doi: 10.1096/fj.11-198853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peterson RT, Fishman MC. Designing zebrafish chemical screens. Methods Cell Biol. 2011;105:525–41. doi: 10.1016/B978-0-12-381320-6.00023-0. [DOI] [PubMed] [Google Scholar]
- 28.Kokel D, Peterson RT. Using the zebrafish photomotor response for psychotropic drug screening. Methods Cell Biol. 2011;105:517–24. doi: 10.1016/B978-0-12-381320-6.00022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.