

ABSTRACT
Reactivation of human cytomegalovirus (CMV) is hazardous to patients undergoing allogeneic cord blood transplantation (CBT), lowering survival rates by approximately 25%. While antiviral treatment ameliorates viremia, complete viral control requires CD8+ T-cell-driven immunity. Mouse studies suggest that cognate antigen-specific CD4+ T-cell licensing of dendritic cells (DCs) is required to generate effective CD8+ T-cell responses. For humans, this was not fully understood. We here show that CD4+ T cells are essential for licensing of human DCs to generate effector and memory CD8+ T-cell immunity against CMV in CBT patients. First, we show in CBT recipients that clonal expansion of CMV-pp65-specific CD4+ T cells precedes the rise in CMV-pp65-specific CD8+ T cells. Second, the elicitation of CMV-pp65-specific CD8+ T cells from rare naive precursors in cord blood requires DC licensing by cognate CMV-pp65-specific CD4+ T cells. Finally, also CD8+ T-cell memory responses require CD4+ T-cell-mediated licensing of DCs in our system, by secretion of gamma interferon (IFN-γ) by pp65-specific CD4+ T cells. Together, these data show that human DCs require licensing by cognate antigen-specific CD4+ T cells to elicit effective CD8+ T-cell-mediated immunity and fight off viral reactivation in CBT patients.
IMPORTANCE Survival rates after stem cell transplantation are lowered by 25% when patients undergo reactivation of cytomegalovirus (CMV) that they harbor. Immune protection against CMV is mostly executed by white blood cells called killer T cells. We here show that for generation of optimally protective killer T-cell responses that respond to CMV, the early elicitation of help from a second branch of CMV-directed T cells, called helper T cells, is required.
INTRODUCTION
Cytomegalovirus (CMV)-seropositive patients who are immunocompromised are at increased risk for developing potentially life-threatening CMV reactivation. Especially after allogeneic cord blood (CB) transplantation (CBT), the first weeks of immune reconstitution are hazardous for developing CMV reactivation, which is associated with decreased survival rates (1, 2). Antiviral treatment can reduce CMV viremia, but effective viral control requires induction of CMV-directed immunity by T lymphocytes. In particular, CD8+ cytotoxic T lymphocytes (CTLs) fulfill a predominant role in protection against CMV disease (2–4). Therefore, strategies that increase early CMV-specific adaptive immune responses after transplantation are currently being explored, which could ultimately help to establish full clearance of, and long-term immunological memory against, CMV. In the human setting, cell-based therapy is being explored, geared toward dendritic cell (DC)-mediated activation of CTLs (5, 6). The elicitation of antigen-specific CTL immunity was in mouse models shown to require cognate CD4+ T-cell licensing (7–10). Furthermore, priming of naive CD8+ T cells requires both the CD4+ T-helper cells and CD8+ T cells to recognize antigen on the same antigen-presenting cell (5, 11, 12). Such CD4+ T-cell help can involve CD40 ligand (CD40L) binding to CD40 on DCs (9, 13, 14). For humans, a requirement for CD4+ T-cell help in DC licensing for formation of effector and memory CTLs has not yet been demonstrated. It is also not yet clear what are the signaling pathways through which CD4+ T cells might execute their licensing.
CMV-specific CD4+ T-cell clones are present in the healthy population, suggesting a role for antigen-specific CD4+ T cells in immunity against CMV, as 50 to 80% of adults experience CMV infection in their lifetime (6, 15). Moreover, effective control of CMV infection was attained in patients when CMV-specific T cells, of which 77% were CD4+ T cells, were infused (16). Further, human DCs loaded with both HLA class I and II/peptide complexes were more effective at generating antigen-specific CTL responses than were those loaded with solely major histocompatibility complex (MHC) class I/peptide complexes (17). In a different setting, not only the absence of antigen-specific CTLs but also the absence of specific CD4+ T-helper cells resulted in higher CMV loads (18). Thus, CD4+ T-helper cells are likely to participate in CMV control.
We set out to clarify the role of human CD4+ T-helper cells in DC licensing for CTL-mediated immunity for both CTL priming and memory CTL activation. First, we show in CBT recipients that clonal expansion of CMV-pp65-specific CD4+ T-helper cells precedes the expansion of primary CMV-pp65-specific CTLs. We clarified that DC licensing is cognate, as expansion of primary CMV-pp65-specific CTLs from naive CB precursors requires the presence of pp65-specific CD4+ T-helper cells, in cocultures. Finally, also DC licensing is required for CTL memory, as DCs licensed by CD4+ T cells, which they do through secretion of gamma interferon (IFN-γ), stimulate much more efficient CMV-pp65-specific CTL memory responses. Together, these data imply that in humans CD4+ T-helper cells are pivotal in DC licensing to elicit CD8+ T-cell immunity during CMV reactivation in CBT patients.
MATERIALS AND METHODS
Patient inclusion and human samples.
Approval for this study was obtained from the ethics committees of the University Medical Centre Utrecht (METC-05-143, METC-11-063, and METC-13-437). Written informed consent was obtained from all participating patients or their legal representatives prior to CBT. In this consent, it was stated that their medical data may be used for research purposes. According to the hospital's standard operating procedures, regular blood samples were taken for viral load detection by quantitative PCR (qPCR) and T-cell number measurements (see below). All children below the age of 18 who received an allogeneic CBT between 2010 and 2013 at the hematopoietic stem cell transplant (HSCT) unit of the Wilhelmina Children's Hospital were evaluated. All patients received a fludarabine- and busulfan-containing regimen with early-given (day −9) antithymoglobulin (19). Eight patients suffered from CMV reactivation post-CBT. These patients were all CMV positive prior to transplantation. Two were excluded because maximum CMV loads did not reach 1,000 copies/ml. From the 6 other patients, CMV loads and CD4+ and CD8+ counts were evaluated and plotted over time. We also included 8 control patients who did not have events that are likely to impact the T-cell counts (T-cell-impacting events), such as viral reactivations of >1,000 copies (cp)/ml (human herpesvirus 6 [HHV-6], CMV, Epstein-Barr virus [EBV], or adenovirus), graft-versus-host disease (GvHD) of ≥grade 2, or graft rejection. CD4+ T-cell counts within the first 3 months after CBT were evaluated using the area under the curve (AUC) trapezoidal method: ∑(cell numbertime y + cell numbertime x)/2) × (timey − timex) (20). Trends in CD4+ T-cell count were evaluated using a Pearson correlation coefficient.
Immune phenotyping.
Immune phenotyping was performed on whole-blood samples every other week once the leukocyte count was >0.4 × 109/liter. Absolute numbers of T cells (CD3+), helper T cells (CD3+ CD4+), and cytotoxic T cells (CD3+ CD8+) were determined using Trucount technology (BD Biosciences). A volume of 20 μl of CD3-fluorescein isothiocyanate (FITC), CD45-peridinin chlorophyll protein (PerCP), and CD19-allophycocyanin (APC) or CD3-FITC, CD8-phycoerythrin (PE), CD45-PerCP, and CD4-APC reagent (Multitest; BD Biosciences) was added to a Trucount tube containing a known quantity of beads, followed by 100 μl of EDTA-treated whole blood and incubated for 15 min at room temperature. Erythrocytes (RBCs) were subsequently lysed for 15 min with 450 μl of fluorescence-activated cell sorting (FACS) lysing solution (BD Biosciences). Samples were acquired using a FACSCalibur cytometer and analyzed with Multiset software (BD Biosciences). Qualitative and subset analyses of T-cell compartments were performed as described previously (21).
Cord blood dendritic cell culture.
CD34+ cells were isolated according to the manufacturer's instructions (Miltenyi Biotec) and expanded using 20 ng/ml interleukin-3 (IL-3) (Invitrogen), 20 ng/ml IL-6 (BD Biosciences), and 50 ng/ml stem cell factor (SCF) and 50 ng/ml FLT3-L (both from Peprotech). For DC culture, 3 × 106 CD34+ cells were cultured in a T25 flask (Thermo) in X-Vivo medium (Lonza) containing 2 mM l-glutamine, 100 U/ml penicillin-streptomycin, and 5% human serum in the presence of 20 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) and 20 ng/ml IL-4 (all from Invitrogen), 20 ng/ml SCF, and 100 ng/ml FLT3-L at 37°C and 5% CO2 for 7 days.
CD8+ T-cell priming assay.
CB CD34+-derived DCs were loaded with 10 μg/ml pp65 (Miltenyi Biotec; purity, >95%; low endotoxin; <10 endotoxin units [EU]/ml), medium, or 10 μg/ml bovine serum albumin (BSA) (Roche; 10,000 DCs in 100 μl 5% X-Vivo plus human serum per well, 96-well plate [Thermo]). Then, 50,000 donor-matched naive CD8+ T cells were added (separated from CD34− fraction using Miltenyi Biotec magnetically activated cell sorting [MACS] beads according to the manufacturer's instructions) together with medium, 50,000 CD4+ T cells (separated using MACS beads), or 10 μg/ml CD40 agonist clone 7 (Bioceros). All were cocultured for 3 weeks at 37°C and 5% CO2 for 7 days. On days 8 and 15, CD34+-derived DCs were loaded with 1 × 10−6.5 M NLV peptide and irradiated with 30 Gy. Ten thousand DCs were plated per well, and the T cells were added for restimulation. Every week at days 2 and 5, IL-7 and IL-15 (Immunotools) were added, both at a final concentration of 5 ng/ml. After 3 weeks, cells were stained with an HLA-A2 pp654–503 pentamer (ProImmune) and positive cells were single-cell sorted and stimulated for several weeks as described under “CD8+ T-cell cloning.” Prior to cryopreservation, a small aliquot of T cells (1 × 105 to 5 × 105) was harvested for T-cell receptor (TCR) sequencing.
TCRβ chain sequencing.
TCRβ chains were sequenced as previously described (22). Briefly, a one-sided anchored reverse transcription-PCR (RT-PCR) was performed in order to amplify TCRβ mRNA. Amplified products were purified from the agarose gel and ligated into a pGEM-T Easy vector (Promega), followed by transformation into chemically competent Escherichia coli DH5α bacteria. Thirty-two bacterial colonies were screened for the presence of a TCR construct and subsequently sequenced via capillary electrophoresis. Sequences were analyzed using web-based software (www.imgt.org) (23), and TCRs were identified using the official ImMunoGeneTics nomenclature as previously shown (24).
Cross-presentation assay.
Cross-presentation essays with monocyte-derived DCs (MoDCs) and CMV peptide NLVPMVATV (NLV)-specific CD8+ T-cell clones were performed as described in reference 25. In addition, stimulation of DCs was performed by coincubation after loading with a range of CD40 antibody (0.1 to 10 μg/ml clone 7; Bioceros), a range of pp655–523-specific CD4+ T cells or donor-matched nonspecific T cells (CD4 MACS; Miltenyi Biotec), or a range of recombinant IFN-γ (Immunotools). Blocking of the CD40-CD40L interaction was done by preincubating CD4+ T cells with 10 μg/ml CD40L antibody for 30 min at 37°C and 5% CO2 (Bioceros). Blocking of the CD80/CD86 (B7-1/B7-2)-CD28/CD152 (CTLA-4) interaction was done by preincubating the loaded DCs with 10 μg/ml anti-CD80/CD86 for 30 min at 37°C and 5% CO2 (10 μg/ml abatacept). Blocking of CD137-CD147L interaction was done by preincubating CD4+ T cells with 10 μg/ml CD137 antibody (clone 4b4-1; BioLegend) (26) for 30 min at 37°C and 5% CO2.
For the supernatant exchange experiment, pp65-loaded HLA-DRB1*01 MoDCs were cocultured with cognate CMV-pp655–523-specific CD4+ T cells overnight. Next, we collected the supernatant, added this in the presence of phosphate-buffered saline (PBS) or 10 μg/ml IFN-γ blocking antibody (BD Biosciences) to new pp65-loaded MoDCs, and incubated the cells for another 24 h.
After incubation, DCs were washed and human CMV (HCMV) pp65-specific CD8+ T cells were cocultured with pp65-loaded DCs for 4 to 6 h in the presence of GolgiStop (1/1,500; BD Bioscience). Cells were subsequently stained for surface markers and the presence of intracellular IFN-γ and tumor necrosis factor (TNF), followed by flow cytometry-based analysis.
CD4+ T-cell cloning.
HCMV-pp65-specific CD4+ T cells were isolated from HLA-DRB1*0101+ peripheral blood mononuclear cells (PBMCs) using the IFN-γ secretion assay (Miltenyi Biotec, Bergisch Gladbach, Germany). Briefly, PBMCs were stimulated with 2 μg/ml pp65-KYQEFFWDANDIYRI peptide (HLA-DRB1*0101 binding pp65 peptide), and after 4 h of stimulation, IFN-γ-secreting CD4+ T cells were isolated using FACS. The CMV-pp65-specific CD4+ T-cell line was stimulated three times weekly with irradiated (30-Gy) allogeneic PBMCs (1 × 106 cells/ml) and 800 ng/ml phytohemagglutinin (PHA; Murex Biotec Limited, Dartford, United Kingdom).
CD8+ T-cell cloning.
An HLA-A*0201-restricted, HCMV-pp65-specific CD8+ T-cell clone was prepared. In brief, T cells from an HLA-A*0201+ donor were stained with HLA-A2/pp654–503 tetramers and subsequently single-cell sorted in a 96-well plate (Thermo) containing irradiated B lymphoblastoid cell line (B-LCL) feeder cells (1 × 105 cells/ml, irradiated with 70 Gy) and PBMCs from 3 healthy donors (1 × 106 cells/ml, irradiated with 30 Gy). One microgram/milliliter leucoagglutinin PHA-L (Sigma-Aldrich) and 120 U/ml of recombinant IL-2 (Immunotools) were added. T-cell clones specific to pp654–503 were selected using tetramer staining. Positive clones were restimulated and expanded during several stimulation cycles and frozen in aliquots that were freshly thawed before each use in an assay.
Monocyte-derived DC culture.
Peripheral blood mononuclear cells (PBMCs) from healthy HLA-A*02.01/HLA-DR*01.01-positive donors were separated from peripheral blood by Ficoll Isopaque density gradient centrifugation (GE Healthcare Bio-Sciences AB) and either were used directly or frozen until further experimentation. For DC induction, PBMCs were incubated at 37°C and 5% CO2 for 1 h with plastic for the monocytes to adhere, in X-Vivo 15 medium (Lonza) containing 2 mM l-glutamine, 100 U/ml penicillin-streptomycin, and 2% human serum (all obtained from Invitrogen). Cells were washed 3 times with PBS (room temperature) and subsequently cultured for 5 days at 37°C and 5% CO2 in X-Vivo 15 medium containing 450 U/ml GM-CSF (Immunotools) and 300 U/ml IL-4 (Immunotools). Cytokines were refreshed after 3 days. DCs were collected for experiments on day 5 by incubation in PBS (4°C) for 1 h.
DC maturation assay.
Day 4-1/2 monocyte-derived DCs (MoDCs) were incubated overnight (O/N) in the presence of medium, pp65 (3 μg/ml), pp65 and CD40 antibody clone 7 (10 μg/ml; Bioceros), pp65 and CD40L antibody clone 5c8 (10 μg/ml; Bioceros), pp65 and 200,000 pp655–523-specific CD4+ T cells, or poly(I·C) (30 μg/ml; Sigma-Aldrich) and lipopolysaccharide (LPS) (100 ng/ml [Sigma-Aldrich]). In mouse DCs, maturation signals elicited via LPS triggering stimulated acquisition of antigen cross-presentation capacity (27). Cells were subsequently harvested and analyzed for costimulatory marker expression using flow cytometry.
Flow cytometry.
For staining, cells were first washed twice in PBS containing 2% fetal calf serum (FCS) (Invitrogen) and 0.1% sodium azide (NaN3; Sigma-Aldrich). Next, antigen nonspecific binding was prevented by prior incubation of cells with 10% mouse serum (Fitzgerald). Cells were next incubated with combinations of Pacific blue, phycoerythrin (PE), fluorescein isothiocyanate (FITC), allophycocyanin (APC), and PE-Cy7-conjugated mouse anti–human antibody (Ab) (CD3, CD4, CD8, CD11c, CD40, CD45, CD69, CD80, CD83, CD86, CD107a, HLA-DR, HLA-ABC, and TRAIL). Where indicated, after surface staining, T cells were washed twice in PBS-2% FCS-0.1% NaN3) and fixed, permeabilized, and intracellularly stained using monoclonal antibodies (MAbs) to IFN-γ and TNF. Cells were acquired on a FACSCanto II cytometer and analyzed using FACS Diva version 6.13 (BD Bioscience) or FlowJo version 7.6.5 software. Data were analyzed using GraphPad Prism 5.
Detection of cytokines in culture supernatant.
Cytokine concentrations were measured by the MultiPlex Core Facility of the Laboratory of Translational Immunology (LTI) using Luminex technology with in-house-developed bead sets and Bio-Plex Manager version 6.1 software (Bio-Rad Laboratories) as previously described (28).
Detection of CMV-specific CD4+ and CD8+ T cells in patient samples.
NLV-specific CD8+ T cells were detected using HLA-A2 pp654–503 pentamer (ProImmune) or HLA-B7 pp654–426 tetramer (produced in-house). Antigen-specific CD4+ and CD8+ T cells were detected with intracellular IFN-γ staining after stimulation with a pp65 and IE-1 15-mer overlapping peptide mix (JPT Peptide Technologies), as described in reference 29.
RESULTS
CD4+ and CD8+ T-cell dynamics after cord blood transplantation in patients with and without CMV reactivation.
We studied early T-cell reconstitution in pediatric patients undergoing complete immune reconstitution through allogeneic cord blood transplantation (CBT), in relation to CMV reactivation. Six CBT recipients experienced CMV reactivation (>1,000 virus copies/ml), and eight control patients were included (without infectious complications). We analyzed the reconstitution of CD4+ and CD8+ T cells and CMV loads (Fig. 1). We observed expansion and contraction of the CD8+ T-cell population as CMV viral load increased and regressed (Fig. 1A). The control patients instead experienced a consistent and gradual increase in CD8+ T-cell numbers during reconstitution (Fig. 1B). The CD4+ T-helper cell numbers also fluctuated more in patients with CMV reactivation than in control patients (Fig. 1C). Such an expansion and contraction pattern for CD4+ T cells was previously observed in reconstitution under viral pressure (30). Of note, while the total numbers of CD4+ T cells were comparable during the first 90 days after CBT (Fig. 1D, measured as area under the curve of CD4+ T-cell measurements during the first 90 days post-SCT), in CMV-reactivating patients the percentage of activated, HLA-DR+/CD38+ CD4+ T cells was increased (Fig. 1E and F).
FIG 1.

T-cell dynamics in CBT recipients. (A and B) T-cell development over time in CBT recipients with (A) or without (B) CMV reactivation (load of >1,000 copies/ml). Red and blue lines represents CD4+ and CD8+ T-cell numbers, respectively (absolute counts). The black dashed line represents CMV loads (copies/ml). The numbers 1, 2, and 3 (in blue circles) correspond with referred time points in panel G. (C) CD4+ T-cell dynamics (R2 of CD4+ trendline) and median in the first 90 days after transplantation in CBT recipients without (dots) or with (squares) CMV reactivation. A high R2 indicates little fluctuation from the predicted trendline. (D) Total CD4+ T-cell numbers (area under the curve [AUC]) and median in the first 90 days after transplantation in CBT recipients without (dots) or with (squares) CMV reactivation. ns, not significant. (E) Mean percentage (+ standard error of the mean [SEM]) of activated CD4+ T cells in CBT recipients with (red line) or without (black line) CMV reactivation (average of 15 days per data point). (F) AUCs and medians of activated CD4+ T cells in the first 90 days. (G) Percentages of pp65-specific CD4+ and CD8+ T cells (tetramer and/or IFN-γ release upon pp65-peptide mix stimulation) before CMV reactivation (1), during CMV reactivation (2), and after CMV clearance to below detection limits (3) (see panel A) in 4 CBT recipients with reactivation (1 to 4) and 2 CBT recipients without reactivation (5 and 6). Red boxes, CD4+ T cells; blue boxes, CD8+ T cells (corresponding to panels A and B). No samples were available during CMV reactivation for patient 3. Significance in panels C, D, and F was determined using a nonparametric Mann-Whitney test.
CMV-specific CD4+ T-helper cells precede primary CMV-specific CTL expansion after CBT.
We hypothesized that CD4+ T cells, through DC licensing, may support CMV-specific CTL responses, as was shown in mouse-based research (31–33). Cognate interaction between CD4+ T-helper cells and DCs would thereby enable DCs to stimulate more effective CTL responses. To first investigate expansion of the primary CMV-specific CTL population in relation to CMV viremia, we analyzed PBMCs from 4 available CBT recipients who exhibited CMV reactivation (Fig. 1G, patients 1 to 4). We measured a sample prior to and during CMV reactivation and after CMV control (time points indicated in Fig. 1A). As control samples, we included samples from two patients, 5 and 6, who carried CMV prior to CBT and yet did not reactivate (Fig. 1G). In all CBT patients who cleared CMV reactivation, we observed expansion of the primary CMV-specific CTL population (Fig. 1G, right column, patients 1 to 4; mean, 127 days post-SCT). These cells were of CB origin, as confirmed by chimerism analyses (data not shown). The two control patients, 5 and 6, did not elicit CMV-specific CD8+ T cells at 120 and 180 days post-SCT. Early on during CMV reactivation, CMV-specific CTLs did not yet expand, except in patient 4 (Fig. 1G). Considering the CMV-specific CD4+ T-helper cell population, we next measured IFN-γ production in CD4+ T cells after stimulation with a CMV-pp65-overlapping peptide mix as described previously (3, 18, 34) (also data not shown). We observed expansion of the primary CMV-specific CD4+ T-helper cell population in all analyzed samples of reactivating patients, early on during CMV reactivation (Fig. 1G, middle column). Finally, in control patient 6, we detected CMV-specific CD4+ T cells at day 180, indicating that CMV-positive patients (IgG positivity prior to SCT) may eventually develop anti-CMV T cells but later than do patients who reactivate. Taken together, recovery of the CMV-pp65-specific CD4+ T-cell population precedes expansion of primary CMV-pp65-specific CTLs, supporting a role of CD4+ T cells in CD8+ T-cell priming.
CD4+ T-cell licensing of DCs is necessary to prime naive CD8+ T cells in vitro.
To address whether cognate CD4+ T cells facilitate DC licensing for CD8+ T-cell priming, we performed cocultures of CB-derived naive CD8+ T cells with donor-matched CD34+-derived DCs, in the presence or absence of polyclonal donor-matched CD4+ T cells. DCs had been preloaded with pp65 protein or BSA, and cocultures were allowed to proceed for 3 weeks of duration. Using HLA-A2 pentamers loaded with the CMV-derived peptide NLVPMVATV (NLV/A2 in short), we identified pp654–503-specific CTLs (Fig. 2A). Only when CD8+ T-cell priming had been performed in the presence of pp65 protein and CD4+ T cells did we observe CD8+ T cells that bound NLV/A2 pentamers (bright fluorescence, >log4 intensity) (Fig. 2B). To confirm that NLV/A2 reactivity represents pp654–503-specific CD8+ T cells, we performed single-cell sorting of events over log4 intensity and derived clones. As a control, we sorted several cells from the cultures with BSA or pp65 without CD4+ T cells (>log3.5 intensity, as not much NLV/A2 reactivity was present). We succeeded in derivation of 5 independent CMV-specific CTL clones but only from DC/CTL cultures supplemented with both pp65 and CD4+ T cells (Fig. 2C). Of note, we derived one CMV-specific CTL clone from DC/CTL cultures supplemented with both pp65 protein antigen and anti-CD40 antibody but no CD4+ T cells. Together, these data show that CD4+ T cells can license DCs to expand a primary CTL population and that such DC licensing can involve CD40-CD40L interaction, as previously observed in mice (Fig. 2D, clone 6) (7, 9, 13, 35, 36).
FIG 2.

CD4+ T-cell licensing of DCs is necessary to prime naive CD8+ T cells in vitro. (A) Gating strategy and pentamer staining of CMV-pp65-specific CD8+ T cells. SSC, side scatter; FSC, forward scatter. (B) Gating strategy and pentamer staining of primed CB-derived CD8+ T cells. DCs were loaded with 10 μg/ml BSA (left graph) or 10 μg/ml pp65 (middle and right graph) and cocultured with donor-matched naive CD8+ T cells in the absence (middle graph) or presence (left and right graphs) of CD4+ T cells. High-level pentamer staining events (>log4 intensity) were single-cell sorted and clonally expanded for 4 to 6 weeks (representative of 6 independent experiments). (C) Representative pentamer staining of two individually derived CD8+ T-cell clones. (D) Characteristics of 6 clones from 3 independent experiments. Shown are mean fluorescence intensity (MFI) values of pentamer stainings and TCR sequences. Clone 6 was produced using a CD40 agonist antibody. TRBV, T-cell receptor beta variable gene; TRBJ, T-cell receptor beta joining gene. (E and F) Phenotype analysis of pp65-specific CD8+ T-cell clones. (G) Cytokine production (IFN-γ, TNF, and IL-2; pg/ml) of CD8+ T cells after PMA/ionomycin stimulation.
We wished to further strengthen the finding that the T-cell clones grown from rare precursor cells within the polyclonal cord blood T-cell population harbor in fact TCRs specific to CMV-derived epitopes, using DNA sequencing of the recombined CDR3β TCR regions as a second method (Fig. 2C and D). We found that our derived CDR3β sequences are frequently shared in CMV-specific CD8+ T cells, supporting CMV specificity (Fig. 2D) (37). CDR3β sequence variants were unique and not yet described (37).
We next investigated the cellular characteristics of CTL clones that were generated from naive CB precursors. All clones had an effector memory phenotype (CD45ROhigh/CCR7−/CD62Lint/CD27int, Fig. 2E) (38). CD5, CD25, and CD127 levels were comparable, while CD28 expression varied between different clones (Fig. 2F), and none of the clones expressed PD-1 or CTLA-4 in a resting state (data not shown). Functionally, we could not detect cytokine production, possibly since clonal expansion ensued for nearly 3 months, inducing T-cell exhaustion. Using phorbol myristate acetate (PMA)/ionomycin, we circumvented this state, now yielding high levels of cytokine production (Fig. 2G), indicating that these clones were able to respond appropriately. In conclusion, we used human CB-derived cocultures of antigen-loaded DCs and naive lymphocytes to show that CD4+ T-cell licensing of DCs is necessary for antigen-specific CD8+ T-cell priming.
Cognate CD4+ T cells induce licensing of DCs for enhanced memory CTL responses.
DCs are not only instrumental during T-cell priming but also important for restimulation of antigen-specific T-cell clones. We therefore next asked whether human cognate CD4+ T cells are necessary to licensing of DCs for CD8+ T-cell memory (10). To this end, we derived human HLA-A2*01+/HLA-DRB1*01+ monocyte-derived DCs and loaded these with pp65 protein antigen for presentation via HLA-DRB1 and HLA-A2. Next, we induced licensing of the DCs by administration of CMV-pp655–523-specific CD4+ T cells recognizing HLA-DRB1*01/KYQEFFWDANDIYRI complexes presented by the DCs (50,000 DCs and increasing numbers of CD4+ T cells) (39). Medium was refreshed to avoid the possibility of CD4+ T-cell-derived cytokines directly stimulating CTL activation. We added 50,000 memory CMV-pp654–503-specific CTLs to the licensed DCs and determined memory CTL activation by intracellular cytokine staining after 4 hours of coculture of clones (25) in the presence of GolgiStop. NLVPMVATV peptide (1 × 10−6 M) was added to DCs as a positive control for CTL activation (Fig. 3B). We found that upon licensing of DCs, CD8+ T-cell activation was enhanced, as determined by percentages of IFN-γ- and TNF-producing cells and surface-expressed LAMP-1 (Fig. 3A and B) or amounts of IFN-γ and TNF produced (Fig. 3C and D and data not shown). Next, is cognate CD4+ T-cell licensing required for DC-mediated CTL memory responses? We repeated the DC licensing experiment using polyclonal DRB1*01+-restricted CD4+ T cells and found that CD4+ T cells needed to be antigen specific to induce DC licensing, as no induction of CTL activation was seen after addition of polyclonal CD4+ T cells (Fig. 3E). Finally, CD4+ T cells enhanced CTL stimulation via DC licensing and not by direct stimulation of the memory CTLs, as coculture of CD4+ T cells, pp65, and CD8+ T cells in the absence of DCs did not yield cytokine production by the memory CTLs (Fig. 3B).
FIG 3.

Cognate CD4+ T cells induce licensing of DCs for enhanced memory CTL responses. (A to D) Summary and representative plots of CD8+ T-cell activation. (A) MoDCs were loaded with HCMV-derived pp65 and cocultured with 50,000 A2/NLVPMVATV-specific CD8+ T cells in the absence (upper graphs) or presence (lower graphs) of HLA-DRB1*01/KYQEFFWDANDIYRI-specific CD4+ T cells. Freshly thawed T cells were gated based on CD3 and CD8 expression and analyzed for activation-induced production of IFN-γ (left) and TNF (middle) and LAMP-1 surface expression (right). (B) Summary (mean + standard error of the mean [SEM]) of HCMV pp654–503 cross-presentation. Bars represent production of IFN-γ after coculture with MoDCs loaded with 3 μg pp65 in the presence of antigen-specific CD4+ T cells (mean, 120.9%; SEM, 15.8%; n = 6). The black bar shows a maximum response after stimulation with NLV peptide-loaded DCs. (C) Mean fluorescence intensity (MFI) of CD8+ T-cell cytokine production (IFN-γ and TNF) and LAMP-1 surface expression after coculture with pp65-loaded DCs with (red bars) or without (white bars) 100,000 antigen-specific CD4+ T cells (mean + SEM, n = 4). (D) MFI of IFN-γ, gated on IFN-γ-producing CD8+ T cells (mean + SEM, n = 4). (E) MoDCs were loaded with HCMV-derived pp65 and cocultured with 50,000 A2/NLVPMVATV-specific CD8+ T cells in the presence of donor-matched polyclonal CD4+ T cells (mean + SEM, n = 4). Significance in all panels was determined using a nonparametric Mann-Whitney test. *, P < 0.05.
CD40-CD40L, CD80/86-CD28, and CD137-CD137L are not involved in CD4+ T-cell-mediated memory CD8+ T-cell activation.
We next asked if CD4+ T cells facilitate licensing of DCs via CD40L molecules, as was suggested in mouse studies (7, 9, 13, 35, 36). We therefore exchanged cognate CD4+ T cells with a stimulating CD40 antibody in our MoDC/CTL cocultures described for Fig. 3. As a negative control, we included an agonist CD40L antibody. We observed only a modest effect on CD8+ T-cell activation (Fig. 4A). The CD40L antibody did not influence CD8+ T-cell activation. We confirmed these data by performing the CD4+ T-cell coincubation experiments in the presence of CD40L-blocking antibodies (Fig. 4B). We similarly tested CD80/CD86 (B7-1/B7-2)-CD28 signaling, considering their importance in DC/T-cell interaction (40) and that CD80/86 blockade using abatacept is used in several autoimmune disorders (41, 42). Abatacept treatment did not inhibit the CD4+ T-cell-induced activation of memory CD8+ T cells (Fig. 4C). CD137-CD137L (4-1BB–4-1BBL) was also recently implicated in DC-mediated T-cell priming (40, 43). We therefore tested whether blocking of CD137L modulates CD4+ T-cell-induced activation of memory CD8+ T cells. This was not the case (Fig. 4D). Taken together, we conclude that CD4+ T-cell-induced licensing of DCs is not attributable to CD40L-, CD28-, or CD137L-mediated interaction.
FIG 4.

CD40-CD40L, CD80/86-CD28, and CD137-CD137L are not involved in CD4+ T-cell-mediated memory CD8+ T-cell activation. (A) IFN-γ production of CD8+ T cells after coculture with pp65-loaded DCs in the presence of a CD40 (dark blue) or CD40L (light blue) agonist. (B to D) IFN-γ production of CD8+ T cells after coculture with pp65-loaded DCs and antigen-specific CD4+ T cells in the presence of CD40-CD40L blocking (B), CD80/CD86 blocking (C), or CD137-CD137L blocking Ab (mean + standard error of the mean [SEM], n = 4). Significance in all panels was determined using a nonparametric Mann-Whitney test. (E and F) Expression of DC maturation markers CD40, CD80, and CD86 (E) and HLA-ABC (F) after stimulation with medium (white bars), pp65 (gray bars), pp65- and antigen-specific CD4+ T cells (red bars), and pp65 and anti-CD40 (black bars) (mean + SEM, n = 4). *, P < 0.05; **, P < 0.01; ns, not significant.
Finally, is enhanced stimulation of memory CTLs by licensed DCs a mere consequence of cognate CD4+ T-cell-mediated upregulation of DC surface molecules (Fig. 4E and F)? This does not seem to be the case, as incubation of pp65-loaded DCs with antigen-specific CD4+ T cells or CD40L did not cause an overt increase of HLA-ABC; costimulatory marker CD40, CD80, or CD86; or HLA-DR (Fig. 4E and F). Instead, cognate CD4+ T-cell licensing of DCs may involve the enhanced stimulation of memory CTLs via increased antigen presentation of HLA-A2/NLVPMVATV complexes.
Identification of candidate soluble CD4+ T-cell-secreted mediators for licensing of DCs.
Cognate CD4+ T cells may exert DC licensing for enhanced CTL memory responses via secretion of soluble mediators. To test this hypothesis, we cocultured pp65-loaded HLA-DRB1*01 MoDCs with cognate CMV-pp655–523-specific CD4+ T cells overnight. Next, we collected the supernatant and added this to new pp65-loaded MoDCs. After overnight incubation, CTL activation was assessed (Fig. 5A). We found that the enhanced DC licensing was transferred via the supernatant, indicating that CD4+-mediated DC licensing occurs at least partly through soluble factors (Fig. 5B).
FIG 5.

Identification of candidate soluble CD4+ T-cell-secreted mediators for licensing of DCs (A) Schematic outline of supernatant exchange experiments. (B) Summary (mean + standard error of the mean [SEM]) of HCMV pp654–503 cross-presentation. Bars represent production of IFN-γ after coculture with loaded MoDCs in the presence of supernatant of pp65-loaded MoDCs (white bar) or pp65-loaded MoDCs cocultured with antigen-specific CD4+ T cells (blue bar). (C to E) Cytokine and chemokine production. MoDCs were loaded with pp65 and cocultured for 12 to 16 h without T cells (white bars) or with antigen-specific (orange and red bars) or polyclonal (gray bars) CD4+ T cells. Shown are amounts (pg/ml) of TNF (80 to 1,270 pg/ml), IL-6 (71 to 176 pg/ml), IFN-γ (0 to 50 pg/ml), CCL3 (6.9 to 31 ng/ml), CCL4 (2.6 to 5.9 ng/ml), and IL-12 measured with multiplex assay (mean + SEM, n = 4). Significance in all panels was determined using a nonparametric Mann-Whitney test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To identify possible proteins involved, we next analyzed culture supernatants of cocultures of pp65-loaded HLA-DRB1*01 MoDCs with cognate CMV-pp655–523-specific CD4+ T cells or polyclonal HLA-DRB1*01-restricted CD4+ T cells (37°C, O/N), by cytokine multiplex array. The supernatants of cognate DC/T-cell cocultures but not DC/polyclonal T-cell cocultures contained increased amounts of IFN-γ, TNF, and IL-6 cytokines and CCL3 and CCL4 chemokines (Fig. 5C and D). Intracellular cytokine staining confirmed that both IFN-γ and TNF are produced by cognate CD4+ T cells when cocultured with medium, pp65-loaded MoDCs, or PMA/ionomycin (Fig. 6A). While IL-12 was proposed as a cytokine produced by human CD1c+ DCs involved in CD8+ T-cell priming (44), we did not detect IL-12 (Fig. 5E) or IL-10 or IL-15 (data not shown) in our human DC/T-cell cocultures.
FIG 6.

IFN-γ produced by cognate CD4+ T cells enhances memory CTL stimulation by licensed DCs. (A) Intracellular staining for IFN-γ gated on CD3- and CD4-positive cells after 12 to 16 h of coculture of antigen-specific CD4+ T cells with medium (left graph), pp65-loaded MoDCs (middle graph), or PMA/ionomycin (right graph). (B) CTL activation after coculture with pp65-loaded MoDCs and supernatant of pp65-loaded MoDCs cocultured with antigen-specific CD4+ T cells in the presence of PBS (white bar) or IFN-γ-blocking antibodies (blue bar). (C) Summary (mean + standard error of the mean [SEM]) of HCMV pp654–503 cross-presentation. Bars represent production of IFN-γ after coculture with MoDCs loaded with 3 μg pp65 in the presence of recombinant IFN-γ (0.15 to 150 ng/ml, mean + SEM, n = 4). (D) MoDCs were loaded with HCMV-derived pp65 and cocultured with 50,000 A2/NLVPMVATV-specific CD8+ T cells in the absence (upper graphs) or presence (lower graphs) of recombinant IFN-γ. Freshly thawed T cells were gated based on CD3 and CD8 expression and analyzed for activation-induced production of IFN-γ (left) and TNF (middle) and LAMP-1 surface expression (right). Significance in all panels was determined using a nonparametric Mann-Whitney test. *, P < 0.05.
IFN-γ produced by cognate CD4+ T cells enhances memory CTL stimulation by licensed DCs.
The multiplex array revealed IFN-γ as a candidate cytokine produced by cognate CD4+ T cells that enhances DC licensing and consequential memory CTL stimulation, mainly since IFN-γ was the only factor produced exclusively by cognate CD4+ T cells and not by DCs (Fig. 5C). We confirmed this by intracellular IFN-γ staining of the stimulated CD4+ T cells (Fig. 6A). To address the possibility that IFN-γ contributes to DC licensing, we again performed supernatant exchange experiments as described above, but only now, we preincubated the supernatant with IFN-γ-blocking antibodies (10 μg/ml) or PBS as a control. We found that the increased DC licensing partly depends on IFN-γ, although other factors are likely to contribute (Fig. 6B).
Next, we added recombinant IFN-γ (ranging from 0.15 to 150 ng/ml) to 50,000 pp65-loaded HLA-A2*01+ MoDCs in the absence of CMV-pp655–523-specific CD4+ T cells. After 12 to 16 h, we added 50,000 memory CMV-pp654–503-specific CTLs and analyzed cells for CTL stimulation by intracellular cytokine staining after 4 hours of coculture in the presence of GolgiStop. NLVPMVATV peptide (1 × 10−6 M) was included as a positive control for CTL activation (Fig. 6C). We found increased CD8+ T-cell stimulation in an IFN-γ dose-dependent manner, as determined by percentages of IFN-γ- and TNF-producing cells and surface-expressed LAMP-1 (Fig. 6C and D) or amounts of IFN-γ and TNF produced. Thus, the coculture of CMV-pp65 antigen-loaded DCs with cognate CD4+ T cells provokes IFN-γ production by these CD4+ T cells, which consequently facilitates the display of CMV-pp65 peptide/A2 complexes to CD8+ T cells. In conclusion, cognate CD4+ T cells enhanced DC licensing for memory CD8+ immunity, in a manner that requires MHC class II-TCR interaction and subsequent release of IFN-γ by CD4+ T cells. Primary antigen-specific CTL expansion also requires cognate CD4+ T cells, but here, DC licensing appears to work via the CD40-CD40L axis, as in mice (7–9). Taken together, these data provide mechanistic support for how early reconstitution of CD4+ T cells in CBT recipients helps early antigen-driven CD8+ T-cell-mediated immune protection against viral reactivation.
DISCUSSION
Infection-related mortality and GvHD are major causes of death after CBT in both adults and pediatric patients (45, 46). Patients are particularly vulnerable to viral reactivation, including reactivation with CMV (1), Epstein-Barr virus (EBV) (47), human herpesvirus 6 (HHV-6) (48), and varicella-zoster virus (VZV) (49). As the immune system is rebuilt from stem cell precursors, immune protective CD8+ T cells are formed, which exhibit antigen-specific receptors that recognize epitopes from viruses, including CMV. It had not been fully understood whether and how virus-specific CD4+ T cells participate in CD8+ T-cell-mediated protection against viral reactivation. From mouse-based research, a role of CD4+ T cells in CD8+ T-cell priming was deduced. For example, effective CTL induction was seen only when CD4+ T cells were present (50–53). At the same time, from SCT studies, there had been speculation that CD4+ T cells may somehow bolster CD8+ T-cell-mediated viral immune protection (54). For example, studies show that not only CMV-specific CD8+ T cells but also CMV-specific CD4+ T-cell numbers can be used to predict the risk for reactivation in patients after allo-SCT (34). Additional support for CD4+ T cells in CMV immunity comes from CBT patients, showing that recovery of CD4+ CD45RA+ T cells is required to clear CMV viremia (55). From mouse-based research, it was learned that the induction of virus antigen-specific CD8+ T cells requires the prior licensing of DCs by interaction with cognate, antigen-specific CD4+ T cells (7–12, 56) (summarized in Fig. 7). It was our aim to show the possible applicability of such studies to viral reactivation after SCT in human patients. We here show that antigen-specific CD4+ T cells precede the rise of antigen-specific CD8+ T cells after CBT, which are necessary to control CMV reactivation. Using a CB-based culture system, we further show that CD4+ T cells are required to prime antigen-specific CD8+ T cells. These results are in line with conclusions based on mouse work.
FIG 7.
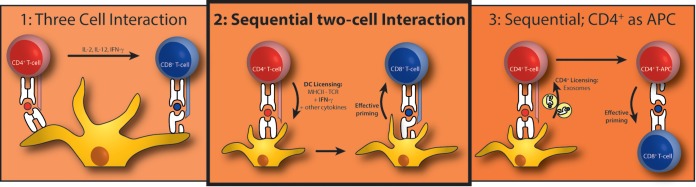
Three suggested mechanisms for CD4+ T-cell-mediated CTL priming. (1) Three-cell interaction, where an antigen-specific CD4+ T cell and CD8+ T cell are in close proximity, interacting through the same DC. The CD4+ T cell directly influences CTL priming by cytokine release. (2) Sequential two-cell interaction, where the DC is licensed by an antigen-specific CD4+ T cell. This licensing happens by a combination of signals 1, 2, and 3; our data support this model best. (3) Sequential; CD4+ as antigen-presenting cell (APC), where the CD4+ T cell acquires antigen-presenting cell capacities by receiving peptide-MHC complexes. One proposed mechanism is via exosomes.
The role of CD4+ T cells in providing help in elicitation of CTL-mediated viral control may be different when using bone marrow or mobilized peripheral stem cells, although in these settings, CD4+ T-cell reconstitution is also correlated with long-term survival (57, 58). When using adult bone marrow or mobilized peripheral stem cells, CMV-specific CD4+ and CD8+ T cells can be detected independently of CMV viremia, but levels of CMV-specific CD4+ (54) or CD8+ (3, 59) T cells are protective in these settings. A major difference is the fact that these patients receive antigen-specific CD8+ T cells from their donor that can clonally expand, circumventing the required priming in the CB setting. Therefore, expansion of antigen-specific CD8+ T cells could be seen as early as 21 days after SCT (59). The mechanism by which CD4+ T cells contribute to survival in the bone marrow or mobilized peripheral stem cell transplantation setting is not fully known, although it has been shown that CD4+ T-cell help is important to maintain CTL effector function in chronic viral infections in mice. Our data presented here on how cognate antigen-specific CD4+ T cells provide DC licensing for effective memory CD8+ T-cell responses by secreting IFN-γ provide experimental support for the described observations (60).
As stated in the introduction, CMV reactivation after CBT correlates with decreased survival rates (61–64). Besides CMV-induced pneumonitis, CMV reactivation is associated with increased risk of GvHD, while GvHD is also a risk factor for viral reactivations (62, 65–68). CMV is the most frequent reactivation, but other viruses also hamper survival. Reactivation of EBV (47), HHV-6 (48), and VZV (49) plays a major role after CBT. We here describe that CMV control coincides with the presence of CMV-specific CD8+ T-cell expansion, which is preceded by the appearance of a CMV-specific CD4+ T-cell population. Using CB-based cocultures, we show the requirement for cognate CD4+ T cells in DC licensing for the expansion of antigen-specific CD8+ T cells. We believe that this is a general mechanism that can be applied broadly to antiviral and possibly even antitumor immune responses. Especially considering the important role of CD8+ T cells in relapse control, this work supports the importance of monitoring the CD4+ T-cell reconstitution early after CBT and paves the road to CD4+ T-cell-based intervention strategies.
ACKNOWLEDGMENTS
We thank members of the Boes laboratory for helpful discussions. We thank Esther Quakkelaar for help with the TCR sequencing.
T.W.H.F., L.S., M.J., D.K., S.N., L.B., D.V.B., M.H.M.H., J.J.B., and M.B. designed the experiments. T.W.H.F., L.S., M.J., D.K., C.D.H., M.P., R.S., and M.M.V.L. conducted the experiments. T.W.H.F. and M.B. wrote the manuscript.
The authors declare no conflict of interest.
This work was financially supported by a KiKa grant to J.J.B.
REFERENCES
- 1.Boeckh M, Ljungman P. 2009. How we treat cytomegalovirus in hematopoietic cell transplant recipients. Blood 113:5711–5719. doi: 10.1182/blood-2008-10-143560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flinsenberg TW, Compeer EB, Boelens JJ, Boes M. 2011. Antigen cross-presentation: extending recent laboratory findings to therapeutic intervention. Clin Exp Immunol 165:8–18. doi: 10.1111/j.1365-2249.2011.04411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tormo N, Solano C, Benet I, Nieto J, de la Cámara R, Lopez J, Garcia Noblejas A, Munoz-Cobo B, Costa E, Clari MA, Hernandez-Boluda JC, Remigia MJ, Navarro D. 2011. Reconstitution of CMV pp65 and IE-1-specific IFN-gamma CD8(+) and CD4(+) T-cell responses affording protection from CMV DNAemia following allogeneic hematopoietic SCT. Bone Marrow Transplant 46:1437–1443. doi: 10.1038/bmt.2010.330. [DOI] [PubMed] [Google Scholar]
- 4.Polic B, Hengel H, Krmpotic A, Trgovcich J, Pavic I, Luccaronin P, Jonjic S, Koszinowski UH. 1998. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J Exp Med 188:1047–1054. doi: 10.1084/jem.188.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen BM, Ohlfest JR. 2012. Increasing the efficacy of tumor cell vaccines by enhancing cross priming. Cancer Lett 325:155–164. doi: 10.1016/j.canlet.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loeth N, Assing K, Madsen HO, Vindelov L, Buus S, Stryhn A. 2012. Humoral and cellular CMV responses in healthy donors; identification of a frequent population of CMV-specific, CD4+ T cells in seronegative donors. PLoS One 7:e31420. doi: 10.1371/journal.pone.0031420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. 1998. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- 8.Ridge JP, Di Rosa F, Matzinger P. 1998. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- 9.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. 1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- 10.Smith CM, Wilson NS, Waithman J, Villadangos JA, Carbone FR, Heath WR, Belz GT. 2004. Cognate CD4(+) T cell licensing of dendritic cells in CD8(+) T cell immunity. Nat Immunol 5:1143–1148. doi: 10.1038/ni1129. [DOI] [PubMed] [Google Scholar]
- 11.Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. 1997. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med 186:65–70. doi: 10.1084/jem.186.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keene JA, Forman J. 1982. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J Exp Med 155:768–782. doi: 10.1084/jem.155.3.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fransen MF, Sluijter M, Morreau H, Arens R, Melief CJ. 2011. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin Cancer Res 17:2270–2280. doi: 10.1158/1078-0432.CCR-10-2888. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen LT, Elford AR, Murakami K, Garza KM, Schoenberger SP, Odermatt B, Speiser DE, Ohashi PS. 2002. Tumor growth enhances cross-presentation leading to limited T cell activation without tolerance. J Exp Med 195:423–435. doi: 10.1084/jem.20010032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF, Cannon MJ. 2006. Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin Infect Dis 43:1143–1151. doi: 10.1086/508173. [DOI] [PubMed] [Google Scholar]
- 16.Einsele H, Roosnek E, Rufer N, Sinzger C, Riegler S, Loffler J, Grigoleit U, Moris A, Rammensee HG, Kanz L, Kleihauer A, Frank F, Jahn G, Hebart H. 2002. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood 99:3916–3922. doi: 10.1182/blood.V99.11.3916. [DOI] [PubMed] [Google Scholar]
- 17.Aarntzen EH, de Vries I, Lesterhuis WJ, Schuurhuis D, Jacobs JF, Bol K, Schreibelt G, Mus R, De Wilt JH, Haanen JB, Schadendorf D, Croockewit A, Blokx WA, Van Rossum MM, Kwok WW, Adema GJ, Punt CJ, Figdor CG. 2013. Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res 73:19–29. doi: 10.1158/0008-5472.CAN-12-1127. [DOI] [PubMed] [Google Scholar]
- 18.Clari MA, Aguilar G, Benet I, Belda J, Gimenez E, Bravo D, Carbonell JA, Henae L, Navarro D. 2013. Evaluation of cytomegalovirus (CMV)-specific T-cell immunity for the assessment of the risk of active CMV infection in non-immunosuppressed surgical and trauma intensive care unit patients. J Med Virol 85:1802–1810. doi: 10.1002/jmv.23621. [DOI] [PubMed] [Google Scholar]
- 19.Lindemans CA, Chiesa R, Amrolia PJ, Rao K, Nikolajeva O, de Wildt A, Gerhardt CE, Gilmour KC, Bierings B, Veys P, Boelens JJ. 2014. Impact of thymoglobulin prior to pediatric unrelated umbilical cord blood transplantation on immune reconstitution and clinical outcome. Blood 123:126–132. doi: 10.1182/blood-2013-05-502385. [DOI] [PubMed] [Google Scholar]
- 20.Bartelink IH, Belitser SV, Knibbe CA, Danhof M, de Pagter AJ, Egberts TC, Boelens JJ. 2013. Immune reconstitution kinetics as an early predictor for mortality using various hematopoietic stem cell sources in children. Biol Blood Marrow Transplant 19:305–313. doi: 10.1016/j.bbmt.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 21.Fujii H, Cuvelier G, She K, Aslanian S, Shimizu H, Kariminia A, Krailo M, Chen Z, McMaster R, Bergman A, Goldman F, Grupp SA, Wall DA, Gilman AL, Schultz KR. 2008. Biomarkers in newly diagnosed pediatric-extensive chronic graft-versus-host disease: a report from the Children's Oncology Group. Blood 111:3276–3285. doi: 10.1182/blood-2007-08-106286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quigley MF, Almeida JR, Price DA, Douek DC. 2011. Unbiased molecular analysis of T cell receptor expression using template-switch anchored RT-PCR. Curr Protoc Immunol Chapter 10:Unit 10.33. doi: 10.1002/0471142735.im1033s94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giudicelli V, Brochet X, Lefranc MP. 2011. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc 2011:695–715. doi: 10.1101/pdb.prot5633. [DOI] [PubMed] [Google Scholar]
- 24.Koning D, Costa AI, Hasrat R, Grady BP, Spijkers S, Nanlohy N, Kesmir C, van Baarle D. 2014. In vitro expansion of antigen-specific CD8 T cells distorts the T-cell repertoire. J Immunol Methods 405:199–203. doi: 10.1016/j.jim.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Flinsenberg TW, Compeer EB, Koning D, Klein M, Amelung FJ, van Baarle D, Boelens JJ, Boes M. 2012. Fcgamma receptor antigen targeting potentiates cross-presentation by human blood and lymphoid tissue BDCA-3+ dendritic cells. Blood 120:5163–5172. doi: 10.1182/blood-2012-06-434498. [DOI] [PubMed] [Google Scholar]
- 26.Salih HR, Kosowski SG, Haluska VF, Starling GC, Loo DT, Lee F, Aruffo AA, Trail PA, Kiener PA. 2000. Constitutive expression of functional 4-1BB (CD137) ligand on carcinoma cells. J Immunol 165:2903–2910. doi: 10.4049/jimmunol.165.5.2903. [DOI] [PubMed] [Google Scholar]
- 27.Palliser D, Ploegh H, Boes M. 2004. Myeloid differentiation factor 88 is required for cross-priming in vivo. J Immunol 172:3415–3421. doi: 10.4049/jimmunol.172.6.3415. [DOI] [PubMed] [Google Scholar]
- 28.de Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. 2007. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis 66:589–598. doi: 10.1136/ard.2006.061853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kern F, Faulhaber N, Frommel C, Khatamzas E, Prosch S, Schonemann C, Kretzschmar I, Volkmer-Engert R, Volk HD, Reinke P. 2000. Analysis of CD8 T cell reactivity to cytomegalovirus using protein-spanning pools of overlapping pentadecapeptides. Eur J Immunol 30:1676–1682. doi:. [DOI] [PubMed] [Google Scholar]
- 30.de Pagter AP, Boelens JJ, Scherrenburg J, Vroom-de BT, Tesselaar K, Nanlohy N, Sanders EA, Schuurman R, van Baarle D. 2012. First analysis of human herpesvirus 6T-cell responses: specific boosting after HHV6 reactivation in stem cell transplantation recipients. Clin Immunol 144:179–189. doi: 10.1016/j.clim.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Matloubian M, Concepcion RJ, Ahmed R. 1994. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 68:8056–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakanishi Y, Lu B, Gerard C, Iwasaki A. 2009. CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature 462:510–513. doi: 10.1038/nature08511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. 1998. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solano C, Benet I, Clari MA, Nieto J, de la Cámara R, Lopez J, Hernandez-Boluda JC, Remigia MJ, Jarque I, Calabuig ML, Garcia-Noblejas A, Alberola J, Tamarit A, Gimeno C, Navarro D. 2008. Enumeration of cytomegalovirus-specific interferon gamma CD8+ and CD4+ T cells early after allogeneic stem cell transplantation may identify patients at risk of active cytomegalovirus infection. Haematologica 93:1434–1436. doi: 10.3324/haematol.12880. [DOI] [PubMed] [Google Scholar]
- 35.Schuurhuis DH, Laban S, Toes RE, Ricciardi-Castagnoli P, Kleijmeer MJ, van der Voort EI, Rea D, Offringa R, Geuze HJ, Melief CJ, Ossendorp F. 2000. Immature dendritic cells acquire CD8(+) cytotoxic T lymphocyte priming capacity upon activation by T helper cell-independent or -dependent stimuli. J Exp Med 192:145–150. doi: 10.1084/jem.192.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toes RE, Schoenberger SP, van der Voort EI, Offringa R, Melief CJ. 1998. CD40-CD40 ligand interactions and their role in cytotoxic T lymphocyte priming and anti-tumor immunity. Semin Immunol 10:443–448. doi: 10.1006/smim.1998.0147. [DOI] [PubMed] [Google Scholar]
- 37.Venturi V, Chin HY, Asher TE, Ladell K, Scheinberg P, Bornstein E, van Bockel D, Kelleher AD, Douek DC, Price DA, Davenport MP. 2008. TCR beta-chain sharing in human CD8+ T cell responses to cytomegalovirus and EBV. J Immunol 181:7853–7862. doi: 10.4049/jimmunol.181.11.7853. [DOI] [PubMed] [Google Scholar]
- 38.Sallusto F, Geginat J, Lanzavecchia A. 2004. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol 22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 39.van Loenen MM, Hagedoorn RS, de Boer R, Falkenburg JH, Heemskerk MH. 2013. Extracellular domains of CD8alpha and CD8ss subunits are sufficient for HLA class I restricted helper functions of TCR-engineered CD4(+) T cells. PLoS One 8:e65212. doi: 10.1371/journal.pone.0065212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, Flies DB. 2013. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keating GM. 2013. Abatacept: a review of its use in the management of rheumatoid arthritis. Drugs 73:1095–1119. doi: 10.1007/s40265-013-0080-9. [DOI] [PubMed] [Google Scholar]
- 42.Rosman Z, Shoenfeld Y, Zandman-Goddard G. 2013. Biologic therapy for autoimmune diseases: an update. BMC Med 11:88. doi: 10.1186/1741-7015-11-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harfuddin Z, Kwajah S, Chong Nyi Sim A, Macary PA, Schwarz H. 2013. CD137L-stimulated dendritic cells are more potent than conventional dendritic cells at eliciting cytotoxic T-cell responses. Oncoimmunology 2:e26859. doi: 10.4161/onci.26859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nizzoli G, Krietsch J, Weick A, Steinfelder S, Facciotti F, Gruarin P, Bianco A, Steckel B, Moro M, Crosti M, Romagnani C, Stolzel K, Torretta S, Pignataro L, Scheibenbogen C, Neddermann P, De Francesco R, Abrignani S, Geginat J. 2013. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood 122:932–942. doi: 10.1182/blood-2013-04-495424. [DOI] [PubMed] [Google Scholar]
- 45.Kurtzberg J, Prasad VK, Carter SL, Wagner JE, Baxter-Lowe LA, Wall D, Kapoor N, Guinan EC, Feig SA, Wagner EL, Kernan NA. 2008. Results of the Cord Blood Transplantation Study (COBLT): clinical outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with hematologic malignancies. Blood 112:4318–4327. doi: 10.1182/blood-2007-06-098020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rocha V, Labopin M, Sanz G, Arcese W, Schwerdtfeger R, Bosi A, Joacbsen N, Ruutu T, de Lima M, Finke J, Frassoni F, Fluckman E. 2004. Transplants of umbilical-cord blood or bone marrow from unrelated donors in adults with acute leukemia. N Engl J Med 351:2276–2285. doi: 10.1056/NEJMoa041469. [DOI] [PubMed] [Google Scholar]
- 47.Rasche L, Kapp M, Einsele H, Mielke S. 2014. EBV-induced post transplant lymphoproliferative disorders: a persisting challenge in allogeneic hematopoietic SCT. Bone Marrow Transplant 49:163–167. doi: 10.1038/bmt.2013.96. [DOI] [PubMed] [Google Scholar]
- 48.de Pagter PJ, Schuurman R, Keukens L, Schutten M, Cornelissen JJ, van Baarle D, Fries E, Sanders EA, Minnema MC, van der Holt BR, Meijer E, Boelens JJ. 2013. Human herpes virus 6 reactivation: important predictor for poor outcome after myeloablative, but not non-myeloablative allo-SCT. Bone Marrow Transplant 48:1460–1464. doi: 10.1038/bmt.2013.78. [DOI] [PubMed] [Google Scholar]
- 49.Vandenbosch K, Ovetchkine P, Champagne MA, Haddad E, Alexandrov L, Duval M. 2008. Varicella-zoster virus disease is more frequent after cord blood than after bone marrow transplantation. Biol Blood Marrow Transplant 14:867–871. doi: 10.1016/j.bbmt.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 50.Gao FG, Khammanivong V, Liu WJ, Leggatt GR, Frazer IH, Fernando GJ. 2002. Antigen-specific CD4+ T-cell help is required to activate a memory CD8+ T cell to a fully functional tumor killer cell. Cancer Res 62:6438–6441 http://cancerres.aacrjournals.org/content/62/22/6438.long. [PubMed] [Google Scholar]
- 51.Ossendorp F, Toes RE, Offringa R, van der Burg SH, Melief CJ. 2000. Importance of CD4(+) T helper cell responses in tumor immunity. Immunol Lett 74:75–79. doi: 10.1016/S0165-2478(00)00252-2. [DOI] [PubMed] [Google Scholar]
- 52.Sun JC, Bevan MJ. 2003. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun JC, Williams MA, Bevan MJ. 2004. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol 5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szabolcs P, Niedzwiecki D. 2008. Immune reconstitution in children after unrelated cord blood transplantation. Biol Blood Marrow Transplant 14:66–72. doi: 10.1016/j.bbmt.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 55.Brown JA, Stevenson K, Kim HT, Cutler C, Ballen K, McDonough S, Reynolds C, Herrera M, Liney D, Ho V, Kao G, Armand P, Koreth J, Alyea E, McAfee S, Attar E, Dey B, Spitzer T, Soiffer R, Ritz J, Antin JH, Boussiotis VA. 2010. Clearance of CMV viremia and survival after double umbilical cord blood transplantation in adults depends on reconstitution of thymopoiesis. Blood 115:4111–4119. doi: 10.1182/blood-2009-09-244145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lanzavecchia A. 1998. Immunology. Licence to kill. Nature 393:413–414. doi: 10.1038/30845. [DOI] [PubMed] [Google Scholar]
- 57.Fedele R, Martino M, Garreffa C, Messina G, Console G, Princi D, Dattola A, Moscato T, Massara E, Spiniello E, Irrera G, Iacopino P. 2012. The impact of early CD4+ lymphocyte recovery on the outcome of patients who undergo allogeneic bone marrow or peripheral blood stem cell transplantation. Blood Transfus 10:174–180. doi: 10.2450/2012.0034-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berger M, Figari O, Bruno B, Raiola A, Dominietto A, Fiorone M, Podesta M, Tedone E, Pozzi S, Fagioli F, Madon E, Bacigalupo A. 2008. Lymphocyte subsets recovery following allogeneic bone marrow transplantation (BMT): CD4+ cell count and transplant-related mortality. Bone Marrow Transplant 41:55–62. doi: 10.1038/sj.bmt.1705870. [DOI] [PubMed] [Google Scholar]
- 59.Cwynarski K, Ainsworth J, Cobbold M, Wagner S, Mahendra P, Apperley J, Goldman J, Craddock C, Moss PA. 2001. Direct visualization of cytomegalovirus-specific T-cell reconstitution after allogeneic stem cell transplantation. Blood 97:1232–1240. doi: 10.1182/blood.V97.5.1232. [DOI] [PubMed] [Google Scholar]
- 60.Kalams SA, Walker BD. 1998. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J Exp Med 188:2199–2204. doi: 10.1084/jem.188.12.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gandhi MK, Khanna R. 2004. Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. Lancet Infect Dis 4:725–738. doi: 10.1016/S1473-3099(04)01202-2. [DOI] [PubMed] [Google Scholar]
- 62.Ljungman P, Perez-Bercoff L, Jonsson J, Avetisyan G, Sparrelid E, Aschan J, Barkholt L, Larsson K, Winiarski J, Yun Z, Ringden O. 2006. Risk factors for the development of cytomegalovirus disease after allogeneic stem cell transplantation. Haematologica 91:78–83 http://www.haematologica.org/content/91/1/78.long. [PubMed] [Google Scholar]
- 63.Ljungman P, Hakki M, Boeckh M. 2010. Cytomegalovirus in hematopoietic stem cell transplant recipients. Infect Dis Clin North Am 24:319–337. doi: 10.1016/j.idc.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 64.Mikulska M, Raiola AM, Bruzzi P, Varaldo R, Annunziata S, Lamparelli T, Frassoni F, Tedone E, Galano B, Bacigalupo A, Viscoli C. 2012. CMV infection after transplant from cord blood compared to other alternative donors: the importance of donor-negative CMV serostatus. Biol Blood Marrow Transplant 18:92–99. doi: 10.1016/j.bbmt.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 65.George B, Pati N, Gilroy N, Ratnamohan M, Huang G, Kerridge I, Hertzberg M, Gottlieb D, Bradstock K. 2010. Pre-transplant cytomegalovirus (CMV) serostatus remains the most important determinant of CMV reactivation after allogeneic hematopoietic stem cell transplantation in the era of surveillance and preemptive therapy. Transpl Infect Dis 12:322–329. doi: 10.1111/j.1399-3062.2010.00504.x. [DOI] [PubMed] [Google Scholar]
- 66.Jaskula E, Dlubek D, Sedzimirska M, Duda D, Tarnowska A, Lange A. 2010. Reactivations of cytomegalovirus, human herpes virus 6, and Epstein-Barr virus differ with respect to risk factors and clinical outcome after hematopoietic stem cell transplantation. Transplant Proc 42:3273–3276. doi: 10.1016/j.transproceed.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 67.Miller W, Flynn P, McCullough J, Balfour HH Jr, Goldman A, Haake R, McGlave P, Ramsay N, Kersey J. 1986. Cytomegalovirus infection after bone marrow transplantation: an association with acute graft-v-host disease. Blood 67:1162–1167. [PubMed] [Google Scholar]
- 68.Pichereau C, Desseaux K, Janin A, Scieux C, Peffault de Latour R, Xhaard A, Robin M, Ribaud P, Agbalika F, Chevret S, Socie G. 2012. The complex relationship between human herpesvirus 6 and acute graft-versus-host disease. Biol Blood Marrow Transplant 18:141–144. doi: 10.1016/j.bbmt.2011.07.018. [DOI] [PubMed] [Google Scholar]