

ABSTRACT
Congenital human cytomegalovirus (HCMV) infection is a leading cause of birth defects, primarily manifesting as neurological disorders. HCMV infection alters expression of cellular microRNAs (miRs) and induces cell cycle arrest, which in turn modifies the cellular environment to favor virus replication. Previous observations found that HCMV infection reduces miR-21 expression in neural progenitor/stem cells (NPCs). Here, we show that infection of NPCs and U-251MG cells represses miR-21 while increasing the levels of Cdc25a, a cell cycle regulator and known target of miR-21. These opposing responses to infection prompted an investigation of the relationship between miR-21, Cdc25a, and viral replication. Overexpression of miR-21 in NPCs and U-251MG cells inhibited viral gene expression, genome replication, and production of infectious progeny, while shRNA-knockdown of miR-21 in U-251MG cells increased viral gene expression. In contrast, overexpression of Cdc25a in U-251MG cells increased viral gene expression and production of infectious progeny and overcame the inhibitory effects of miR-21 overexpression. Three viral gene products—IE1, pp71, and UL26—were shown to inhibit miR-21 expression at the transcriptional level. These results suggest that Cdc25a promotes HCMV replication and elevation of Cdc25a levels after HCMV infection are due in part to HCMV-mediated repression of miR-21. Thus, miR-21 is an intrinsic antiviral factor that is modulated by HCMV infection. This suggests a role for miR-21 downregulation in the neuropathogenesis of HCMV infection of the developing CNS.
IMPORTANCE Human cytomegalovirus (HCMV) is a ubiquitous pathogen and has very high prevalence among population, especially in China, and congenital HCMV infection is a major cause for birth defects. Elucidating virus-host interactions that govern HCMV replication in neuronal cells is critical to understanding the neuropathogenesis of birth defects resulting from congenital infection. In this study, we confirm that HCMV infection downregulates miR-21 but upregulates Cdc25a. Further determined the negative effects of cellular miRNA miR-21 on HCMV replication in neural progenitor/stem cells and U-251MG glioblastoma/astrocytoma cells. More importantly, our results provide the first evidence that miR-21 negatively regulates HCMV replication by targeting Cdc25a, a vital cell cycle regulator. We further found that viral gene products of IE1, pp71, and UL26 play roles in inhibiting miR-21 expression, which in turn causes increases in Cdc25a and benefits HCMV replication. Thus, miR-21 appears to be an intrinsic antiviral factor that represents a potential target for therapeutic intervention.
INTRODUCTION
Human cytomegalovirus (HCMV) infects 50 to 90% of the population worldwide, with extremely high seroprevalence in China (over 90%). This virus is medically important, causing congenital infection with lifelong disabilities resulting from neurological damage (1–3), as well as significant life-threatening disease in immunocompromised individuals (4). Productive infection occurs in a wide range of cell types in vivo and in vitro, including fibroblasts and epithelial and endothelial cells. More interestingly, HCMV also replicates in neural cells of glia, immature neurons, and neural progenitor/stem cells (NPCs), as well as in glioblastoma and neuroblastoma cell lines (5–9). Increasingly, observational studies have indicated that HCMV is associated with glioblastoma (10–13), and recent reports find that this virus establishes persistent/latent infection in T98G glioblastoma cells (14, 15). The brain and auditory system (16–18) are the end-organ sites where damage manifests as sensorineural hearing loss (SNHL), mental retardation, and developmental delays (3, 19–23). However, the neuropathogenesis of congenital HCMV infections, including regulation of virus replication in neural cells, is poorly understood.
HCMV, a member of the β-herpesvirus subfamily, contains a linear double-stranded DNA genome of 235 kb. During productive infection viral genes are expressed temporally and classified into immediate-early (IE), early, and late kinetic classes (24). To establish a beneficial environment for virus replication, HCMV has developed ways to alter cell cycle and inhibit cell death pathways. The virus utilizes cellular machinery to express viral genes and initiate virus replication, especially during the IE phase. Infection also alters expression of cellular gene products involved in cell cycle regulation, DNA damage-repair, and intrinsic defense mechanisms (25–30). For example, cellular cyclin A2-CDK inhibits HCMV IE gene expression (31, 32) and promotes quiescent infection (33), while p53, a cell cycle regulator, is required for efficient HCMV replication (28, 34).
Viral gene expression starts immediately after the viral genome enters the nucleus. A subsequent complex network of virus-cell interactions regulates the cascade of viral gene expression. MicroRNAs (miRs) are one family of cellular factors that regulate viral replication. These 20- to 24-nucleotide (nt) RNAs contribute to the regulation of organogenesis, cell proliferation, differentiation and apoptosis, determination of stem cell fate, virus infection, and antiviral immune responses (35–38). miRs cause degradation of target mRNAs through perfect and imperfect complementation between a seed sequence in the miR and target sequences, typically located within the 3′UTR or sometimes the 5′UTR of the target mRNA (39). Some miRs directly target viral mRNAs, while others act indirectly via cellular genes that in turn impact viral gene expression (40–43). For example, miR-155 inhibits Epstein-Bar virus (EBV) lytic infection and plays a role in maintenance of latency (44), miR-498 and miR-330d reduce Kaposi's sarcoma-associated herpesvirus (KSHV) replication by targeting the viral RTA (43, 45), and miR-100 and miR-101 attenuate HCMV replication via the mTOR pathway (41).
Cellular miRs are also altered in response to viral infection. For example, miR-155 is upregulated by EBV infection (40, 46), miR-21 is upregulated by EBV (40, 47) and hepatitis C virus infection (48), and miR-17, miR-20, miR-96, miR-182, and miR-183 are upregulated, while miR-21, miR-99, miR-100, and miR-101 are downregulated by HCMV infection (41, 49–51).
In previous work we found that miR-21 is downregulated during HCMV infection of NPCs (50) or fibroblasts (41). miR-21 is important in regulating cell cycle and for controlling tumor growth and inducing apoptosis (52). The tumor suppressors PDCD4, PTEN, and TPM1 and cancer-related genes such as Cdc25a, an oncogene, are all targets of miR-21 regulation (53–56). Cdc25a is a member of the CDC25 phosphatase family and participates in G1-to-S transition (57, 58). Since HCMV infection causes cell cycle arrest at G1/S and G2/M (59, 60), understanding the interplay of miR-21 and Cdc25a during HCMV infection in neural cells may provide insights into neuropathogenesis.
In the present study, we show that miR-21 impedes HCMV replication by reducing viral protein expression and expression of cellular Cdc25a. Consistent with a role for Cdc25a in promoting virus replication, overexpression of Cdc25a reversed the negative effects of miR-21 overexpression on HCMV replication. Elevation of Cdc25a levels during HCMV infection is due in part to HCMV-mediated repression of miR-21. Thus, miR-21 has intrinsic antiviral effects that could be exploited for antiviral development.
MATERIALS AND METHODS
Ethics statement.
The Wuhan Institute of Virology Institutional Review Board approved (WIVH10201202) the isolation of primary human embryonic lung fibroblasts (HELs) and NPCs from postmortem fetal embryo tissue and waived the need for consent. The original source of the postmortem fetal embryo tissue was Zhongnan Hospital (61, 62).
Cells and cell culture.
NPCs were propagated using growth medium (GM; Dulbecco modified Eagle medium [DMEM]/F-12 supplemented with 2 mM GlutaMAX, penicillin-streptomycin [100 U/ml and 100 μg/ml], 1.5 μg/ml amphotericin B, 50 μg/ml gentamicin [Gibco/Life Technology], 10% BIT9500 [Stem Cell Technologies], and 20 ng/ml EGF and 20 ng/ml bFGF [Prospect]) as described previously (6, 50, 61). Human U-251MG (human neuronal glioblastoma/astrocytoma, CLS 300385) and HEK293T (American Type Culture Collection [ATCC], CRL-321) cells were cultured in DMEM (Gibco/Life Technology); HELs were cultured in minimal essential medium (MEM; Gibco/Life Technology). Both DMEM and MEM were supplemented with 10% fetal bovine serum (Gibco/Life Technology), glutamine (2 mM, Gibco/Life Technology), and penicillin-streptomycin (100 U/ml and 100 μg/ml). Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Virus and virus infection.
HCMV strain Towne (ATCC-VR977) was propagated in HELs as described previously (61–63). To prevent fetal bovine serum-induced NPC differentiation, the virus used for NPC infection was concentrated and resuspended in GM as described previously (61). UV-inactivated HMCV was prepared and used as described previously (64). NPCs (3 × 106 cells/100-mm dish) were infected with HCMV at a multiplicity of infection (MOI) of 3 without synchronization (50). U-251MG cells were synchronized in G0 by serum starvation for 48 h and then treated with trypsin and reseeded onto poly-d-lysine-coated dishes (106 cells/100-mm dish). Cells were infected with HCMV after attachment for 1 h. Infected cells were harvested by trypsinization, counted, pelleted, snap-frozen in liquid nitrogen, and stored at −80°C until infection time course completed.
Plasmid construction.
The primers used for plasmid construction are listed in Table 1; plasmid applications are presented in Table 2.
TABLE 1.
Primers used for plasmid construction
| Plasmid | Primer sequence (5′-3′) |
|
|---|---|---|
| Forward | Reverse | |
| pCDH-miR-21-GFP | TCTAGACCTTTAGGAGCATTATGAGCAT | GGATCCAGACTATCCCCATTTCTCCAT |
| pCDH-Cdc25a-GFP | GCTAGCAGTCGTGTTTGTGTTTGACCC | GGATCCTCAGAGCTTCTTCAGACGACTG |
| pCDH-Cdc25a+WT-3′UTR-GFP | GCTAGCAGTCGTGTTTGTGTTTGACCC | GGATCCCTGCAACATTCCAGCACTGAG |
| pCDH-Cdc25a+MT-3′UTR-GFP | GCTAGCAGTCGTGTTTGTGTTTGACCC | GGATCCCTGCAACATTCCAGCACTGAG |
| pCDH-IE1-GFP | GGATCCATGGAGTCCTCTGCCAAGA | GGATCCTTACTGGTCAGCCTTGCTTCT |
| pCDH-IE2-GFP | GGATCCATGGAGTCCTCTGCCAAGA | GGATCCCTGAGACTTGTTCCT CAG |
| pCDH-pp65-GFP | GAATCCATGGAGTCGCGCGGTCGCCGTT | GAATTCTCAACCTCGGTGCTTTTTGGGCGTC |
| pCDH-pp71-GFP | TCTAGAATGTCTCAGGCATCGTCCTCG | GGATCCCTAGATGCGGGGTCGACTGCG |
| pCDH-flag-UL26-GFP | ATGGACTACAAGGACGACGATGATAAGATGACGAGCAGGCGCG | GGATCCTTACGGCAACAGCGCTGATGGC |
| pGL3-miPPR21 | GGTACCTGTATTCTGGGTAAGAAGGAGCT | CTCGAGCTTACCACCTGGACTCAAAAGG |
| pGL3cM-Cdc25a-3′UTR | GGTACCATACTGCCATTCTAGGTAGGGT | GAGCTCCTGCAACATTCCAGCACTGAG |
| pGL3cM-CCNE2-3′UTR | GGTACCAAGATTGGGTAGAACTGGTATCACT | CTCGAGCTATACAGAAGACTTCATACCGTAA |
| pLKO.1-shRNA-21-1 | CCGGAAACATTACCCAGCATCATTGCTCGAGCAATGATGCTGGGTAATGTTTTTTTTG | AATTCAAAAAAAACATTACCCAGCATCATTGCTCGAGCAATGATGCTGGGTAATGTTT |
| pLKO.1-shRNA-21-2 | CCGGATCCATATCCAATGTTCTCATTCTCGAGAATGAGAACATTGGATATGGATTTTTTG | AATTCAAAAAATCCATATCCAATGTTCTCATTCTCGAGAATGAGAACATTGGATATGGAT |
| pLKO.1-shRNA-21-3 | CCGGGACTGATGTTGACTGTTGAATCTCGAGATTCAACAGTCAACATCAGTCTTTTTG | AATTCAAAAAGACTGATGTTGACTGTTGAATCTCGAGATTCAACAGTCAACATCAGTC |
| pLKO.1-scramble | CCGGCCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGGTTTTTG | AATTCAAAAACCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG |
| pcDNA3.0-UL83 | GAATCCATGGAGTCGCGCGGTCGCCGTT | GAATTCTCAACCTCGGTGCTTTTTGGGCGTC |
| pcDNA3.0-GAPDH | CGCGGATCCATGGGGAAGGTGAAGGTCGGA | CCGCTCGAGTACTCCTTGGAGGCCATGTG |
TABLE 2.
Plasmids and their applications
| Plasmid | Applications |
|---|---|
| pCDH-miR-21-GFP | Ectopic expression of miR-21 |
| pCDH-miR-UL22A-GFP | Ectopic expression of miR-UL22A |
| pCDH-Cdc25a-GFP | Ectopic expression of Cdc25a |
| pCDH-Cdc25a+WT-3′UTR-GFP | Ectopic expression of Cdc25a; impact of miR-21 on Cdc25a expression |
| pCDH-Cdc25a+MT-3′UTR-GFP | Impact of miR-21 on Cdc25a expression |
| pCDH-IE1-GFP | Ectopic expression of IE1; impact of IE1 on miR-21 |
| pCDH-IE2-GFP | Ectopic expression of IE2; impact of IE2 on miR-21 |
| pCDH-pp65-GFP | Ectopic expression of pp65; impact of pp65 on miR-21 |
| pCDH-pp71-GFP | Ectopic expression of pp71; impact of pp71 on miR-21 |
| pCDH-flag-UL26-GFP | Ectopic expression of UL26; impact of UL26 on miR-21 |
| pGL3-miPPR-21 | Impact of HCMV proteins on luciferase expression from the miR-21 promoter |
| pGL3cM-Cdc25a-3′UTR | Impact of miR-21 on luciferase expression with the 3′UTR from Cdc25a |
| pGL3cM-CCNE2-3′UTR | Impact of miR-21 on luciferase expression with a negative control 3′UTR |
| pLKO.1-shRNA-21 | miR-21 knockdown |
| pcDNA3.0-UL83 | Standard for viral genome quantification by qPCR |
| pcDNA3.0-GAPDH | Standard for cellular genome quantification by qPCR |
Lentiviral vectors.
Lentiviruses were constructed using the pCDH-CMV-MCS-EF1-copGFP system (System Biosciences). Cellular sequences were amplified from cDNA produced from NPCs. Viral sequences were amplified from cDNA produced from HCMV-infected (strain Towne) HELs. Plasmid pCDH-miR-21-GFP contains a 421-bp miR-21 fragment that includes the 72-nt miR-21 hairpin; pCDH-miR-UL22A-GFP, described previously (62), was used as a control. Plasmid pCDH-Cdc25a-GFP contains only the Cdc25a open reading frame (ORF; without its 3′ untranslated region [3′UTR]), while plasmid pCDH-Cdc25a+WT-3′UTR-GFP contains the Cdc25a ORF and the native 1,765-nt 3′UTR. A mutation in the miR-21 binding site within the 1,765-nt 3′UTR was made by overlapping PCR, and the resulting construct was named pCDH-Cdc25a+MT-3′UTR-GFP. Plasmids pCDH-IE1-GFP, pCDH-IE2-GFP, pCDH-pp65-GFP, and pCDH-pp71-GFP contain full-length ORFs encoding viral proteins IE1 (UL123), IE2 (UL122), pp65 (UL83), and pp71 (UL82), respectively. Plasmid pCDH-flag-UL26-GFP contains a modified UL26 ORF that encodes an N-terminal flag epitope-UL26 fusion protein by incorporating the appropriate sequences into the forward PCR primer.
Lentiviral vectors with shRNAs.
Lentiviruses to knock down miR-21 were constructed using lentiviral vector pLKO.1 puro (Addgene). Three short hairpin RNAs (shRNAs) that specifically target the primary miR-21 sequence and a scrambled shRNA that does not target any human or virus genes were designed (http://jura.wi.mit.edu/bioc/siRNAext/), synthesized, and inserted into AgeI/EcoRI-digested pLKO.1 puro to make pLKO.1-shRNA-21-1, pLKO.1-shRNA-21-2, pLKO.1-shRNA-21-3, and pLKO.1-scramble, respectively. shRNA-21-1, shRNA-21-2, and shRNA-21-3 target the primary miR-21 sequence at positions 296 to 317, 271 to 293 and 195 to 215, respectively.
Quantitative PCR (qPCR) standards.
The HCMV UL83 ORF was PCR amplified from HCMV (strain Towne) DNA. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was amplified from cellular DNA. UL83 or GAPDH PCR products were cloned into pcDNA3.0 to make plasmids pcDNA3.0-UL83 and pcDNA3.0-GAPDH, respectively.
Reporter and effector constructs.
Plasmid pGL3-miPPR21 was produced by inserting a 712-nt region of the miR-21 promoter upstream of the luciferase ORF in pGL3-Basic (Promega). Plasmids pGL3cM-Cdc25a-3′UTR and pGL3cM-CCNE2-3′UTR were constructed by inserting a 1,765-nt region of the Cdc25a 3′UTR (containing the predicted miR-21 target site) or a 1,213-nt region of the CCNE2 3′UTR (lacking mR-21 target sequences) 3′ of the luciferase expression cassette in pGL3cM (Promega) (65).
Lentivirus preparation and transduction.
Defective-lentivirus stocks were prepared as described previously (66). In brief, 1.5 × 106 HEK293T cells were seeded in 100-mm dishes. On the following day, 15 μg of pCDH-CMV-MCS-EF1-copGFP (empty vector, here abbreviated as pCDH-GFP) or lentiviral vector plasmids (described above) were cotransfected with 12 μg of pML-Δ8.9 and 8 μg of pVSV-G (System Biosciences) via CaPO4 precipitation. The cells were refed 24 h posttransfection with fresh DMEM containing 10% fetal bovine serum, and the transfection efficiency was monitored by green fluorescent protein (GFP) detection. Lentiviruses released into the culture media were harvested at 48 or 72 h posttransfection, clarified of cell debris by centrifugation, and frozen at −80°C. Stocks were titrated by transducing HEK293T cells with 10-fold serial dilutions in 96-well plates and counting GFP-positive cells at 48 h posttransduction (hpt).
U-251MG cells were transduced at an MOI of 10, and NPCs were transduced at an MOI of 1. Medium was replaced with fresh medium at 3 (NPCs) or 24 (U251 MG cells) hpt. Cultures in which >90% of cells were GFP positive at 48 to 72 hpt were evaluated for transgene expression by qRT-PCR or Western blotting prior to HCMV infection. For shRNA knockdown of miR-21, HEK293T or U-251MG cells were transduced with lentiviruses (MOI = 10) derived from pLKO.1-shRNA-21-1, -2, -3, or -scramble, and the miR-21 levels were measured by qRT-PCR.
qPCR.
HCMV-infected synchronized U-251MG cells or asynchronous NPCs were harvested at 8, 24, 48, 72, 96, and 120 h postinfection (hpi). Cell pellets were processed for DNA extraction using a genome extraction kit (Tiangen Biotech) according to the manufacturer's instructions. DNA concentrations were determined using a NanoDrop ND-1000 (Thermo Scientific, USA). Real-time qPCR was conducted using a CFX-96 Connect system (Bio-Rad) with iQ SYBR green Supermix (Bio-Rad). Then, 20-μl PCRs included 20 ng of DNA, 10 μl of 2× qPCR mix, and 250 nM concentrations (each) of forward (F) and reverse (R) primers. UL83-CN F and R primers were used to quantitate HCMV DNA and GAPDH-CN F and R primers were used to quantitate cellular DNA (Table 3). Tenfold serial dilutions of plasmids pcDNA3.0-UL83 and pcDNA3.0-GAPDH were used to generate standard curves. Reactions were denatured at 95°C for 3 min, followed by 40 two-step cycles at 95°C for 10 s and 60°C for 30 s. Viral genome copy numbers were normalized to GAPDH copies to produce viral genome copies/cell. All reactions were conducted in triplicate. The results reported are means ± the standard deviations (SD) from three independent experiments.
TABLE 3.
Primers for qPCR and qRT-PCR
| Primer | Orientationa | Sequence (5′-3′) |
|---|---|---|
| Specific RT primers | ||
| miR-21 stem-loop | GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACTCAACAGTC | |
| U6 | GAATTTGCGTGTCATCCTTG | |
| Real-time primers | ||
| Mature miR-21 | F | TGCGTGTCGTGGAGTC |
| R | CGGGTAGCTTATCAGACTGA | |
| U6 | F | GCTTCGGCAGCACATATACTAAAAT |
| R | CGCTTCACGAATTTGCGTGTCAT | |
| Cdc25a | F | AGAGTCAACTAATCCAGAGAAGG |
| R | AGCAACTGTATGAAAGAGATAACC | |
| GAPDH | F | GAGTCAACGGATTTGGTCGT |
| R | GACAAGCTTCCCGTTCTCAG | |
| UL83 (CN) | F | GCGAGACCGTGGAACTGC |
| R | TTGCCCTGGATGCGATACTG | |
| GAPDH-(CN) | F | GACCCCTTCATTGACCTCAACTA |
| R | TCCTGGAAGATGGTGATGGG |
F, forward; R, reverse.
Quantitative reverse transcriptase PCR (qRT-PCR).
Total RNA was extracted from cell samples using TRIzol reagent according to the manufacturer's protocol (Invitrogen). DNA was removed with 10 U of RNase-free DNase I (Fermentas) in a reaction mixture containing 10 μg of RNA and 1 U of RNase inhibitor (Fermentas) at 37°C for 30 min. The quality of RNA samples was examined by electrophoresis and detected using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). To examine mature miR-21 levels, 2 μg of RNA from each sample was reverse transcribed with miR-21-specific stem-loop primers or U6-specific primers using RevertAid H Minus M-MuLV reverse transcriptase (Fermentas) by a method modified from a previous description (67). In brief, miR-21 RT primers, U6 RT primers were mixed with 2 μg of RNA and incubated at 70°C for 10 min, then cooled on ice for 2 min. Reaction buffer, deoxynucleoside triphosphate mix, RNase inhibitor and M-MuLV reverse transcriptase were added to a final 20-μl reaction volume. The reverse transcription (RT) reaction mixtures were incubated at 16°C for 30 min, 42°C for 60 min, and 70°C for 5 min. For the detection of target gene expression, 500 ng of RNA was reverse transcribed using PrimeScript II RTase (Tarkara) according to the manufacturer's instructions. Real-time PCR (qPCR) was conducted on a CFX-96 Connect system (Bio-Rad) with the iQ SYBR green Supermix (Bio-Rad). First, 20-μl PCRs included 2 μl of RT reaction product, 10 μl of 2× qPCR mix, and 250 nM concentrations of F and R primers. The reaction mixtures were denatured at 95°C for 3 min, followed by 40 two-step cycles of 95°C for 10 s and 60°C for 30 s. All reactions were run in triplicate for each experiment. The results were obtained from three independent experiments and are presented as means ± 1 SD. miR-21 expression levels were normalized to U6 expression, and GAPDH was used as an internal standard for target gene expression levels. The primers used in qRT-PCR are listed in Table 3.
Luciferase assays.
HEK293T cells were seeded in 12-well plates (2.5 × 105 cells/well). The next day cells were transfected via CaPO4 precipitation. The medium was changed 8 h posttransfection. To evaluate the effect of miR-21 on its Cdc25a target sequence, 1 μg of pGL3cM-Cdc25a-3′UTR or pGL3cM-CCNE2-3′UTR was cotransfected with 10 ng of pCMV-SPORT-β-gal (kindly provided by J. A. Melendez at the College of Nanoscale Sciences and Engineering, Albany, NY) and 1 μg of pCDH-miR-21-GFP or vector control pCDH-GFP. To assess the effects of viral proteins on miR-21 promoter activity, 1.5 μg of pCDH-IE1-GFP, pCDH-IE2-GFP, pCDH-pp65-GFP, pCDH-pp71-GFP, pCDH-flag-UL26-GFP, or the vector control pCDH-GFP was cotransfected with 10 ng of pCMV-SPORT-β-gal and 500 ng of pGL3-miPPR-21. The cells were harvested with reporter lysis buffer at 48 h posttransfection, and the luciferase activities were determined using a luciferase assay kit (Promega, catalog no. E4036) as described previously (68). The β-galactosidase activities were measured using a β-galactosidase enzyme assay system (Promega). To control for variations in transfection efficiencies, the luciferase activities were normalized to the β-galactosidase activities. The relative luciferase activities were then determined by normalization to results from vector controls. All reactions were conducted in triplicate. The results reported are means ± the SD from three independent experiments.
Immunofluorescence assay.
Synchronized lentivirus-transduced U-251MG cells were seeded onto uncoated dishes containing poly-d-lysine-coated coverslips, allowed to attach for 1 h, and then infected with HCMV at an MOI of 0.5 or 5. At the indicated time points, the coverslips were harvested and fixed with 3% formaldehyde, and the HCMV protein expression was detected using specific antibodies as described previously (69). More than 300 cells in at least six random fields were counted per group. Images were obtained with a Nikon Eclipse 80i fluorescence microscope equipped with a Nikon DS-Ri1 camera and NIS-Elements F3.0 software. Each experiment was repeated three times.
Western blotting.
Cell pellets were lysed in radioimmunoprecipitation assay buffer, and cell lysates were prepared as described elsewhere (28). After sonication, protein concentrations were determined using a BCA protein assay kit (Beyotime). Equal protein quantities were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore). Membranes were probed with antibodies as previously described (61). Primary antibodies included mouse monoclonal antibodies against HCMV-IE1 (clone p63-27; IgG2a) and HCMV-gB (clone 27-156; IgG2b), against HCMV-IE1/2 (clone Ch16), HCMV-UL44, and HCMV-pp65 (IgG1; Virusys), against HCMV-Cdc25a and HCMV-β-actin (IgG; Santa Cruz Biotechnology), against HCMV-flag (IgG; Sigma-Aldrich Co., LLC), and against goat polyclonal antibody to pp71 (Santa Cruz Biotechnology). Secondary antibodies included horseradish peroxidase-conjugated sheep anti-mouse IgG (Amersham Bioscience), donkey anti-rabbit IgG (Amersham Bioscience), or donkey anti-goat IgG (Proteintech Group). Signals were quantitated by densitometry using the ImageJ software package.
Statistical analyses.
Each experiment was performed in triplicate. The data were averaged from three independent experiments and analyzed by one-way analysis of variance. The results were represented as means ± the SD. Differences were considered to be significant when P values were <0.05.
RESULTS
miR-21 and Cdc25a are inversely affected by HCMV infection.
Previous work demonstrated that HCMV can replicate in NPCs, NPC-derived glia and neurons, as well as certain neural cell lines, including the malignant glioblastoma cell line U-251MG and the neuroblastoma cell line SH-SY5Y (6, 15, 70). To confirm that U-251MG cells can be productively infected, synchronized U-251MG cultures were infected with HCMV and stained for IE1 protein expression 24 hpi. All cells were IE1-positive after infection at an MOI of 5, confirming efficient viral entry and IE gene expression in these cells. Expression of viral proteins and production of infectious progeny were further determined by Western blotting and plaque assay. Viral proteins representing IE (IE1/IE2), early (UL44), and late (gB) phases of replication were detected in infected cells, and levels of infectious viral progeny in the culture supernatants increased with time, indicating that U-251MG cells are fully permissive to HCMV replication (data not shown).
Previous studies indicated that expression of miR-21 is downregulated by HCMV in NPCs (50). Cdc25a, a confirmed target of miR-21, is required for promoting cells from G1 to S phase, which is believed to benefit HCMV replication (27, 30, 56). To determine the levels of miR-21 and Cdc25a during HCMV infection of neural cells, NPCs and U-251MG cells were infected with HCMV, and then the miR-21 and Cdc25a mRNA levels were quantitated by qRT-PCR at different times postinfection. In both cell types, miR-21 levels decreased as early as 2 hpi and declined to ca. 60% of uninfected controls by 96 hpi (Fig. 1A and B). In contrast, Cdc25a mRNA levels increased upon HCMV infection. In the first 12 hpi modest increases of 1.2- to 1.4-fold occurred in both cell types. Cdc25a mRNA levels in NPCs increased abruptly (4-fold) at 24 hpi and then declined gradually but remained >3-fold elevated (Fig. 1A). In U-251MG cells Cdc25a mRNA levels increased gradually throughout infection to reach a maximal induction of 8.4-fold at 96 hpi (Fig. 1B). Cdc25a protein levels also increased after HCMV infection of both cell types (Fig. 1C and D). These results demonstrate that expression of miR-21 and Cdc25a are differentially modulated by HCMV infection, which led us to speculate whether miR-21 and Cdc25a may play a different role in influencing HCMV replication.
FIG 1.
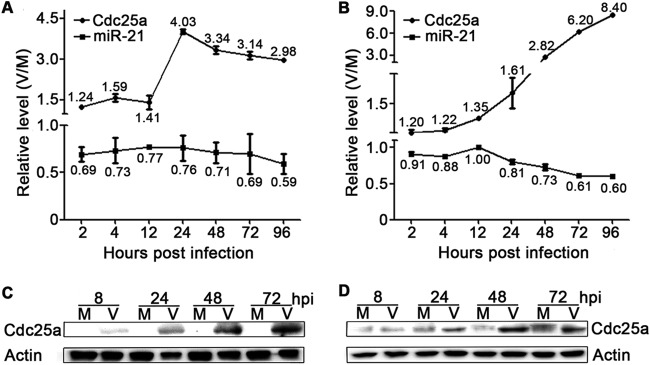
Expression of miR-21 and Cdc25a in NPCs and U-251MG cells during HCMV infection. NPC (A) or U-251MG (B) cultures were infected with HCMV at MOIs of 3 or 5, respectively. Mock-infected (M) or virus-infected (V) cells were harvested at the indicated times, and the miR-21 and Cdc25a mRNA levels were quantified using qRT-PCR. The data were normalized to levels measured in mock-infected cultures to provide relative levels (V/M). The results shown are means ± 1 SD of data from three independent experiments, each conducted in triplicate. Replicate NPC (C) or U-251MG (D) cultures were evaluated by Western blotting for Cdc25a protein expression. Actin serves as a loading control.
Virus replication is suppressed by overexpression of miR-21 in neural cells.
To evaluate the effects of miR-21 on HCMV infection, U-251MG cells and NPCs were transduced with lentivirus expressing miR-21 (pCDH-miR-21-GFP), a control miRNA of miR-UL22A (pCDH-miR-UL22A-GFP) or an empty vector (pCDH-GFP). HCMV miR-UL22A, which has no effect on HCMV replication (62), was used as a control miRNA. U-251MG cells and NPCs were transduced at an MOI of 10 and 1, respectively, since the cell morphology of NPCs were affected during transduction with MOI of 10. Lentivirus transduction efficiencies were monitored by determining the percentage of GFP-positive cells. At 72 hpt, 100% of the resulting U-251MG cells (miR-21-U-251MG, Fig. 2A, left) and ca. 70% of NPCs were GFP positive (data not shown). miR-21 expression levels were quantitated by qRT-PCR and normalized to control cells. A 4-fold level of miR-21 overexpression was achieved in U-251MG cells at 48 hpt (Fig. 2A, right) and 1.5-fold in NPCs (data not shown).
FIG 2.

miR-21 attenuates HCMV replication in U-251MG cells. (A) U-251MG cells were transduced at an MOI of 10 with lentiviruses expressing miR-21 (21) or a control miRNA, miR-UL22A (Ctrl).Cellular morphology was monitored by phase-contrast microscopy, and the transduction efficiency was determined by the detection of lentivirus-encoded GFP at 48 hpt (left). The miR-21 levels at 48 hpt were quantified by qRT-PCR and expressed as fold differences relative to Ctrl (right). (B) Transduced U-251MG cells were infected with HCMV at an MOI of 1, and viral and cellular proteins were detected by Western blotting at the indicated times postinfection. Protein levels relative to Ctrl were determined by densitometry and are indicated below each blot. Actin serves as a loading control. (C) HCMV DNA copy number per cell was determined by qRT-PCR and standardized to cellular DNA (GAPDH) copy number. (D) Titers of infectious virus in the culture supernatants at 72 and 96 hpi were determined by plaque assay. The fold differences are indicated. miR-21, HCMV genome copy number, and virus titer results are means ± 1 SD of data from three independent experiments, each conducted in triplicate. *, P < 0.05; **, P < 0.01.
The resulting miR-21 expressing U-251MG cells or NPCs were then infected with HCMV at an MOI of 1, and samples from replicate cultures were harvested at 8, 24, 48, and 72 hpi. The levels of viral proteins (IE1, UL44, and gB) and the cellular Cdc25a protein levels were determined by Western blotting, intracellular HCMV genome copy numbers were determined by qPCR, and the release of infectious progeny was evaluated by plaque assay. Significant decreases were observed in HCMV protein expression, DNA synthesis, and infectious virus yield. Compared to control-transduced U-251MG cells, miR-21-U-251MG cells contained smaller amounts of IE1 at 8, 24, and 48 hpi, less UL44 at 24, 48, and 72 hpi, and less gB at 48 and 72 hpi. Accordingly, Cdc25a protein levels increased progressively during infection of both miR-21-U-251MG cells and control cells, Cdc25a levels in miR-21-U-251MG cells were clearly lower than in controls at 24 and 48 hpi (Fig. 2B). Accompanying the changes of viral proteins and Cdc25a protein, HCMV genome copy numbers were significantly reduced in infected miR-21-U-251MG cells at 48, 72, and 96 hpi (Fig. 2C), and the supernatants contained 7.5- and 4.0-fold less infectious virus than those of control cells at 72 and 96 hpi, respectively (Fig. 2D). Similar attenuation of HCMV replication was observed in miR-21-overexpressing NPCs (data not shown). Together, these results are consistent with miR-21 attenuation of HCMV replication in both NPCs and U-251 MG cells.
Knockdown of miR-21 enhances Cdc25a and HCMV gene expression.
To further assess the effects of miR-21 on HCMV replication, miR-21 expression was knocked down in U-251MG cells by lentiviral transduction. Three lentiviruses expressing different shRNAs (sh21-1, sh21-2, and sh21-3) targeting miR-21 or a control lentivirus expressing a scrambled shRNA sequence (Scram) were used to transduce HEK293T or U-251MG cells. The results from U-251MG cells are shown in Fig. 3; similar results were obtained in HEK293T (not shown). The lentivirus expressing sh21-1 reduced miR-21 expression most efficiently (Fig. 3A) and was used for subsequent studies. Compared to Scram control transduced cells, transduction of U-251MG cells with the sh21-1-expressing lentivirus reduced miR-21 levels by 60% and increased Cdc25a mRNA levels by 33%. Cdc25a protein increased 3-fold (Fig. 3B). Transduced U-251MG cells were infected with HCMV at an MOI of 0.5. miR-21 knockdown resulted in 1.37- to 4.2-fold increases in IE1 and UL44 protein levels at 24 and 48 hpi and significant increases (2.0- to 6.6-fold) in Cdc25a at both 24 and 48 hpi (Fig. 3C).
FIG 3.
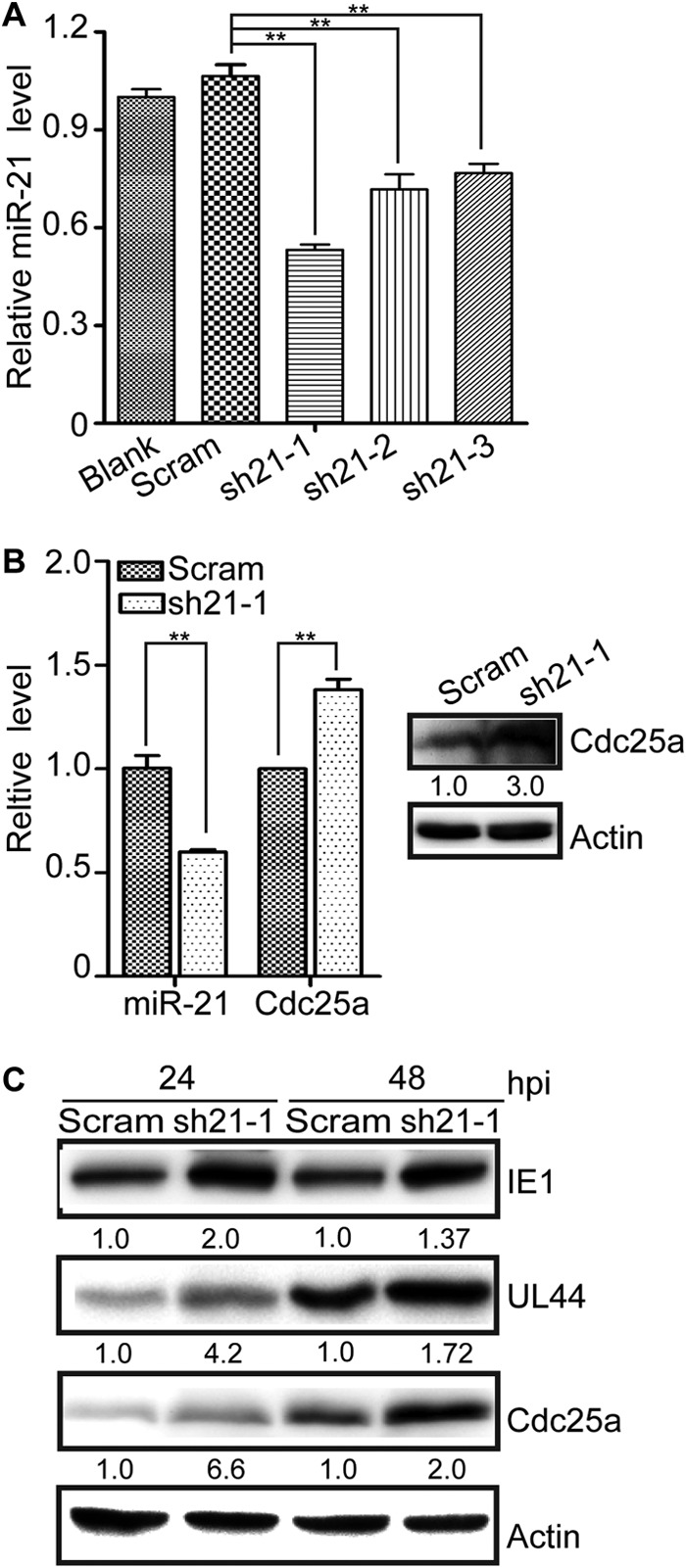
miR-21 knockdown enhances the expression of Cdc25a and viral proteins. (A) U-251MG cells were untreated (Blank) or transduced with lentiviruses expressing three different shRNAs targeting miR-21 (sh21-1, -2, and -3) or expressing a scrambled sequence control shRNA (Scram). miR-21 levels were quantified by qRT-PCR and expressed as fold differences relative to Scram. (B) miR-21 and Cdc25a mRNA levels were quantified by qRT-PCR in U-251MG cells transduced with the sh21-1 lentivirus and expressed as fold differences relative to Scram lentivirus-transduced cells (left). Cdc25a protein levels in replicate cultures were determined by Western blotting (right). Relative protein levels were determined by densitometry and are indicated below each blot. Actin serves as a loading control. (C) Transduced U-251MG cells were infected with HCMV at an MOI of 0.5, and viral and cellular proteins were detected by Western blotting at 24 and 48 hpi. The protein levels relative to Scram-transduced cells were determined by densitometry and are indicated below each blot. Actin serves as a loading control. qRT-PCR results are means ± 1 SD of data from three independent experiments, each conducted in triplicate. **, P < 0.01.
Overexpression of Cdc25a enhances HCMV replication and counteracts inhibition by miR-21.
Cdc25a is a host factor involved in cell cycle regulation and promotes G1 to S transition (57, 58). However, in HCMV-infected fibroblasts the upregulation of Cdc25a does not result in cell cycle progression, but rather, the cells are arrested at G1/S (59, 60). To confirm that similar arrest occurs in U-251MG cells, the impact of HCMV infection on cell cycle in U-251MG cells was studied. At 24 h after release from serum starvation only 28% ± 0.4% of mock-infected cells remained in G0/G1 and 52.6% ± 12% had progressed to S phase, whereas 62% ± 0.9% of infected cells remained in G0/G1 and only 26.2% ± 1.3% had progressed to S phase (data not shown). Thus, as in fibroblasts, HCMV infection of U-251MG cells also results in G1/S arrest. That HCMV upregulates Cdc25a suggests that, despite the context of cell cycle arrest, increased Cdc25a levels may somehow benefit HCMV replication. Since HCMV infection arrests cell cycle at G1/S and upregulates Cdc25a (71), the increase in Cdc25a induced by HCMV infection may be beneficial for HCMV replication. Moreover, the observation that HCMV downregulates miR-21 and Cdc25a is a target of miR-21 (72) further suggests that upregulation of Cdc25a may be, at least in part, a consequence of reduced miR-21 levels. Conversely, inhibition of HCMV replication by overexpression of miR-21 may, in part, result from impaired induction of Cdc25a.
To address these questions, miR-21 inhibition of Cdc25a expression was first confirmed using transient-transfection assays. Figure 4A shows miR-21 with its seed sequence aligned with the target sequence from the 3′UTR of Cdc25a. HEK293T cells were transfected with one of two reporter plasmids: one encoding luciferase with a 3′UTR from Cdc25a that contains the miR-21 target sequence (pGL3cM-Cdc25a-3′UTR), and the other encoding luciferase with a control 3′UTR lacking miR-21 target sequences (pGL3cM-CCNE2-3′UTR). The reporter plasmids pGL3cM-Cdc25a-3′UTR or pGL3cM-CCNE2-3′UTR were cotransfected with one of two effector plasmids: (i) pCDH-miR-21-GFP, a miR-21-expressing plasmid, or (ii) pCDH-miR-UL22A, a plasmid expressing control microRNA miR-UL22A. The expression of miR-21 specifically and significantly reduced luciferase expression from pGL3cM-Cdc25a-3′UTR (which contains the Cdc25a 3′UTR) but not from the control plasmid pGL3cM-CCNE2-3′UTR (which contains the control 3′UTR) (Fig. 4B). In a similar experiment, Cdc25a expression was measured by Western blotting after transfection of HEK293T cells with a reporter plasmid encoding Cdc25a with its native 3′UTR (pCDH-Cdc25a+WT-3′UTR-GFP). When cells were cotransfected with either the miR-21-expressing plasmid (pCDH-miR-21-GFP) or a plasmid expressing miR-UL22A as a control miRNA (pCDH-miR-UL22A), miR-21 expression resulted in significantly less expression of Cdc25a protein (Fig. 5C, left). To further confirm miR-21 regulates Cdc25a expression through its 3′UTR, the miR-21 target seed sequence in WT-3′UTR of plasmid pCDH-Cdc25a+WT-3′UTR-GFP was mutated as shown in Fig. 4A to make plasmid pCDH-Cdc25a+MT-3′UTR-GFP. HEK293T cells were cotransfected with pCDH-miR-21-GFP, along with either pCDH-Cdc25a+WT-3′UTR-GFP or pCDH-Cdc25a+MT-3′UTR-GFP. Cdc25a protein expression was significantly increased by the mutation in the 3′UTR (Fig. 4C, right). These experiments confirm that Cdc25a is downregulated by miR-21 and show that the 3′UTR from Cdc25a is necessary in cis for miR-21 inhibition.
FIG 4.
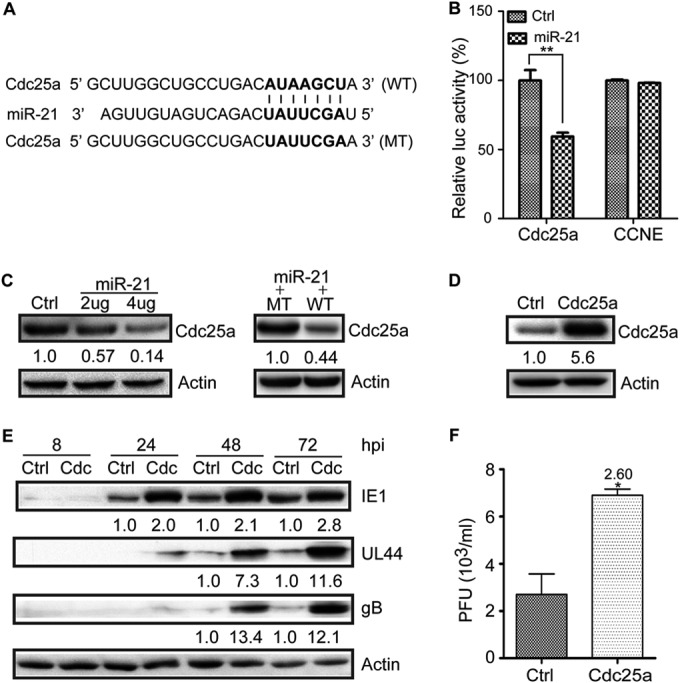
Overexpression of Cdc25a enhances HCMV replication in U-251MG cells. (A) Potential base pairing is indicated by vertical lines between the seed sequence of miR-21 (bold) and its target sequences within the wild-type (WT) 3′UTR of Cdc25a. Mutation (MT) of the miR-21 target sequence is predicted to eliminate miR-21-mediated repression. (B) HEK293T cells were cotransfected with plasmids expressing miR-21 or control miRNA miR-UL22A, along with reporter plasmids encoding luciferase, followed either by the Cdc25a 3′UTR (Cdc25a) or a control 3′UTR (CCNE2). The luciferase activities were measured at 48 h posttransfection and are expressed as percent differences relative to cells cotransfected with the CCNE2 and miR-UL22A-expressing control plasmids. (C) For the left panel, HEK293T cells were cotransfected with a plasmid encoding Cdc25a, followed by its native 3′UTR with either a miR-UL22A-expressing control plasmid (4 μg) or a miR-21-expressing plasmid (2 or 4 μg). For the right panel, HEK293T cells were cotransfected with miR-21-expressing plasmid (4 μg) with either pCDH-Cdc25a+WT-3′UTR-GFP (2 μg) or pCDH-Cdc25a+MT-3′UTR-GFP (2 μg). Cdc25a protein levels were determined by Western blotting. Protein levels relative to miR-UL22A-transfected control cells were determined by densitometry and are indicated below each blot. Actin serves as a loading control. (D) U-251MG cells were transduced with lentiviruses expressing Cdc25a without its native 3′UTR (Cdc25a) or empty vector control (Ctrl, pCDH-GFP) and Cdc25a was detected by Western blotting. Protein levels relative to Ctrl-transduced cells were determined by densitometry and are indicated below each blot. Actin serves as a loading control. (E) Lentivirus-transduced U-251MG cells (Ctrl and Cdc25a) were infected with HCMV at an MOI of 1, and viral and cellular proteins were detected by Western blotting at the indicated times postinfection. Protein levels relative to Ctrl were determined by densitometry and are indicated below each blot. Actin serves as a loading control. (F) Titers of infectious virus in the culture supernatants at 72 hpi were determined by plaque assay. The fold differences are indicated. Luciferase and virus titer results are means ± 1 SD of data from three independent experiments, each conducted in triplicate. *, P < 0.05; **, P < 0.01.
FIG 5.
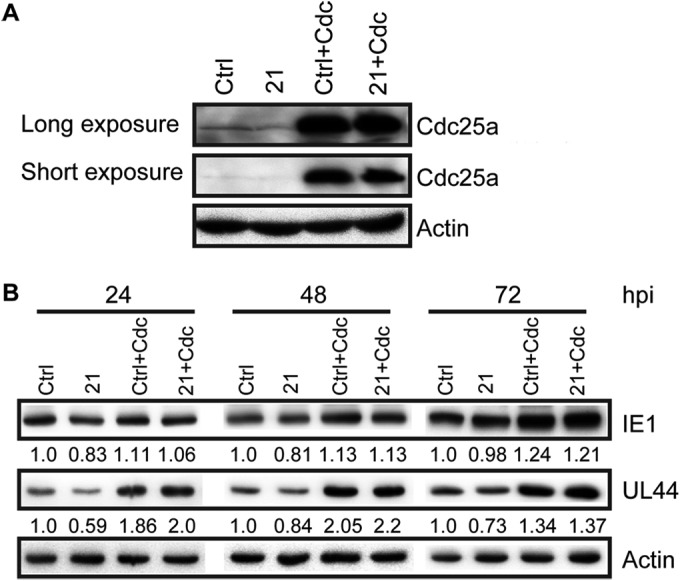
Cdc25a overexpression counteracts the negative effects of miR-21 on HCMV gene expression. (A) U-251MG cells were transduced with lentiviruses that overexpress miR-21 (21) or miR-UL22A (Ctrl). Portions of these cells were further transduced with a lentivirus encoding Cdc25a without its 3′UTR (Ctrl+Cdc and 21+Cdc). Cdc25a protein levels were determined by Western blotting; short and long exposures of the same blot are shown. (B) Transduced cells were infected with HCMV at an MOI of 0.5, and viral and cellular proteins were detected by Western blotting at the indicated times postinfection. Actin serves as a loading control.
To evaluate the impact of Cdc25a on HCMV replication, U-251MG cells were transduced with an empty vector control lentivirus (pCDH-GFP) or a lentivirus expressing Cdc25a without the 3′UTR. Cells transduced with the latter exhibited 5.6-fold-higher levels of Cdc25a protein compared to control-transduced cells (Fig. 4D). Control- and Cdc25a-U-251MG cells were next infected with HCMV at an MOI of 1 and viral protein levels and infectious virus yield were determined at different times after infection. IE1, UL44, and gB levels were 2- to 13.4-fold higher in HCMV-infected Cdc25a-U-251MG cells than in control cells (Fig. 4E). At 72 hpi the culture supernatant of HCMV-infected Cdc25a-U-251MG cells contained 2.6-fold-larger amounts of infectious virus than that of control cells (Fig. 4F). These results confirm that increased amounts of Cdc25a promote HCMV replication.
If the hypothesis that miR-21 inhibits HCMV replication by targeting Cdc25a is correct, then cells expressing Cdc25a from an ectopic (lentiviral transduced) gene cassette lacking the 3′UTR miR-21 target sequence (pCDH-Cdc25a-GFP) should be insensitive to miR-21-mediated downregulation of Cdc25a, and such cells should not exhibit significant inhibitory effects on HCMV replication associated with the overexpression of miR-21. To test this prediction, control cells or U-251MG cells overexpressing miR-21 were further transduced with pCDH-Cdc25a-GFP, resulting in Ctrl+Cdc and 21+Cdc cells. Western blotting results confirmed upregulation of Cdc25a that was insensitive to miR-21 (Fig. 5A). When cells were infected with HCMV, Cdc25a overexpression resulted in increased levels of IE1 and UL44 at 24, 48, and 72 hpi. However, while overexpression of miR-21 inhibited IE1 and UL44 expression at 24 hpi, these effects were overcome in the context of Cdc25a overexpression; Cdc25a-transduced cells exhibited higher levels of IE1 and UL44 at all time points, and concomitant overexpression of miR-21 had no obvious inhibitory effects (Fig. 5B). Taken together, these results support the hypothesis that miR-21 inhibits HCMV replication by targeting Cdc25a; this, in turn, implies that downregulation of miR-21 at early times of infection serves to promote HCMV replication by increasing Cdc25a levels.
Overexpression of miR-21 impairs HCMV entry.
High levels of miR-21 could impair IE protein expression either directly by influencing the efficiency of IE transcription/translation, or indirectly by reducing the number of cells successfully infected by HCMV. HCMV entry can be measured indirectly by the number of IE-positive cells at early times after infection (61). To determine whether miR-21 overexpression impacts virus entry, synchronous miR-21 expressing U-251MG cells (miR-21-U-251MG cells, 100% GFP positive) or miR-UL22A-expressing control cells (100% GFP positive) were infected with HCMV at an MOI of 0.5 or 5 and stained for IE1 expression at 12, 24, and 48 hpi (Fig. 6A). Total and IE1-positive cells were counted, and the percentages of positive cells were calculated. At both MOIs, overexpression of miR-21 resulted in significantly fewer IE1-positive cells at 24 and 48 hpi (P < 0.05) (Fig. 6B).
FIG 6.

miR-21 impairs viral entry. (A) U-251MG cells were transduced with lentiviruses that overexpress miR-21 or miR-UL22A (Ctrl) and infected with HCMV at MOIs of 0.5 or 5. Cells were stained for IE1 and counterstained with Hoechst (to show nuclei) at the times postinfection indicated. Scale bar, 30 μm. (B) Total cells and IE1-positive cells were counted in over six random fields from each experiment. (C) Transduced U-251MG cells were infected with HCMV at an MOI of 5 or 10, washed with PBS, harvested at 1 hpi, and assayed by Western blotting for cell-associated pp65. Protein levels relative to Ctrl were determined by densitometry and are indicated below each blot. Actin serves as a loading control. Cell count results are means ± 1 SD of data from three independent experiments, each conducted in triplicate. *, P < 0.05.
To confirm that these results are a consequence of entry and not postentry inhibition of IE1 expression, viral entry was further assessed by measuring levels of cell-associated pp65. This protein is an abundant component of virions and can be detected in cells immediately upon viral entry even in the absence of viral gene expression. Synchronous miR-21-U-251MG cell cultures or miR-UL22A-expressing control cells were infected for 1 h with HCMV at MOIs of 5 and 10 and then washed extensively three times with phosphate-buffered saline (PBS) to remove unattached viral particles. The levels of cell-associated pp65 at 1 hpi were determined by Western blotting. As shown in Fig. 6C, at either MOI the amount of pp65 deposited into cells by entering viral particles was ca. 40% lower in miR-21-U-251MG cells compared to controls. These results indicate that overexpression of miR-21 reduces the efficiency of HCMV entry.
HCMV IE1, pp71, and UL26 proteins downregulate miR-21.
The above studies indicate that miR-21 negatively impacts HCMV replication and that from very early times of infection HCMV actively downregulates miR-21 levels. Possible viral effectors include the IE gene products such as IE1 and IE2, which are abundantly expressed and localize to the nucleus within 2 h of infection, or virion components such as pp65, pp71, and UL26, which are carried into cells by infecting virions. To determine whether IE1, IE2, pp65, pp71, or UL26 can impact miR-21 levels, U-251MG cells were transduced with lentiviruses expressing each protein. Since no UL26 antibody was available, the UL26 ORF was modified to include a flag tag epitope. At 48 hpt, >95% of cells in transduced cultures were GFP positive (not shown). The expression of each transgene was confirmed by Western blotting (Fig. 7A). Mature miR-21 levels in transduced cell populations were quantitated by qRT-PCR. IE2 and pp65 had no effect on miR-21 levels, whereas IE1, pp71, and UL26 suppressed miR-21 levels by 23, 20, and 46%, respectively (Fig. 7B).
FIG 7.
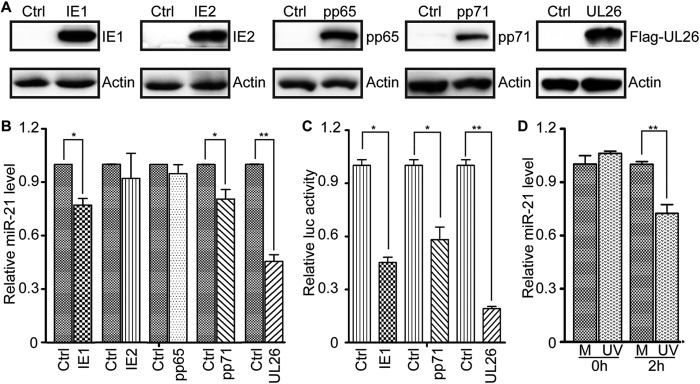
miR-21 is downregulated by HCMV proteins IE1, pp71, and UL26. (A) U-251MG cells were transduced with an empty vector control lentivirus (Ctrl) or lentiviruses expressing IE1, IE2, pp65, pp71, or UL26. The levels of each protein were determined by Western blotting. Actin serves as a loading control. (B) miR-21 levels in transduced cells were quantified by qRT-PCR and expressed as fold differences relative to Ctrl-transduced cells. (C) HEK293T cells were cotransfected with a reporter plasmid encoding luciferase under the control of the miR-21 promoter with plasmids expressing IE1, IE2, pp71, or UL26. The luciferase activities were measured at 48 h posttransfection and are expressed as fold differences relative to IE2-transfected cells. (D) U-251MG cells were mock treated (M) or exposed to UV-inactivated HCMV equivalent to an MOI of 10 (UV). miR-21 levels were determined by qRT-PCR immediately (0 h) or after 2 h of exposure and expressed as fold differences relative to mock-treated cells. Luciferase and qRT-PCR results are means ± 1 SD of data from three independent experiments, each conducted in triplicate. *, P < 0.05; **, P < 0.01.
To determine whether these effects were mediated at the transcriptional level, HEK293T cells were cotransfected with a reporter plasmid encoding luciferase under the control of the miR-21 promoter paired with effector plasmids expressing IE1, IE2, pp71, or UL26. Luciferase activities were determined at 48 h posttransfection when cultures were >90% GFP positive. Cotransfection with plasmids encoding IE1, pp71, or UL26 reduced luciferase activities by 55, 42, and 80%, respectively, relative to cotransfection with the IE2-expressing plasmid (Fig. 7C). These results suggest that downregulation of miR-21 at the early stages of HCMV infection may be attributable to viral IE1, pp71, or UL26 proteins.
To further determine whether a virion component induced downregulation of miR-21, synchronized U251-MG cells were exposed to UV-inactivated HCMV equivalent to an MOI of 10, and mature miR-21 levels were determined by qRT-PCR immediately and 2 h later. As shown in Fig. 7D, the exposure of cells to UV-inactivated HCMV for 2 h reduced miR-21 to 70% of that in mock-treated cells, indicating that virion components participate in miR-21 downregulation, at least at very early times after infection.
DISCUSSION
HCMV is a ubiquitous pathogen that establishes a lifelong infection in the host after primary infection. Both primary and recurrent infection can lead to congenital infection, and both result in significant birth defects in newborns. Outcomes are usually more severe when congenital infection is acquired from primary maternal infection during pregnancy. This multifaceted infection uses multiple regulation pathways to modulate viral replication.
Many viruses use viral or cellular miRs to modify cell cycle control and other pathways important for viral replication (41, 73–75). For example, miR-BART2, encoded by EBV, inhibits EBV lytic replication by targeting the viral DNA polymerase, BALF5 (76); the KSHV-encoded miRs miR-K12-7-5p, miR-K1, and miR-K3 play roles in the regulation of viral replication by targeting RTA, the viral transcriptional activator (77–79); and HCMV-encoded miR-UL112-1 suppresses the expression of HCMV IE proteins, especially IE1, which in turn inhibits HCMV replication (80). HCMV-encoded miR-US25-1 targets cell cycle regulators, including cyclin E2 (39), and inhibits the replication of HCMV (81). HCMV miRs UL112-1, US5-1, and US5-2 target host secretory pathways to favor virion assembly (82). Cellular miRs such as miR-100 and miR-101 regulate virus infections (48, 75, 83) via interaction with mTOR/raptor, a component of the mTOR pathway (41). Our present study provides the first demonstration that the cellular miRNA miR-21 attenuates HCMV infection by targeting Cdc25a, a known cell cycle regulator.
Previous studies using whole-genome expression or miRNA microarray analysis found that miR-21 is downregulated by HCMV infection in both NPCs and fibroblasts (41, 50). In the present study quantitative methods were used to determine expression levels of miR-21 in both neural origin cells (NPCs) and U-251MG cells before and after HCMV infection. The results confirmed the previous reports and further showed that in both cell types miR-21 is downregulated immediately upon infection (2 hpi) and remains suppressed throughout infection (Fig. 1). It is possible that miR-21 downregulation is an indirect consequence of HCMV perturbation of cell cycle, but the rapidity of downregulation (2 hpi) suggests this is unlikely. In NPCs miR-21 downregulation is rapid, whereas in U-251MG cells miR-21 declines more gradually. The latter results may reflect the delayed/protracted replication of HCMV in these cells. From these results we inferred that miR-21 may have an inhibitory effect on HCMV replication. This was confirmed by the overexpression of miR-21, which decreased viral gene expression, viral genome replication, and the production of infectious progeny in both NPCs and U-251MG cells (Fig. 2). Conversely, knockdown of miR-21 increased the levels of viral proteins IE1 and UL44 (Fig. 4), further supporting the hypothesis that miR-21 inhibits viral replication.
In contrast, Cdc25a expression levels increased during HCMV infection (Fig. 1, 2, and 3) and upon knockdown of miR-21 expression (Fig. 3). Both overexpression of ectopic Cdc25a and increased endogenous Cdc25a expression by miR-21 knockdown augmented HCMV replication, further suggesting that, upon infection, HCMV increases Cdc25a levels to promote more efficient viral replication (Fig. 4). Given that Cdc25a and miR-21 are inversely regulated during HCMV infection and that Cdc25a is a known miR-21 target (56), it seems that miR-21's inhibitory effects on HCMV replication are mediated at least in part by its ability to downregulate expression of Cdc25a. Consistent with this, the negative effects of miR-21 expression on viral gene expression were overcome by overexpression of Cdc25a (Fig. 5). Thus, HCMV inhibits miR-21 as a means to enhance Cdc25a levels, which may benefit viral replication by promoting cells to enter G1/S transition. However, miR-21 also targets other cell cycle regulators such as PDCD4 and PTEN (53–56), and their roles in HCMV replication have not been evaluated.
Manipulation of cell cycle benefits HCMV replication at later stages (e.g., enhancing viral DNA synthesis in the nucleus). As shown in Fig. 2C, significant differences in viral DNA replication were observed at times coincident with increases in CDC25a levels. However, overexpression of miR-21 prior to HCMV infection had a significant effect on IE1 protein expression as early as 8 hpi (Fig. 2B), and miR-21 was downregulated as early as 2 h after HCMV infection. This prompted evaluation of the effects of miR-21 overexpression on HCMV entry. We observed that overexpression of miR-21 resulted in fewer IE1-positive cells and reduced amounts of cell-associated pp65 shortly (1 hpi) after HCMV infection (Fig. 6). These results indicate that miR-21 also affects viral entry. Whether miR-21's impact on HCMV entry is also associated with Cdc25a perturbation of cell cycle regulation, or other pathways, remains to be determined.
The effects of miR-21 overexpression on HCMV gene expression and genome replication manifest at early times of infection and are largely gone by 96 hpi (Fig. 2). This suggests that the repressive effects of endogenous and even ectopic miR-21 can in time be overcome by a counteracting viral mechanism. The rapidity of miR-21 downregulation in infected NPCs implies that this may be mediated by virion-associated factors deposited into the cells upon entry or by viral IE proteins that are rapidly expressed after infection. Using transient transfections, we found that the viral IE1 protein and two tegument proteins, pp71 and UL26, have the capacity to downregulate miR-21 expression. Furthermore, the exposure of cells to UV-inactivated HCMV confirmed that virion-associated factors can rapidly reduce miR-21 levels (Fig. 7). However, that this downregulation is not as complete as that induced by live virus infection at later times suggests that de novo-synthesized factors expressed during viral replication also play a role in miR-21 downregulation. Such mechanisms require further elucidation.
Proposed interactions between HCMV, miR-21, and Cdc25a are summarized in Fig. 8. HCMV infection results in the deposition of virion-associated pp71 and UL26, as well as de novo synthesis of IE1. These and perhaps other viral factors contribute to downregulation of miR-21 at very early times; decreased miR-21 levels result in increased expression of Cdc25a, which in turn modulates the cell cycle to enhance HCMV replication (Fig. 8A). In cells overexpressing miR-21, Cdc25a levels are depressed and viral entry is inefficient; at later times of infection, decreased Cdc25a levels may also contribute to a cellular environment that is unfavorable for HCMV replication (Fig. 8B). How Cdc25a promotes HCMV replication and whether miR-21 targets viral genes to directly regulate HCMV infection remain to be further investigated.
FIG 8.
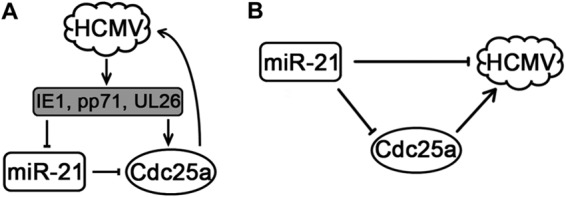
Regulatory pathways involving HCMV, miR-21, and Cdc25a. (A) HCMV proteins downregulate miR-21, relieving miR-21-mediated repression of Cdc25a, which promotes replication of HCMV. (B) Overexpression of miR-21 reduces Cdc25a levels, which impairs replication of HCMV.
HCMV infections during fetal development often result in devastating neural damage and neural developmental disorders (16–18). Although the pathogenesis of HCMV in this setting is not well understood, damage is presumed to occur from active viral replication in neuronal cells, including NPCs. The data presented here expand our understanding of HCMV replication in cell types relevant to neuropathogenesis and could potentially lead to novel therapeutic interventions targeting viral replication.
In addition to congenital disease in newborns, accumulating evidence suggests that HCMV DNA and a subset of viral proteins are present in malignant glioblastoma in adults and young children (84, 85). Whether viral replication occurs in these tumors has not been firmly established, although recent clinical evidence suggests that antiviral therapy targeting viral replication helps to prolong glioblastoma patient survival (86). The studies presented here, conducted in the glioma cell line U-251MG, may have implications toward further understanding HCMV's role in malignant glioblastoma. For example, Cdc25a, an oncoprotein, promotes G1-to-S progression (57, 58) and is overexpressed in many cancers (56–58, 87), including colon cancer (56) and glioma (87). HCMV proteins such as IE1, which is frequently detected in glioblastoma tissues, can inhibit miR-21 expression and thereby presumably increase Cdc25a levels. These results suggest one potential mechanism by which HCMV might contribute to gliomagenesis or pathogenesis.
Taken together, our findings indicate that HCMV downregulates miR-21 to amplify Cdc25a and hence modifies the intracellular environment to benefit its replication. Virion-associated factors and perhaps the viral proteins IE1, pp71, and UL26 function to downregulate miR-21 at early times of infection. Thus, miR-21 appears to be an intrinsic antiviral molecule that also targets Cdc25a, an oncogene, and may represent a new therapeutic target for HCMV-associated tumors.
ACKNOWLEDGMENTS
This study was supported by the National Program on Key Basic Research Project (973 program 2011CB504804 and 2012CB519003), the National Natural Science Foundation of China (81071350, 81271850, and 31170155), the Scientific Innovation Project of Chinese Academy of Sciences (XDB02050500, XDA0104107), and a seed grant from the University of Idaho (YDP-764) to M.-H.L.
REFERENCES
- 1.Boppana SB, Ross SA, Shimamura M, Palmer AL, Ahmed A, Michaels MG, Sanchez PJ, Bernstein DI, Tolan RW Jr, Novak Z, Chowdhury N, Britt WJ, Fowler KB. 2011. Saliva polymerase-chain-reaction assay for cytomegalovirus screening in newborns. N Engl J Med 364:2111–2118. doi: 10.1056/NEJMoa1006561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Misono S, Sie KC, Weiss NS, Huang ML, Boeckh M, Norton SJ, Yueh B. 2011. Congenital cytomegalovirus infection in pediatric hearing loss. Arch Otolaryngol Head Neck Surg 137:47–53. doi: 10.1001/archoto.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stagno S, Pass RF, Cloud G, Britt WJ, Henderson RE, Walton PD, Veren DA, Page F, Alford CA. 1986. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA 256:1904–1908. [PubMed] [Google Scholar]
- 4.Mercorelli B, Lembo D, Palu G, Loregian A. 2011. Early inhibitors of human cytomegalovirus: state-of-art and therapeutic perspectives. Pharmacol Ther 131:309–329. doi: 10.1016/j.pharmthera.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui X, Lee R, Adler SP, McVoy MA. 2013. Antibody inhibition of human cytomegalovirus spread in epithelial cell cultures. J Virol Methods 192:44–50. doi: 10.1016/j.jviromet.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo MH, Schwartz PH, Fortunato EA. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J Virol 82:9994–10007. doi: 10.1128/JVI.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryckman BJ, Chase MC, Johnson DC. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc Natl Acad Sci U S A 105:14118–14123. doi: 10.1073/pnas.0804365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sinzger C, Jahn G. 1996. Human cytomegalovirus cell tropism and pathogenesis. Intervirology 39:302–319. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J Virol 79:10330–10338. doi: 10.1128/JVI.79.16.10330-10338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cobbs CS. 2013. Cytomegalovirus and brain tumor: epidemiology, biology and therapeutic aspects. Curr Opin Oncol 25:682–688. doi: 10.1097/CCO.0000000000000005. [DOI] [PubMed] [Google Scholar]
- 11.Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ. 2002. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 62:3347–3350. [PubMed] [Google Scholar]
- 12.Miller G. 2009. Brain cancer A viral link to glioblastoma? Science 323:30–31. doi: 10.1126/science.323.5910.30. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, Sampson JH. 2008. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol 10:10–18. doi: 10.1215/15228517-2007-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duan YL, Ye HQ, Zavala AG, Yang CQ, Miao LF, Fu BS, Seo KS, Davrinche C, Luo MH, Fortunato EA. 2014. Maintenance of large numbers of virus genomes in human cytomegalovirus-infected T98G glioblastoma cells. J Virol 88:3861–3873. doi: 10.1128/JVI.01166-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo MH, Fortunato EA. 2007. Long-term infection and shedding of human cytomegalovirus in T98G glioblastoma cells. J Virol 81:10424–10436. doi: 10.1128/JVI.00866-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boppana SB, Fowler KB, Vaid Y, Hedlund G, Stagno S, Britt WJ, Pass RF. 1997. Neuroradiographic findings in the newborn period and long-term outcome in children with symptomatic congenital cytomegalovirus infection. Pediatrics 99:409–414. doi: 10.1542/peds.99.3.409. [DOI] [PubMed] [Google Scholar]
- 17.Boppana SB, Pass RF, Britt WJ, Stagno S, Alford CA. 1992. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J 11:93–99. doi: 10.1097/00006454-199202000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Dahle AJ, Fowler KB, Wright JD, Boppana SB, Britt WJ, Pass RF. 2000. Longitudinal investigation of hearing disorders in children with congenital cytomegalovirus. J Am Acad Audiol 11:283–290. [PubMed] [Google Scholar]
- 19.Conboy TJ, Pass RF, Stagno S, Britt WJ, Alford CA, McFarland CE, Boll TJ. 1986. Intellectual development in school-aged children with asymptomatic congenital cytomegalovirus infection. Pediatrics 77:801–806. [PubMed] [Google Scholar]
- 20.Grosse SD, Ross DS, Dollard SC. 2008. Congenital cytomegalovirus (CMV) infection as a cause of permanent bilateral hearing loss: a quantitative assessment. J Clin Virol 41:57–62. doi: 10.1016/j.jcv.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Pass RF, Stagno S, Myers GJ, Alford CA. 1980. Outcome of symptomatic congenital cytomegalovirus infection: results of long-term longitudinal follow-up. Pediatrics 66:758–762. [PubMed] [Google Scholar]
- 22.Revello MG, Zavattoni M, Furione M, Fabbri E, Gerna G. 2006. Preconceptional primary human cytomegalovirus infection and risk of congenital infection. J Infect Dis 193:783–787. doi: 10.1086/500509. [DOI] [PubMed] [Google Scholar]
- 23.Schachtele SJ, Mutnal MB, Schleiss MR, Lokensgard JR. 2011. Cytomegalovirus-induced sensorineural hearing loss with persistent cochlear inflammation in neonatal mice. J Neurovirol 17:201–211. doi: 10.1007/s13365-011-0024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spector DH. 1996. Activation and regulation of human cytomegalovirus early genes. Intervirology 39:361–377. [DOI] [PubMed] [Google Scholar]
- 25.Fehr AR, Yu D. 2013. Control the host cell cycle: viral regulation of the anaphase-promoting complex. J Virol 87:8818–8825. doi: 10.1128/JVI.00088-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hertel L, Mocarski ES. 2004. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of pseudomitosis independent of US28 function. J Virol 78:11988–12011. doi: 10.1128/JVI.78.21.11988-12011.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jault FM, Jault JM, Ruchti F, Fortunato EA, Clark C, Corbeil J, Richman DD, Spector DH. 1995. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J Virol 69:6697–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo MH, Rosenke K, Czornak K, Fortunato EA. 2007. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J Virol 81:1934–1950. doi: 10.1128/JVI.01670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J Virol 80:3863–3871. doi: 10.1128/JVI.80.8.3863-3871.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salvant BS, Fortunato EA, Spector DH. 1998. Cell cycle dysregulation by human cytomegalovirus: influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J Virol 72:3729–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bogdanow B, Weisbach H, von Einem J, Straschewski S, Voigt S, Winkler M, Hagemeier C, Wiebusch L. 2013. Human cytomegalovirus tegument protein pp150 acts as a cyclin A2-CDK-dependent sensor of the host cell cycle and differentiation state. Proc Natl Acad Sci U S A 110:17510–17515. doi: 10.1073/pnas.1312235110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oduro JD, Uecker R, Hagemeier C, Wiebusch L. 2012. Inhibition of human cytomegalovirus immediate-early gene expression by cyclin A2-dependent kinase activity. J Virol 86:9369–9383. doi: 10.1128/JVI.07181-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zydek M, Hagemeier C, Wiebusch L. 2010. Cyclin-dependent kinase activity controls the onset of the HCMV lytic cycle. PLoS Pathog 6:e1001096. doi: 10.1371/journal.ppat.1001096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. 2006. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol 80:8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skalsky RL, Cullen BR. 2010. Viruses, microRNAs, and host interactions. Annu Rev Microbiol 64:123–141. doi: 10.1146/annurev.micro.112408.134243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ambros V. 2004. The functions of animal microRNAs. Nature 431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 37.Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 38.Kloosterman WP, Plasterk RH. 2006. The diverse functions of microRNAs in animal development and disease. Dev Cell 11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Grey F, Tirabassi R, Meyers H, Wu G, McWeeney S, Hook L, Nelson JA. 2010. A viral microRNA downregulates multiple cell cycle genes through mRNA 5′UTRs. PLoS Pathog 6:e1000967. doi: 10.1371/journal.ppat.1000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cameron JE, Fewell C, Yin Q, McBride J, Wang X, Lin Z, Flemington EK. 2008. Epstein-Barr virus growth/latency III program alters cellular microRNA expression. Virology 382:257–266. doi: 10.1016/j.virol.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang FZ, Weber F, Croce C, Liu CG, Liao X, Pellett PE. 2008. Human cytomegalovirus infection alters the expression of cellular microRNA species that affect its replication. J Virol 82:9065–9074. doi: 10.1128/JVI.00961-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buggele WA, Krause KE, Horvath CM. 2013. Small RNA profiling of influenza A virus-infected cells identifies miR-449b as a regulator of histone deacetylase 1 and interferon beta. PLoS One 8:e76560. doi: 10.1371/journal.pone.0076560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan Q, Li W, Tang Q, Yao S, Lv Z, Feng N, Ma X, Bai Z, Zeng Y, Qin D, Lu C. 2013. Cellular microRNAs 498 and 320d regulate herpes simplex virus 1 induction of Kaposi's sarcoma-associated herpesvirus lytic replication by targeting RTA. PLoS One 8:e55832. doi: 10.1371/journal.pone.0055832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forte E, Luftig MA. 2011. The role of microRNAs in Epstein-Barr virus latency and lytic reactivation. Microbes Infect 13:1156–1167. doi: 10.1016/j.micinf.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Catrina Ene AM, Borze I, Guled M, Costache M, Leen G, Sajin M, Ionica E, Chitu A, Knuutila S. 2014. MicroRNA expression profiles in Kaposi's sarcoma. Pathol Oncol Res 20:153–159. doi: 10.1007/s12253-013-9678-1. [DOI] [PubMed] [Google Scholar]
- 46.Godshalk SE, Bhaduri-McIntosh S, Slack FJ. 2008. Epstein-Barr virus-mediated dysregulation of human microRNA expression. Cell Cycle 7:3595–3600. doi: 10.4161/cc.7.22.7120. [DOI] [PubMed] [Google Scholar]
- 47.Yang GD, Huang TJ, Peng LX, Yang CF, Liu RY, Huang HB, Chu QQ, Yang HJ, Huang JL, Zhu ZY, Qian CN, Huang BJ. 2013. Epstein-Barr virus-encoded LMP1 upregulates microRNA-21 to promote the resistance of nasopharyngeal carcinoma cells to cisplatin-induced apoptosis by suppressing PDCD4 and Fas-L PLoS One 8:e78355. doi: 10.1371/journal.pone.0078355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y, Chen J, Wang H, Shi J, Wu K, Liu S, Liu Y, Wu J. 2013. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PLoS Pathog 9:e1003248. doi: 10.1371/journal.ppat.1003248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li SQ, Zhu JG, Zhang WL, Chen YR, Zhang K, Popescu LM, Ma XL, Lau WB, Rong R, Yu XQ, Wang BB, Li YF, Xiao CS, Zhang MM, Wang SY, Yu LP, Chen AF, Yang XC, Cai J. 2011. Signature microRNA expression profile of essential hypertension and its novel link to human cytomegalovirus infection. Circulation 124:175–U154. doi: 10.1161/CIRCULATIONAHA.110.012237. [DOI] [PubMed] [Google Scholar]
- 50.Luo MH, Hannemann H, Kulkarni AS, Schwartz PH, O'Dowd JM, Fortunato EA. 2010. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J Virol 84:3528–3541. doi: 10.1128/JVI.02161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stark TJ, Arnold JD, Spector DH, Yeo GW. 2012. High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J Virol 86:226–235. doi: 10.1128/JVI.05903-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krichevsky AM, Gabriely G. 2009. miR-21: a small multi-faceted RNA. J Cell Mol Med 13:39–53. doi: 10.1111/j.1582-4934.2008.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. 2008. MicroRNA-21 (miR-21) posttranscriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 27:2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 54.Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH. 2010. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin Chim Acta 411:846–852. doi: 10.1016/j.cca.2010.02.074. [DOI] [PubMed] [Google Scholar]
- 55.Zhu SM, Si ML, Wu HL, Mo YY. 2007. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 282:14328–14336. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 56.Wang P, Zou F, Zhang X, Li H, Dulak A, Tomko RJ Jr, Lazo JS, Wang Z, Zhang L, Yu J. 2009. microRNA-21 negatively regulates Cdc25A and cell cycle progression in colon cancer cells. Cancer Res 69:8157–8165. doi: 10.1158/0008-5472.CAN-09-1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ray D, Kiyokawa H. 2008. CDC25A phosphatase: a rate-limiting oncogene that determines genomic stability. Cancer Res 68:1251–1253. doi: 10.1158/0008-5472.CAN-07-5983. [DOI] [PubMed] [Google Scholar]
- 58.Boutros R, Lobjois V, Ducommun B. 2007. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer 7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 59.Dittmer D, Mocarski ES. 1997. Human cytomegalovirus infection inhibits G1/S transition. J Virol 71:1629–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu M, Shenk T. 1996. Human cytomegalovirus infection inhibits cell cycle progression at multiple points, including the transition from G1 to S. J Virol 70:8850–8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pan X, Li XJ, Liu XJ, Yuan H, Li JF, Duan YL, Ye HQ, Fu YR, Qiao GH, Wu CC, Yang B, Tian XH, Hu KH, Miao LF, Chen XL, Zheng J, Rayner S, Schwartz PH, Britt WJ, Xu J, Luo MH. 2013. Later passage neural progenitor cells from neonatal brain are more permissive for human cytomegalovirus infection. J Virol 87:10968–10979. doi: 10.1128/JVI.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen ZZ, Pan X, Miao LF, Ye HQ, Chavanas S, Davrinche C, McVoy M, Luo MH. 2014. Comprehensive analysis of human cytomegalovirus microRNA expression during lytic and quiescent infection. PLoS One 9:e88531. doi: 10.1371/journal.pone.0088531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tamashiro JC, Hock LJ, Spector DH. 1982. Construction of a cloned library of the EcoRI fragments from the human cytomegalovirus genome (strain AD169). J Virol 42:547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fortunato EA, Dell'Aquila ML, Spector DH. 2000. Specific chromosome 1 breaks induced by human cytomegalovirus. Proc Natl Acad Sci U S A 97:853–858. doi: 10.1073/pnas.97.2.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Su H, Yang JR, Xu T, Huang J, Xu L, Yuan Y, Zhuang SM. 2009. MicroRNA-101, downregulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res 69:1135–1142. doi: 10.1158/0008-5472.CAN-08-2886. [DOI] [PubMed] [Google Scholar]
- 66.Tiscornia G, Singer O, Verma IM. 2006. Production and purification of lentiviral vectors. Nat Protoc 1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- 67.Lou Y, Yang X, Wang F, Cui Z, Huang Y. 2010. MicroRNA-21 promotes the cell proliferation, invasion and migration abilities in ovarian epithelial carcinomas through inhibiting the expression of PTEN protein. Int J Mol Med 26:819–827. [DOI] [PubMed] [Google Scholar]
- 68.Chen C, Okayama H. 1987. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7:2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duan Y, Miao L, Ye H, Yang C, Fu B, Schwartz PH, Rayner S, Fortunato EA, Luo MH. 2012. A faster immunofluorescence assay for tracking infection progress of human cytomegalovirus. Acta Biochim Biophys Sinica 44:597–605. doi: 10.1093/abbs/gms041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang HT, Wang B, Liu ZJ, Bai ZQ, Li L, Liu HY, Qian DM, Yan ZY, Song XX. 2009. Effect of human cytomegalovirus infection on nerve growth factor expression in human glioma U251 cells. Biomed Environ Sci 22:354–358. doi: 10.1016/S0895-3988(09)60068-4. [DOI] [PubMed] [Google Scholar]
- 71.Gaspar M, Shenk T. 2006. Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc Natl Acad Sci U S A 103:2821–2826. doi: 10.1073/pnas.0511148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Oliveira PE, Zhang L, Wang Z, Lazo JS. 2009. Hypoxia-mediated regulation of Cdc25A phosphatase by p21 and miR-21. Cell Cycle 8:3157–3164. doi: 10.4161/cc.8.19.9704. [DOI] [PubMed] [Google Scholar]
- 73.Li C, Wang Y, Wang S, Wu B, Hao J, Fan H, Ju Y, Ding Y, Chen L, Chu X, Liu W, Ye X, Meng S. 2013. Hepatitis B virus mRNA-mediated miR-122 inhibition upregulates PTTG1-binding protein, which promotes hepatocellular carcinoma tumor growth and cell invasion. J Virol 87:2193–2205. doi: 10.1128/JVI.02831-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang S, Qiu L, Yan X, Jin W, Wang Y, Chen L, Wu E, Ye X, Gao GF, Wang F, Chen Y, Duan Z, Meng S. 2012. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G1-modulated P53 activity. Hepatology 55:730–741. doi: 10.1002/hep.24809. [DOI] [PubMed] [Google Scholar]
- 75.Wang D, Cao L, Xu Z, Fang L, Zhong Y, Chen Q, Luo R, Chen H, Li K, Xiao S. 2013. miR-125b reduces porcine reproductive and respiratory syndrome virus replication by negatively regulating the NF-κB pathway. PLoS One 8:e55838. doi: 10.1371/journal.pone.0055838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barth S, Pfuhl T, Mamiani A, Ehses C, Roemer K, Kremmer E, Jaker C, Hock J, Meister G, Grasser FA. 2008. Epstein-Barr virus-encoded microRNA miR-BART2 downregulates the viral DNA polymerase BALF5. Nucleic Acids Res 36:666–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. 2010. Regulation of NF-κB inhibitor IκBα and viral replication by a KSHV microRNA. Nat Cell Biol 12:193–199. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin X, Liang D, He Z, Deng Q, Robertson ES, Lan K. 2011. miR-K12-7-5p encoded by Kaposi's sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS One 6:e16224. doi: 10.1371/journal.pone.0016224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu CC, Li Z, Chu CY, Feng J, Sun R, Rana TM. 2010. MicroRNAs encoded by Kaposi's sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep 11:784–790. doi: 10.1038/embor.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grey F, Meyers H, White EA, Spector DH, Nelson J. 2007. A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathog 3:e163. doi: 10.1371/journal.ppat.0030163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stern-Ginossar N, Saleh N, Goldberg MD, Prichard M, Wolf DG, Mandelboim O. 2009. Analysis of human cytomegalovirus-encoded microRNA activity during infection. J Virol 83:10684–10693. doi: 10.1128/JVI.01292-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T, Hancock M, Landais I, Jeng S, McWeeney S, Britt W, Nelson JA. 2014. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 15:363–373. doi: 10.1016/j.chom.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen Y, Shen A, Rider PJ, Yu Y, Wu K, Mu Y, Hao Q, Liu Y, Gong H, Zhu Y, Liu F, Wu J. 2011. A liver-specific microRNA binds to a highly conserved RNA sequence of hepatitis B virus and negatively regulates viral gene expression and replication. FASEB J 25:4511–4521. doi: 10.1096/fj.11-187781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harkins L, Volk AL, Samanta M, Mikolaenko I, Britt WJ, Bland KI, Cobbs CS. 2002. Specific localization of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 360:1557–1563. doi: 10.1016/S0140-6736(02)11524-8. [DOI] [PubMed] [Google Scholar]
- 85.Michaelis M, Doerr HW, Cinatl J. 2009. The story of human cytomegalovirus and cancer: increasing evidence and open questions. Neoplasia 11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stragliotto G, Rahbar A, Solberg NW, Lilja A, Taher C, Orrego A, Bjurman B, Tammik C, Skarman P, Peredo I, Soderberg-Naucler C. 2013. Effects of valganciclovir as an add-on therapy in patients with cytomegalovirus-positive glioblastoma: a randomized, double-blind, hypothesis-generating study. Int J Cancer 133:1204–1213. doi: 10.1002/ijc.28111. [DOI] [PubMed] [Google Scholar]
- 87.Yamashita Y, Kasugai I, Sato M, Tanuma N, Sato I, Nomura M, Yamashita K, Sonoda Y, Kumabe T, Tominaga T, Katakura R, Shima H. 2010. CDC25A mRNA levels significantly correlate with Ki-67 expression in human glioma samples. J Neurooncol 100:43–49. doi: 10.1007/s11060-010-0147-3. [DOI] [PubMed] [Google Scholar]