

ABSTRACT
Viruses frequently combine multiple activities into one polypeptide to conserve coding capacity. This strategy creates regulatory challenges to ascertain that the combined activities are compatible and do not interfere with each other. The papillomavirus E1 protein, as many other helicases, has the intrinsic ability to form hexamers and double hexamers (DH) that serve as the replicative DNA helicase. However, E1 also has the more unusual ability to generate local melting by forming a double trimer (DT) complex that can untwist the double-stranded origin of DNA replication (ori) DNA in preparation for DH formation. Here we describe a switching mechanism that allows the papillomavirus E1 protein to form these two different kinds of oligomers and to transition between them. We show that a conserved regulatory module attached to the E1 helicase domain blocks hexamer and DH formation and promotes DT formation. In the presence of the appropriate trigger, the inhibitory effect of the regulatory module is relieved and the transition to DH formation can occur.
IMPORTANCE This study provides a mechanistic understanding into how a multifunctional viral polypeptide can provide different, seemingly incompatible activities. A conserved regulatory sequence module attached to the AAA+ helicase domain in the papillomavirus E1 protein allows the formation of different oligomers with different biochemical activities.
INTRODUCTION
DNA helicases are important enzymes that catalyze the separation of the two cDNA strands in preparation for processes that require exposure of single-stranded DNA (ssDNA), such as DNA replication, repair, and recombination (for reviews, see references 1 and 2). Hexameric DNA helicases are ring-shaped enzymes that function by encircling one of the DNA strands while displacing the other in an ATP-dependent manner. These enzymes are highly processive, and the key to the processivity is believed to be the encirclement of the DNA strand by the hexameric ring, which prevents disassociation of the enzyme from the substrate. Consistent with such a mode of action, a common feature of hexameric helicases is that the presence of ssDNA induces formation of hexamers or stabilizes existing hexamers (1).
The viral E1 protein from the papillomavirus family represents a subgroup of hexameric helicases. These DNA helicases, which belong to the SF3 helicase family (3), are not dedicated helicases but serve many different functions in the viral life cycle (for a review, see reference 4). The E1 proteins are termed initiator proteins because their primary function is to assist in the initiation of replication of the viral genome. The E1 protein takes part in initiation of DNA replication in at least four different ways. E1 binds cooperatively with the viral E2 transcription factor to the origin of DNA replication (ori), which contains binding sites (BS) for both proteins, forming a highly sequence-specific E1-E2 complex (5–15). In this complex, E1 uses its specific DNA binding domain (DBD) to bind to two of the four E1 binding sites present in ori as a head-to-head dimer (7, 8, 16, 17). The interaction between the E1 helicase domain and the E2 transactivation domain has two distinct effects on DNA binding (10, 18–23). The cooperativity in the interaction contributes to the affinity of binding, and the interaction also blocks a nonspecific DNA binding activity present in the E1 helicase domain, contributing to the specificity of DNA binding (12).
In the next step, through processes that are not well understood, E2 is displaced and the E1-E2 complex is converted into an E1 double-trimer (DT) complex on the ori (24–28). In the DT, E1 is bound to the ori as two head-to-head trimers, encircling double-stranded DNA (dsDNA) (24, 29). Formation of this complex depends on binding of the E1 DBD to the four E1 BS in the ori. However, a nonspecific DNA binding activity present in the helicase domain, which makes non-sequence-specific contacts with the DNA flanking the E1 BS, is also critical for formation of the DT (24, 25, 27, 30). This complex is capable of local melting of the ori using an ATP hydrolysis-dependent untwisting process (25).
The locally melted template, with the two head-to-head trimers constituting the DT, now becomes the substrate for additional E1 molecules forming the E1 double hexamer (DH), which is composed of two head-to-head hexamers (31). Each hexamer encircles ssDNA and functions as a helicase with the ability to unwind the dsDNA bidirectionally in front of the replication forks (32–36). At this point, the enzymes involved in the replication process such as topoisomerase I, DNA polymerase α, and replication protein A (RPA) are recruited to the ori through interaction with E1 (21, 23, 37, 38). Although formation of the DH requires a DT precursor, hexamers of E1 can readily form directly if ssDNA is provided (31, 36). Both full-length E1 and shorter C-terminal fragments containing the E1 helicase domain are able to form such ssDNA-induced hexamers by utilizing the intrinsic ability of the oligomerization domain and helicase domain to bind ssDNA and form hexamers (36, 39, 40).
The need to utilize one protein to provide multiple functions presents a challenge not only in that multiple activities have to be incorporated into one polypeptide, but also because mechanisms that prevent the different activities from interfering with each other have to be built in. As described above, some of the switches in activity that E1 undergoes are related to changes in oligomerization status. Because E1 has the intrinsic ability to form hexamers and double hexamers, an obvious requirement is to prevent hexamer formation when complexes other than hexamers and DH (such as the E1-E2 and the DT complexes) are required. Conversely, a block to hexamer and DH formation has to be conditional and regulated such that the hexamer and DH can form when so required. Consequently, the transition from E1-E2 to DT to DH must involve regulatory steps that control the ability of E1 to oligomerize.
Structural analyses provide an important part of how we understand E1 oligomerization. Multiple crystal structures containing the helicase and oligomerization domains of E1 have been solved, providing excellent information about the overall structure of these helicases, how they form hexamers, how they bind nucleotide, and how they associate with ssDNA (34, 41, 42). In two of these structures, a 28-residue C-terminal peptide was deemed to be protease sensitive and therefore removed from the helicase domain to facilitate crystallization (34, 41). In the third structure, even though the peptide was present, it was not ordered in the structure and therefore not visible, indicating that this peptide is not an integral part of the helicase domain (42).
Here we show that the short C-terminal peptide has critical functions in the biochemical activities of the E1 protein. The 28-residue peptide contains two separate elements, an acidic region and a C-terminal tail (C tail), which together regulate E1 oligomerization. The acidic region is a negative regulator of oligomerization and promotes DT formation. The C tail is required for DH formation by counteracting the effect of the acidic region. Using these two elements, E1 can undertake the orderly transition between the DT complex, which melts the ori, and the DH, which unwinds the viral DNA in front of the replication fork, providing a glimpse into how multifunctional initiator proteins transition from one activity to another.
MATERIALS AND METHODS
Expression and purification of E1 and E2.
Full-length E1 and E1 fragments from bovine papillomavirus type 1 (BPV-1) were expressed in Escherichia coli as N-terminal glutathione S-transferase (GST) fusions and purified by glutathione agarose affinity chromatography. The GST portion was removed by digestion with thrombin and the material further purified by ion-exchange chromatography. E2 was expressed without a tag and purified by mono-S and mono-Q ion-exchange chromatography (43).
EMSA.
Four percent acrylamide gels (39:1, acrylamide/bisacrylamide) containing 0.5× Tris-borate-EDTA (TBE) lacking EDTA were used for all electrophoretic mobility shift assays (EMSAs). E1 was added to 32P-labeled probe (∼2 fmol) in 10 μl binding buffer (BB; 20 mM HEPES [pH 7.5], 70 mM NaCl, 0.7 mg/ml bovine serum albumin [BSA], 0.1% NP-40, 5% glycerol, 5 mM dithiothreitol [DTT], 5 mM MgCl2) and 2 mM ATP or ADP. After incubation at room temperature (RT) for 1 h, the samples were loaded and run for 2 h at 9 V/cm (24).
Probes.
The 84-bp ori probe for EMSA was generated by PCR using one primer labeled with 32P at the 5′ end with polynucleotide kinase and [γ-32P]ATP with a cloned fragment containing the BPV-1 ori sequence as a template. The probe contains the sequences between nucleotides (nt) 7906 and 48 in the BPV-1 genome. To generate the wild-type (wt) 84-bp probe (see Fig. 6), four staggered oligonucleotides were generated, which after phosphorylation and ligation generated the 84-bp probe with perfect complementarity. To generate the bubble probe (see Fig. 6), one of the four oligonucleotides was replaced with an oligonucleotide that after annealing generated a 6-bp single-stranded region between nt 7928 and 7933 in the BPV-1 genome.
FIG 6.

The C-terminal module interacts with the E1 oligomerization domain. (A) E1308–577 and luciferase were translated in vitro in a reticulocyte lysate in the presence of [35S]methionine, used in pulldown experiments with GST, GST544–577 579R/583R/586R, or GST544–605, and analyzed by SDS-PAGE followed by autoradiography. In the input lane, 1% of input was loaded. (B) The same gel as that described for panel A was stained with Coomassie brilliant blue (CBB) to show the presence of the GST fusion proteins. (C) E1308–410 was translated in vitro in a reticulocyte lysate in the presence of [35S]methionine, used in pulldown experiments with GST, GST544–577 579R/583R/586R, or GST544–577, and analyzed by SDS-PAGE followed by autoradiography. (D) The same gel as for panel C was stained with CBB to show the presence of the GST fusion proteins. (E) The C tail is required to sense the presence of ssDNA. Three quantities (2.5, 5, or 10 ng) of wt E1 or the F594A or C596A mutant were used in EMSA using two different probes. Wild-type E1 was incubated with the wt 84-bp ori probe in the presence of ADP or ATP. Wild-type E1 or the F594A or C596A mutant was incubated with a probe that differed from the wt probe by a 6-bp mismatched region (bubble probe) in the presence of ADP. Migration of the DT and DH complexes is indicated.
Glycerol gradient sedimentation.
Glycerol gradients (5 ml) were generated in 20 mM HEPES (pH 7.5), 70 mM NaCl, and 5 mM DTT. In the experiment whose results are shown in Fig. 1D and E, 1 μg of E1 protein was incubated with 200 ng of a 41-nt oligonucleotide labeled with 32P and then loaded onto a 15 to 30% glycerol gradient, which was run for 16 h at 49,000 rpm in an SW 55 rotor. For the experiment whose results are shown in Fig. 1F and 2D, 100 μg of E1308–577 and E1308–597, respectively, was loaded onto a 5 to 30% glycerol gradient, which was run for 23 h at 49,000 rpm in an SW55 rotor. The gradients were fractionated into 150-μl fractions, and the peaks were localized by Bradford assays (for marker proteins and for E1308–577 and E1308–597) or by counting in a scintillation counter to detect 32P in DNA-containing samples. Marker proteins were purchased from Sigma-Aldrich and run in parallel gradients.
FIG 1.

A C-terminal 28-aa peptide is important for complex formation by E1308–605. (A) The C-terminal regions from 6 different papillomavirus E1 proteins and simian virus 40 (SV40) T-ag were aligned. Highly conserved residues are highlighted, and the acidic region is bracketed. (B) A 41-mer ssDNA probe was incubated with three levels (8, 16, and 32 ng) of E1308–605 (lanes 2 to 4) or E1308–577 (lanes 6 to 8) in the presence of ADP and subjected to EMSA. In lanes 1 and 5, no protein was added. The position of the E1 hexamer complex is indicated. (C) Two femtomoles of helicase substrate was incubated with four quantities (8, 16, 32, and 64 ng) of full-length E11–605 (lanes 3 to 6), E1308–605 (lanes 7 to 10), and E1308–577 (lanes 11 to 14) in the presence of ATP. Lane 1 contained probe alone, while lane 2 contained boiled probe. The migrations of dsDNA and ssDNA are indicated. (D) Binding reactions for EMSA with E1308–605, as shown in panel B, were scaled up and sedimented in a 15 to 30% glycerol gradient. The radioactivity in the fractions was quantitated and plotted. The peak fractions were loaded onto an EMSA gel to verify that the gradient peak corresponded to the complex that we observe by EMSA (inset). Arrows indicate the sedimentation of the marker proteins bovine serum albumin (BSA), alcohol dehydrogenase (ADH), and β-amylase. (E) Binding reactions for EMSA with E1308–577, as shown in panel B, were scaled up and sedimented in a 15 to 30% glycerol gradient. The radioactivity in the fractions was quantitated and plotted. The last fraction plotted corresponds to the bottom portion of the tube, which was cut off and quantified. (F) One hundred micrograms of E1308–577 was sedimented in a 5 to 30% glycerol gradient. The protein was detected by Bradford assays and compared to the sedimentation of the marker proteins carbonic anhydrase and BSA.
FIG 2.
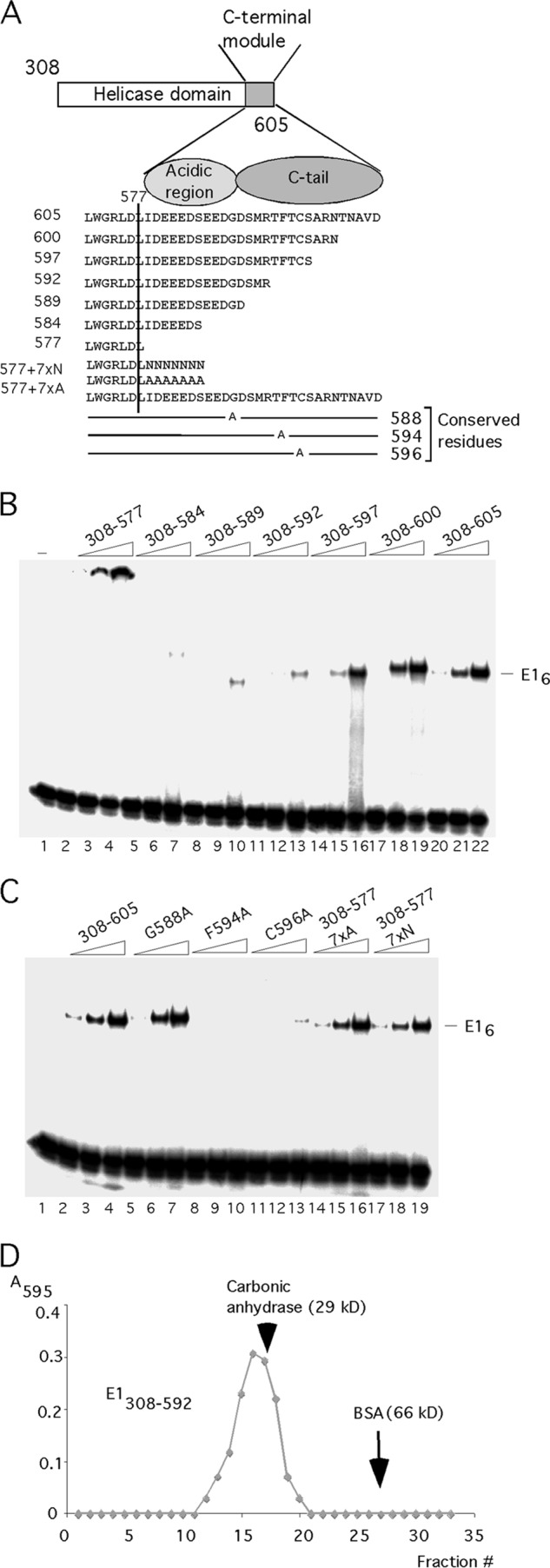
The C-terminal 28 residues regulate E1 hexamer formation. (A) Cartoon showing the sequence of the C-terminal module with deletions and point mutations in the context of E308–605. (B) The deletion mutants in the C-terminal module were tested for hexamer formation in the context of E1308–605 using an ssDNA probe. Two, 4, and 8 ng each of the proteins E1308–577, E1308–584, E1308–589, E1308–592, E1308–597, E1308–600, and E1308–605 were incubated with a 41-mer ssDNA probe (∼2 fmol) in the presence of ADP and tested for hexamer formation by EMSA. The migration of the E1 hexamer is indicated. (C) Two, 4, and 8 ng each of the proteins with point mutations G588A, F594A, and C596A and the deletion mutants E1308–577 7×A and E1308–577 7×N were incubated with a 41-mer ssDNA probe in the presence of ADP and tested for hexamer formation by EMSA. (D) One hundred micrograms of E1308–592 was sedimented in a 5 to 30% glycerol gradient. The protein was detected by Bradford assays and its sedimentation was compared to that of the marker proteins carbonic anhydrase and bovine serum albumin (BSA).
DNA helicase assays.
Short helicase substrates were generated by annealing an end-labeled 41-mer oligonucleotide to a 59-mer oligonucleotide with partial complementarity, generating a molecule with 26 bp of dsDNA, 15 nt of 5′ ssDNA, and 33 nt of 3′ ssDNA. After gel purification, the substrate (∼2 fmol) was incubated with E1 in BB containing 2 mM ATP but lacking NaCl for 15 min at 37°C. After the incubation, SDS was added to 0.1% and the sample was loaded onto 10% acrylamide gels (29:1, acrylamide/bisacrylamide).
The long helicase substrates were generated by annealing a primer to M13 mp18 and extending with Klenow DNA polymerase in the presence of 50 μM dATP, dGTP, and dTTP, 10 μM dCTP, 80 μM ddATP, and 40 μCi [α-32P]dCTP. The average length of the extension products is determined by the relative levels of ddATP. The helicase assays were carried out as described above, except that 12 μg/ml of E. coli ssDNA binding protein (SSB) was included in the reaction mixture. The samples were treated with proteinase K (200 μg/ml) and analyzed by agarose gel electrophoresis.
Unwinding assays.
Unwinding assays were performed by incubating 2 fmol of probe with E1 at 32°C for 30 min under EMSA conditions (see above) but in the presence of 10 μg/ml E. coli SSB. Before loading samples on the EMSA gel, Sarkosyl was added to 0.1% to disrupt E1-DNA complexes without affecting ssDNA-SSB complexes (see Fig. 5D).
FIG 5.

The C-terminal module is required for unwinding and DNA replication in vitro. (A) The 84-bp ori probe was incubated with three levels (2.5, 5, and 10 ng) of E11–584, E11–589, E11–592, E11–597, E11–600, and E11–605 in the presence of ATP and E. coli SSB and analyzed by EMSA. ssDNA + SSB complexes are indicated. (B) The 84-bp ori probe was incubated with three levels (2.5, 5, and 10 ng) of E11–605, E11–605 G588A, E11–605 M591A, E11–605 T593A, E11–605 F594A, E11–605 C596A, and E11–577 7×N in the presence of ATP and E. coli SSB and analyzed by EMSA. (C) The 84-bp ori probe was incubated with four levels (2.5, 5, 10 and 20 ng) of E11–605, E11–605 586R, E11–605 579R/583R, E11–605 579R/583R/586R, and E11–577 in the presence of ATP and E. coli SSB and analyzed by EMSA. (D) Low levels of Sarkosyl disrupt E1 complexes with ssDNA and dsDNA but do not affect complexes between E. coli SSB and ssDNA. The double-stranded 84-bp ori probe was incubated with E1 in the presence of ADP (lanes 2 and 3). Immediately before loading onto an EMSA gel, one sample (lane 3) was treated with 0.1% Sarkosyl. The ssDNA probe was incubated with E1 in the presence of ADP (lanes 5 and 6). Immediately before loading onto an EMSA gel, one sample (lane 6) was treated with 0.1% Sarkosyl. The ssDNA probe was incubated with E. coli SSB (lanes 7 and 8). Immediately prior to loading onto the EMSA gel, one sample (lane 8) was treated with 0.1% Sarkosyl. (E) Deletions and point mutations in the acidic region and C tail were expressed and purified from E. coli and tested for DNA replication in vitro as described in Materials and Methods. Two hundred nanograms of each mutant protein was incubated in replication extract in the presence of [α-32P]dCTP and analyzed by agarose gel electrophoresis. (F) The arginine substitutions in the acidic region were tested for DNA replication in vitro. Three quantities (100, 200, and 400 ng) of wt E1 and the E1 579R/583R, 586R, 579R/583R/586R mutants and E11–577 were used in cell-free replication reactions in the presence of [α-32P]dCTP and analyzed by agarose gel electrophoresis. (G) Inactivation of the acidic region in the context of the E1308–605 C596A restores E1 hexamer formation and ATPase activity to wt levels. Mutants in the C-terminal module were tested for hexamer formation in the context of E1308–605 using an ssDNA probe. Two, 4, and 8 ng each of the proteins E1308–605, E1308–605 C596A, and E1308–605 C596A D579R/D573RE586R were incubated with a 41-mer ssDNA probe (∼2 fmol) in the presence of ADP and tested for hexamer formation by EMSA. The migration of the E1 hexamer is indicated. The altered mobility of the complexes formed with E1308–605 C596A D579R/D573RE586R is caused by the E586R substitution. (H) Mutants in the C-terminal module in the context of E1308–605 were tested for ATPase activity. The proteins E1308–605, E1308–605 C596A, andE1308–605 C596A D579R/D573RE586R were incubated with [γ-32P]ATP and ssDNA, unless otherwise indicated, and tested for ATPase activity as described in Materials and Methods. The migration of ATP and free phosphate is indicated, and the level of hydrolyzed ATP is shown below the lanes.
ATPase assays.
ATPase assays were performed in a 20-μl reaction mixture containing 30 mM HEPES (pH 7.5), 30 mM NaCl, 1 mM DTT, 7 mM MgCl2, 100 μg of bovine serum albumin/ml, 100 μM ATP, and 40,000 cpm of [γ-32P]ATP and E1. Reaction mixtures were incubated for 1 h at room temperature, and the reactions were stopped by the addition of EDTA to a final concentration of 10 mM. Then, 2-μl portions of the reaction mixtures were spotted onto a polyethyleneimine (PEI)-cellulose plate, and the plates were then developed in 1 M formic acid and 0.5 M LiCl2 for 40 min. After drying, each plate was exposed to a Fuji imaging plate, and the level of free phosphate was determined by scanning the plate using a Fuji BAS imager.
Pulldown assays.
Pulldown assays were performed as described previously (44). E1308–577 and E1308–410 were subcloned into the pET 11 C vector and in vitro transcribed and translated using a TNT coupled rabbit reticulocyte kit (Promega) according to the manufacturer's instructions. Two hundred nanograms of GST fusion protein (GST, GST E1544–605, or GST E1544–605 3 × R) was incubated with 1 μl of in vitro translation mixture in 20 μl of a solution containing 20 mM HEPES (pH 7.9), 50 mM KCl, 2.5 mM MgCl2, 10% glycerol, 1 mM DTT, 0.2% NP-40, and 3 μl of fetal bovine serum. After 2 h of incubation, 5 ml of glutathione agarose beads was added, and the mixture was incubated for a further 30 min. The beads were washed repeatedly with Tris-buffered saline (TBS) containing 0.1% NP-40, and the samples were analyzed by SDS-PAGE.
In vitro DNA replication.
In vitro DNA replication assays were performed essentially as described previously (9, 11). In vitro replication was performed in 25-μ1 reaction mixtures containing 40 mM HEPES-KOH (pH 7.5), 8 mM MgCl2, 0.5 mM DTT, 3 mM ATP, 0.2 mM (each) GTP, UTP, and CTP, 0.1 mM (each) dATP, dGTP, and dTTP, 10 μM [32P]dCTP (2 μCi; 3,000 Ci/mmol), 40 mM creatine phosphate, 400 ng creatine kinase, 10 μl S100 extract, and 0.5 μl high-salt nuclear extract from H293 cells. The concentration of template in the in vitro reaction mixtures was 2 ng/μl. Reaction mixtures were incubated for 60 min at 37°C, the reactions were stopped by addition of SDS to 1% and EDTA to 10 mM, and the mixtures were treated with proteinase K followed by phenol-chloroform extraction and precipitation with ethanol and ammonium acetate. The products were analyzed by electrophoresis on 1% agarose gels in Tris-acetate-EDTA (TAE) buffer.
RESULTS
A 28-aa C-terminal peptide in the papillomavirus E1 protein controls the formation of hexamers on ssDNA.
An alignment of the 30 to 35 C-terminal amino acids (aa) from multiple E1 proteins shows the presence of a short ∼10-aa stretch of largely acidic residues (acidic region [Fig. 1A]) followed by a 13- to 18-residue-long sequence, which we have termed the C tail (Fig. 1A). The presence of multiple aspartic and glutamic acid residues is well conserved in the acidic region, although the exact sequence is not. The C tail contains three highly conserved residues, G588, F594, and C596, but otherwise shows limited conservation. The acidic region and the C tail clearly are not integral parts of the E1 helicase domain structure. These peptides were removed prior to crystallization in two structures of the E1 helicase domain (34, 41); in the third structure, these peptides were present in the crystallized fragment but were not visible in the structure, demonstrating that they were not sufficiently ordered to be resolved (42).
To address the function of the acidic region and the C tail, we expressed and purified E1308–605, which includes these two peptides, and E1308–577, which lacks these peptides, from E. coli and tested the biochemical properties of these proteins. E1308–605 formed a robust complex on ssDNA (Fig. 1B, lanes 1 to 4). In contrast, E1308–577 formed a complex that did not enter the gel, indicating a very high molecular mass (Fig. 1B, lanes 5 to 8). To examine the nature of the complexes formed on ssDNA, we analyzed them by glycerol gradient sedimentation (Fig. 1C). E1308–605 in the presence of radiolabeled ssDNA and ATP formed a discrete complex sedimenting slightly faster than β-amylase, indicating an apparent molecular mass of approximately 200 kDa, consistent with a hexamer. In contrast, under the same conditions E1308–577 formed a complex that pelleted in the gradient, indicating a much greater molecular mass (Fig. 1D).
To determine whether this large complex was formed because the E1308–577 protein was aggregated, we sedimented E1308–577 in a glycerol gradient in the absence of ssDNA. E1308–577 formed a discrete peak with an apparent molecular mass of ∼29 kDa, indicating that it is monomeric and not aggregated (Fig. 1E). This indicates that it is the presence of ssDNA that induces the formation of the high-molecular-mass complex observed for E1308–577. We next tested the E1308–605 and E1308–577 proteins for DNA helicase activity using a short partially double-stranded oligonucleotide. The two proteins had similar levels of helicase activity (Fig. 1F, compare lanes 3 to 6 to lanes 7 to 10), which was surprising since the E1308–577 protein failed to form a hexamer with ssDNA.
To address the function of the C-terminal 28 residues, we generated small deletions in this region (Fig. 2A). We generated the proteins E1308–600, E1308–597, E1308–592, and E1308–589, which affect the C-terminal tail, and the deletion protein E1308–584, which also affects the acidic region. In addition, we generated alanine substitutions in the conserved residues G588, F594, and C596 in the C-terminal tail. We first tested these proteins for the ability to form the E1 hexamer on ssDNA (Fig. 2B and C). As observed in Fig. 1B, E1308–577 formed a complex that did not enter the gel (Fig. 2B, lanes 2 to 4), while E1308–605 formed a robust hexamer (Fig. 2B, lanes 20 to 22). E1308–600 (Fig. 2B, lanes 17 to 19) behaved like E1308–605, but the rest of the proteins with deletions, E1308–584, E1308–589, E1308–592, and E1308–597, all showed severe defects for hexamer formation (Fig. 2B, lanes 5 to 7, 8 to 10, 11 to 13, and 14 to 16, respectively). Similarly, proteins with the point mutations F594A and C596A in the C-terminal tail failed to form the E1 hexamer (Fig. 2C, lanes 8 to 10 and 11 to 13), while G588A had no obvious effect on E1 hexamer formation (Fig. 2C, lanes 5 to 7). These results demonstrate that the majority of the mutations in the C-terminal module affect E1 hexamer formation.
We were surprised that none of the proteins with deletions or point mutations, although defective for hexamer formation, exhibited the same phenotype as E1308–577, which forms large complexes in the well. After examining the structure of E1308–577, we considered the possibility that the deletion to residue 577 could cause structural issues in the folding of the helicase domain, possibly because the presence of residues immediately C terminal to residue 577 is important. To address this possibility, we added either seven alanines (7 × N) or seven asparagines (7 × A) to the endpoint at residue 577, generating E1308–577 7×N and E1308–577 7×A, respectively, and tested these for hexamer formation in the presence of ssDNA. As shown in Fig. 2C, lanes 14 to 19, these additions restored fully the ability to form the E1 hexamer. This result was surprising since it demonstrates that the acidic region and the C tail are not per se required for hexamer formation by the E1 helicase domain, contrary to expectation. We were also intrigued by the fact that E1308–577 7×N and E1308–577 7×A (Fig. 2C, lanes 14 to 19) formed discrete hexamers at wt levels while E1308–584 had a severe defect for hexamer formation (Fig. 2B, lanes 5 to 7), although these proteins all have seven residues added to the C terminus of E1308–577. This difference demonstrates that the 7 residues of the acidic region present in E1308–584 have a direct negative effect on E1 hexamer formation and that the function of the acidic region therefore appears to be to inhibit hexamer formation. In the presence of the C tail, this block to hexamer formation on ssDNA can be bypassed.
To ascertain that deletions in the C-terminal module did not cause gross structural defects, we performed sedimentation analysis with one of these mutants. As shown in Fig. 2D, E1308–592 sedimented as a discrete peak similar to the marker protein carbonic anhydrase (molecular mass, 29 kDa), demonstrating that it remains a discrete monomer.
As results displayed in Fig. 1E show, both E1308–605 and E1308–577 had helicase activity when tested using short partially double-stranded oligonucleotides. For a more stringent helicase assay, more relevant to the role of E1 as a replicative DNA helicase, we generated substrates with much longer double-stranded regions by extending a primer annealed to a single-stranded M13 template in the presence of 32P-labeled nucleotide and low levels of dideoxynucleotide to generate a substrate with a double-stranded region ranging between 1 and 5 kb in length. Such a substrate can provide an estimate of the processivity of the helicase (Fig. 3A to D). E1308–577 failed to unwind this substrate, as did the deletion proteins E1308–584, E1308–589, and E1308–592 (Fig. 3A, lanes 3 to 15). E1308–597 showed a very low level of unwinding (Fig. 3B, lane 5), while E1308–600 and E1308–605 showed robust helicase activity (Fig. 3B, lanes 7 to 12). Addition of 7 × N or 7 × A restored helicase activity to E1308–577 (Fig. 3C, lanes 7 to 12). Proteins with the point mutations in the conserved residues in the C-terminal tail (F594 and C596; Fig. 3D, lanes 10 to 15) showed a very low level of activity for DNA helicase activity, while mutation of the conserved G588 showed no defect. (Fig. 3D, lanes 7 to 9). These results show that all mutants that could form the hexamer well were also able to unwind the long helicase substrate while mutations defective for hexamer formation were unable to do so.
FIG 3.

Mutations in the C-terminal module affect the helicase and ATPase activity of E1. A helicase substrate was generated by annealing a primer to M13 ssDNA followed by extension with a mixture of deoxy- and dideoxynucleoside triphosphates (dNTPs and ddNTPs) and Klenow DNA polymerase to generate partially double-stranded templates with long (0.2- to 5-kb) radiolabeled strands. In each set, 2 fmol of the substrate was incubated with 3 levels (4, 8, and 16 ng) of the proteins. Proteins E1308–577, E1308–584, E1308–589, and E1308–592 (A), E1308–597, E1308–600, and E1308–605 (B), E1308–577, E1308–577 7×A, E1308–577 7×N, and E1308–605 (C), and E1308–605, E1308–605 G588A, E1308–605 F594A, and E1308–605 C596A (D) were tested. In each panel, a size marker (first lane), a boiled probe (B), and a no-E1 (−) sample were included. (E) Point mutants and deletions in the C-terminal module in the context of E1308–605 were tested for ATPase activity. Samples were incubated with [γ-32P]ATP in the presence of ssDNA except where otherwise indicated, followed by separation of free phosphate from ATP by thin-layer chromatography. The level of hydrolysis is shown below each lane.
ATP hydrolysis by the E1 protein and other hexameric helicases is dependent on the ATP binding and hydrolysis site formed by two adjacent subunits in the hexamer (34, 42, 45). Because hexamer formation is greatly stimulated by the presence of ssDNA, consequently ATPase is greatly stimulated by ssDNA. We tested the ability of our C-terminal mutants to hydrolyze [γ-32P]ATP (Fig. 3E). In the absence of ssDNA, E1308–605 had a low level of ATPase activity (Fig. 3E, lane 1), which was greatly stimulated (6-fold) in the presence of ssDNA (Fig. 3E, lane 2). The remaining samples were tested in the presence of ssDNA. E1308–577 showed a low level of ATPase activity (Fig. 3E, lane 4), while the deletion mutants E1308–584, E1308–589, and E1308–592 showed only a trace of ATPase activity (Fig. 3E, lanes 5 to 7). E1308–597 had a significantly higher activity, and activity was restored completely for E1308–600 (Fig. 3E, lanes 8 to 9). As observed with the helicase activity, E1308–577 7×A and E1308–577 7×N both showed a fully restored ATPase activity (Fig. 3E, lanes 11 to 12). Two of the three proteins with point mutations in the C-terminal tail, G588A (Fig. 3E, lane 13) and C596A (Fig. 3E, lane 15), showed a modest reduction of activity, while that with mutation F594A (lane 14) lacked activity altogether. These results are consistent with the ability of the different mutants to form hexamers on ssDNA.
The results shown in Fig. 2 and 3 clearly demonstrate that for the activities associated with the C-terminal half of E1 (hexamer formation on ssDNA, DNA helicase activity, and DNA-dependent ATPase activity) the acidic region and the C tail are largely dispensable. However, when present, the acidic region clearly has a negative effect on all of these activities, likely because the acidic region inhibits E1 hexamer formation. In the presence of the C tail, the acidic region is no longer inhibitory for hexamer formation.
The acidic region and the C tail affect DT and DH formation by full-length E1.
Because the acidic region and the C tail appear to be dispensable for the activities found in the E1 oligomerization and helicase domains, we generated the same deletions and mutations as those that we had tested in E1308–605 in the context of full-length E1 (Fig. 4). In contrast to E1308–605, full-length E1 forms several discrete types of complexes with dsDNA. E1 can form an E12E22 complex together with the viral transcription factor E2. This complex serves to recognize the ori site specifically (5–15). E1 also forms a DT complex on the ori; this complex generates local melting of the ori and serves as a precursor for the E1 DH, which is the viral replicative DNA helicase (24, 25, 27, 32, 33). We first tested the C-terminal deletions mutants for DT formation (Fig. 4B and C). All the deletion mutants, with the exception of E11–577, formed the DT well (Fig. 4B, lanes 5 to 16). Addition of 7 × N or 7 × A to E11–577 did not restore the ability to form the DT (Fig. 4C, lanes 5 to 10), and the five alanine substitutions in the C tail had no effect on DT formation (Fig. 4C, lanes 11 to 25). These results establish two important facts. First, there is a specific requirement for the acidic region for DT formation, since E11–584 forms the DT (Fig. 4B, lanes 14 to 16) while E11–577 7×N does not (Fig. 4C, lanes 5 to 7). Second, the C tail is clearly not required for DT formation.
FIG 4.

The acidic region is required for DT formation, and the C-terminal tail is required for DH formation by E11–605. (A) Cartoon showing the deletions and point mutations in the C-terminal module in the context of E11–605. (B) The 84-bp ori probe was incubated with three levels (2.5, 5, and 10 ng) of the deletion mutants E11–597, E11–592, E11–589, E11–584, and E11–577 in the presence of ADP and tested for DT formation by EMSA. The mobility of the DT complex is indicated. (C) The 84-bp ori probe was incubated with three levels (2.5, 5, and 10 ng) of the mutants E11–577 7 × N, E11–577, E11–605 G588A, E11–605 M591A, E11–605 T593A, E11–605 F594A, and E11–605 C596A in the presence of ADP and tested for DT formation. The mobility of the DT complex is indicated. (D) The 84-bp ori probe was incubated with three levels (2.5, 5, and 10 ng) of the mutants E11–577 7×N, E11–577, E11–584, E11–589, E11–592, E11–597, and E11–600 in the presence of ATP and tested for DH formation. The mobility of the DH complex is indicated. (E) The 84-bp ori probe was incubated with three levels (2.5, 5, and 10 ng) of the mutant proteins E11–577 7×A, E11–577, E11–605 G588A, E11–605 M591A, E11–605 T593A, E11–605 F594A, and E11–605 C596A and tested for DH formation in the presence of ATP by EMSA. (F) The 84-bp ori probe was incubated with three levels (2.5, 5, and 10 ng) of wt E1 or the mutants E11–605 579R/583R and E11–577 and tested for DT formation in the presence of ADP by EMSA. For the mutants E11–605 586R, E11–605 579R/583R/586R, and E11–605 579R/583R/586R/587R, four levels of E1 (1.2, 2.5, 5 and 10 ng) were used. (G) To determine whether the arginine substitutions in the acidic region caused structural defects, these mutants were tested for the ability to form the E12E22 complex: 1.2 and 2.5 ng of wt E1 or the mutant E11–605 586R, E11–605 579R/583R, E11–605 579R/583R/586R, or E11–605 579R/583R/586R/587R were incubated with the 84-bp ori probe and 2 ng of E2 and tested for E12E22 complex formation by EMSA. In lane 12, 1.2 ng of E11–577 was tested for E12E22 complex formation.
We next examined the mutant proteins for their ability to form the E1 DH by EMSA (Fig. 4D and E). When E1 is incubated with probe in the presence of ATP, the DT is slowly converted into a DH, in a process that requires local melting of the dsDNA (24, 25, 27). Interestingly, only one of the deletions, E11–600, was still able to form the DH, demonstrating that the C tail is critically important for DH formation (Fig. 4D, lanes 20 to 22). Results consistent with this finding were observed with point mutations in the C tail; proteins with mutations G588A, M591A, and T593A formed the DH (Fig. 4E, lanes 11 to 19). However, mutation of the conserved residues F594A (Fig. 4E, lanes 20 to 22) and C596A (Fig. 4E, lanes 23 to 25) resulted in failure to form the DH. Together, these results show that the C tail plays a specific role in the formation of the DH but plays no role in DT formation, while the acidic region is specifically required for DT formation.
To ascertain that the acidic region plays a role for DT formation also in the context of the intact E11–605, we introduced arginine substitutions into the acidic region (Fig. 4F). Because the acidic region contains many acidic residues and the alignment with other E1 proteins indicates that considerable sequence variation is allowed as long as the acidic nature is maintained, we generated radical substitutions in multiple positions. We generated the proteins with mutations D586R, D579R/D583R, D579R/D583R/E586R, and D579R/D583R/E586R/D587R in the context of E11–605. After expression and purification, we tested these mutants for DT formation by EMSA. As expected, the single mutation (E586R) had little effect on DT formation (Fig. 4F, lanes 8 to 11). The double mutant (D579R/D583R) could still form the DT but now also formed a larger complex (Fig. 4F, lanes 5 to 7), something that we never observed with wt E1. The triple and quadruple mutations were shown to cause severe defects for DT formation, generating complexes similar to those generated by E11–577 (Fig. 4F, compare lanes 12 to 19 and 20 to 22). These results demonstrate that the acidic region in the context of full-length E1 is necessary for formation of the DT.
To verify that the defects for DT formation were not caused by structural effects of the arginine substitutions, we tested the mutants for formation of a different complex that does not rely on the ability of E1 to oligomerize (Fig. 4G). E1 can together with the viral E2 protein form an E12E22 complex on the ori. Formation of this complex depends on site-specific DNA binding by both proteins as well as interactions between the E1 and E2 DBDs and between the E1 helicase domain and the activation domain of E2 (6, 9, 10, 41, 46). Clearly, all the arginine substitution mutants were capable of forming the E12E22 complex (Fig. 4G, lanes 4 to 11) as was E11–577, ruling out serious structural defects for these proteins.
The acidic region and the C tail are required for unwinding and in vitro DNA replication.
The EMSA whose results are displayed in Fig. 4 showed that mutation of the acidic region results in a defect for DT and DH formation. To provide functional analysis of mutations affecting the acidic region and the C tail, we utilized an unwinding assay. The E1 DT formed on the ori can locally melt the DNA, allowing formation of the E1 DH, which then unwinds the ori DNA and generates ssDNA (24, 25). We can detect the ssDNA generated in this process by the addition of E. coli SSB, which forms a complex with ssDNA that can be detected by EMSA. As expected, the deletion mutants E11–584, E11–589, and E11–592 (Fig. 5A, lanes 3 to 11), which fail to form the DH, lacked any detectable unwinding activity. E11–597 showed a trace of unwinding (lanes 12 to 14), while E11–600 and full-length E1 (E11–605) showed substantial activity (lanes 15 to 20). We next tested point mutations in the C-terminal tail and the acidic region for unwinding (Fig. 5B and C). Similar to the results for DH formation, G588A and M591A mutants showed wt levels of unwinding (Fig. 5B, compare lanes 3 to 5 to lanes 6 to 11). However, proteins with the point mutations T593A, F594A, and C596A showed only a faint trace of activity (Fig. 5B, lanes 12 to 20). E11–577 7×N (Fig. 5B, lanes 21 to 23) lacked activity altogether. The arginine substitutions in the acidic region had more-modest effects on unwinding. D586R showed a 2- to 4-fold reduction in unwinding (Fig. 5C, lanes 7 to 10), while D579R/D583R/D586R showed a 4-8-fold reduction effect for unwinding (Fig. 5C, compare lanes 3 to 6 to lanes 15 to 18). With the results shown in Fig. 5D, we demonstrated that the Sarkosyl treatment used to disrupt E1 complexes in panels A to C does not affect SSB-ssDNA complexes (compare lanes 2 and 3, 5 and 6, and 7 and 8).
As a final step in measuring the activity of the mutations in the C terminus of E1, we measured cell-free DNA replication in human cell extracts. As shown in Fig. 5E, E11–600 had close to wt activity (compare lanes 2 and 3) while all the remaining deletion mutants, E11–597, E11–592, E11–589, E11–584, and E11–577 (Fig. 5E, lanes 4 to 8) as well as the two F594A and C596A mutants (Fig. 5E, lanes 12 and 13) lacked activity altogether. Of the remaining point mutation proteins, the T593A mutant had a reduced activity (Fig. 5E, lane 11) while the G588A and M591A mutants (Fig. 5E, lanes 9 and 10) had close to wt activity.
As shown in Fig. 5F, we tested the activities of the arginine substitutions in the acidic region for in vitro replication. The E586R mutant had partial activity (Fig. 5F, lanes 8 to 10) while the D579R/D583R (Fig. 5F, lanes 5 to 7) and D579R/D583R/E586R (Fig. 5F, lanes 11 to 13) mutants lacked detectable replication activity. These results are generally consistent with the defects observed for complex formation and unwinding and show that the acidic region and the C tail are essential for viral DNA replication.
Our ability to knock out the acidic region with multiple point mutations indicated to us that it might be possible to test whether the C tail becomes dispensable when the acidic region is inactive. Such a result would strongly support the notion that the function of the C tail is to counteract the activity of the acidic region. We therefore generated the constructs E1308–605 579R 583R 586R, in which the acid region is knocked out, and E1308–605 579R 583R 586R C596A, in which both the acidic region and the C tail are knocked out. Surprisingly, we failed to purify E1308–605 579R 583R 586R due to poor solubility; however, E1308–605 579R 583R 586R C596A could readily be purified, and we tested this protein for the formation of hexamer on ssDNA as well as for ATPase activity (Fig. 5G). Strikingly, knocking out the acidic region in the context of E1308–605 C596A, which has a severe defect for hexamer formation (Fig. 5G, lanes 5 to 7), restores hexamer formation to wt levels (Fig. 5G, compare lanes 2 to 4 and 8 to 10). Similarly, E1308–605 C596A, which has a significant defect for ATPase activity, can be restored to wt activity by knocking out the acidic region (Fig. 5H, compare lanes 3, 4, and 5). Although these results are not completely conclusive since we were unable to measure that activity of the acidic region mutant alone, they clearly suggest that the defects of the mutants in the C tail are caused by the exposure of the acidic region.
The acidic region interacts with the oligomerization domain.
The results in Fig. 2 and 3 demonstrate that the acidic region is a negative regulator of E1 hexamer formation. An obvious possibility is that the acidic peptide can interact with the oligomerization domain and prevent the interaction between the oligomerization domains that is required for hexamer formation. To determine whether we could detect such an interaction, we performed pulldown assays using a GST fusion containing a C-terminal fragment from E1 including the acidic region and the C tail (GSTE1544–605). This protein interacted well with the in vitro-translated oligomerization and helicase domain fragment E1308–577 (Fig. 6A, lane 3). The interaction was entirely dependent on the acidic region as illustrated by the fact that the triple arginine substitution (D579R/D583R/E586R) in the acidic region caused loss of the ability to bring down the fragment (GST 544–605 3 × R [Fig. 6A, lane 2]). We observed a similar result using an in vitro-translated oligomerization domain fragment (E1308–410) (Fig. 6C, compare lanes 2 and 3), demonstrating that the oligomerization domain by itself is sufficient for the interaction with the acidic region.
ssDNA is a trigger for DH formation.
E1308–605 forms hexamers on ssDNA in the presence of either ADP or ATP, demonstrating that nucleotide hydrolysis is not necessary for hexamer formation (36, 42). DH formation with E11–605, however, requires ATP likely because the generation of ssDNA, which is required for DH formation, is dependent on ATP hydrolysis by the DT (24, 25). We reasoned that it might be possible to bypass the ATP dependence if we could bypass the local melting step. We therefore generated an ori probe containing a 6-bp single-stranded region at the position in the ori (nt 7928 to 7933) where the E1 helicase domain interacts with and melts the dsDNA (25–27). We then tested this “bubble probe” for complex formation in parallel with a completely double-stranded probe. On the completely double-stranded probe, wt E1 forms a DT in the presence of ADP, but formation of DH requires ATP (Fig. 6E, lanes 2 to 7). Strikingly, using the bubble probe, we could generate a DH complex also in the presence of ADP (Fig. 6E, lanes 9 to 11). This result indicates that in the presence of ssDNA, ATP hydrolysis is no longer necessary for DH formation, consistent with an ability of E1308–605 to form hexamers on ssDNA without ATP hydrolysis.
Our data indicate that the acidic region is required for DT formation because it inhibits hexamer formation. This inhibition clearly is conditional since prolonged incubation of the DT in the presence of ATP results in the transition to a DH. The timing of the first appearance of DH (∼10 to 12 min) correlates with the time required to generate local melting, indicating that the trigger for DH formation likely is ssDNA (25). Consistent with this idea, the DH forms instantly (in <1 min) on the bubble probe, where melting is not required (Fig. 6E, lanes 9 to 11).
Our data show that the C tail in the presence of ssDNA can counteract the inhibitory effect that the acidic region exerts on oligomerization, and a logical conclusion is therefore that it is the recognition of ssDNA by the C tail that triggers DH formation. We therefore tested whether the C tail C594A and F596A mutants could form the DH on the bubble probe in the presence of ADP (Fig. 6E, lanes 12 to 17). Interestingly, both of these mutants failed to form the DH, demonstrating that without the C tail, ssDNA no longer triggers DH formation. Although this result does not prove that the C tail recognizes ssDNA, it is clearly consistent with such a possibility. These data together provide strong evidence that ssDNA is the trigger for DH formation, likely through recognition by the C tail.
DISCUSSION
The E1 initiator protein combines multiple activities that are required for initiation of papillomavirus DNA replication. These activities are deployed sequentially and result in recognition of the ori by an E1-E2 complex, local melting of the ds viral DNA by an E1 DT complex, formation of an E1 DH helicase that encircles ssDNA, and eventually unwinding of the dsDNA. We have identified a short peptide in the C terminus of E1 that functions as a regulatory module in this process. The peptide contains a negative regulator of oligomerization, the acidic region, which promotes DT formation. Another part of the peptide, the C tail, can counteract the negative regulator and therefore promotes E1 oligomerization and DH formation.
We were initially interested in the function of the E1 C-terminal peptide because it is fairly well conserved but not visible in the crystal structures, indicating that it is not an integral part of the helicase domain. This prediction appears to be correct, because the acidic region and the C tail can be functionally replaced by 7 × N or 7 × A; the acidic region and the C tail clearly represent add-ons that do not affect the structure of the helicase domain.
Our mutational analysis of the C-terminal peptide in the context of the oligomerization and helicase domain (E1308–605) resulted in several surprising observations. The intact E1308–605 fragment readily formed the expected hexamer with ssDNA and had robust helicase and ATPase activities (Fig. 2 and 3). The shorter E1308–577 fragment, which corresponds to the fragment used for structure determination of the E1 hexamer in complex with ssDNA (34), failed to form a discrete complex and had poor helicase activity on long substrates as well as reduced ATPase activity. Strikingly, although this defect is due to the absence of the C-terminal module, the defect is not specific since addition of 7 × A or 7 × N at residue 577 restored wt activity to the fragment. We believe that the deletion to residue 577 may compromise folding in some way. This folding defect must be fairly subtle, however, since E1308–577 remains monomeric and does not aggregate in the absence of DNA (Fig. 1). Also, under crystallization conditions this protein clearly forms hexamers (34).
Because the C-terminal module is not visible in the E1 hexamer structure, Whelan et al. (49) performed small-angle X-ray scattering to determine the position of the C-terminal module in the hexamer. The authors found that it occupied a cleft between adjacent subunits in the ring, and based on electrostatic potential calculations the authors suggested that the C-terminal module may bridge adjacent subunits. Comparison of E1299–605 and E1292–579 showed that a 26-aa deletion that removes the C-terminal module resulted in a protein that was less capable of forming hexamers with ssDNA, had reduced ATPase activity, and especially on longer substrates had poor helicase activity compared to the intact protein. Based on these data, the authors suggested that the C-terminal module functions as a brace that holds the hexamer together during unwinding of dsDNA. In the hexameric T7 gene 4 helicase, a 31-aa N-terminal tail crosses over from one subunit and interacts with the adjacent subunit (47, 48).
The data that we present here clearly contradict a model in which the C-terminal module functions as a brace. The restoration of ATPase, DNA binding, and DNA helicase activities that can be accomplished by replacing the 28-residue C-terminal module with short oligopeptides (7 × N or 7 × A) clearly shows that neither the sequence nor the length of the peptide is critical for restoration of activity; and it is hard to imagine a brace without sequence dependence. Our interpretation, as stated above, is that the deletion of the whole C-terminal module results in folding problems that can be rescued by increasing the length of the polypeptide slightly.
Overall, the deletion mutant (E1299–579) used by Whelan et al. (49) showed defects similar to those that we observe in E1308–577, including a reduced ability to form hexamers, reduced processivity of the helicase, and reduced ATPase activity. The slight differences that do exist, e.g., E1299–579 forming a hexamer poorly while E1308–577 forms a complex in the well is likely due to subtle differences in the constructs and/or expression and purification procedures. In our hands, a mutant with a slightly smaller deletion than that of E1308–577 (E1308–581) produces a mixture of hexamers and complexes that stick in the well (data not shown).
We observe excellent correlation between hexamer formation by E1308–605 on ssDNA and DH formation on dsDNA by E11–605, indicating that these processes are closely related and that E1 oligomerizes in the same way in the hexamer and the DH (Fig. 7A). The critical component for forming the hexamer is the oligomerization domain, which is positioned immediately N terminal to the helicase domain and forms a tight structure that holds the hexamer together (34, 42). In the DH, the oligomerization domain likely adopts the same structure. We have less information about the structure of the DT complex, but based on biochemical analysis the DT is critically dependent on template length, while the formation of the DH is not. This is consistent with formation of planar rings in the hexamer and DH. In contrast, in the DT, E1 is likely to bind in a helical arrangement (24, 30, 34, 42). Because the oligomerization arrangement is unlikely to be shared between the hexamer and the DT, it is plausible that blocking of the oligomerization domain to prevent formation of hexamers and DHs would still allow the DT to form.
FIG 7.

Effects of mutations in the C-terminal module on E1 complex formation and model of control of oligomerization of E1 by the C-terminal module. (A) Summary of how point mutations and deletions in the C-terminal module affect hexamer (H), double trimer (DT), and double hexamer (DH) formation in the context of E1308–605 and E11–605. (B) Model for how the acidic region and C tail control oligomerization of E1. See the text for details.
Function of the acidic region and C tail in E1308–605.
We were surprised that small deletions from the C terminus (E1308–584, E1308–589, E1308–592, and E1308–597) resulted in proteins that behaved differently from either E1308–577, E1308–577 7×N, or E1308–605. The small deletions resulted in severe defects for hexamer formation and ATPase and helicase activity (Fig. 2 and 3). However, the defects were not due to formation of larger complexes (as in E1308–577) but were caused by a failure to form any complex, indicative of a defect in ssDNA binding and/or oligomerization. Comparison of the two proteins E1308–584 and E1308–577 7×N, which both have seven residues added after residue 577, shows that E1308–577 7×N in all respects behaved as E1308–605, while E1308–584 was defective for all the activities that reside in the helicase/oligomerization domain fragment (which are all dependent on hexamer formation) (Fig. 2 and 3). This result demonstrates, first, that the acidic region and the C tail are not required for hexamer formation in the context of E1308–605. Second, when present alone, the acidic region has a strong negative effect on hexamer formation. When both the acidic region and the C-terminal tail are present (as in E1308–605), this negative effect of the acidic region is neutralized, indicating that the C tail is a modulator of the acidic region and that the acidic region and the C tail likely function together. This notion is supported by the results in Fig. 5G and H, which indicate that mutation of the acidic region restores the activity to the C tail C596A mutant, consistent with a role for the C tail in blocking the activity of the acidic region.
Function of the acidic region and C tail in E11–605.
Because the acidic region and the C tail do not serve a specific function in the context of E1308–605, we examined mutations in these two regions in full-length E11–605. In the presence of ds ori DNA, which is the substrate for E11–605, we observed striking effects of removal of the acidic region and the C tail. E11–577 7 × N could still bind DNA but no longer formed discrete complexes (Fig. 4). Restoration of the acidic region, e.g., E11–584, E11–589, E11–592, and E11–597, resulted in formation of the discrete DT complex on dsDNA. This result establishes that the acidic region has to be present to form the DT. Based on the strong inhibitory effect of the acidic region on hexamer formation in the context of E1308–605, it is likely that the way the acidic region promotes DT formation is by inhibiting hexamer formation. We believe that the acidic region inhibits oligomerization by directly interacting with the oligomerization domain (Fig. 6A to D). Because the monomer of the E1 helicase domain in solution may look quite different from the monomer extracted from the hexamer structure, we cannot predict with any accuracy how such an interaction comes about.
Inactivation of the C tail also produces a very clear phenotype in E11–605. Point mutations or deletions in the C tail result in failure to form the DH complex, although DT formation is unaffected. In analogy with the function of the C tail in E1308–605, where the C tail clearly counteracts the inhibitory effect of the acidic region and promotes hexamer formation, the C tail in full-length E1 likely promotes DH formation by counteracting the acidic region. We can clearly rule out any direct effect of the C tail in hexamer formation, since the whole C-terminal module can be removed without affecting hexamer formation.
We can combine the data obtained from E1308–605 and from E11–605 and describe a comprehensive model for the function of the C-terminal module in viral DNA replication (Fig. 7B). Our data show that two short peptides, the acidic region and the C tail, together form a regulatory module, which is attached to the C terminus of the helicase domain. This module is not required for hexamer formation on ssDNA. In the absence of this module, E1308–577 7×N can use the intrinsic ability of the oligomerization and helicase domains to bind ssDNA and to form hexamers, an ability that is common to many helicases (Fig. 7B, row I). In full-length E1, however, the C-terminal module plays an essential role. The ability of E1 to perform local melting of dsDNA is critically dependent on the DT, which forms only in the presence of the acidic region. In the absence of the acidic region, ssDNA cannot be generated, and therefore the DH cannot form either.
If the C tail is removed or mutated in E1308–605, where the acidic region is present, we observe a strong inhibitory effect of the acidic region on hexamer formation with ssDNA (Fig. 7B, row II). Because we can detect a physical interaction between the acidic region and the oligomerization domain and because the oligomerization domain is required for hexamer formation, we believe that the acidic region inhibits oligomerization directly. If the C tail is removed or mutated in E11–605, where the acidic region is present, E1 is fully competent to form the DT complex. However, the DT cannot progress to DH since the C tail is not available to reverse the block to oligomerization for which the exposed acidic region is responsible.
When both the acidic region and the C tail are present in the context of E1308–605, the inhibitory effect of the acidic region is no longer observed, and in the presence of ssDNA, the hexamer is formed (Fig. 7B, row III). We believe that ssDNA triggers the C tail to counteract the inhibitory effect that the acidic region has on oligomerization. Full-length E1 first forms a DT, demonstrating that the acidic region, which promotes DT formation, is active. As local melting proceeds and ssDNA becomes available, the C tail recognizes the ssDNA and triggers DH formation.
In conclusion, our data clearly show that the C-terminal module, which is present in all papillomavirus E1 proteins, is an essential part of the machinery required for viral DNA replication. This module is an add-on to the helicase domain that modifies the activity of the E1 protein. The net result is that the acidic region and the C tail extend the functionality of E1, since in addition to forming the hexamer and DH helicases, which are intrinsic to the helicase and oligomerization domains, the presence of the acidic region allows the formation of the DT complex, which can melt dsDNA locally. The transition to the DH then requires the C tail. The critical importance of these two short peptides in the C-terminal module raises the possibility that these peptides could represent targets for antiviral compounds.
ACKNOWLEDGMENT
This work was supported by grant RO1 AI072345 to A.S.
REFERENCES
- 1.Patel SS, Picha KM. 2000. Structure and function of hexameric helicases. Annu Rev Biochem 69:651–697. doi: 10.1146/annurev.biochem.69.1.651. [DOI] [PubMed] [Google Scholar]
- 2.Singleton MR, Dillingham MS, Wigley DB. 2007. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem 76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 3.Neuwald AF, Aravind L, Spouge JL, Koonin EV. 1999. AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9:27–43. [PubMed] [Google Scholar]
- 4.Bergvall M, Melendy T, Archambault J. 2013. The E1 proteins. Virology 445:35–56. doi: 10.1016/j.virol.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blitz IL, Laimins LA. 1991. The 68-kilodalton E1 protein of bovine papillomavirus is a DNA binding phosphoprotein which associates with the E2 transcriptional activator in vitro. J Virol 65:649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillitzer E, Chen G, Stenlund A. 2000. Separate domains in E1 and E2 proteins serve architectural and productive roles for cooperative DNA binding. EMBO J 19:3069–3079. doi: 10.1093/emboj/19.12.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen G, Stenlund A. 2001. The E1 initiator recognizes multiple overlapping sites in the papillomavirus origin of DNA replication. J Virol 75:292–302. doi: 10.1128/JVI.75.1.292-302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen G, Stenlund A. 1998. Characterization of the DNA-binding domain of the bovine papillomavirus replication initiator E1. J Virol 72:2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sedman J, Stenlund A. 1995. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J 14:6218–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berg M, Stenlund A. 1997. Functional interactions between papillomavirus E1 and E2 proteins. J Virol 71:3853–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang L, Li R, Mohr IJ, Clark R, Botchan MR. 1991. Activation of BPV-1 replication in vitro by the transcription factor E2. Nature 353:628–632. doi: 10.1038/353628a0. [DOI] [PubMed] [Google Scholar]
- 12.Stenlund A. 2003. E1 initiator DNA binding specificity is unmasked by selective inhibition of non-specific DNA binding. EMBO J 22:954–963. doi: 10.1093/emboj/cdg091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seo YS, Muller F, Lusky M, Gibbs E, Kim HY, Phillips B, Hurwitz J. 1993. Bovine papilloma virus (BPV)-encoded E2 protein enhances binding of E1 protein to the BPV replication origin. Proc Natl Acad Sci U S A 90:2865–2869. doi: 10.1073/pnas.90.7.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lusky M, Hurwitz J, Seo YS. 1994. The bovine papillomavirus E2 protein modulates the assembly of but is not stably maintained in a replication-competent multimeric E1-replication origin complex. Proc Natl Acad Sci U S A 91:8895–8899. doi: 10.1073/pnas.91.19.8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lusky M, Fontane E. 1991. Formation of the complex of bovine papillomavirus E1 and E2 proteins is modulated by E2 phosphorylation and depends upon sequences within the carboxyl terminus of E1. Proc Natl Acad Sci U S A 88:6363–6367. doi: 10.1073/pnas.88.14.6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Enemark EJ, Stenlund A, Joshua-Tor L. 2002. Crystal structures of two intermediates in the assembly of the papillomavirus replication initiation complex. EMBO J 21:1487–1496. doi: 10.1093/emboj/21.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Enemark EJ, Chen G, Vaughn DE, Stenlund A, Joshua-Tor L. 2000. Crystal structure of the DNA binding domain of the replication initiation protein E1 from papillomavirus. Mol Cell 6:149–158. doi: 10.1016/S1097-2765(05)00016-X. [DOI] [PubMed] [Google Scholar]
- 18.Yasugi T, Benson JD, Sakai H, Vidal M, Howley PM. 1997. Mapping and characterization of the interaction domains of human papillomavirus type 16 E1 and E2 proteins. J Virol 71:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sarafi TR, McBride AA. 1995. Domains of the BPV-1 E1 replication protein required for origin-specific DNA binding and interaction with the E2 transactivator. Virology 211:385–396. doi: 10.1006/viro.1995.1421. [DOI] [PubMed] [Google Scholar]
- 20.Muller F, Sapp M. 1996. Domains of the E1 protein of human papillomavirus type 33 involved in binding to the E2 protein. Virology 219:247–256. doi: 10.1006/viro.1996.0242. [DOI] [PubMed] [Google Scholar]
- 21.Park P, Copeland W, Yang L, Wang T, Botchan MR, Mohr IJ. 1994. The cellular DNA polymerase alpha-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proc Natl Acad Sci U S A 91:8700–8704. doi: 10.1073/pnas.91.18.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Titolo S, Pelletier A, Sauve F, Brault K, Wardrop E, White PW, Amin A, Cordingley MG, Archambault J. 1999. Role of the ATP-binding domain of the human papillomavirus type 11 E1 helicase in E2-dependent binding to the origin. J Virol 73:5282–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masterson PJ, Stanley MA, Lewis AP, Romanos MA. 1998. A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase alpha-primase p68 subunit. J Virol 72:7407–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuck S, Stenlund A. 2005. Assembly of a double hexameric helicase. Mol Cell 20:377–389. doi: 10.1016/j.molcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 25.Schuck S, Stenlund A. 2011. Mechanistic analysis of local ori melting and helicase assembly by the papillomavirus E1 protein. Mol Cell 43:776–787. doi: 10.1016/j.molcel.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuck S, Stenlund A. 2007. ATP-dependent minor groove recognition of TA base pairs is required for template melting by the E1 initiator protein. J Virol 81:3293–3302. doi: 10.1128/JVI.02432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Schuck S, Stenlund A. 2007. Adjacent residues in the E1 initiator beta-hairpin define different roles of the beta-hairpin in Ori melting, helicase loading, and helicase activity. Mol Cell 25:825–837. doi: 10.1016/j.molcel.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Sanders CM, Stenlund A. 1998. Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalysed by a transcription factor. EMBO J 17:7044–7055. doi: 10.1093/emboj/17.23.7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sedman J, Stenlund A. 1996. The initiator protein E1 binds to the bovine papillomavirus origin of replication as a trimeric ring-like structure. EMBO J 15:5085–5092. [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X, Schuck S, Stenlund A. 2010. Structure-based mutational analysis of the bovine papillomavirus E1 helicase domain identifies residues involved in the nonspecific DNA binding activity required for double trimer formation. J Virol 84:4264–4276. doi: 10.1128/JVI.02214-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fouts ET, Yu X, Egelman EH, Botchan MR. 1999. Biochemical and electron microscopic image analysis of the hexameric E1 helicase. J Biol Chem 274:4447–4458. doi: 10.1074/jbc.274.7.4447. [DOI] [PubMed] [Google Scholar]
- 32.Seo YS, Muller F, Lusky M, Hurwitz J. 1993. Bovine papilloma virus (BPV)-encoded E1 protein contains multiple activities required for BPV DNA replication. Proc Natl Acad Sci U S A 90:702–706. doi: 10.1073/pnas.90.2.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang L, Mohr I, Fouts E, Lim DA, Nohaile M, Botchan M. 1993. The E1 protein of bovine papilloma virus 1 is an ATP-dependent DNA helicase. Proc Natl Acad Sci U S A 90:5086–5090. doi: 10.1073/pnas.90.11.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Enemark EJ, Joshua-Tor L. 2006. Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 35.Lee SJ, Syed S, Enemark EJ, Schuck S, Stenlund A, Ha T, Joshua-Tor L. 2014. Dynamic look at DNA unwinding by a replicative helicase. Proc Natl Acad Sci U S A 111:E827–E835. doi: 10.1073/pnas.1322254111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sedman J, Stenlund A. 1998. The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J Virol 72:6893–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clower RV, Fisk JC, Melendy T. 2006. Papillomavirus E1 protein binds to and stimulates human topoisomerase I. J Virol 80:1584–1587. doi: 10.1128/JVI.80.3.1584-1587.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han Y, Loo YM, Militello KT, Melendy T. 1999. Interactions of the papovavirus DNA replication initiator proteins, bovine papillomavirus type 1 E1 and simian virus 40 large T antigen, with human replication protein A. J Virol 73:4899–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castella S, Burgin D, Sanders CM. 2006. Role of ATP hydrolysis in the DNA translocase activity of the bovine papillomavirus (BPV-1) E1 helicase. Nucleic Acids Res 34:3731–3741. doi: 10.1093/nar/gkl554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanders CM. 2008. A DNA-binding activity in BPV initiator protein E1 required for melting duplex ori DNA but not processive helicase activity initiated on partially single-stranded DNA. Nucleic Acids Res 36:1891–1899. doi: 10.1093/nar/gkn041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbate EA, Berger JM, Botchan MR. 2004. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes Dev 18:1981–1996. doi: 10.1101/gad.1220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanders CM, Kovalevskiy OV, Sizov D, Lebedev AA, Isupov MN, Antson AA. 2007. Papillomavirus E1 helicase assembly maintains an asymmetric state in the absence of DNA and nucleotide cofactors. Nucleic Acids Res 35:6451–6457. doi: 10.1093/nar/gkm705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sedman T, Sedman J, Stenlund A. 1997. Binding of the E1 and E2 proteins to the origin of replication of bovine papillomavirus. J Virol 71:2887–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maiyar AC, Leong ML, Firestone GL. 2003. Importin-alpha mediates the regulated nuclear targeting of serum- and glucocorticoid-inducible protein kinase (Sgk) by recognition of a nuclear localization signal in the kinase central domain. Mol Biol Cell 14:1221–1239. doi: 10.1091/mbc.E02-03-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, Stenlund A. 2010. Mutations in Sensor 1 and Walker B in the bovine papillomavirus E1 initiator protein mimic the nucleotide-bound state. J Virol 84:1912–1919. doi: 10.1128/JVI.01756-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen G, Stenlund A. 2000. Two patches of amino acids on the E2 DNA binding domain define the surface for interaction with E1. J Virol 74:1506–1512. doi: 10.1128/JVI.74.3.1506-1512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singleton MR, Sawaya MR, Ellenberger T, Wigley DB. 2000. Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell 101:589–600. doi: 10.1016/S0092-8674(00)80871-5. [DOI] [PubMed] [Google Scholar]
- 48.Sawaya MR, Guo S, Tabor S, Richardson CC, Ellenberger T. 1999. Crystal structure of the helicase domain from the replicative helicase-primase of bacteriophage T7. Cell 99:167–177. doi: 10.1016/S0092-8674(00)81648-7. [DOI] [PubMed] [Google Scholar]
- 49.Whelan F, Stead JA, Shkumatov AV, Svergun DI, Sanders CM, Antson AA. 2012. A flexible brace maintains the assembly of a hexameric replicative helicase during DNA unwinding. Nucleic Acids Res 40:2271–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]