

ABSTRACT
Measles virus (MV) infection is undergoing resurgence and remains one of the leading causes of death among young children worldwide despite the availability of an effective measles vaccine. MV infects its target cells by coordinated action of the MV H and the fusion (F) envelope glycoprotein; upon receptor engagement by H, the prefusion F undergoes a structural transition, extending and inserting into the target cell membrane and then refolding into a postfusion structure that fuses the viral and cell membranes. By interfering with this structural transition of F, peptides derived from the heptad-repeat (HR) regions of F can potently inhibit MV infection at the entry stage. We show here that specific features of H's interaction with its receptors modulate the susceptibility of MV F to peptide fusion inhibitors. A higher concentration of inhibitory peptides is required to inhibit F-mediated fusion when H is engaged to its nectin-4 receptor than when H is engaged to its CD150 receptor. Peptide inhibition of F may be subverted by continued engagement of receptor by H, a finding that highlights the ongoing role of H-receptor interaction after F has been activated and that helps guide the design of more potent inhibitory peptides. Intranasal administration of these peptides results in peptide accumulation in the airway epithelium with minimal systemic levels of peptide and efficiently prevents MV infection in vivo in animal models. The results suggest an antiviral strategy for prophylaxis in vulnerable and/or immunocompromised hosts.
IMPORTANCE Measles virus (MV) infection causes an acute illness that may be associated with infection of the central nervous system (CNS) and severe neurological disease. No specific treatment is available. We have shown that parenterally delivered fusion-inhibitory peptides protect mice from lethal CNS MV disease. Here we show, using established small-animal models of MV infection, that fusion-inhibitory peptides delivered intranasally provide effective prophylaxis against MV infection. Since the fusion inhibitors are stable at room temperature, this intranasal strategy is feasible even outside health care settings, could be used to protect individuals and communities in case of MV outbreaks, and could complement global efforts to control measles.
INTRODUCTION
Infection by measles virus (MV) remains one of the leading causes of death among young children worldwide (1) despite the availability of an effective measles vaccine. MV was considered to be eliminated in the United States in 2000 (defined as interruption of continuous transmission lasting ≥12 months) (2) but to be a problem in developing countries (3). In 2001, several global partners—the American Red Cross, United Nations Foundation, U.S. Centers for Disease Control and Prevention, UNICEF, and World Health Organization—launched the Measles Initiative with the aim of interrupting MV transmission in large geographic areas and reducing measles deaths by 90% before 2010 by increasing vaccination coverage (1, 4). However, while about 71% reduction in mortality resulting from measles was achieved between 2000 and 2011 globally, in 2012 there were 122,000 measles deaths, and we have experienced a resurgence of measles disease in developed countries. MV is periodically imported into the United States, leading to local outbreaks (5). During 2011, 222 measles cases and 17 measles outbreaks were reported in the United States (1, 6–8), and the situation has deteriorated since then, causing a significant economic burden on health institutions (9). The 2014 MV outbreak was the biggest to occur in the United States since 1996 (10).
The outbreaks in developed countries are often attributed to lack of vaccination; however, a significant number of cases occur in previously vaccinated people (11, 12). The current MV vaccine is effective; however, the immune response to it varies significantly. Up to 10% of people do not develop adequate protective antibodies after the recommended two doses of vaccine, in contrast to the long-standing immunity elicited by the natural infection. Vaccine-elicited immunity may also wane over time (13–15); of the 98 cases of MV in a 2011 outbreak in Canada, over one-half had received two doses of MV vaccine (5, 12). In the absence of natural infection, the population of highly immunized countries may be susceptible to larger outbreaks (5, 16).
MV infection starts in the respiratory tract, where alveolar macrophages and dendritic cells are the primary CD150-expressing targets (17–20). The binding of the MV receptor-binding protein hemagglutinin (H) to CD150, accompanied by membrane fusion mediated by the MV fusion (F) protein, leads to infection of these cells. The first MV-infected cells then transmit the virus to bronchus-associated lymphoid tissues and/or draining lymph nodes, where the virus proliferates in B and T lymphocytes (which also express CD150), and viremia ensues (17, 21). The adherent junction protein PVRL4 (or nectin 4) (22–28) MV receptor on the basolateral surface of the respiratory epithelial cells has been implicated in viral transmission at later stages of pathogenesis but is likely not involved at the early stages (21, 27).
Coordinated action of the MV H and F envelope glycoproteins is essential for viral entry. The trimeric F structure is kinetically trapped in a metastable conformation, primed for fusion activation upon engagement of H by a cell surface receptor, either CD150 (SLAM) or nectin 4 (22–28). After receptor engagement by H, the prefusion F undergoes a structural transition, extending and inserting its hydrophobic “fusion peptide” into the target cell membrane. From this point on, the entry process is thought to be driven by the refolding of F into a “trimer of hairpin” postfusion structure that brings together and fuses the viral and cell membranes (29–36). Peptides derived from the heptad repeat (HR) regions of F can potently inhibit MV infection at this early stage (37–39) by interfering with this structural transition of F.
Peptide therapeutics are generally not transported from the blood to the brain, failing to cross the brain capillary endothelial wall that makes up the blood-brain barrier (BBB) in vivo (40), and this lack of brain distribution has precluded potential benefits, for example, in the treatment of HIV-1 dementia and viral encephalitis (41). For HIV-1, the peptide fusion inhibitor enfuvirtide is used as a salvage therapy for multidrug-resistant HIV-1-infected patients; however, its clinical use is limited by a short half-life, poor biodistribution properties, and the parenteral route of delivery (39). We have recently shown that MV F-derived cholesterol-conjugated C-terminal heptad repeat (HRC) peptides have an extended in vivo half-life and can cross the BBB after subcutaneous (s.q.) injection (42). The modified MV peptide inhibitors presented in this paper protect against severe MV infection and its complications in SLAM transgenic mice, an established model for MV encephalitis. Simultaneous s.q. and intranasal (i.n.) administration of peptides was highly effective at blocking MV infection (42). While peptidic compounds would not be ideal candidates for development as anti-MV therapeutic agents if they need to be given parenterally, we propose that simple intranasal delivery of peptides blocks MV infection at its earliest stages. Using two different animal models of infection, we demonstrate that i.n. delivered peptides prevent MV infection in vivo. Peptides cross the pseudostratified epithelium of the lungs to reach the basolateral aspect of the cells and thus reach the site of nectin 4, which is the receptor involved in MV shedding. We speculate that interaction with MV at the nectin-4 site may be responsible for decreasing transmission of MV.
Since these modified MV fusion inhibitors are stable at room temperature, intranasal administration of the agents will make the proposed strategy feasible even outside health care settings: in the field or at home. As a complement to vaccination in global efforts to eradicate measles, this intranasal strategy would be used to protect individuals and communities in case of MV outbreaks.
MATERIALS AND METHODS
Plasmids and reagents.
The genes of MV G954 wild-type (wt) H and wt F were codon optimized, synthesized, and subcloned into the mammalian expression vector pCAGGS. The constructs for SLAM and nectin 4 were commercially acquired. Anti-MV hemagglutinin cl.55 neutralizing antibody, which blocks interaction between H and CD150, has been previously described (43–45).
Peptide synthesis.
All peptides were produced by standard 9-fluorenylmethoxy carbonyl (Fmoc) solid-phase methods. The cholesterol moieties were attached to the peptides via chemoselective reaction between the thiol group of an extra cysteine residue, added C terminally to the sequence, and a bromoacetyl derivative of cholesterol or a bis-maleimide functionalized cholesterol core, as previously described (58–60). Peptides were stored as powder, and before use they were dissolved in dimethyl sulfoxide (DMSO) to 5 mM stock solutions that were kept at −80°C for the in vitro experiment. For the in vivo experiments in cotton rats (CR), peptides were dissolved in DMSO to 96 mg/ml and then diluted in water for injection (Hospira, Inc., Lake Forest, IL, USA) to 12 mg/ml and stored at −80°C. For the experiment in mice, peptides were dissolved in DMSO to 50 mg/ml and stored at −80°C. Peptides were diluted in water for injection.
Protease sensitivity of MV- and HPIV3-derived peptides.
For the trypsin digestion, 1 μg of each peptide was treated with 0.1 μg or 0.5 μg of trypsin in 10 μl of 1× phosphate-buffered saline (PBS). Peptide solutions were then incubated at 0°C, 22°C, or 37°C for 30 min. Following incubation, 10 μl of Laemmli's SDS reducing buffer was added to each solution. Samples were boiled for 10 min at 99°C and then run on a 4 to 20% Tris-glycine (TG) gel at 120 V. The gel was allowed to fix overnight in 0.0125% glutaraldehyde in PBS. Gels were stained using a Pierce silver stain kit (catalog number 24600).
Transient expression of H and F genes.
Transfections were performed in 293T cells according to the Lipofectamine 2000 manufacturer's protocols (Invitrogen).
Cells and viruses.
293T (human kidney epithelial), BHK (baby hamster kidney), and Vero-SLAM (African green monkey kidney) cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco, Invitrogen) supplemented with 10% fetal bovine serum (FBS) and antibiotics in 5% CO2. The Vero-SLAM culture medium was supplemented with Geneticin. Wild-type MV strain G954 (genotype B3.2) was isolated in Gambia in 1993 (27) (BB-0033-00053). Recombinant MV IC323, expressing enhanced green fluorescent protein (EGFP) (MV-IC323-EGFP) (28), was kindly provided by Y. Yanagi (Kyushu university, Fukuoka, Japan). The WTFb strain for cotton rat infection was described in reference 46. All virus strains were propagated and titrated in Vero-SLAM cells.
Viral entry assay.
Vero-SLAM cell monolayers were incubated with 40 to 50 PFU of wild-type MV G954 in the presence of various concentrations of peptides. After 90 min, 2× minimal essential medium containing 10% FBS was mixed with 1% Avicell and added to the dishes. The plates were then incubated at 37°C for 72 h. After removing the medium overlay, the cells were immunostained for plaque detection. The number of plaques in the control (no peptide) and experimental wells were counted under a dissecting stereoscope.
HAE cultures.
The EpiAirway AIR-100 system (MatTek Corporation) used in these experiments consists of normal, human-derived tracheobronchial epithelial cells that have been cultured to form a pseudostratified, highly differentiated mucociliary epithelium. Upon receipt from the manufacturer, human airway epithelium (HAE) cultures were handled as previously described (47–49); we have used HAE to test several entry inhibitors (48–52). To assess the potential for delivery of the HRC4 peptides via the airway to the basolateral compartment, 20 μl of 250 μM peptide inhibitor was applied to the apical surface. At the indicated time point, aliquots of the basolateral supernatant fluid were collected to quantify the amount of peptide delivered to the basolateral reservoir. The 20 μl of 250 μM in the 1 ml of reservoir would have had an upper-limit final concentration of 5 μM.
β-Gal complementation-based fusion assay.
The beta-galactosidase (β-Gal) complementation-based fusion assay was performed as described previously (53, 54). Briefly, 293T cells transiently transfected with SLAM and the omega reporter subunit were incubated with cells coexpressing viral glycoproteins (MV H and MV F) and the alpha reporter subunit.
ELISA.
For biodistribution studies, each organ was weighed and mixed in PBS (1:1, wt/vol) using an Ultra-Turrax homogenizer. Samples were then treated with acetonitrile–1% trifluoroacetic acid (TFA), 1:4 (vol/vol), for 1 h on a rotor at 4°C and then centrifuged for 10 min at 8,000 rpm. Supernatant fluids were collected, and peptide concentration was determined using an enzyme-linked immunosorbent assay (ELISA). Maxisorp 96-well plates (Nunc) were coated overnight with purified rabbit anti-MV F HRC antibodies (5 μg/ml) in carbonate/bicarbonate buffer, pH 7.4. Plates were washed twice using PBS followed by incubation with 3% bovine serum albumin (BSA) in PBS (blocking buffer) for 30 min. The blocking buffer was replaced with 2 dilutions of each sample in 3% PBS–BSA in duplicate, and the mixture was incubated for 90 min at room temperature (RT). After multiple washes in PBS, the peptide was detected using a horseradish peroxidase (HRP)-conjugated rabbit custom-made anti-MV F HRC antibody (1:1,500) in blocking buffer for 2 h at RT. HRP activity was recorded as absorbance at 492 nm on the Sigmafast OPD substrate system (Sigma-Aldrich) after adding the stop solution. Standard curves were established for each peptide (using the same ELISA conditions as for the test samples), and the detection limit was determined to be 0.15 nM.
Determination of MV-specific antibodies in murine serum by ELISA.
Sera were taken from infected mice at the end of the protocol by intracardiac puncture in EDTA Vacutainer tubes and tested for anti-MV antibodies by ELISA as previously described (45). Briefly, MV nucleoprotein obtained from baculovirus-infected insect cells and purified was coated onto 96-well ELISA plates overnight (1 μg/well). Plates were blocked using 1× PBS–0.05% Tween–5% milk, and sera were tested in several dilutions by use of goat anti-mouse horseradish peroxidase-conjugated antibody (Ab) (A3673; Sigma) and revealed with o-phenylenediamine substrate, measuring absorbance at 490 to 650 nm after stopping in 12% H2SO4. The titer of N-specific antibodies in each serum sample was determined using a standard curve established with sera from mice immunized with MV in complete Freund adjuvant and expressed in relative units. The limit of detection of the test was close to 1:100,000 in the positive control.
Immunofluorescence/immunohistochemistry.
Seven-day-old suckling SLAM TG mice (on a C57/BL6 background) received MV HRC4 (6 mg/kg of body weight) i.n. in 20 μl of diluent. The animals were sacrificed 8 h posttreatment, and lungs were collected and frozen with cold isopentane on dry ice. The cryosections were dried for 30 min and fixed in a 4% formalin solution. After multiple washes in PBS, saturation was performed using in PBS–4% FBS (30 min at RT) before incubation with specific rabbit anti-MV HRC antibody (overnight at 4°C) in PBS–4% FBS. After multiple washes, tissue sections were incubated with the secondary goat anti-rabbit Ab conjugated with Alexa Fluor 555 in PBS–4% FBS (2 h at RT). The nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). After multiple washes in PBS, mounting was performed using Fluoroprep (bioMérieux). Lung sections were analyzed using an Axioplan 2 imaging microscope (Zeiss).
Infection of mice.
SLAM transgenic (tg) mice (45, 55), on a C57/BL6 background, were bred at the institute's animal facility (Plateau de Biologie Expérimentale de la Souris [PBES], ENS-Lyon) as heterozygotes for SLAM transgenes. One-week-old mice were infected i.n. with 10 μl of MV G954 in both nares (500 PFU of MV/mouse). SLAM tg mice were given 6 mg/kg of MV HRC4 peptide i.n. 24 h before infection and on the day of infection. For parenteral delivery, mice were injected daily s.q. with 6 mg/kg of MV HRC4 peptide, from 24 h before infection up to 7 days postinfection.
SLAM tg mice crossed in an alpha/beta interferon (IFN-α/β) receptor (IFNAR)-deficient background; SLAM × IFNAR mice (55, 56) were bred at the institute's animal facility (PBES, ENS-Lyon) as heterozygotes for SLAM transgenes. Mice were infected i.n. with 10 μl of MV G954 in both nares (20,000 PFU of MV/mouse, ∼50 50% lethal doses [LD50]) and were given 6 mg/kg of MV HRC4 peptide i.n. 24 h and 6 h before infection. Control mice received parallel administrations of the diluent. All animals were observed daily for 28 days, and those showing clinical signs (neurological symptoms, ataxia, lethargy) were euthanized. The protocol was approved by the Regional Ethical Committee (CECCAPP protocol N° ENS-2011-003 and ENS-2012-041).
Infection of cotton rats and virus titer determination and treatment.
Inbred cotton rats (Sigmodon hispidus) were purchased from Harlan Laboratories, Inc., Indianapolis, IN. Both male and female cotton rats at the age of 5 to 7 weeks were used.
For i.n. infection, 105 50% tissue culture infective dose (TCID50) of MV WTFb was inoculated in phosphate-buffered saline to isoflurane-anesthetized cotton rats in a volume of 100 μl. To evaluate the effect of HRC peptides, animals were inoculated i.n. with peptide (5 mg/kg in 100 μl of water) 24 h and 12 h before infection. Four days after infection, the animals were asphyxiated using CO2 and their lungs were collected and weighed. Lung tissue was minced with scissors and Dounce homogenized with a glass homogenizer. Serial 10-fold dilutions of supernatant fluids were assessed for the presence of infectious virus in 48-well plates, using cytopathic effect (CPE) in Vero-SLAM cells as the endpoint. Plates were scored for CPE microscopically after 7 days. The amount of virus in inocula was expressed as fold dilutions that resulted in the infection of 50% of inoculated tissue culture monolayers (TCID50). The TCID50 was calculated as described previously (57). The animal experiments were approved by the Institutional Animal Care and Use Committee of Ohio State University.
Biodistribution analysis.
SLAM TG mice, on the background of C57/BL6, received MV HRC4 (6 mg/kg) i.n. in 20 μl of diluent. After 8 h, blood was collected by intracardiac puncture in EDTA Vacutainer tubes, and sera were conserved at −20°C until their use in ELISA.
For biodistribution experiments in cotton rats, the animals received the indicated peptides (6 mg/kg) i.n. in 100 μl of diluent. After 8 h, blood was collected by intracardiac puncture in EDTA Vacutainer tubes, and sera were conserved at −20°C until their use in ELISA. Organs from each animal were collected and conserved at −80°C. The protocol was approved by the Regional Ethical Committee (CECCAP protocol N°ENS-2012-042).
Statistical analysis.
Data are expressed as means and standard deviations (SD). Statistical analyses were performed using a Mann-Whitney U test and a Mantel-Cox test and GraphPad Prism 5 software.
RESULTS
HPIV3 F-derived HRC peptides inhibit wt MV entry.
We have shown that HRC peptides derived from human parainfluenza virus type 3 (HPIV3) F inhibit Nipah virus (NiV) infection and NiV-mediated fusion between infected cells (51) and that conjugating these peptides to a cholesterol moiety increases their potency. In a previous report, HPIV3-derived peptides were also shown to have a 50% inhibitory concentration (IC50) of ∼2 μM against MV in vitro (42). Here we tested whether HPIV3 HRC-derived peptides with and without lipid moieties inhibit MV viral entry (Table 1 and references 51, 58, and 59). The inhibitory activities of VG, VGPEG4-chol, [VG-PEG4]2-chol, and VIKIPEG4-chol (Table 1) against wt MV G954 were assessed in plaque reduction assays (Fig. 1). VG peptides containing the cholesterol moiety (VGPEG4-chol and [VGPEG4]2-chol) and VIKIPEG4-chol performed better than the unconjugated peptide (VG). While for the unconjugated peptide the IC50s were ∼6 μM (VG), for the cholesterol-conjugated peptides the IC50s were all 100-fold lower and similar to each other: the IC50 for VGPEG4-chol was ∼0.04 μM, that for [VG-PEG4]2-chol was ∼0.02 μM, and that for VIKIPEG4-chol was ∼0.01 μM. Similar results showing that the cholesterol moiety increased potency of the HPIV3-derived peptides were obtained with strain WTFb (data not shown). MV viral entry in cell lines expressing SLAM was blocked by HPIV3-derived peptides.
TABLE 1.
Sequences and modifications of HPIV3 and MV HRC-derived peptides
| Peptide | Sequence and modifications |
|---|---|
| MV HRC1a | Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(CH2CONH2)-NH2 |
| MV HRC2a | Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(PEG4-Chol)-NH2 |
| MV HRC3a | [Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(MAL-PEG11)-NH2]2 |
| MV HRC4a | [Ac-PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG-C-(MAL-PEG4)NH2]2-Chol |
| VGb | Ac-VALDPIDISIVLNKAKSDLEESKEWIRRSNGKLDSI-GSGSG-C-(CH2CONH2)-NH2 |
| VGPEG4-cholb | Ac-VALDPIDISIVLNKAKSDLEESKEWIRRSNGKLDSI-GSGSG-C-(PEG4-Chol)-NH2 |
| [VGPEG11]2b | [Ac-VALDPIDISIVLNKAKSDLEESKEWIRRSNGKLDSI-GSGSG-C-(MAL-dPEG11)-NH2]2 |
| [VGPEG4]2-cholb | [Ac-VALDPIDISIVLNKAKSDLEESKEWIRRSNGKLDSI-GSGSG-C-(MAL-PEG4)-NH2]2-Chol |
| VIKIPEG4-cholc | Ac-VALDPIDISIVLNKIKSDLEESKEWIRRSNKILDSI-GSGSG-C-(PEG4-Chol)-NH2 |
FIG 1.
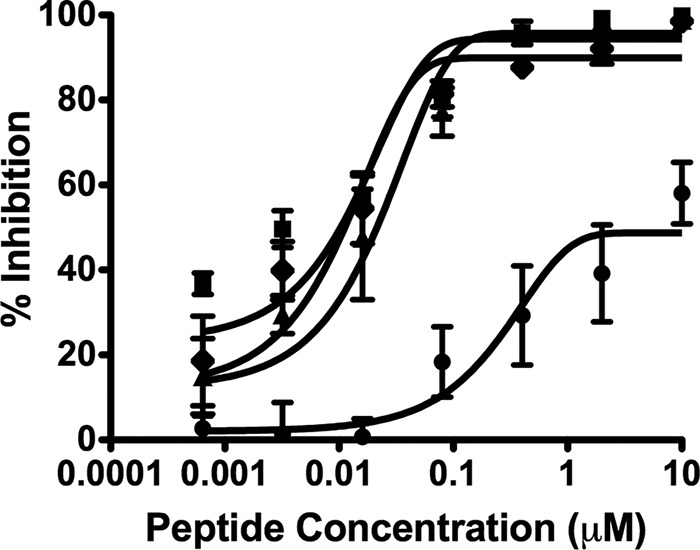
Inhibition of MV entry by HPIV3 F-derived HRC peptides. Vero-SLAM cell monolayers were infected with wild-type MV G954 in the presence of peptides VG (circles), VGPEG4-chol (squares), [VGPEG4]2-chol (triangles), and VIKIPEG4-chol (diamonds) at the indicated concentrations. Viral entry was assessed by plaque assay. Results are presented as percent reduction in plaque number (y axis) compared to the absence of treatment, as a function of compound concentration (x axis). Each point represents the mean (± standard error) of results from four experiments.
HPIV3 F-derived HRC peptides inhibit wt MV H/F-mediated fusion.
In vitro, we previously assessed whether MV-derived peptides block MV fusion over time at 2, 4, and 6 h. Only the dimeric peptide with cholesterol (MV HRC 4) was inhibitory at the 6 h time point, suggesting that it may have greater antiviral potential (42). HPIV3-derived peptides were tested here for their ability to inhibit fusion using a beta-galactosidase (β-Gal) complementation assay (54). This assay measures the fusion of cells that express viral envelope glycoproteins (MV G954 H/F) with cells that express the MV receptor SLAM (Fig. 2). The HPIV3 peptides inhibited fusion at early time points (1 h) (Fig. 2A, B, and C); less inhibition was seen later (4 h) (Fig. 2D, E, and F) for all peptide concentrations used (0.1 μM, 1 μM, and 10 μM). Almost no inhibition was observed at 6 h (Fig. 2G, H, and I), with the exception of the dimeric [VGPEG4]2-chol, which inhibits fusion by 50% at the concentration of 10 μM (Fig. 2I).
FIG 2.

Inhibition of MV H/F-mediated fusion by HPIV3 F-derived HRC peptides. Fusion of MV H/F-coexpressing cells with SLAM-bearing cells in the presence of VG, VGPEG4-chol, [VGPEG4]2-chol, and VIKIPEG4-chol as indicated at 0.1 μM (A, D, and G), 1 μM (B, E, and H), and 10 μM (C, F, and I) was quantitated at 1 h (A, B, and C), 4 h (D, E, and F), or 6 h (G, H, and I), using a β-galactosidase complementation assay. Results are presented as percent reduction in luminescence (y axis) compared with no treatment. Each point is the mean (± standard error) of results from 3 experiments.
Protease sensitivity of cholesterol-tagged peptides.
A decrease in fusion inhibitory potency over time was observed for the MV peptides as well (42). We considered the possibility that the observed loss of fusion inhibition was due to degradation of the peptides over time during the experiment. To assess the relative protease sensitivity of HPIV3- and MV-derived peptides, we subjected the peptides to trypsin digestion, followed by SDS-PAGE analysis of the reaction products. For the data shown in Fig. 3, VGPEG4-chol, [VG-PEG4]2-chol, VIKIPEG4-chol, MV HRC2, and MV HRC4 peptides were either incubated in the absence of protease or treated with protease at 0°C, 22°C, or 37°C. Incubation with either 0.1 μg/10 μl (Fig. 3A) or 0.5 μg/10 μl (Fig. 3B) trypsin at 0°C resulted in significant degradation for all the peptides, with the exception of HPIV3 F-derived VIKIPEG4-chol, which appears to be resistant to degradation under these conditions. At 22°C and 37°C, all the peptides were degraded completely except for the VIKIPEG4-chol peptide, which was still present at 22°C though significantly degraded. No significant differences were observed between the monomeric and dimeric forms. The loss of fusion inhibition over time observed in Fig. 2 was thus not due to differences in protease degradation since all the peptides are degraded similarly, with the exception of VIKIPEG4-chol, and VIKIPEG4-chol—despite being the least susceptible to protease degradation—did not block fusion (Fig. 2). MV HRC4 (42) (Table 2) maintains fusion inhibition over time, despite the fact that it is degraded by protease as much as the other peptides (Fig. 3).
FIG 3.
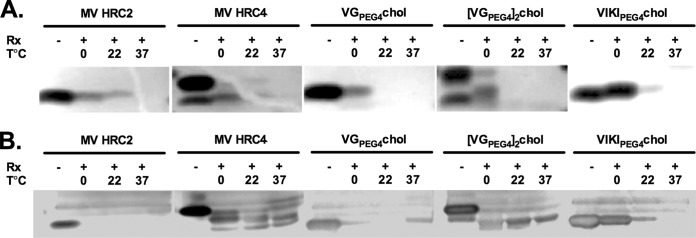
Protease sensitivity of MV- and HPIV3-derived peptides. The indicated peptides were incubated in the absence (−) or presence (+) of trypsin, at either 0.01 μg/μl (A) or 0.05 μg/μl (B) for 1 h at 0°C, 22°C, or 37°C. The products of the reaction were subjected to reducing SDS-PAGE gels and silver stained as described in Materials and Methods.
TABLE 2.
HPIV3- and MV HRC-derived peptides' in vitro efficacy
| Peptide | Viral entry inhibition |
Fusion inhibition (at 6 h) |
||||
|---|---|---|---|---|---|---|
| IC50 (μM) | IC90 (μM) | SLAM |
Nectin 4 |
|||
| IC50 (μM) | IC90 (μM) | IC50 (μM) | IC90 (μM) | |||
| MV HRC1 | 6.9a | >10a | ∼1 | ∼9 | ∼9.4 | >10 |
| MV HRC2 | 2.2 × 10−3a | 26 × 10−3a | ∼0.06 | ∼2 | ∼4.5 | >10 |
| MV HRC3 | 0.285a | 2.837a | NDb | ND | ND | ND |
| MV HRC4 | <1 × 10−3a | 2.7 × 10−3a | ∼0.001 | ∼0.044 | ∼0.06 | ∼0.3 |
| VG | ∼6.58 | >10 | >10 | >10 | ND | ND |
| VGPEG4-chol | ∼0.04 | ∼0.3 | >10 | >10 | ND | ND |
| [VGPEG11]2 | ND | ND | ND | ND | ND | ND |
| [VGPEG4]2-chol | ∼0.02 | ∼0.3 | ∼6.3 | >10 | ND | ND |
| VIKIPEG4-chol | ∼0.01 | ∼1.2 | >10 | >10 | ND | ND |
Data from reference 42.
ND, not determined.
Anti-H neutralizing antibody complements peptide fusion inhibitors.
For HPIV3, the neuraminidase activity of the hemagglutinin-neuraminidase (HN) promotes detachment of the HN from its receptor. Mutations that confer neuraminidase deficiency result in an HN that remains constitutively receptor engaged. Since receptor engagement promotes F's fusion activity, constant receptor engagement may permit an HN/F pair to resist inhibition by fusion-inhibitory peptides (60). Interaction of measles virus H with its receptor is dynamic, with on and off events between individual molecules (61), while the affinity of the overall complex is maintained (27, 62). We hypothesized that reducing the number of MV H attachment proteins interacting with CD150 receptor molecules would increase the fusion inhibition efficacy of peptides. To test this notion, we used the monoclonal neutralizing antibody HA55, which blocks H binding to SLAM (43–45), to compete with the SLAM receptor. In the experiment whose results are shown in Fig. 4, MV cells coexpressing MV G954 H and F were overlaid with Vero-SLAM cells with VGPEG4-chol peptide present at 1 μM (black bars) or without peptide (white bars). At the indicated time points (0, 30, 60, and 90 min), monoclonal antibody HA55 (1:10) was added. After 4 h, inhibition of fusion was assessed with the β-Gal complementation assay. Adding the monoclonal antibody at time zero resulted in complete inhibition, as expected since it blocks interaction of H with its receptor. When the antibody was added alone at later time points, fusion proceeded as in the control samples, and no inhibition was observed (Fig. 4, white bar). When the monoclonal antibody was added to the sample that had been treated with peptide at time zero, fusion was completely inhibited. Adding the antibody to peptide-treated cells 30 min after overlaying the Vero-SLAM cells resulted in >80% inhibition of fusion. At later time points, the inhibitory activity of the peptides when combined with antibody wanes (Fig. 4, black bars). These results suggest that decreasing H-receptor interaction can potentiate the antiviral effects of fusion-inhibitory peptides.
FIG 4.
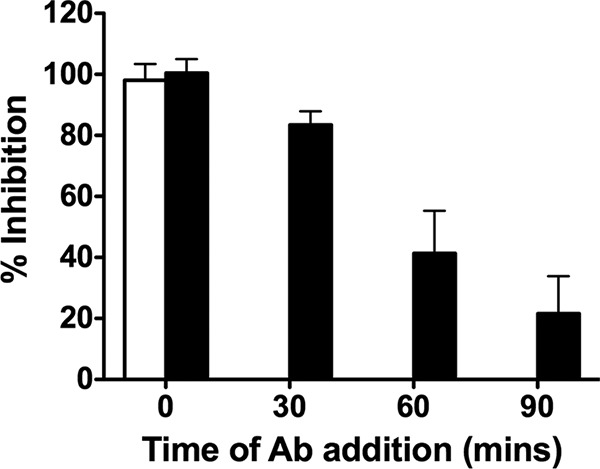
Inhibition of cell fusion in the presence of anti-MV H antibodies. Fusion inhibition (y axis) was assessed in the presence of HA55 neutralizing antibody added at the indicated times (x axis) in the absence (white bar) or presence (black bars) of 1 μM of VGPEG4-chol peptide. Data represent averages (± standard deviation) of data from triplicate wells from a representative experiment.
Peptides inhibit fusion of wt MV with nectin 4-bearing cells.
The data shown in Fig. 4 implicate the engagement of H to its receptor as a factor that influences peptide efficacy. Since MV H binds to nectin 4 with higher affinity than to SLAM (28, 62–64), we determined whether peptides are less effective in nectin 4-bearing cells than in SLAM-bearing cells.
MV HRC1, MV HRC2, and MV HRC4 were used to block fusion between G954 H- and F-expressing cells overlaid with cells that were transfected with either nectin 4 (Fig. 5A) or SLAM (Fig. 5B). Cholesterol-conjugated peptides (MV HRC2 and MV HRC4) were superior to the unconjugated peptide (MV HRC1); however, peptide efficacy was lower with the nectin 4-transfected cells (Fig. 5A) than with the SLAM-transfected cells (Fig. 5B). For the unconjugated peptide MV HRC1, the IC50s were ∼9.4 μM (with nectin 4 cells) and ∼1 μM (with SLAM cells); for the MV HRC2 cholesterol-conjugated peptides, the IC50s were ∼4.5 μM with nectin 4-bearing cells and ∼0.06 μM with SLAM-bearing cells (Fig. 5). The dimeric peptide with cholesterol MV HRC4 outperformed the other peptides over a wide range of concentrations and was the only one for which 100% inhibition was observed with nectin 4 cells. Nevertheless, MV HRC4 showed a significant increase in IC50: ∼0.06 μM for nectin 4-bearing cells but >0.001 μM with SLAM cells.
FIG 5.
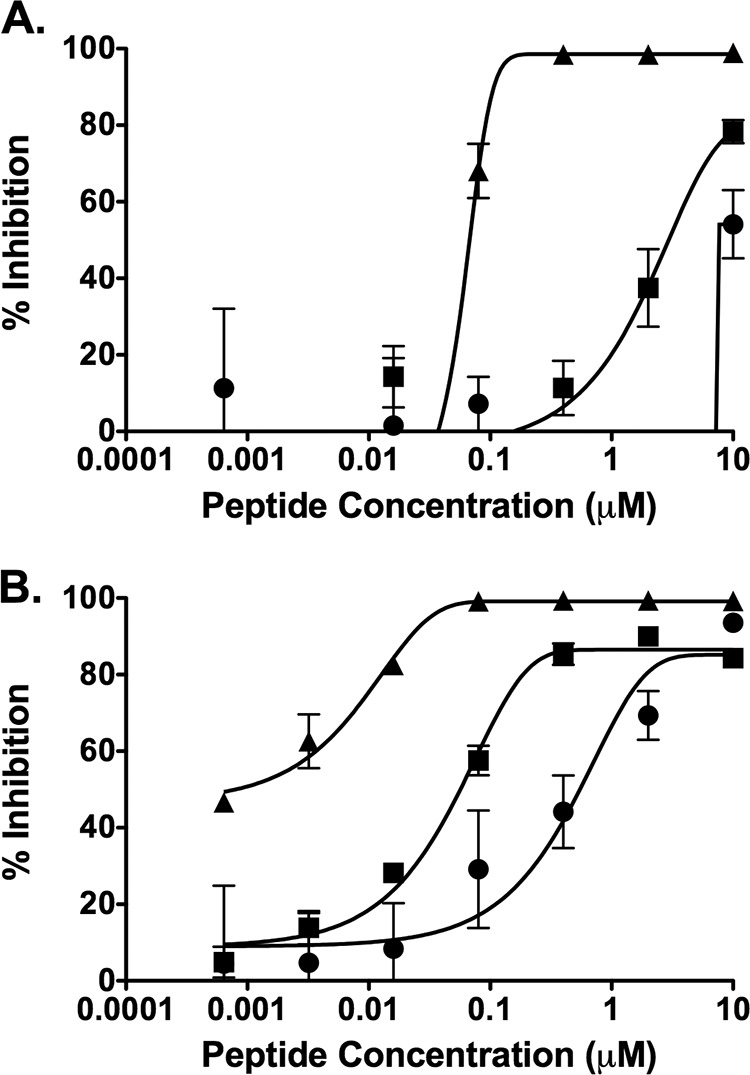
Inhibition of cell fusion with cells bearing nectin 4 or SLAM. Fusion of MV H/F-coexpressing cells with cells transfected with nectin 4 (A) or SLAM (B) in the presence of MV HRC1 (circles), MV HRC2 (squares), or MV HRC4 (triangles) was quantitated after 6 h by a β-galactosidase complementation assay. Results are presented as percent reduction in luminescence (y axis) compared with no treatment, as a function of compound concentration (x axis). Each point is the mean (± standard error) of results from 3 experiments.
Peptide transits throughout the human airway epithelium.
Since the expression of nectin 4 at the basolateral aspect of pulmonary epithelial cells plays a major role in MV shedding (65), an intranasally delivered peptide that reaches the basolateral side of the epithelium might decrease viral shedding. We first explored this idea using a pseudostratified human airway epithelium (HAE) ex vivo model of human lung airway (47, 48, 65–69). MV HRC2, MV HRC4, and VIKIPEG4-chol peptides were added at the apical side of the HAE to determine whether they could be found at the basolateral aspect after a time lapse (Fig. 6A). The amount of peptide (20 μl at 250 μM) was sufficient to reach concentrations of 5 μM if added directly to the basolateral reservoir (which contains 1 ml of medium). The presence of peptide basolaterally was detected by ELISA at 8 h, 24 h, 48 h, and 72 h after treatment, with peak concentrations for MV HRC2 (∼0.03 μM) attained at 48 h and for MV HRC4 (∼0.02 μM) at 24 h, indicating that both peptides reached the basolateral reservoir. VIKIPEG4-chol levels were the lowest of the three peptides tested (∼0.01 μM). The results indicate that only 0.4% of the apically delivered peptide reached the basolateral reservoir; however, based on the data in Fig. 5 this basolateral aspect concentration may be sufficient to block fusion promoted by H bound to nectin 4-bearing cells.
FIG 6.
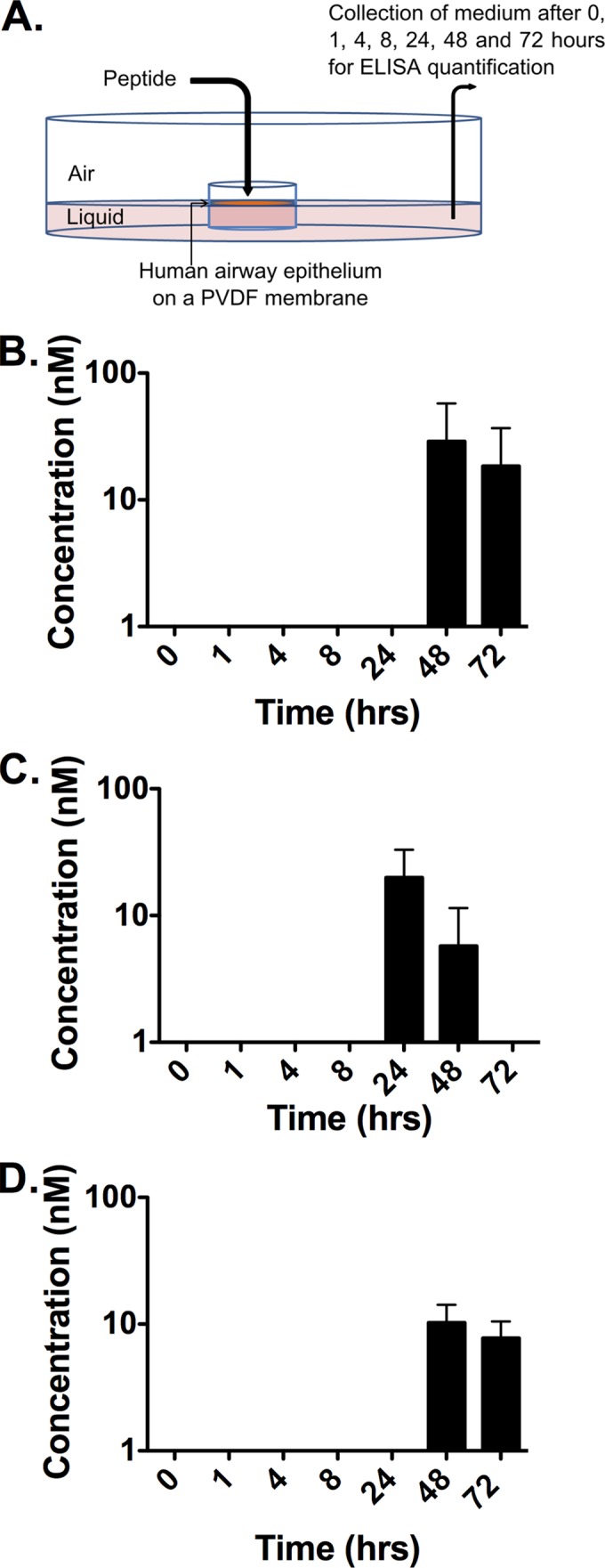
Peptides cross the human airway epithelium (HAE). (A) Drops containing peptides settle at the apical side (air interface) of the HAE. Medium (liquid interface) was collected at each time point for quantification of peptide by ELISA. PVDF, polyvinylidene difluoride. (B to D) Kinetic analysis (ELISA) of the peptide concentration in the liquid reservoir of the culture, reflecting minimal transit through the HAE (MV HRC2 in panel B, MV HRC4 in panel C, and VIKIPEG4-chol in panel D).
Intranasally delivered cholesterol-conjugated dimer peptides inhibit MV infection in cotton rats.
MV infection in cotton rats (CR) mimics natural infection (70–83). MV replicates in CR lung tissue with peak titers occurring on day 4 or 5, and infection is overcome by day 8. We have previously shown that MV HRC4 peptide delivered i.n. remains in the lungs (42). Here we assessed the biodistribution of HPIV3-derived VIKIPEG4-chol (chosen because it was the most resistant to protease degradation, as shown in Fig. 3) and of MV HRC2 and MV HRC4 peptides in the CR. Peptides were delivered i.n. (6 mg/kg, 3 animals per peptide), and the animals were euthanized 8 h after administration. Serum, lungs, and brain were collected for each animal, and peptide concentrations were measured by ELISA (42) (Table 3). MV HRC2 and MV HRC4 concentrated in the lungs after intranasal delivery (MV HRC2, 0.57 μM; MV HRC4, 0.44 μM) and reached lower concentrations in the serum (0.04 μM and 0.03 μM). Thus, MV HRC2 and HRC4 cross the airway epithelium of the CR in vivo (as well as human airway epithelium ex vivo). These peptides also reach the brain, with the monomeric HRC2 attaining a higher concentration in the brain (0.01 μM) than the dimeric MV HRC4 (0.002 μM). The HPIV3-derived VIKIPEG4-chol, in contrast, remains concentrated in the lungs (1.3 μM) (Table 3). The finding that most of the administered peptide remains localized in the respiratory tract may suggest that this peptide could provide i.n. prophylaxis against viruses transmitted via the respiratory route.
TABLE 3.
Bioavailability of MV HRC-derived peptides in cotton rats and mice 8 h after intranasal delivery
| Animal and MV peptide | Mean bioavailability ± SD (μM) in: |
||
|---|---|---|---|
| Brain | Lungs | Serum | |
| Cotton rat | |||
| MV HRC2 | 0.01 ± 0.004 | 0.57 ± 0.04 | 0.04 ± 0.02 |
| MV HRC4 | 0.002 ± 0.001 | 0.44 ± 0.25 | 0.03 ± 0.01 |
| VIKIPEG4-chol | >0.001 | 1.3 ± 0.3 | >0.001 |
| Mouse | |||
| MV HRC4 | 0.004 ± 0.003 | 0.25 ± 0.025 | 0.04 ± 0.02 |
We assessed the prophylactic efficacy of the MV- and HPIV3-derived peptides after intranasal delivery in the CR model of MV infection. We tested the three peptides whose biodistribution was assessed as described above (VIKIPEG4-chol, MV HRC2, and MV HRC4), and we added MV HRC1, MV HRC3, VG, [VG-PEG11]2, VGPEG4-chol, and [VG-PEG4]2-chol peptides as additional controls (51, 58). The animals were given the fusion-inhibitory peptides i.n. twice (5 mg/kg each) at 24 and 12 h before infection (Fig. 7). Four days after infection, the viral titer in the lungs of untreated animals was 104 TCID50/g lung tissue (±1 × 103); the HPIV3 peptide-treated animals had viral titers with no significant differences from those of the untreated animals (Fig. 7). Interestingly, the animals treated with VIKIPEG4-chol (which in the biodistribution experiment reached 3 times the lung concentration of MV HRC2 and HRC4) showed only a small reduction in viral titer. After prophylaxis with the MV HRC 1-derived peptides, the titer was reduced to 9 × 103 TCID50/g lung tissue (±2 × 103); and after prophylaxis with the other MV-derived peptides (MV HRC2, MV HRC3, and MV HRC4), no MV virus at all was detected in the lungs. Statistical analysis revealed a strong significance of the MV peptides' inhibitory effect using a Mann-Whitney U test (P < 0.001) compared to either the untreated group or the group treated with HPIV3-derived peptides. The intranasally delivered MV peptides (with the exception of the monomeric untagged MV HRC1) efficiently blocked MV infection in the CR.
FIG 7.
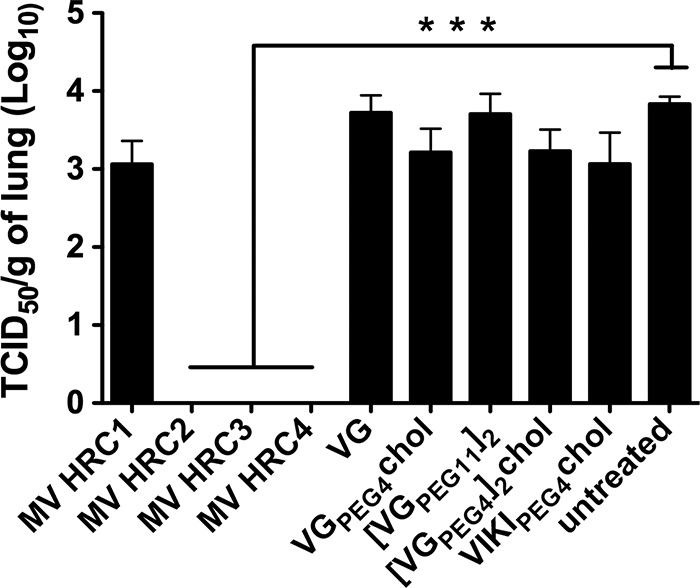
Intranasal administration of MV-derived peptides protects cotton rats from MV infection. Cotton rats (n = 4) were infected i.n. with MV 24 h after the first peptide treatment and were euthanized 4 days postinfection. MV titration of lung homogenates showed that MV HRC2, MV HRC3, and MV HRC4 block infection in CR (***, P = 0.001 in Mann-Whitney U test). The limit of viral detection was 102 PFU/gram.
Intranasally delivered cholesterol-conjugated dimer peptides inhibit MV infection in mice.
We previously showed that simultaneous s.q. and i.n. administration of cholesterol-tagged MV HRC4 protected mice from a lethal acute MV neurological syndrome (42). In the present study, we assessed whether i.n. delivery of MV HRC4 peptides alone is sufficient to protect from fatal measles encephalitis. In order to better understand the importance of each route of administration (s.q. versus i.n.), we assessed the ability of the two methods of delivery to prevent fatal measles encephalitis in SLAM transgenic mice. Previously, we found that i.n. delivery of peptides in mice did not result in demonstrable serum levels after 4 h in mice (42); however, here we detected peptide in the circulation and in the brain after 8 h, suggesting that entry of the fusion inhibitors into the circulation via the airway epithelium requires time (Table 3). Additionally, we show that after i.n. delivery, a therapeutic concentration of peptides is maintained in the respiratory tract for several hours in mice (Fig. 8A to C and Table 3) (see also reference 42). Thus, limited i.n. administration might be sufficient for attaining a concentration higher than the IC90 of the peptide, justifying an in vivo experiment comparing i.n. protection with s.q. administration.
FIG 8.

Intranasal administration of MV HRC4 protects mice from lethal MV encephalitis. (A to C) Immunofluorescent staining of MV HRC4 (in red) in the lungs of mice after i.n. administration. Nuclei were counterstained using DAPI. While peptide staining is absent from untreated animals (A), a high concentration of MV HRC4 (in red) is present in the lungs of treated mice (B), and magnification (C) reveals the presence of the fusion inhibitors in the bronchioles as well as in the parenchyma. One-week-old SLAM transgenic mice (n = 5) were challenged with intranasal MV (G954 strain) 24 h after the first MV-HRC peptide treatment and were monitored for 5 weeks. Control animals (mock) were injected with vehicle alone. (D) Peptide was administered subcutaneously daily from day −1 to day +7 after infection. (E) Mice received only 2 intranasal (i.n.) administrations of MV HRC peptides, 24 h before and 2 h after infection. Statistical significance of the difference between the MV HRC4 and untreated groups in E (*, P = 0.0334) was analyzed using the Mantel-Cox test. (F) Production of anti-NP antibodies in the sera of infected (as indicated on the x axis) SLAM transgenic mice. (G) Four- to 5-week-old SLAM transgenic/IFNAR knockout mice (n = 10) were challenged with intranasal MV (50 LD50 of strain G954) 24 h after the first MV HRC peptide treatment and were monitored for 4 weeks. Control animals (mock; n = 10) were injected with vehicle alone. Mice received only two i.n. administrations of MV HRC peptides, 24 h and 6 h before infection. Statistical significance of the differences between the MV HRC4 and untreated groups (***, P < 0.0001) was analyzed using the Mantel-Cox test.
We next compared the efficiency levels of the peptides at protecting suckling mice from a lethal MV challenge. Daily s.q. administration from the day before to the 7th day after infection protected 60% of the animals (Fig. 8D), whereas two i.n. administrations of MV HRC4 (6 mg/kg) at 24 h before and 2 h after infection significantly improved survival of the mice, protecting 80% of them (P = 0.03 in the Mantel-Cox test) (Fig. 8E). This result suggests that i.n. administration protects SLAM transgenic suckling mice from fatal central nervous system (CNS) infection. We detected no anti-MV antibodies in surviving treated animals, in contrast to the untreated survivor, confirming that MV HRC4 blocks initial infection at an early step (Fig. 8F).
Since infection is halted at an early stage, we hypothesized that the peptide antiviral strategy may be effective in immunodeficient hosts (55). SLAM transgenic mice crossed into an IFN type I receptor-deficient background (SLAM × IFNAR mice) (56, 84) received 2 i.n. administrations of MV HRC4 (6 mg/kg) at 24 h and 6 h before infection. Figure 8G shows the survival curve of a total of 20 mice (10 treated and 10 untreated) infected with 50 LD50. All untreated animals died by day 20 postinfection. Of the treated animals, 90% were still alive after 28 days, confirming a significant survival advantage in this group (P < 0.0001 in the Mantel-Cox test) (Fig. 8G). This result suggests that i.n. administration protects immunocompromised hosts from fatal CNS infection.
DISCUSSION
Measles virus (MV) induces a respiratory infection and can cause a profound suppression of the immune system that may permit opportunistic infection and contribute to morbidity and mortality (97, 98). CNS complications of MV infection may occur soon after infection in the case of acute encephalomyelitis, or years after infection, as a result of viral persistence in subacute sclerosing panencephalitis (SSPE) and progressive infectious encephalitis or measles inclusion body encephalitis (MIBE). There are no specific therapies for acute complications of MV or for persistent MV infections (85–88). Since we expect our proposed antiviral strategy to be host factor independent, it will fill a specific need for immunocompromised people at risk for MV infection, who cannot be vaccinated or do not respond adequately to vaccine.
We have previously shown that HPIV3 HRC-derived peptides are effective inhibitors of viral entry for other paramyxoviruses as well, including Hendra virus, Nipah virus, and simian virus 5 (SV5 or PIV5) (51, 58, 59), raising the possibility of broad-spectrum antivirals. We therefore tested their efficacy against the MV fusion machinery in vitro and against MV infection in vivo. Here we report that the HPIV3 HRC-derived peptides inhibit wt MV entry but do not inhibit fusion mediated by the MV H and F glycoproteins. While MV HRC peptides prevent MV infection in vivo, HPIV3 HRC peptides—even the ones effective in vivo against Nipah virus (51, 58)—are not effective at blocking MV infection in vivo.
In our work on HPIV3, we showed that fusion requires engagement of the receptor binding protein beyond initial triggering of the F protein and that engagement of the receptor by the HPIV3 receptor binding protein (HN) is essential for F's function until membrane merger occurs (60). Consistent with this information, for MV it has been shown that simple engagement of H with a soluble form of receptor was not sufficient to promote F-mediated fusion (89). The current model for paramyxovirus fusion, which informs strategies to counteract viral resistance, suggests that fusion inhibition cannot be countered by mutations in the receptor-binding protein (or domain) but only by mutations in the fusion protein. We have shown, however, that HN variants that are more efficient at activating F may promote partial resistance to standard peptide inhibitors (60, 90). We show here that an antibody that targets the measles virus H-receptor interaction enhances the efficacy of fusion-inhibitory peptides (Fig. 4). The affinity of H for its receptor is thus another factor that influences the potency of antifusion antivirals (Fig. 6). These data are consistent with our model for paramyxovirus fusion, in which ongoing activation by receptor-engaged H is required in order for fusion to proceed. Another consequence of this model would be that ongoing activation of the fusion process by receptor-engaged H may eventually override inhibition by an F-targeted peptide. This mechanism could explain the differences observed between HPIV3 peptide efficacy against viral entry and efficacy against viral fusion (Fig. 1 and 2). Irrespective of the model for viral fusion, the results reported here have relevance for understanding the mechanism of resistance to entry/fusion inhibitors and for designing effective inhibitors, since it may be advantageous to target both the H and F proteins.
We previously showed that dimerization combined with cholesterol conjugation to an MV HRC F-derived peptide generated a peptide that efficiently protected SLAM transgenic mice from MV fatal CNS infection when administered both i.n. and s.q. (42). The HPIV3-derived dimeric peptide [VGPEG4]2-chol, despite being the most potent of the HPIV3-derived peptides at inhibiting MV fusion, did not block infection in vivo. These results suggest that, unlike NiV and HPIV3 infections, which can both be inhibited by the same HPIV3 HRC peptides (51, 58), inhibition of MV requires a sequence specifically derived from its own F protein. In the future, “cocktails” of antiviral peptides may be a useful approach to achieving a broad-spectrum coverage. In the present experiment (as in our previous study), we detected no anti-MV antibody in the serum of surviving SLAM TG mice (treated i.n. with MV HRC4 peptides [Fig. 8E]) and no virus in cotton rats treated with MV HRCs (Fig. 7). These findings suggest that the prophylactic treatment led to complete inhibition of the virus at a very early stage of the infection, before induction of an antibody response.
An orally available nucleoside inhibitor of the morbillivirus polymerase has been assessed in work by others for a different morbillivirus, canine distemper virus (CDV), in ferrets. Prophylaxis with the nucleoside inhibitor reduced the viral titer in vivo and prolonged survival; however, all 9 animals—those treated before infection and those with continued treatment throughout infection—eventually died (91). Surprisingly, efficacy was 100% in the 3 animals who were treated only 3 days postinfection (without prophylaxis) (91). To explain these results, the authors posit that the postinfection treatment was effective because it allowed for a boost in immunity elicited by viral replication during the first 3 days but then prevented depletion of T and B lymphocytes (17, 21, 28, 65). However, this explanation would imply that preexposure prophylactic treatment interferes with postinfection treatment and simply delays the fatal outcome.
In healthy individuals, MV infection elicits a very strong MV-specific immune response and also transient immune suppression (17, 21, 28, 65). Viremia is brought under control by cellular immunity within 2 weeks of infection, but viral genomes persist for several months after infection, even in the face of humoral immunity (17, 92, 93). If the explanation offered for the paradoxical effect of the polymerase inhibitor is accurate, then individuals with impaired innate immunity would not be candidates for therapy with the nucleoside inhibitor of morbillivirus polymerase. Our proposed antiviral strategy targets a different stage in the viral cycle and was highly effective prophylactically in two animal models of MV infection. If the CDV data were to be established for MV and explained, and/or if a polymerase inhibitor that is effective for MV is identified, then the two strategies could be complementary.
Nectin 4 has been recently shown to serve as an entry receptor for MV (27, 28). This receptor, expressed at the basolateral aspect of the epithelial cells in the lungs, is responsible for viral shedding in cynomolgus monkey tracheas, suggesting that it may play a major role in the human-to-human spread of MV (94). In the present study, we show that MV HRC4 efficiently blocks fusion with nectin 4-bearing cells and can cross the pulmonary epithelium not only in HAE culture but also in vivo, suggesting that it can reach the basolateral side of the cells, where it may prevent spread. The hypothesis that this treatment may prevent host-to-host transmission will be tested in the future. In recent work by others, ferrets infected with CDV bearing an H that is unable to bind nectin 4 underwent transient immunosuppression but did not show clinical disease (95). It is conceivable that blockage of nectin 4-mediated fusion by peptides may also reduce clinical symptoms. Simple i.n. peptide administration may protect vulnerable people during an MV outbreak. A similar strategy of i.n. peptide delivery has recently been proposed for preventing the spread of respiratory syncytial virus infection to the lung (96), suggesting that this approach may be extended to other respiratory infections. The efficacy of i.n. peptides for MV prophylaxis opens a new avenue for rapid protection against highly contagious MV infection in a world experiencing a resurgence of this disease.
ACKNOWLEDGMENTS
We are grateful for the Friedman Research Scholar Award to M.P.
We are grateful to Lawrence Golub for his support and to Dan and Nancy Paduano for support of innovative research projects. We thank the members of the “Immunobiology of viral infections” group (CIRI, Lyon) for their help with this study.
The work was supported by grants from NIH (NINDS) NS073781 and (NIAID) AI 109050 to M.P., (NIAID) R21/R33AI101333 and R01 AI031971 to A.M., and INSERM and the French Foundation for Medical Research (FRM) to B.H.
REFERENCES
- 1.Moss WJ, Griffin DE. 2012. Measles. Lancet 379:153–164. doi: 10.1016/S0140-6736(10)62352-5. [DOI] [PubMed] [Google Scholar]
- 2.Katz SL, Hinman AR. 2004. Summary and conclusions: measles elimination meeting, 16–17 March 2000. J Infect Dis 189(Suppl 1):S43–S47. doi: 10.1086/377696. [DOI] [PubMed] [Google Scholar]
- 3.Hutchins SS, Bellini WJ, Coronado V, Jiles R, Wooten K, Deladisma A. 2004. Population immunity to measles in the United States, 1999. J Infect Dis 189(Suppl 1):S91–S97. doi: 10.1086/377713. [DOI] [PubMed] [Google Scholar]
- 4.Simons E, Ferrari M, Fricks J, Wannemuehler K, Anand A, Burton A, Strebel P. 2012. Assessment of the 2010 global measles mortality reduction goal: results from a model of surveillance data. Lancet 379:2173–2178. doi: 10.1016/S0140-6736(12)60522-4. [DOI] [PubMed] [Google Scholar]
- 5.Poland GA, Jacobson RM. 2012. The re-emergence of measles in developed countries: time to develop the next-generation measles vaccines? Vaccine 30:103–104. doi: 10.1016/j.vaccine.2011.11.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Limb M. 2012. Outbreaks among travellers linked to almost twofold rise in measles cases. BMJ 345:e5797. doi: 10.1136/bmj.e5797. [DOI] [PubMed] [Google Scholar]
- 7.Kantele A, Mattila L, Ott K, Davidkin I, Siikamaki H. 2012. Fever with rash in patients returning from popular tourist resort Phuket, Thailand: dengue–or measles? J Travel Med 19:317–319. doi: 10.1111/j.1708-8305.2012.00639.x. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention. 2012. Measles outbreak associated with an arriving refugee Los Angeles County, California, August-September 2011. MMWR Morb Mortal Wkly Rep 61:385–389 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6121a1.htm?s_cid=mm6121a1_w. [PubMed] [Google Scholar]
- 9.Ortega-Sanchez IR, Vijayaraghavan M, Barskey AE, Wallace GS. 2014. The economic burden of sixteen measles outbreaks on United States public health departments in 2011. Vaccine 32:1311–1317. doi: 10.1016/j.vaccine.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Gastanaduy PA, Redd SB, Fiebelkorn AP, Rota JS, Rota PA, Bellini WJ, Seward JF, Wallace GS. 2014. Measles—United States, January 1-May 23, 2014. MMWR Morb Mortal Wkly Rep 63:496–499 http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6322a4.htm?s_cid=mm6322a4_w. [PMC free article] [PubMed] [Google Scholar]
- 11.Seward JF, Orenstein WA. 2012. Editorial commentary: a rare event: a measles outbreak in a population with high 2-dose measles vaccine coverage. Clin Infect Dis 55:403–405. doi: 10.1093/cid/cis445. [DOI] [PubMed] [Google Scholar]
- 12.De Serres G, Boulianne N, Defay F, Brousseau N, Benoit M, Lacoursiere S, Guillemette F, Soto J, Ouakki M, Ward BJ, Skowronski DM. 2012. Higher risk of measles when the first dose of a 2-dose schedule of measles vaccine is given at 12-14 months versus 15 months of age. Clin Infect Dis 55:394–402. doi: 10.1093/cid/cis439. [DOI] [PubMed] [Google Scholar]
- 13.Haralambieva IH, Ovsyannikova IG, O'Byrne M, Pankratz VS, Jacobson RM, Poland GA. 2011. A large observational study to concurrently assess persistence of measles specific B-cell and T-cell immunity in individuals following two doses of MMR vaccine. Vaccine 29:4485–4491. doi: 10.1016/j.vaccine.2011.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen CJ, Lee PI, Hsieh YC, Chen PY, Ho YH, Chang CJ, Liu DP, Chang FY, Chiu CH, Huang YC, Lee CY, Lin TY. 2012. Waning population immunity to measles in Taiwan. Vaccine 30:6721–6727. doi: 10.1016/j.vaccine.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 15.Kontio M, Jokinen S, Paunio M, Peltola H, Davidkin I. 2012. Waning antibody levels and avidity: implications for MMR vaccine-induced protection. J Infect Dis 206:1542–1548. doi: 10.1093/infdis/jis568. [DOI] [PubMed] [Google Scholar]
- 16.Fox A, Hung TM, Wertheim H, Hoa le NM, Vincent A, Lang B, Waters P, Ha NH, Trung NV, Farrar J, Van Kinh N, Horby P. 2013. Acute measles encephalitis in partially vaccinated adults. PLoS One 8:e71671. doi: 10.1371/journal.pone.0071671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Vries RD, Mesman AW, Geijtenbeek TB, Duprex WP, de Swart RL. 2012. The pathogenesis of measles. Curr Opin Virol 2:248–255. doi: 10.1016/j.coviro.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira CS, Frenzke M, Leonard VH, Welstead GG, Richardson CD, Cattaneo R. 2010. Measles virus infection of alveolar macrophages and dendritic cells precedes spread to lymphatic organs in transgenic mice expressing human signaling lymphocytic activation molecule (SLAM, CD150). J Virol 84:3033–3042. doi: 10.1128/JVI.01559-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avota E, Koethe S, Schneider-Schaulies S. 2013. Membrane dynamics and interactions in measles virus dendritic cell infections. Cell Microbiol 15:161–169. doi: 10.1111/cmi.12025. [DOI] [PubMed] [Google Scholar]
- 20.Koethe S, Avota E, Schneider-Schaulies S. 2012. Measles virus transmission from dendritic cells to T cells: formation of synapse-like interfaces concentrating viral and cellular components. J Virol 86:9773–9781. doi: 10.1128/JVI.00458-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemon K, de Vries RD, Mesman AW, McQuaid S, van Amerongen G, Yuksel S, Ludlow M, Rennick LJ, Kuiken T, Rima BK, Geijtenbeek TB, Osterhaus AD, Duprex WP, de Swart RL. 2011. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog 7:e1001263. doi: 10.1371/journal.ppat.1001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yanagi Y, Ono N, Tatsuo H, Hashimoto K, Minagawa H. 2002. Measles virus receptor SLAM (CD150). Virology 299:155–161. doi: 10.1006/viro.2002.1471. [DOI] [PubMed] [Google Scholar]
- 23.Yanagi Y, Takeda M, Ohno S, Hashiguchi T. 2009. Measles virus receptors. Curr Top Microbiol Immunol 329:13–30. doi: 10.1007/978-3-540-70523-9_2. [DOI] [PubMed] [Google Scholar]
- 24.Hashiguchi T, Maenaka K, Yanagi Y. 2011. Measles virus hemagglutinin: structural insights into cell entry and measles vaccine. Front Microbiol 2:247. doi: 10.3389/fmicb.2011.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. The cellular receptor for measles virus: SLAM (CDw 150). Uirusu 50:289–296. (In Japanese.) [PubMed] [Google Scholar]
- 26.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 27.Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A 102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamb RA, Paterson RG, Jardetzky TS. 2006. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology 344:30–37. doi: 10.1016/j.virol.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrison SC. 2008. Viral membrane fusion. Nat Struct Mol Biol 15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang A, Dutch RE. 2012. Paramyxovirus fusion and entry: multiple paths to a common end. Viruses 4:613–636. doi: 10.3390/v4040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plemper RK, Brindley MA, Iorio RM. 2011. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog 7:e1002058. doi: 10.1371/journal.ppat.1002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol 43:189–219. doi: 10.1080/10409230802058320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sapir A, Avinoam O, Podbilewicz B, Chernomordik LV. 2008. Viral and developmental cell fusion mechanisms: conservation and divergence. Dev Cell 14:11–21. doi: 10.1016/j.devcel.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambert DM, Barney S, Lambert AL, Guthrie K, Medinas R, Davis DE, Bucy T, Erickson J, Merutka G, Petteway SR Jr. 1996. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc Natl Acad Sci U S A 93:2186–2191. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eckert DM, Kim PS. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem 70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 39.Berkhout B, Eggink D, Sanders RW. 2012. Is there a future for antiviral fusion inhibitors? Curr Opin Virol 2:50–59. doi: 10.1016/j.coviro.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Price RW, Parham R, Kroll JL, Wring SA, Baker B, Sailstad J, Hoh R, Liegler T, Spudich S, Kuritzkes DR, Deeks SG. 2008. Enfuvirtide cerebrospinal fluid (CSF) pharmacokinetics and potential use in defining CSF HIV-1 origin. Antivir Ther 13:369–374. [PMC free article] [PubMed] [Google Scholar]
- 41.Liner KJ II, Ro MJ, Robertson KR. 2010. HIV, antiretroviral therapies, and the brain. Curr HIV/AIDS Rep 7:85–91. doi: 10.1007/s11904-010-0042-8. [DOI] [PubMed] [Google Scholar]
- 42.Welsch JC, Talekar A, Mathieu C, Pessi A, Moscona A, Horvat B, Porotto M. 2013. Fatal measles virus infection prevented by brain-penetrant fusion inhibitors. J Virol 87:13785–13794. doi: 10.1128/JVI.02436-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masse N, Ainouze M, Neel B, Wild TF, Buckland R, Langedijk JP. 2004. Measles virus (MV) hemagglutinin: evidence that attachment sites for MV receptors SLAM and CD46 overlap on the globular head. J Virol 78:9051–9063. doi: 10.1128/JVI.78.17.9051-9063.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giraudon P, Wild TF. 1981. Monoclonal antibodies against measles virus. J Gen Virol 54:325–332. doi: 10.1099/0022-1317-54-2-325. [DOI] [PubMed] [Google Scholar]
- 45.Sellin CI, Davoust N, Guillaume V, Baas D, Belin MF, Buckland R, Wild TF, Horvat B. 2006. High pathogenicity of wild-type measles virus infection in CD150 (SLAM) transgenic mice. J Virol 80:6420–6429. doi: 10.1128/JVI.00209-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rima BK, Earle JA, Baczko K, ter Meulen V, Liebert UG, Carstens C, Carabana J, Caballero M, Celma ML, Fernandez-Munoz R. 1997. Sequence divergence of measles virus haemagglutinin during natural evolution and adaptation to cell culture. J Gen Virol 78(Part 1):97–106. [DOI] [PubMed] [Google Scholar]
- 47.Xu R, Palmer SG, Porotto M, Palermo LM, Niewiesk S, Wilson IA, Moscona A. 2013. Interaction between the hemagglutinin-neuraminidase and fusion glycoproteins of human parainfluenza virus type III regulates viral growth in vivo. mBio 4:e00803-13. doi: 10.1128/mBio.00803-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palmer SG, Porotto M, Palermo LM, Cunha LF, Greengard O, Moscona A. 2012. Adaptation of human parainfluenza virus to airway epithelium reveals fusion properties required for growth in host tissue. mBio 3:e00137-12. doi: 10.1128/mBio.00137-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moscona A, Porotto M, Palmer S, Tai C, Aschenbrenner L, Triana-Baltzer G, Li QX, Wurtman D, Niewiesk S, Fang F. 2010. A recombinant sialidase fusion protein effectively inhibits human parainfluenza viral infection in vitro and in vivo. J Infect Dis 202:234–241. doi: 10.1086/653621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farzan SF, Palermo LM, Yokoyama CC, Orefice G, Fornabaio M, Sarkar A, Kellogg GE, Greengard O, Porotto M, Moscona A. 2011. Premature activation of the paramyxovirus fusion protein before target cell attachment with corruption of the viral fusion machinery. J Biol Chem 286:37945–37954. doi: 10.1074/jbc.M111.256248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Porotto M, Rockx B, Yokoyama CC, Talekar A, Devito I, Palermo LM, Liu J, Cortese R, Lu M, Feldmann H, Pessi A, Moscona A. 2010. Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog 6:e1001168. doi: 10.1371/journal.ppat.1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palermo L, Porotto M, Yokoyama C, Palmer S, Mungall B, Greengard O, Niewiesk S, Moscona A. 2009. Human parainfluenza virus infection of the airway epithelium: the viral hemagglutinin-neuraminidase regulates fusion protein activation and modulates infectivity. J Virol 83:6900–6908. doi: 10.1128/JVI.00475-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porotto M, Fornabaio M, Kellogg GE, Moscona A. 2007. A second receptor binding site on human parainfluenza virus type 3 hemagglutinin-neuraminidase contributes to activation of the fusion mechanism. J Virol 81:3216–3228. doi: 10.1128/JVI.02617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moosmann P, Rusconi S. 1996. Alpha complementation of LacZ in mammalian cells. Nucleic Acids Res 24:1171–1172. doi: 10.1093/nar/24.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sellin CI, Horvat B. 2009. Current animal models: transgenic animal models for the study of measles pathogenesis. Curr Top Microbiol Immunol 330:111–127. doi: 10.1007/978-3-540-70617-5_6. [DOI] [PubMed] [Google Scholar]
- 56.Sellin CI, Jegou JF, Renneson J, Druelle J, Wild TF, Marie JC, Horvat B. 2009. Interplay between virus-specific effector response and Foxp3 regulatory T cells in measles virus immunopathogenesis. PLoS One 4:e4948. doi: 10.1371/journal.pone.0004948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pfeuffer J, Puschel K, Meulen V, Schneider-Schaulies J, Niewiesk S. 2003. Extent of measles virus spread and immune suppression differentiates between wild-type and vaccine strains in the cotton rat model (Sigmodon hispidus). J Virol 77:150–158. doi: 10.1128/JVI.77.1.150-158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pessi A, Langella A, Capito E, Ghezzi S, Vicenzi E, Poli G, Ketas T, Mathieu C, Cortese R, Horvat B, Moscona A, Porotto M. 2012. A general strategy to endow natural fusion-protein-derived peptides with potent antiviral activity. PLoS One 7:e36833. doi: 10.1371/journal.pone.0036833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Porotto M, Yokoyama CC, Palermo LM, Mungall B, Aljofan M, Cortese R, Pessi A, Moscona A. 2010. Viral entry inhibitors targeted to the membrane site of action. J Virol 84:6760–6768. doi: 10.1128/JVI.00135-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Porotto M, Devito I, Palmer SG, Jurgens EM, Yee JL, Yokoyama CC, Pessi A, Moscona A. 2011. Spring-loaded model revisited: paramyxovirus fusion requires engagement of a receptor binding protein beyond initial triggering of the fusion protein. J Virol 85:12867–12880. doi: 10.1128/JVI.05873-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Navaratnarajah CK, Vongpunsawad S, Oezguen N, Stehle T, Braun W, Hashiguchi T, Maenaka K, Yanagi Y, Cattaneo R. 2008. Dynamic interaction of the measles virus hemagglutinin with its receptor signaling lymphocytic activation molecule (SLAM, CD150). J Biol Chem 283:11763–11771. doi: 10.1074/jbc.M800896200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mateo M, Navaratnarajah CK, Syed S, Cattaneo R. 2013. The measles virus hemagglutinin beta-propeller head beta4-beta5 hydrophobic groove governs functional interactions with nectin-4 and CD46 but not those with the signaling lymphocytic activation molecule. J Virol 87:9208–9216. doi: 10.1128/JVI.01210-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mateo M, Navaratnarajah CK, Cattaneo R. 2014. Structural basis of efficient contagion: measles variations on a theme by parainfluenza viruses. Curr Opin Virol 5:16–23. doi: 10.1016/j.coviro.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hashiguchi T, Ose T, Kubota M, Maita N, Kamishikiryo J, Maenaka K, Yanagi Y. 2011. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat Struct Mol Biol 18:135–141. doi: 10.1038/nsmb.1969. [DOI] [PubMed] [Google Scholar]
- 65.Leonard VH, Sinn PL, Hodge G, Miest T, Devaux P, Oezguen N, Braun W, McCray PB Jr, McChesney MB, Cattaneo R. 2008. Measles virus blind to its epithelial cell receptor remains virulent in rhesus monkeys but cannot cross the airway epithelium and is not shed. J Clin Invest 118:2448–2458. doi: 10.1172/JCI35454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thompson CI, Barclay WS, Zambon MC, Pickles RJ. 2006. Infection of human airway epithelium by human and avian strains of influenza A virus. J Virol 80:8060–8068. doi: 10.1128/JVI.00384-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang L, Bukreyev A, Thompson CI, Watson B, Peeples ME, Collins PL, Pickles RJ. 2005. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J Virol 79:1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mellow TE, Murphy PC, Carson JL, Noah TL, Zhang L, Pickles RJ. 2004. The effect of respiratory synctial virus on chemokine release by differentiated airway epithelium. Exp Lung Res 30:43–57. doi: 10.1080/01902140490252812. [DOI] [PubMed] [Google Scholar]
- 69.Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. 2002. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J Virol 76:5654–5666. doi: 10.1128/JVI.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim D, Niewiesk S. 2014. Synergistic induction of interferon alpha through TLR-3 and TLR-9 agonists stimulates immune responses against measles virus in neonatal cotton rats. Vaccine 32:265–270. doi: 10.1016/j.vaccine.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim MY, Shu Y, Carsillo T, Zhang J, Yu L, Peterson C, Longhi S, Girod S, Niewiesk S, Oglesbee M. 2013. hsp70 and a novel axis of type I interferon-dependent antiviral immunity in the measles virus-infected brain. J Virol 87:998–1009. doi: 10.1128/JVI.02710-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim D, Niewiesk S. 2013. Synergistic induction of interferon alpha through TLR-3 and TLR-9 agonists identifies CD21 as interferon alpha receptor for the B cell response. PLoS Pathog 9:e1003233. doi: 10.1371/journal.ppat.1003233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Green MG, Huey D, Niewiesk S. 2013. The cotton rat (Sigmodon hispidus) as an animal model for respiratory tract infections with human pathogens. Lab Anim (NY) 42:170–176. doi: 10.1038/laban.188. [DOI] [PubMed] [Google Scholar]
- 74.Oglesbee M, Niewiesk S. 2011. Measles virus neurovirulence and host immunity. Future Virol 6:85–99. doi: 10.2217/fvl.10.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Niewiesk S. 2009. Current animal models: cotton rat animal model. Curr Top Microbiol Immunol 330:89–110. doi: 10.1007/978-3-540-70617-5_5. [DOI] [PubMed] [Google Scholar]
- 76.Pueschel K, Tietz A, Carsillo M, Steward M, Niewiesk S. 2007. Measles virus-specific CD4 T-cell activity does not correlate with protection against lung infection or viral clearance. J Virol 81:8571–8578. doi: 10.1128/JVI.00160-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moeller-Ehrlich K, Ludlow M, Beschorner R, Meyermann R, Rima BK, Duprex WP, Niewiesk S, Schneider-Schaulies J. 2007. Two functionally linked amino acids in the stem 2 region of measles virus haemagglutinin determine infectivity and virulence in the rodent central nervous system. J Gen Virol 88:3112–3120. doi: 10.1099/vir.0.83235-0. [DOI] [PubMed] [Google Scholar]
- 78.Streif S, Pueschel K, Tietz A, Blanco J, Meulen VT, Niewiesk S. 2004. Effector CD8+T cells are suppressed by measles virus infection during delayed type hypersensitivity reaction. Viral Immunol 17:604–608. doi: 10.1089/vim.2004.17.604. [DOI] [PubMed] [Google Scholar]
- 79.Moll M, Pfeuffer J, Klenk HD, Niewiesk S, Maisner A. 2004. Polarized glycoprotein targeting affects the spread of measles virus in vitro and in vivo. J Gen Virol 85:1019–1027. doi: 10.1099/vir.0.19663-0. [DOI] [PubMed] [Google Scholar]
- 80.Carsillo T, Carsillo M, Niewiesk S, Vasconcelos D, Oglesbee M. 2004. Hyperthermic pre-conditioning promotes measles virus clearance from brain in a mouse model of persistent infection. Brain Res 1004:73–82. doi: 10.1016/j.brainres.2003.12.041. [DOI] [PubMed] [Google Scholar]
- 81.Schlereth B, Buonocore L, Tietz A, ter Meulen V, Rose J, Niewiesk S. 2003. Successful mucosal immunization of cotton rats in the presence of measles virus-specific antibodies depends on degree of attenuation of vaccine vector and virus dose. J Gen Virol 84:2145–2151. doi: 10.1099/vir.0.19050-0. [DOI] [PubMed] [Google Scholar]
- 82.Niewiesk S, Prince G. 2002. Diversifying animal models: the use of cotton rats (Sigmodon hispidus) in infectious diseases. Lab Anim 36:357–372. doi: 10.1258/002367702320389026. [DOI] [PubMed] [Google Scholar]
- 83.Weidinger G, Ohlmann M, Schlereth B, Sutter G, Niewiesk S. 2001. Vaccination with recombinant modified vaccinia virus Ankara protects against measles virus infection in the mouse and cotton rat model. Vaccine 19:2764–2768. doi: 10.1016/S0264-410X(00)00531-4. [DOI] [PubMed] [Google Scholar]
- 84.Ohno S, Ono N, Seki F, Takeda M, Kura S, Tsuzuki T, Yanagi Y. 2007. Measles virus infection of SLAM (CD150) knockin mice reproduces tropism and immunosuppression in human infection. J Virol 81:1650–1659. doi: 10.1128/JVI.02134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O'Donnell LA, Rall GF. 2010. Blue moon neurovirology: the merits of studying rare CNS diseases of viral origin. J Neuroimmune Pharmacol 5:443–455. doi: 10.1007/s11481-010-9200-4. [DOI] [PubMed] [Google Scholar]
- 86.Young VA, Rall GF. 2009. Making it to the synapse: measles virus spread in and among neurons. Curr Top Microbiol Immunol 330:3–30. doi: 10.1007/978-3-540-70617-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Makhortova NR, Askovich P, Patterson CE, Gechman LA, Gerard NP, Rall GF. 2007. Neurokinin-1 enables measles virus trans-synaptic spread in neurons. Virology 362:235–244. doi: 10.1016/j.virol.2007.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reuter D, Schneider-Schaulies J. 2010. Measles virus infection of the CNS: human disease, animal models, and approaches to therapy. Med Microbiol Immunol 199:261–271. doi: 10.1007/s00430-010-0153-2. [DOI] [PubMed] [Google Scholar]
- 89.Brindley MA, Takeda M, Plattet P, Plemper RK. 2012. Triggering the measles virus membrane fusion machinery. Proc Natl Acad Sci U S A 109:E3018–E3027. doi: 10.1073/pnas.1210925109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Porotto M, Yokoyama CC, Orefice G, Kim HS, Aljofan M, Mungall BA, Moscona A. 2009. Kinetic dependence of paramyxovirus entry inhibition. J Virol 83:6947–6951. doi: 10.1128/JVI.00416-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krumm SA, Yan D, Hovingh ES, Evers TJ, Enkirch T, Reddy GP, Sun A, Saindane MT, Arrendale RF, Painter G, Liotta DC, Natchus MG, von Messling V, Plemper RK. 2014. An orally available, small-molecule polymerase inhibitor shows efficacy against a lethal morbillivirus infection in a large animal model. Sci Transl Med 6:232ra252. doi: 10.1126/scitranslmed.3008517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Griffin DE, Lin WH, Pan CH. 2012. Measles virus, immune control, and persistence. FEMS Microbiol Rev 36:649–662. doi: 10.1111/j.1574-6976.2012.00330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lin WH, Kouyos RD, Adams RJ, Grenfell BT, Griffin DE. 2012. Prolonged persistence of measles virus RNA is characteristic of primary infection dynamics. Proc Natl Acad Sci U S A 109:14989–14994. doi: 10.1073/pnas.1211138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Frenzke M, Sawatsky B, Wong XX, Delpeut S, Mateo M, Cattaneo R, von Messling V. 2013. Nectin-4-dependent measles virus spread to the cynomolgus monkey tracheal epithelium: role of infected immune cells infiltrating the lamina propria. J Virol 87:2526–2534. doi: 10.1128/JVI.03037-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sawatsky B, Wong XX, Hinkelmann S, Cattaneo R, von Messling V. 2012. Canine distemper virus epithelial cell infection is required for clinical disease but not for immunosuppression. J Virol 86:3658–3666. doi: 10.1128/JVI.06414-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bird GH, Boyapalle S, Wong T, Opoku-Nsiah K, Bedi R, Crannell WC, Perry AF, Nguyen H, Sampayo V, Devareddy A, Mohapatra S, Mohapatra SS, Walensky LD. 2014. Mucosal delivery of a double-stapled RSV peptide prevents nasopulmonary infection. J Clin Invest 124:2113–2124. doi: 10.1172/JCI71856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Auwaerter PG, Rota PA, Elkins WR, Adams RJ, DeLozier T, Shi Y, Bellini WJ, Murphy BR, Griffin DE. 1999. Measles virus infection in rhesus macaques: altered immune responses and comparison of the virulence of six different virus strains. J Infect Dis 180:950–958. doi: 10.1086/314993. [DOI] [PubMed] [Google Scholar]
- 98.Druelle J, Sellin CI, Waku-Kouomou D, Horvat B, Wild FT. 2008. Wild type measles virus attenuation independent of type I IFN. Virol J 5:22. doi: 10.1186/1743-422X-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]