

ABSTRACT
Measles virus (MeV), a morbillivirus within the paramyxovirus family, expresses two envelope glycoproteins. The attachment (H) protein mediates receptor binding, followed by triggering of the fusion (F) protein, which leads to merger of the viral envelope with target cell membranes. Receptor binding by members of related paramyxovirus genera rearranges the head domains of the attachment proteins, liberating an F-contact domain within the attachment protein helical stalk. However, morbillivirus glycoproteins first assemble intracellularly prior to receptor binding, raising the question of whether alternative protein-protein interfaces are involved or whether an entirely distinct triggering principle is employed. To test these possibilities, we generated headless H stem mutants of progressively shorter length. Conformationally restricted H stems remained capable of intracellular assembly with a standard F protein and a soluble MeV F mutant. Proteolytic maturation of F, but not the altered biochemical conditions at the cell surface, reduces the strength of glycoprotein interaction, readying the complexes for triggering. F mutants stabilized in the prefusion conformation interact with H intracellularly and at the cell surface, while destabilized F mutants interact only intracellularly, prior to F maturation. These results showcase an MeV entry machinery that functionally varies conserved motifs of the proposed paramyxovirus infection pathway. Intracellular and plasma membrane-resident MeV glycoprotein complexes employ the same protein-protein interface. F maturation prepares for complex separation after triggering, and the H head domains in prereceptor-bound conformation prevent premature stalk rearrangements and F activation. Intracellular preassembly affects MeV fusion profiles and may contribute to the high cell-to-cell fusion activity characteristic of the morbillivirus genus.
IMPORTANCE Paramyxoviruses of the morbillivirus genus, such as measles, are highly contagious, major human and animal pathogens. MeV envelope glycoproteins preassemble intracellularly into tightly associated hetero-oligomers. To address whether preassembly reflects a unique measles virus entry strategy, we characterized the protein-protein interface of intracellular and surface-exposed fusion complexes and investigated the effect of the attachment protein head domains, glycoprotein maturation, and altered biochemical conditions at the cell surface on measles virus fusion complexes. Our results demonstrate that measles virus functionally varies conserved elements of the paramyxovirus entry pathway, providing a possible explanation for the high cell-to-cell fusion activity of morbilliviruses. Insight gained from these data affects the design of effective broad-spectrum paramyxovirus entry inhibitors.
INTRODUCTION
Measles virus (MeV) is a highly contagious member of the paramyxovirus family that infects cells through fusion of the viral envelope with cellular membranes. The virus is one of the most infectious pathogens identified to date and remains responsible for major human morbidity and mortality worldwide despite the presence of an effective live attenuated vaccine (1). As is characteristic for the entry strategy of all members of the Paramyxovirinae subfamily, two envelope glycoprotein complexes are required for viral entry. The attachment (H, HN, or G, depending on the Paramyxovirinae genus) protein mediates receptor binding and, subsequently, stimulates major conformational changes of the fusion (F) protein that ultimately result in membrane merger and fusion pore formation (2, 3).
The physiological oligomer of the MeV H protein is a tetramer, which is composed of a stalk domain that connects the transmembrane domains and short luminal tails to the globular head domains harboring the receptor binding sites (4–6). While the structure of the MeV H stalk remains to be determined, partial structures of the related parainfluenza virus 5 (PIV5) and Newcastle disease virus (NDV) HN protein stalks revealed four-helix bundle (4HB) conformations (7–9). Addition of a tetrameric helix bundle of Saccharomyces cerevisiae GCN-derived leucine zippers stabilized a headless MeV H stalk in a bioactive conformation (10), indicating that the 4HB-like stalk conformation is most likely conserved among the Paramyxovirinae and extends to MeV H. Signaling lymphocyte activation molecule (SLAM) and nectin-4 serve as cognate receptors for all MeV strains (11–13). In addition, some laboratory-adapted strains are capable of gaining cell entry through the regulator of complement activation CD46 (14, 15). Chimeric attachment proteins, mutational analyses, and structural modeling have revealed that the Paramyxovirinae attachment protein stalk contains the docking site for specific interaction with the homotypic F protein complex (16–23).
Assembling into a homotrimer, the F protein is first synthesized as an inactive F0 precursor, which, in the case of MeV and most other Paramyxovirinae F proteins, is cleaved into membrane-integral F1 and disulfide-linked F2 subunits by Golgi-resident furin proteases (1, 2). Proteolysis liberates the amino termini of the membrane attack domains or fusion peptides and is thus mandatory for F bioactivity. Unlike the precedence established by, for instance, influenza virus hemagglutinin (HA) proteins, which are subject to major conformational rearrangements upon cleavage activation (24), crystal structures of cleaved and uncleaved paramyxovirus F complexes in the prefusion conformation revealed only minor structural changes (25, 26).
The current mechanistic model of the paramyxovirus entry machinery is driven by crystal structures of Newcastle disease virus (NDV) and parainfluenza virus 5 (PIV5) attachment protein ectodomains in which the globular head domains either back-fold pairwise onto the 4HB stalks or assemble into tetramers (7–9, 27). In the former configuration (termed “heads down”), the head domains are considered to mask the F binding sites in the attachment protein stalks, consequently preventing direct protein-protein interactions and triggering of F protein refolding (9, 23, 28, 29). Then, receptor binding is thought to induce the assembly of head domain tetramers in a “heads-up” state, which exposes the F binding sites in the stalk domain and enables direct glycoprotein interactions for F triggering. In support of this model, several studies demonstrated that head-truncated PIV5, MeV, NDV, mumps virus, and Nipah virus (NiV) attachment protein fragments remain capable of efficient and specific, albeit unregulated, F triggering (10, 22, 23, 30). Furthermore, intracellular retention of one of the PIV5 envelope glycoprotein complexes through added endoplasmic reticulum (ER) localization domains failed to coretain the homotypic partner (31), and PIV5 glycoproteins do not coimmunoprecipitate (22, 28).
While formal proof is pending, tantalizing experimental evidence suggests that rearrangements of the attachment protein heads relative to the stalk domains upon receptor binding may, indeed, present a conserved theme for initiation of the Paramyxovirinae entry cascade (29) that is also employed by members of the morbillivirus genus. For instance, biochemical analyses of native MeV H protein complexes revealed discrete conformational changes after receptor binding (32), MeV H head domains crystallized in two distinct configurations when complexed with SLAM receptor (5), and probing of the NiV attachment protein using monoclonal antibodies spotlighted a series of conformational changes required for fusion triggering (23).
However, envelope glycoprotein complexes of morbilliviruses such as MeV and canine distemper virus (CDV) engage in strong intracellular interactions and efficiently coimmunoprecipitate (16, 33, 34), and MeV glycoproteins effectively coretain the homotypic wild-type partner when engineered with ER-targeting domains (35). It was recently suggested that these findings may reflect the presence of two discrete protein-protein interfaces between morbillivirus H and F and Nipah virus G and F complexes, one preceding receptor binding and possibly involving contacts between the F and attachment protein head domains and the other forming after receptor binding and engaging F and the attachment protein stalk domain (29) (schematically illustrated in Fig. 1A). In contrast to this model, MeV H mutants with major α-helical stalk elongations inserted downstream of the predicted F contact zone remain F-binding- and triggering-competent and support efficient virus replication (16). Rather than two distinct interfaces, the stalk-extended MeV H constructs suggest the presence of a single H/F protein-protein interface formed by a section of the H stalk and a corresponding contact domain in prefusion F. Since H and F preassemble intracellularly into a prefusion hetero-oligomer, why does F not prematurely refold into a postfusion conformation upon furin-mediated cleavage activation in the Golgi compartment if there is only one protein-protein interface, and how does receptor binding regulate morbillivirus entry (Fig. 1B)?
FIG 1.
MeV glycoprotein oligomerization models. Prior to receptor binding, the attachment protein (H) is shown in red in a hypothetical heads-down conformation. (A and C) As proposed in Jardetzky and Lamb (29), the head domains may physically impede interaction of the F0 trimer with the H stalk, and intracellular H/F0 complex preassembly is mediated through an alternative interface between the F and H head domains (A). Receptor binding induces an H heads-up conformation, allowing productive interaction of H with the F protein, resulting in F triggering (C). (B) Alternatively, F0 may directly interact with the H stalk domain even if the H heads are in heads-down mode. While the model shown in panel A necessitates a protein-protein interface switch, the model shown in panel B raises the question of how premature F triggering is prevented. Note that evidence for different head arrangements was reported for MeV H (5); but the depicted heads-up and heads-down conformations are hypothetical, and cartoons are based on structural data obtained for NDV and PIV5 HN proteins and the subsequently proposed universal Paramyxovirinae fusion models (29).
To address the question of whether morbillivirus envelope glycoprotein complexes engage in distinct interactions prior to and after receptor binding, we generated a series of head-truncated MeV H stalk proteins of progressively shorter length and evaluated their ability to physically interact with F intracellularly and after transport to the plasma membrane. Having defined the role of the H stalk domain in the intracellular assembly of MeV fusion complexes, we tested the effect of F proteolytic maturation on the stability of the preassembled MeV glycoprotein hetero-oligomers. Our study spotlights an MeV entry mechanism in which conserved themes of the paramyxovirus membrane fusion machinery, such as receptor-induced attachment protein rearrangement, are combined with features unique to the morbillivirus genus, such as tightly assembled intracellular fusion complexes, resulting in highly efficient viral entry.
MATERIALS AND METHODS
Cell lines and transfections.
HEK293T cells (CRL-11268; ATCC), MDCK (Madin-Darby canine kidney) cells (CCL-34; ATCC), and Vero (African green monkey kidney epithelial) cells (CCL-81; ATCC) stably expressing human SLAM (Vero-SLAM cells) (36) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 7.5% (vol/vol) fetal bovine serum (FBS) at 37°C and 5% CO2. At every third passage, Vero-SLAM cells were exposed to G418 selection to maintain SLAM expression. GeneJuice (Millipore) was used for all transient-transfection reactions.
Molecular biology.
The basis for all H and F expression constructs was previously described; pCG-HFlag and pCG-FHA plasmids encoding H proteins with a triple Flag epitope tag added to the cytosolic H amino terminus (HFlag) (4) or F proteins with a double HA epitope tag added to the cytosolic F carboxy terminus (FHA) (33), respectively, were used. GCN4 tandem stop cassettes were introduced through PCR-based modification as described before (10); H stem mutants truncated at stalk residue 133 (H-133stemGCN4a7A) or 122 (H-122stemGCNΔ15) were previously described (10) and are termed here H-stem133-GCN and H-stem122-GCN, respectively, for simplicity. Equivalent PCR-based mutagenesis strategies using appropriate primers were employed to generate foldon domain-stabilized soluble F (solFfoldon). A QuikChange protocol (Stratagene) was employed for all subsequent site-directed mutagenesis steps. The integrity of all constructs was verified through DNA sequencing.
SDS-PAGE and antibodies.
Total cell lysates and immunoprecipitated material were resuspended in urea buffer (200 mM Tris, pH 6.8, 8 M urea, 5% sodium dodecyl sulfate [SDS], 0.1 mM EDTA, 0.03% bromphenol blue, 1.5% dithiothreitol). Denatured (25 min at 50°C) lysates were fractionated by gel electrophoresis on 4 to 20% NuPAGE gels (H-stem samples; Life Technologies) or 10% Tris-glycine gels (F samples) and transferred to polyvinylidene difluoride (PVDF) membranes (GE Healthcare), and protein material was detected through decoration with specific antibodies directed against the Flag (M2; Sigma) or HA (16b12; Covance) epitope tag. Immunoblots were developed using mouse IgG light-chain-specific horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson) and a ChemiDoc digital imaging system (Bio-Rad). Representative results of two to three independent repeats of each experiment are shown.
Envelope glycoprotein coimmunoprecipitation.
Cells (6 × 105 per well in a six-well plate format) were cotransfected with 1.5 μg each of plasmid DNA encoding the MeV H and F mutants specified in the figures and incubated in the presence of 75 μM fusion-inhibitory peptide (FIP; Bachem) for 36 h. After samples were washed in phosphate-buffered saline (PBS), cells were lysed in immunoprecipitation buffer (50 mM Tris-Cl, pH 7.4, 150 mM sodium chloride, 1 mM EDTA, 1% Triton X-100) containing protease inhibitors and 1 mM phenylmethylsulfonyl fluoride (PMSF). Lysates were cleared by centrifugation for 30 min at 20,000 × g and 4°C, followed by incubation with antibodies directed against the Flag epitope and absorption to matrix-immobilized protein G. Precipitated material was washed in buffer 1 (100 mM Tris, pH 7.6, 500 mM lithium chloride, 0.1% Triton X-100) and then in buffer 2 (20 mM HEPES, pH 7.2, 2 mM EGTA, 10 mM magnesium chloride, 0.1% Triton X-100), denatured in urea buffer, and subjected to SDS-PAGE and immunoblotting.
Envelope glycoprotein cross-linking.
Cells were transfected and incubated as described above, washed in PBS, and treated with 3,3′-dithiobis(sulfosuccinimidylpropionate) (DTSSP; Pierce) cross-linker at a 1 mM final concentration, followed by quenching and lysis in radioimmunoprecipitation assay (RIPA) buffer (1% sodium deoxycholate, 1% NP-40, 150 mM NaCl, 50 mM Tris-Cl, pH 7.2, 10 mM EDTA, 50 mM sodium fluoride, protease inhibitors [Roche], 1 mM phenylmethylsulfonyl fluoride). Cleared lysates were subjected to immunoprecipitation using anti-Flag antibodies, and washed precipitates were subjected to SDS-PAGE and immunoblotting. Coprecipitated F protein material was visualized as outlined above.
Cellular surface immunoprecipitations.
Vero-SLAM cells transfected with MeV H- and F-expressing plasmids as specified in the figures were extensively washed at 36 h posttransfection and subjected to surface immunoprecipitations by incubating intact monolayers at 4°C in the presence of a monoclonal antibody (MAb) directed against an epitope in the MeV H ectodomain (Millipore) and F in the prefusion (anti-prefusion F) (37) or postfusion (anti-triggered F) (38) conformation. Antibodies were used at a 1:1,000 (anti-MeV H) or 1:2,000 (anti-MeV F) dilution in DMEM. Unbound antibody was then removed through extensive washing, cells were lysed in RIPA buffer, and cleared lysates were subjected to precipitation of immunocomplexes with immobilized protein G-Sepharose and SDS-PAGE analysis, as described above.
Surface biotinylation.
Protein surface expression was determined as described before (39) with the following modifications. Cells (6 × 105 per well in a six-well plate format) were transfected with 1.5 μg of plasmid DNA encoding the MeV H and F constructs specified in the figures in the presence of FIP. Washed cells were biotinylated with 0.5 mg/ml sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate (Pierce), quenched, and subjected to precipitation using immobilized streptavidin (GE Healthcare) after lysis in RIPA buffer. Washed precipitates were denatured in urea buffer and subjected to SDS-PAGE analysis.
Cell-to-cell fusion microphotographs.
Fluorescence microscopy was performed with a Zeiss Axio Observer D.1 inverted microscope. For phase-contrast images, a Nikon Diaphot 200 inverted microscope was employed. In both cases, a 10× objective was used.
Dual split-protein cell content-mixing assay.
293T effector cells were transfected with plasmids encoding the Dsp1–7 dual split-protein component (40), H, or H-stem122-GCN and MeV F at different ratios as specified in the figures. 293T target cells received plasmid DNA encoding the Dsp8–11 subunit. Premature effector cell fusion was suppressed through addition of 75 μM FIP. Cells were washed at 30 h posttransfection, both populations were combined at equal ratios, and mixed cells were transferred to solid-wall 96-well plates. Reconstitution of Renilla luciferase as a marker for cell content mixing was assessed using ViviRen (Promega) live-cell substrate and a Synergy H1 (BioTek) multifunction microplate reader with the gain set at 135 and 250. Data sets were normalized for values obtained for each H mutant at a ratio of 0.5 of H, and F-encoding plasmids were transfected. Per H construct analyzed, four individual data sets were generated, each assessed at either gain level. The values shown represent the averages of the normalized values of all eight resulting data sets.
Statistical analysis.
Statistical significance of differences between measurements was assessed by unpaired two-tailed t tests. Experimental uncertainties are identified by standard deviation (SD) or standard error of the mean (SEM) as specified in the figure legends.
RESULTS
In recent work, we developed a headless MeV H construct that retained intracellular transport competence and F-triggering activity when stabilized through a terminal tetramerization domain derived from the leucine zipper of the yeast transcription factor GCN4 (10). The addition of the membrane-impermeable chemical cross-linker DTSSP to cells coexpressing F and this H-stem122-GCN, which consists of the cytosolic tail, transmembrane domain, and H ectodomain stalk up to residue 122, resulted in efficient covalent linking of surface-exposed, proteolytically matured H/F1+2 fusion complexes, underscoring that physical docking of F to the attachment protein stalk is a prerequisite for F triggering.
Testing the H/F0 protein-protein interface.
The H-stem122-GCN construct presents a suitable tool to address the question of whether intracellular assembly of MeV H/F0 complexes relies on a discrete protein-protein interface that involves direct contacts between the uncleaved F0 precursor protein and residues in the H head domain. In these intracellular prefusion complexes, the H heads must naturally be in a prereceptor-bound conformation, which for simplicity we will refer to in the following as a hypothetical MeV H heads-down configuration (Fig. 1) in analogy to the PIV5 and NDV HN structural data. We coexpressed standard MeV F with MeV H, head-truncated H-stem122-GCN, or derivatives thereof harboring a phenylalanine-to-alanine substitution of H residue 111 that we have previously shown to substantially reduce DTSSP-mediated H/F1+2 cross-linking (16). Gel fractionation and immunoblotting of whole-cell lysates revealed an abundance of cleaved F1+2 material at steady state, confirming efficient proteolytic maturation of F0 complexes in the presence of the different H proteins (Fig. 2A). DTSSP cross-linking of surface-displayed complexes followed by immunoprecipitation of H under biochemically stringent buffer conditions and detection of coprecipitated F material confirmed efficient linking of H/F1+2 and H-stem122-GCN/F1+2 oligomers (Fig. 2B). Cross-linking was greatly reduced or completely abolished in the presence of the F111A point mutation, as previously reported (10).
FIG 2.

Distinct effects of H stalk mutations and head truncation on H/F0 and H/F1+2 complex assembly. (A) Whole-cell lysates (WCL) of cells expressing MeV HFlag mutants and FHA were analyzed by immunoblotting (IB) using epitope tag-specific antibodies. (B) Cells from the experiment shown in panel A were subjected to coimmunoprecipitation (IP) of F with H or surface cross-linking using DTSSP to stabilize the H-F1+2 interaction, in both cases followed by detection of coprecipitated F antigenic material. (C) Cells transfected with MeV HFlag mutants and MeV or PIV3 FHA were analyzed as described for panels A and B. (D) MeV receptor-negative MDCK cells were transfected with MeV FHA and HFlag mutants and analyzed for intracellular interaction as outlined above. α, anti.
To test intracellular assembly of these envelope glycoprotein complexes, we subjected whole-cell lysates of equally transfected cells to H protein immunoprecipitation under mild buffer conditions, followed by immunoblotting for coprecipitated F material. Remarkably, full-length H proteins interacted very efficiently with F0 complexes, regardless of the presence or absence of the F111A substitution (Fig. 2B). Equally noteworthy, the H-stem122-GCN mutant tested was unable to form a biochemically stable intracellular complex with F0, despite DTSSP-mediated cross-linking of H-stem122-GCN/F1+2 on the cell surface and the demonstrated ability of the H-stem122-GCN to productively trigger F refolding for membrane fusion (10). Intracellular interaction of the H-stem122-GCN(F111A) mutant with F0 was likewise substantially reduced compared to full-length H(F111A) but was still detectable, suggesting that the presence of the F111A mutation may slightly improve the intracellular interaction of the H stem with F0.
Control experiments confirmed, first, that both DTSSP-mediated H-stem122-GCN/F1+2 cross-linking and H-stem122-GCN/F0 coimmunoprecipitation are based on specific protein-protein interactions requiring homotypic glycoprotein pairs; coexpression of the different MeV H protein variants with heterotypic human parainfluenzavirus 3-derived F proteins did not reveal appreciable coimmunoprecipitation or cross-linking (Fig. 2C). Second, MeV receptors present in the secretory system of the host cell had no effect on the differential coimmunoprecipitation of intracellular F0 trimers by H-stem122-GCN and H(F111A) or standard H since we obtained equivalent results when we expressed the envelope glycoproteins in MeV receptor-negative MDCK cells (Fig. 2D).
At first sight, these results appeared consistent with the hypothesis that intracellular H/F0 and surface-expressed, fusion-competent H/F1+2 complexes may engage distinct protein-protein interfaces, the former possibly involving F0 to H head contacts in a hypothetical heads-down-like conformation of the H tetramer, which the H-stem122-GCN mutant could not establish. However, why do F0 complexes in the absence of the H head domains not simply interact with the exposed F-binding sites present in the stalk of H-stem122-GCN proteins, in essence, short-circuiting any receptor-induced H-head rearrangement step? In fact, we would anticipate such a direct interaction to take place since PIV5 F-based structural data most likely extend to MeV and since proteolytic maturation of the MeV F trimer is also anticipated to result in only minimal structural changes (25).
Effect of H stalk length on intracellular interaction with F0.
To address this MeV glycoprotein interaction conundrum, we generated a series of progressively shorter H stem constructs ranging from H-stem110-GCN to H-stem72-GCN, all stabilized by a carboxy-terminal GCN4 tetramerization domain according to our previously described strategy (10). With the exception of H-stem110-GCN, none of these H stem constructs was capable of triggering F for cell-to-cell fusion (Fig. 3A). However, compared to the highly active H-stem122-GCN or standard H, H-stem110-GCN also induced only minimal cell-to-cell fusion, which in quantitative assays did not result in luciferase reporter activities above background levels (Table 1).
FIG 3.
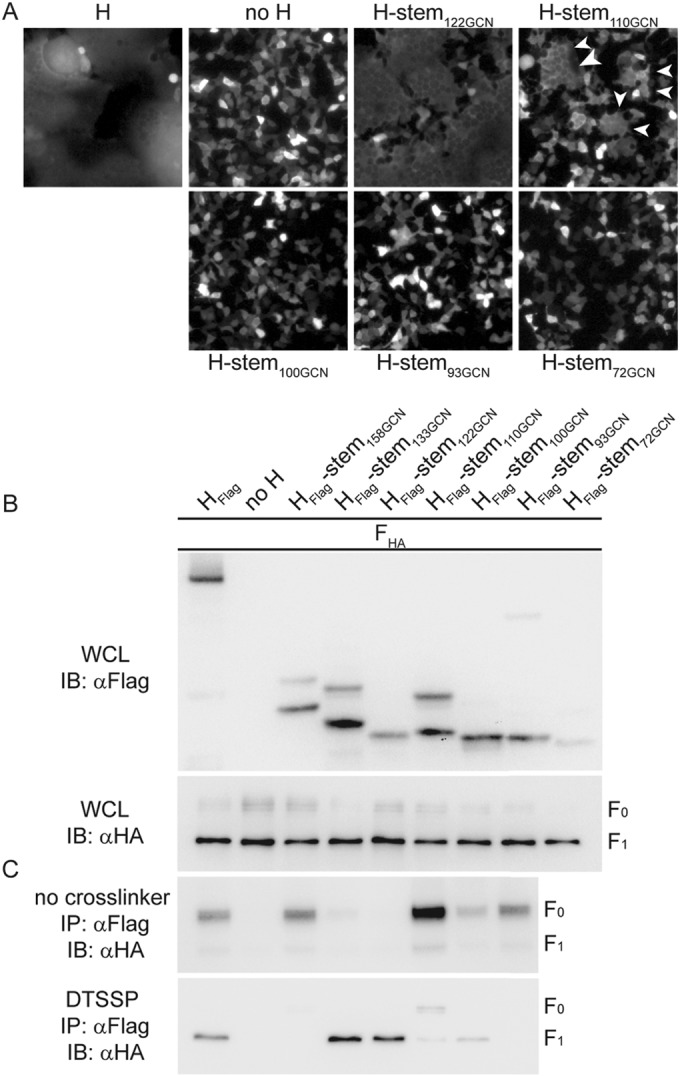
MeV H/F0 assembly is based on interaction of F with the H stalk domain. (A) Cell-to-cell fusion of cells transfected with MeV F and the newly generated GCN domain-stabilized H stem mutants of progressively shorter length. For comparison, the previously reported bioactive H-stem122-GCN was included. In the case of cells expressing H-stem110-GCN and F, arrowheads highlight individual syncytia. (B and C) Analysis of cell lysates and coprecipitated F material as outlined in the legend of Fig. 2A and B after pulldown with the specified H mutants in the presence or absence of DTSSP.
TABLE 1.
Intracellular transport competence and F-triggering activity of progressively shortened, GCN-stabilized MeV H-stem mutants
| H mutant | Relative cell surface expressiona (% of H-stem122-GCN) | Relative bioactivityb (% of H-stem122-GCN) |
|---|---|---|
| H-stem122-GCN | 100 | 100 |
| H-stem158-GCNc | 7 ± 6d | ND |
| H-stem133-GCNc | 37 ± 13d | 85e |
| H-stem110-GCN | 137 ± 30f | Unquantifiable |
| H-stem100-GCN | 123 ± 22f | ND |
| H-stem93-GCN | 38 ± 12f | ND |
| H-stem72-GCN | 0 ± 7f | ND |
Determined through surface biotinylation.
Determined through reporter-based cell-to-cell fusion assays. Quantitative assays were performed only when some syncytia were microscopically detectable after cotransfection of the H mutant with MeV F. ND, not determined.
H-stem mutants and data as reported in Brindley et al. (10).
Results represent means of three experiments ± SD and are expressed relative to H-stem122-GCN.
Results are based on a logistic regression model fitted to kinetic fusion curves as detailed in Brindley et al. (10). The highest overall activities provide the basis for the normalizations.
Results represent means of five experiments ± SD and are expressed relative to H-stem122-GCN.
Nevertheless, all newly generated stem constructs except H-stem72-GCN were readily detectable in immunoblots of whole-cell lysates (Fig. 3B), and surface biotinylation-based quantitation of intracellular transport competence revealed plasma membrane steady-state levels of at least 38% that of bioactive H-stem122-GCN, which served as a reference (Table 1). The exception was the shortest H-stem72-GCN mutant, which remained undetectable at the cell surface. When subjected to coimmunoprecipitation or chemical cross-linking, the different H stems segregated into two classes based on distinct phenotypes. H stems 122-GCN and 133-GCN could be efficiently cross-linked with F1+2 at the cell surface but did not coprecipitate F0. In contrast, previously described H-stem158-GCN and the new stems 110-GCN, 100-GCN, and 93-GCN appreciably—albeit with different efficiencies—coprecipitated F0 complexes but did not cross-link with F1+2 (H-stem158-GCN and H-stem93-GCN), or cross-linking with F1+2 was greatly reduced (H-stem110-GCN and H-stem100-GCN) (Fig. 3C). H-stem72-GCN is not shown in this assay due to its overall very low surface steady-state level.
These data illuminate two features of the MeV H-F interaction pathway: first, intracellular assembly of hetero-oligomers consisting of unprocessed F0 trimers with H tetramers does not require a new interface between F and the H head domains; and, second, tightly preassembled MeV F0/H oligomers are not a mandatory precursor for the generation of functional fusion complexes through proteolytic maturation of F. However, why are triggering-competent H stems 133-GCN and 122-GCN unable to tightly interact with F0 trimers, and what is the role of the MeV H head domains in fusion regulation if they do not mask F binding sites?
Interaction of disulfide bond-stabilized H stems with F0 complexes.
Efficient MeV F triggering reportedly depends on structural flexibility in the central section of the H stalk domain (32, 41, 42). We therefore hypothesized that the MeV H head domains in the prereceptor-bound, hypothetical heads-down conformation may clamp the H stalk in a pretriggered, F-binding-competent form. Of all MeV H stem constructs generated, H-stem133-GCN and H-stem122-GCN may shuttle between pre- and posttriggered conformations, preventing the formation of biochemically detectable intracellular complexes with F0 trimers. If this is correct, we would expect to restore the intracellular assembly of fusion complexes involving H-stem133-GCN or H-stem122-GCN by locking these H stems into a pretriggered conformation. To test this idea, we introduced different cysteine substitutions into the H-stem122-GCN construct, which were all previously implicated in the formation of covalent MeV tetramers and/or an, in most cases, reversible block of F-triggering activity (32, 42). Expression of the resulting H-stem122-GCN mutants with standard F followed by coimmunoprecipitation analysis revealed that several of the substitutions indeed enabled some intracellular interaction of the H stems with F0; a D101C, T112C, or D113C exchange in particular resulted in efficient coimmunoprecipitation of F0 trimers with the H stem (Fig. 4A). However, DTSSP-mediated cross-linking of H-stem122-GCN with F1+2 was reduced in the presence of these mutations, possibly reflecting that covalently rigidifying the H stalk may become less favorable once F is processed.
FIG 4.

Conformational H stalk stabilization restores intracellular stalk interaction of headless H-stem122-GCN with F. (A) Analysis of cell lysates and coprecipitated F material as outlined in the legend of Fig. 2A and B after pulldown with the specified H mutants in the presence or absence of DTSSP. (B) Schematic of the foldon domain-stabilized solFHA-foldon mutant. (C) Immunodetection of solFHA-foldon in whole-cell lysates (WCL) and culture supernatants after immunoprecipitation. (D and E) Analysis of the effect of H-stalk-stabilizing mutations D101C, T112C, and D113C from the constructs shown in panel A introduced into full-length H (D) or H-stem122-GCN (E) on intracellular interaction with solFHA-foldon.
To address the question of whether coimmunoprecipitation predominantly monitors H and F ectodomain interactions or is substantially influenced by transmembrane domain and/or cytosolic tail contacts, we generated a soluble form of MeV F in which the carboxy-terminal transmembrane and cytosolic residues are replaced with the T4 bacteriophage fibritin-derived foldon trimerization domain (43). The resulting solFfoldon (Fig. 4B) was intracellular transport competent and efficiently secreted from transfected cells (Fig. 4C). Coexpression of solFfoldon either with standard H or disulfide bond-restricted full-length H variants H(D101C), H(T112C), and H(D113C), followed by coimmunoprecipitation analysis, revealed an interaction pattern closely resembling that observed for standard F (Fig. 4D); unchanged H and all of the H mutants tested efficiently interacted with solFfoldon. When we subjected the H stems to this assay, we likewise noted efficient intracellular interaction of the disulfide bond-stabilized H-stem122-GCN(D101C), H-stem122-GCN(T112C), and H-stem122-GCN(D113C) with solFfoldon, whereas the original H-stem122-GCN was also unable to interact with soluble F (Fig. 4E). Equally noteworthy, the even shorter H-stem110-GCN construct, which we had found capable of efficient coprecipitation of membrane-integral standard F0 complexes, also formed biochemically detectable hetero-oligomers with solFfoldon.
These results indicate that protein-protein interfaces governing hetero-oligomerization are located in the MeV glycoprotein ectodomains and, in the case of H, apparently involve microdomains posited to be membrane-proximal to a previously identified candidate F-contact zone spanning H stalk residues 111 to 118 (16, 17).
DTSSP cross-linking targets functional fusion complexes in a prefusion conformation.
This model does not address, however, why intracellular MeV H/F0 complexes tightly interact, whereas H/F1+2 hetero-oligomers do not coimmunoprecipitate efficiently without DTSSP cross-linking, even if membrane fusion is pharmaceutically suppressed by peptidic (44) or small-molecule (45) fusion inhibitors or if proteins are expressed in MeV receptor-negative cells (Fig. 2D). Three alternative explanations for this phenotype are conceivable: DTSSP cross-linking could simply target postfusion rather than functional prefusion H and F complexes; the altered biochemical conditions experienced by fusion complexes located on the cell surface instead of in the secretory system could destabilize the H and F interaction; or proteolytic maturation may induce slight conformational changes within the F trimer that reduce the strength of interaction with H, readying the complex for triggering.
To distinguish between these possibilities, we launched a two-pronged approach. First, we generated two F mutants that were previously suggested either to be locked into a prefusion conformation and unable to induce membrane fusion [F(L325D)] (46) or to show reduced conformational stability in the metastable prefusion form and readily assume a postfusion fold [F(L457A-V459A)] (41).
Indeed, coexpression of F(L325D) with MeV H did not result in any microscopically appreciable cell-to-cell fusion, whereas the F(L457A-V459A) protein showed some bioactivity (Fig. 5A). Using a surface immunoprecipitation protocol and pre- and postfusion F-specific monoclonal antibodies that were previously characterized (41), we next assessed the conformational state of F(L325D) and F(L457A-V459A) complexes when coexpressed with standard MeV H in receptor-positive cells. Under these conditions, standard F complexes were present in both pre- and postfusion conformations on the cell surface (Fig. 5B). As expected, F(L325D) trimers remained exclusively in the prefusion conformation, whereas essentially all F(L457A-V459A) complexes had refolded into the postfusion form. Addition of DTSSP prior to lysis of transfected cells demonstrated efficient cross-linking of proteolytically matured F1+2(L325D) trimers with both the standard H and H-stem122-GCN (Fig. 5C). In contrast, conformationally less stable F(L457A-V459A) coprecipitated with H prior to F maturation, but the extent of DTSSP cross-linking of cell surface-expressed H and F1+2(L457A-V459A) complexes was marginal. These results confirm that DTSSP covalently links proteolytically matured fusion complexes that are in a prefusion conformation.
FIG 5.

DTSSP cross-linking stabilizes surface-exposed MeV glycoprotein complexes in prefusion conformation. (A) Cell-to-cell fusion of cells transfected with MeV H and F mutant F(L325D) or F(L457A-V459A). (B) F surface immunoprecipitation (SF IP) after incubation of intact cells transfected with MeV H and F(L325D) or F(L457A-V459A) with conformation-dependent antibodies directed against the F trimer in the prefusion (αPre F) or postfusion (αTrig F) conformation. (C) Analysis of cell lysates and coprecipitated F mutants F(L325D) and F(L457A-V459A) after pulldown with the specified H mutants in the presence or absence of DTSSP as outlined in the legend of Fig. 2A and B.
Effect of F protein maturation on the stability of envelope glycoprotein hetero-oligomers.
To test whether the different biochemical conditions in the secretory system versus the extracellular environment or F maturation destabilizes the H/F1+2 hetero-oligomers, we destroyed the furin protease consensus motif through a K111N substitution in the F protein to create nonactivatable F0 trimers. The intracellular transport rate of the F(K111N) mutant was reduced compared to that of standard F, but the surface-expressed mutant material remained predominantly in the F0 state (Fig. 6A). A subpopulation with an F1-like migration pattern in gel electrophoresis was detectable in the F(K111N) preparations, which most likely results from postextraction cleavage by nonfurin proteases since cell-to-cell fusion activity of F(K111N) coexpressed with standard H was completely abolished (Fig. 6B). Uncleaved F(K111N) interacted efficiently with H intracellularly and after transport to the plasma membrane (Fig. 6C).
FIG 6.

Uncleaved F0 complexes remain stably associated with H on the cell surface. (A) Cell-to-cell fusion microphotographs of cells transfected with MeV H and F mutant F(K111N), disrupting the furin proteolysis motif. (B) Surface biotinylation of cells transfected with F(K111N) or standard F. The F1-like material observed with F(K111N)-transfected cells represents nonproductive, most likely postextraction, F proteolysis. (C and D) Analysis of coprecipitated F(K111N) after pulldown with standard H (C) or the specified H mutants (D) in the presence or absence of DTSSP as outlined in the legend of Fig. 2A and B. (E) Fusion profiles of MeV H/F and H-stem122GCN/F pairs using a quantitative fusion assay based on the reconstitution of dual-split luciferase/enhanced green fluorescent protein fusion proteins. F-encoding plasmid DNA was kept constant for all samples, and the relative amount of H-encoding plasmid DNA was progressively reduced. Values represent mean relative luciferase units ± SEM of four independent experiments, each normalized (norm.) for a relative H-to-F ratio of 0.5. To determine the statistical significance of differences between the standard and mutant H-based data sets, unpaired two-tailed t tests were performed (*, P < 0.05; **P < 0.01; ***P < 0.001; NS not significant). SF bio, surface biotinylation.
Coexpression of F(K111N) with standard H, H(F111A), and H-stem122-GCN followed by DTSSP cross-linking revealed efficient interaction of cell surface-exposed F0(K111N) trimers also with the H(F111A) mutant (Fig. 6D). This result stands in contrast to our initial observation of abrogated cross-linking of mature F1+2 with H(F111A), suggesting that F0 cleavage into F1+2 trimers rather than the altered biochemical conditions of the extracellular milieu must be predominantly responsible for the reduced strength of the interaction of plasma membrane-resident MeV fusion complexes. These findings corroborate the hypothesis that the F111A substitution in the H stalk may induce a postreceptor-bound conformation of H as previously proposed (32), which leads to complete physical separation of the H and F complexes immediately after F proteolytical maturation in the Golgi compartment.
Contribution of intracellular glycoprotein assembly to MeV fusion profiles.
Unlike morbillivirus H and F, the envelope glycoproteins of most other Paramyxovirinae family members do not tightly assemble in the secretory system of the host cell. Having generated triggering-competent but intracellular assembly-deficient MeV H stem mutants, we asked whether tight preassembly of H and F complexes in the secretory system has an appreciable effect on the MeV membrane fusion profile. We cotransfected cells with different relative ratios of H and F or of H-stem122-GCN and F expression plasmids and determined fusion activity in a quantitative cell-to-cell fusion assay that monitors cell content mixing (10, 32). The standard H/F system was capable of buffering changes in relative glycoprotein levels since fusion activities did not change over a broad ratio range when F plasmid levels were kept constant and H plasmids were successively reduced (Fig. 6E). We noticed only at extreme ratios a steep drop of fusion activity from this plateau to background levels. In contrast, the equivalent titration of intracellular assembly-incompetent H-stem122-GCN-encoding plasmids relative to F plasmid levels lacked the initial plateau phase of fusion activity. Rather, activity decreased more gradually with the reduction in H stem plasmid levels, translating into significantly different fusion profiles of intracellular assembly-competent and -incompetent MeV H mutants.
DISCUSSION
By biochemically characterizing the evolution of the MeV H and F protein interactions as the envelope glycoproteins advance through the secretory system of the host cell, we dissect conserved motifs of the paramyxovirus F-triggering machinery from those exclusive to the morbillivirus entry system (Fig. 7). Upon membrane insertion, MeV glycoproteins hetero-oligomerize intracellularly into tightly assembled H/F0 complexes, engaging an F-docking site that is located in the H stalk. Proteolytic maturation of F0 in Golgi compartments substantially reduces the avidity of the H-F interaction, readying the complexes for physical separation after F triggering. In the prereceptor-bound state, the H head domains suppress spontaneous reorganization of the central H stalk section into the trigger-active configuration. Engagement of receptor lifts this restriction, clearing the path for H-stalk unwinding and triggering of the associated F trimer. We show that these unique features may contribute to the very high cell-to-cell fusion activity that is characteristic of morbillivirus-derived fusion complexes (1). Four major lines of evidence support these conclusions.
FIG 7.
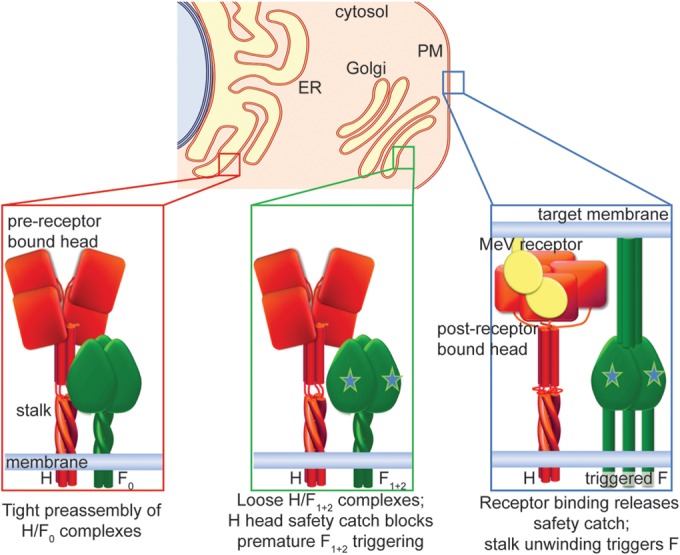
(Top) Three-step safety-catch model of maturation and activation of the MeV entry machinery as the envelope proteins advance from the endoplasmic reticulum (ER) through the Golgi apparatus (Golgi) to the plasma membrane (PM). (Left) Hetero-oligomer preassembly. MeV H and F0 proteins tightly assemble in the ER, engaging a protein-protein interface between the prefusion F head and H stalk domain. The H head domains in a hypothetical, prereceptor-bound heads-down conformation do not prevent assembly of the H/F0 complex. (Middle) Readying the fusion complex. Proteolytic F maturation (symbolized by stars) in late Golgi compartments greatly reduces the strength of hetero-oligomer interaction, preparing the complexes for physical separation after triggering to accommodate F refolding. The H head domains in heads-down conformation act as a safety catch, preventing conformational changes of the H stalk and blocking premature F triggering. (Right) Triggering and hetero-oligomer separation. Receptor binding induces rearrangements of the H head domains (safety catch off), allowing unwinding of the central stalk section, which triggers the F refolding cascade leading to H and F1+2 separation and, ultimately, membrane fusion.
First, MeV glycoprotein complexes tightly assemble in the secretory system of the host cell, engaging an H stalk/F protein-protein interface. In previous work, we have demonstrated that intracellularly retained MeV H or F proteins are able to coretain their transport-competent homotypic counterpart (35). In contrast, PIV5 glycoproteins, for instance, neither coimmunoprecipitate nor coretain each other in equivalent experiments (31). Our observation that headless MeV H stem constructs of different stalk lengths remain capable of interacting with F0 when appropriately stabilized conformationally confirmed that the H head domains are not mandatory for intracellular assembly. These data spotlight that both intracellular H/F0 and cell surface-exposed H/F1+2 complexes are based upon interaction of F with the H stalk domain rather than upon a hypothetical second interface between the F and H head domains. Expansion of our study to efficient coimmunoprecipitation of a soluble F mutant with membrane-integral full-length H and H stems further demonstrated that the interface domains driving hetero-oligomerization are located in the H and F ectodomains and not in the transmembrane sections or cytosolic tails.
Structural studies support the idea that the MeV H heads also rearrange relative to each other upon receptor binding (5). We cannot completely exclude the possibility that the H heads may initially cover stalk residues 111 to 118 in a prereceptor-bound heads-down-like conformation, while the interaction with F is mediated by stalk residues membrane distal to the domain of residues 111 to 118. Receptor binding could then result in movement of the H heads, exposure of residues 111 to 118, and subsequent triggering of the associated F. However, we consider this model unlikely based on our previous finding that substantially (approximately 50% of the original length) stalk-extended H mutants remained F-triggering competent, provided the extensions are inserted membrane distal of stalk residue 118 (16). Since it is difficult to reconcile how the raised head domains (10) of these H mutants could still reach and cover residues 111 to 118, we conclude that the stalk exposure activation model proposed for paramyxovirus HN attachment proteins (29) most likely does not apply equally to the morbillivirus system.
Second, the MeV H head domains appear to act as a safety catch preventing premature H stalk reorganization and triggering of the associated F protein. We along with others have demonstrated that structural flexibility within the central region of the MeV H stalk is instrumental for F triggering (32, 34, 42). In particular, several H mutants carrying disulfide bonds engineered into the H stalk lost F-triggering activity but could be reactivated by the restoration of stalk flexibility through bond reduction. Remarkably, we noticed that the F-triggering-competent H-stem122-GCN mutants required the presence of an engineered additional disulfide bond in the H stalk for interaction with intracellular F0. This finding indicates that the intracellular assembly of tightly associated MeV H/F0 complexes mandates a conformational restriction of the H stalk domain to initiate hetero-oligomerization or prevent premature glycoprotein separation. As evidenced by the retained partial triggering competence of H-stem122-GCN and H-stem133-GCN (10), tight intracellular interaction of H and F is not a prerequisite for fusion activity. Most likely, spontaneous and premature stalk rearrangements may reduce the affinity of these H stems for F or compromise the protein-protein interface. We cannot definitively address at present whether these H stalk rearrangements are reversible and fusion is based on spontaneous encounter of the H stem mutants with unaffiliated F complexes in the plasma membrane or whether a small, biochemically undetectable proportion of intracellularly preassembled H stem/F complexes carries the fusion phenotype.
Importantly, however, of all triggering-competent H constructs, only the headless H stems were dependent on the presence of additional disulfide bonds for this conformational restriction. We therefore conclude that the head domains exercise the stabilizing effect in standard H. Receptor binding-induced head domain rearrangements within MeV H could very plausibly mimic the heads-up and heads-down configuration reported for PIV5 HN and NDV HN. However, in the case of MeV H, these rearrangements do not enable glycoprotein interaction (MeV H and F complexes are already preassembled) or directly trigger F but, rather, appear to lift a mechanical restriction locking the central H stalk part in a pretriggering conformation. Interestingly, a multiple-step fusion-triggering cascade was recently proposed also for the related Nipah virus F and G glycoprotein complexes, which, like morbillivirus H and F, tightly assemble into biochemically appreciable hetero-oligomers (23). Members of different Paramyxovirinae genera may have evolved to reconfigure or mechanistically reassign conserved functional modules in the attachment protein to spatially and temporally regulate their entry machinery.
Third, F proteolytic maturation destabilizes the tightly associated H/F0 complexes, converting the hetero-oligomers into loosely assembled, functional H/F1+2 fusion complexes. In previous work, we have demonstrated that the strength of the H and F interaction varies to some degree with the strain origin of the glycoproteins (33). MeV Edmonston strain-derived glycoprotein pairs appeared particularly loosely assembled, explaining why we could observe in the present study plasma membrane-resident H/F1+2 hetero-oligomers only after cross-linking with DTSSP. Using F conformation-dependent antibodies, we confirmed that DTSSP indeed covalently stabilizes H/F1+2 fusion complexes in the prefusion conformation. This result spotlights that MeV H and F hetero-oligomers progress from tightly associated to loosely interacting as they advance through the secretory system and reach the plasma membrane, where cell-to-cell fusion or incorporation into progeny viral particles takes place. Stalks of full-length H proteins harboring the F111A change appear permanently locked into a postreceptor-bound conformation, which may lead to immediate physical separation of H and F once the H/F association is decreased through F cleavage. Readying preassembled fusion complexes for refolding through releasing the tight association supports the hypothesis that the major conformational changes of F mandate physical separation of the glycoprotein oligomers. Our previous observation that the strength of the interaction of the MeV glycoproteins is, within a reasonable range, inversely correlated to the extent of fusion activity (33) is likewise consistent with this mandatory transition to loosely assembled, fusion-primed complexes. Similar inverse correlations were also reported for henipavirus G and F protein pairs (47).
Based on all available evidence, destabilization of the H/F0 complexes through F proteolysis appears to result from very subtle structural changes in F since crystal structures of cleaved and uncleaved paramyxovirus PIV5 F trimers in prefusion conformation demonstrated only minor rearrangements (25, 48). However, previous epitope mapping studies have indicated biochemically appreciable changes within PIV5 F upon proteolysis (49). Even comparably small conformational changes upon F maturation may thus exude quite a dramatic effect on the strength of MeV glycoprotein hetero-oligomer interaction.
Last, being able to tightly assemble in the secretory system directly affects MeV glycoprotein fusion profiles. The F-triggering-competent headless H stem mutants provided a system in which the strength of the intracellular interaction between H and F0 in the secretory system was dramatically reduced, whereas the amount of plasma membrane-resident H/F1+2 prefusion complexes remained largely unaffected. In the absence of preassembly, one would expect lateral diffusion in the plasma membrane for fusion complex assembly to become rate limiting for fusion when the abundance of individual H and F complexes is reduced. Fusion profiles of tightly preassembled MeV glycoprotein complexes were indeed resilient to changes in relative protein ratios over a wide range, while loosely assembled hetero-oligomers showed a more direct correlation between the reduction of relative protein ratios and fusion activity. One should assume that assembly of physically separated paramyxovirus HN and F complexes into functional hetero-oligomers requires a lateral rearrangement step in the plane of the plasma membrane once the F-docking sites in the HN stalk open up after receptor binding. In contrast, our results demonstrate that MeV glycoproteins have no need for reorganization after receptor binding to form functional fusion complexes. Since proteolytically matured H/F1+2 complexes reach the plasma membrane and are immediately fusion ready, we consider the high cell-to-cell fusion activity of the morbilliviruses a possible physiological consequence of intracellular H and F0 glycoprotein preassembly.
ACKNOWLEDGMENTS
We thank B. Horvat and M. Ehnlund for generously providing MAbs directed against the MeV F protein, N. Kondo and Z. Matsuda for split luciferase reporter plasmids, and A.L. Hammond for critical reading of the manuscript.
This study was supported, in part, by Public Health Service grant AI083402 from the NIH/NIAID (to R.K.P.).
We declare that we have no competing interests.
M.A.B., S.C., and R.K.P. contributed to conception and design of the study and planned the experiments; M.A.B. and S.C. performed the experiments; M.A.B., S.C., and R.K.P. analyzed the data; and R.K.P. wrote the manuscript.
REFERENCES
- 1.Griffin DE. 2007. Measles virus, p 1551–1585 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Lamb RA, Parks GD. 2007. Paramyxoviridae: the viruses and their replication, p 1449–1496 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Plemper RK. 2011. Cell entry of enveloped viruses. Curr Opin Virol 1:92–100. doi: 10.1016/j.coviro.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brindley MA, Plemper RK. 2010. Blue native PAGE and biomolecular complementation reveal a tetrameric or higher-order oligomer organization of the physiological measles virus attachment protein H. J Virol 84:12174–12184. doi: 10.1128/JVI.01222-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hashiguchi T, Ose T, Kubota M, Maita N, Kamishikiryo J, Maenaka K, Yanagi Y. 2011. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat Struct Mol Biol 18:135–141. doi: 10.1038/nsmb.1969. [DOI] [PubMed] [Google Scholar]
- 6.Zhang X, Lu G, Qi J, Li Y, He Y, Xu X, Shi J, Zhang CW, Yan J, Gao GF. 2013. Structure of measles virus hemagglutinin bound to its epithelial receptor nectin-4. Nat Struct Mol Biol 20:67–72. doi: 10.1038/nsmb.2432. [DOI] [PubMed] [Google Scholar]
- 7.Bose S, Welch BD, Kors CA, Yuan P, Jardetzky TS, Lamb RA. 2011. Structure and mutagenesis of the parainfluenza virus 5 hemagglutinin-neuraminidase stalk domain reveals a four-helix bundle and the role of the stalk in fusion promotion. J Virol 85:12855–12866. doi: 10.1128/JVI.06350-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan P, Swanson KA, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc Natl Acad Sci U S A 108:14920–14925. doi: 10.1073/pnas.1111691108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welch BD, Yuan P, Bose S, Kors CA, Lamb RA, Jardetzky TS. 2013. Structure of the parainfluenza virus 5 (PIV5) hemagglutinin-neuraminidase (HN) ectodomain. PLoS Pathog 9:e1003534. doi: 10.1371/journal.ppat.1003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brindley MA, Suter R, Schestak I, Kiss G, Wright ER, Plemper RK. 2013. A stabilized headless measles virus attachment protein stalk efficiently triggers membrane fusion. J Virol 87:11693–11703. doi: 10.1128/JVI.01945-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 12.Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. 2011. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog 7:e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, Gerlier D. 1993. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol 67:6025–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorig RE, Marcil A, Chopra A, Richardson CD. 1993. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 75:295–305. doi: 10.1016/0092-8674(93)80071-L. [DOI] [PubMed] [Google Scholar]
- 16.Paal T, Brindley MA, St Clair C, Prussia A, Gaus D, Krumm SA, Snyder JP, Plemper RK. 2009. Probing the spatial organization of measles virus fusion complexes. J Virol 83:10480–10493. doi: 10.1128/JVI.01195-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JK, Prussia A, Paal T, White LK, Snyder JP, Plemper RK. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J Biol Chem 283:16561–16572. doi: 10.1074/jbc.M801018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deng R, Wang Z, Mirza AM, Iorio RM. 1995. Localization of a domain on the paramyxovirus attachment protein required for the promotion of cellular fusion by its homologous fusion protein spike. Virology 209:457–469. doi: 10.1006/viro.1995.1278. [DOI] [PubMed] [Google Scholar]
- 19.Tsurudome M, Kawano M, Yuasa T, Tabata N, Nishio M, Komada H, Ito Y. 1995. Identification of regions on the hemagglutinin-neuraminidase protein of human parainfluenza virus type 2 important for promoting cell fusion. Virology 213:190–203. doi: 10.1006/viro.1995.1559. [DOI] [PubMed] [Google Scholar]
- 20.Tanabayashi K, Compans RW. 1996. Functional interaction of paramyxovirus glycoproteins: identification of a domain in Sendai virus HN which promotes cell fusion. J Virol 70:6112–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng R, Mirza AM, Mahon PJ, Iorio RM. 1997. Functional chimeric HN glycoproteins derived from Newcastle disease virus and human parainfluenza virus-3. Arch Virol Suppl 13:115–130. [DOI] [PubMed] [Google Scholar]
- 22.Bose S, Zokarkar A, Welch BD, Leser GP, Jardetzky TS, Lamb RA. 2012. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc Natl Acad Sci U S A 109:E2625–E2634. doi: 10.1073/pnas.1213813109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, Stone JA, Bradel-Tretheway B, Dabundo J, Benavides Montano JA, Santos-Montanez J, Biering SB, Nicola AV, Iorio RM, Lu X, Aguilar HC. 2013. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced Nipah virus fusion and cell entry. PLoS Pathog 9:e1003770. doi: 10.1371/journal.ppat.1003770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. 1998. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95:409–417. doi: 10.1016/S0092-8674(00)81771-7. [DOI] [PubMed] [Google Scholar]
- 25.Welch BD, Liu Y, Kors CA, Leser GP, Jardetzky TS, Lamb RA. 2012. Structure of the cleavage-activated prefusion form of the parainfluenza virus 5 fusion protein. Proc Natl Acad Sci U S A 109:16672–16677. doi: 10.1073/pnas.1213802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinhauer DA, Plemper RK. 2012. Structure of the primed paramyxovirus fusion protein. Proc Natl Acad Sci U S A 109:16404–16405. doi: 10.1073/pnas.1214903109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan P, Thompson TB, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS. 2005. Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13:803–815. doi: 10.1016/j.str.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 28.Bose S, Heath CM, Shah PA, Alayyoubi M, Jardetzky TS, Lamb RA. 2013. Mutations in the parainfluenza virus 5 fusion protein reveal domains important for fusion triggering and metastability. J Virol 87:13520–13531. doi: 10.1128/JVI.02123-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jardetzky TS, Lamb RA. 2014. Activation of paramyxovirus membrane fusion and virus entry. Curr Opin Virol 5:24–33. doi: 10.1016/j.coviro.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bose S, Song AS, Jardetzky TS, Lamb RA. 2014. Fusion activation through attachment protein stalk domains indicates a conserved core mechanism of paramyxovirus entry into cells. J Virol 88:3925–3941. doi: 10.1128/JVI.03741-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paterson RG, Johnson ML, Lamb RA. 1997. Paramyxovirus fusion (F) protein and hemagglutinin-neuraminidase (HN) protein interactions: intracellular retention of F and HN does not affect transport of the homotypic HN or F protein. Virology 237:1–9. doi: 10.1006/viro.1997.8759. [DOI] [PubMed] [Google Scholar]
- 32.Brindley MA, Takeda M, Plattet P, Plemper RK. 2012. Triggering the measles virus membrane fusion machinery. Proc Natl Acad Sci U S A 109:E3018–E3027. doi: 10.1073/pnas.1210925109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plemper RK, Hammond AL, Gerlier D, Fielding AK, Cattaneo R. 2002. Strength of envelope protein interaction modulates cytopathicity of measles virus. J Virol 76:5051–5061. doi: 10.1128/JVI.76.10.5051-5061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ader N, Brindley MA, Avila M, Origgi FC, Langedijk JP, Orvell C, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger f protein refolding for membrane fusion. J Biol Chem 287:16324–16334. doi: 10.1074/jbc.M112.342493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plemper RK, Hammond AL, Cattaneo R. 2001. Measles virus envelope glycoproteins hetero-oligomerize in the endoplasmic reticulum. J Biol Chem 276:44239–44246. doi: 10.1074/jbc.M105967200. [DOI] [PubMed] [Google Scholar]
- 36.Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J Virol 75:4399–4401. doi: 10.1128/JVI.75.9.4399-4401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malvoisin E, Wild F. 1990. Contribution of measles virus fusion protein in protective immunity: anti-F monoclonal antibodies neutralize virus infectivity and protect mice against challenge. J Virol 64:5160–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheshberadaran H, Chen SN, Norrby E. 1983. Monoclonal antibodies against five structural components of measles virus. I. Characterization of antigenic determinants on nine strains of measles virus. Virology 128:341–353. [DOI] [PubMed] [Google Scholar]
- 39.Plemper RK, Compans RW. 2003. Mutations in the putative HR-C region of the measles virus F2 glycoprotein modulate syncytium formation. J Virol 77:4181–4190. doi: 10.1128/JVI.77.7.4181-4190.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kondo N, Miyauchi K, Matsuda Z. 2011. Monitoring viral-mediated membrane fusion using fluorescent reporter methods. Curr Protoc Cell Biol Chapter 26:Unit 26.9. doi: 10.1002/0471143030.cb2609s50. [DOI] [PubMed] [Google Scholar]
- 41.Ader N, Brindley M, Avila M, Orvell C, Horvat B, Hiltensperger G, Schneider-Schaulies J, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2013. Mechanism for active membrane fusion triggering by morbillivirus attachment protein. J Virol 87:314–326. doi: 10.1128/JVI.01826-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Navaratnarajah CK, Kumar S, Generous A, Apte-Sengupta S, Mateo M, Cattaneo R. 2014. The measles virus hemagglutinin stalk: structures and functions of the central fusion activation and membrane-proximal segments. J Virol 88:6158–6167. doi: 10.1128/JVI.02846-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tao Y, Strelkov SV, Mesyanzhinov VV, Rossmann MG. 1997. Structure of bacteriophage T4 fibritin: a segmented coiled coil and the role of the C-terminal domain. Structure 5:789–798. doi: 10.1016/S0969-2126(97)00233-5. [DOI] [PubMed] [Google Scholar]
- 44.Epand RM, Epand RF, Richardson CD, Yeagle PL. 1993. Structural requirements for the inhibition of membrane fusion by carbobenzoxy-d-Phe-Phe-Gly. Biochim Biophys Acta 1152:128–134. doi: 10.1016/0005-2736(93)90239-V. [DOI] [PubMed] [Google Scholar]
- 45.Plemper RK, Erlandson KJ, Lakdawala AS, Sun A, Prussia A, Boonsombat J, Aki-Sener E, Yalcin I, Yildiz I, Temiz-Arpaci O, Tekiner B, Liotta DC, Snyder JP, Compans RW. 2004. A target site for template-based design of measles virus entry inhibitors. Proc Natl Acad Sci U S A 101:5628–5633. doi: 10.1073/pnas.0308520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Apte-Sengupta S, Negi S, Leonard VH, Oezguen N, Navaratnarajah CK, Braun W, Cattaneo R. 2012. Base of the measles virus fusion trimer head receives the signal that triggers membrane fusion. J Biol Chem 287:33026–33035. doi: 10.1074/jbc.M112.373308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aguilar HC, Matreyek KA, Filone CM, Hashimi ST, Levroney EL, Negrete OA, Bertolotti-Ciarlet A, Choi DY, McHardy I, Fulcher JA, Su SV, Wolf MC, Kohatsu L, Baum LG, Lee B. 2006. N-glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J Virol 80:4878–4889. doi: 10.1128/JVI.80.10.4878-4889.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A 102:9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dutch RE, Hagglund RN, Nagel MA, Paterson RG, Lamb RA. 2001. Paramyxovirus fusion (F) protein: a conformational change on cleavage activation. Virology 281:138–150. doi: 10.1006/viro.2000.0817. [DOI] [PubMed] [Google Scholar]