

ABSTRACT
Many coxsackievirus B (CVB) isolates bind to human decay-accelerating factor (DAF) as well as to the coxsackievirus and adenovirus receptor (CAR). However, the virus does not interact with murine DAF. To understand why CVB3 binds specifically to human DAF, we constructed a series of chimeric molecules in which specific regions of the human DAF molecule were replaced by the corresponding murine sequences. We found that replacement of human short consensus repeat 2 (SCR2) with murine SCR2 ablated virus binding to human DAF, as did deletion of human SCR2. Although replacement of human SCR4 had a partial inhibitory effect, deletion of SCR4 had no effect. Within human SCR2, replacement of serine 104 (S104) with the proline residue found in murine DAF eliminated virus binding. On the basis of the structure of the CVB3-DAF complex determined by cryo-electron microscopy, DAF S104 is in close contact with a viral capsid residue, a threonine at VP1 position 271. Replacement of this capsid residue with larger amino acids specifically eliminated virus attachment to human DAF but had no effect on attachment to CAR or replication in HeLa cells. Taken together, these results support the current model of virus-DAF interaction and point to a specific role for VP1 T271 and DAF S104 at the virus-DAF interface.
IMPORTANCE The results of the present study point to a specific role for VP1 T271 and DAF S104 at the interface between CVB3 and DAF, and they demonstrate how subtle structural changes can dramatically influence virus-receptor interactions. In addition, the results support a recent pseudoatomic model of the CVB3-DAF interaction obtained by cryo-electron microscopy.
INTRODUCTION
Many coxsackievirus B (CVB) isolates bind to human decay-accelerating factor (DAF) as well as to the coxsackievirus and adenovirus receptor (CAR) (1). Virus interaction with DAF is important for in vitro infection of polarized epithelial monolayers, in which CAR is not expressed on the apical cell surface (2, 3); because CVB infection is transmitted by the fecal-oral route, virus interaction with DAF on intestinal epithelium may be important for the early events of infection in vivo.
DAF is a 70-kDa membrane protein that functions to protect cells from lysis by autologous complement. It is composed of four short consensus repeat (SCR) domains at the amino terminus and a heavily glycosylated serine- and threonine-rich domain at the carboxyl terminus linked to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor. A recent pseudoatomic model of the CVB3-DAF interaction, obtained by cryo-electron microscopy (cryo-EM) and X-ray crystallography, suggests that the primary contacts involve SCR2 (approximately 77% of the total buried surface area), with additional contacts involving SCR3 (19%) and possibly SCR4 (4%) (4). The model is consistent with earlier data indicating that deletion of SCR2 ablates virus attachment, whereas deletion of SCR 1, 3, or 4 has only minor effects (5). Additional support for the model comes from a mutational analysis of DAF-binding and nonbinding isolates, which revealed that capsid residues VP3 234Q and VP2 138D, both of which are predicted to interact with SCR2, are required for CVB3 attachment to DAF (6).
Unlike human DAF, the murine DAF homologue does not bind CVB3 (7). Overall, the sequences of murine and human DAF are approximately 51% identical, with the greatest identity (59%) being seen in SCRs 2 and 4 (Fig. 1). To understand the structural basis for the specific interaction between CVB3 and human DAF, we have studied a series of human-murine chimeric DAF molecules to identify the specific human sequences required for virus attachment. The results demonstrate that virus attachment depends on human sequences within human SCR2; further, the presence of a serine residue at position 104, rather than the proline residue present in murine DAF, is essential for virus attachment, most likely because of its interaction with the viral capsid residue VP1 271.
FIG 1.

DAF structure and sequence. (A) Mature DAF consists of four SCR domains, with a serine and threonine (ST)-rich domain being bound to the membrane by a GPI anchor. (B) The sequence of the mature human DAF (h) aligned with the sequence of GPI-anchored murine DAF (m); the numbering is consistent with the N-terminal peptide sequence determined previously (9) and differs by −2 residues from the numbering used previously (4). Hyphen, the murine residue is identical to the corresponding human residue; equals sign, a gap in the alignment. Individual SCR domains are as defined elsewhere (16). Cleavage of human DAF after residue 319 removes a C-terminal peptide and permits attachment to the GPI anchor (17); it is not known whether the GPI anchoring of murine DAF occurs at the same site. Residues in contact with the viral capsid, as determined previously (4), are highlighted in gray; SCR2 residue S104, found in this study to be essential for virus attachment, is shown on boldface.
MATERIALS AND METHODS
Cells.
HeLa cells and CHO cells stably transfected with DAF, CAR, or the pcDNA3.1 vector control (CHO-DAF, CHO-CAR, and CHO-pcDNA, respectively) were maintained as described previously (6). CHO-K1 cells were maintained in Ham's F-12 medium supplemented with 10% fetal bovine serum and penicillin-streptomycin.
Murine-human DAF chimeras.
Splice-overlap extension PCR (8) was used to introduce murine DAF sequences into human DAF cDNA (9) cloned in the expression vector pcDNA3.1 or human sequences into a pcDNA3.1 clone of GPI-anchored murine DAF (10, 11). In chimeras mHmH and mHmm (consisting of SCRs 1 to 4, where human SCRs are indicated by H and murine SCR domains (SCRs) are indicated by m), human SCR domains were introduced into murine DAF; in these two constructs, the serine and threonine (ST)-rich domain is of murine origin. In all other chimeras, mouse SCR domains were introduced into the human molecule, so that the ST-rich domain is of human origin. Plasmids were transfected into CHO-K1 cells, using polyethyleneimine Max (Polysciences) (12), and protein expression and virus binding were assessed 48 h later.
Cell surface expression was measured by flow cytometry with monoclonal antibodies (MAbs). Constructs containing human SCR1 were detected with murine MAb IA10 and fluorescein isothiocyanate (FITC)-conjugated antibody specific for murine IgG; those with murine SCR1 were detected with hamster MAb RIKO-3 and FITC-conjugated antibody specific for hamster IgG. To account for possible differences in the intensity of staining (mean cell fluorescence [MCF]) with the two SCR1 antibodies, we also stained selected constructs with MAb IF7, specific for human SCR2: for construct mHmm, the MCFs determined with RIKO-3 and with IF7 were equal (MCFIF7/MCFRIKO-3 = 1.03), and for HHmH, the MCF determined with IF7 was slightly higher than that determined with IA10 (MCFIF7/MCFIA10 = 1.19) and the MCF values obtained for the other constructs were adjusted accordingly. For each sample, a corrected mean cell fluorescence (C-MCF) was calculated by subtracting from the measured MCF the background fluorescence measured for cells transfected with the pcDNA3.1 expression vector alone.
Viral capsid mutants.
PCR was used to generate specific substitutions at VP1 T171 in a cDNA clone encoding full-length CVB3-RD downstream of the T7 promoter in the vector pSport1 (6). To produce virus, viral RNA was transcribed in vitro and introduced into HeLa cells as previously described (6).
Virus binding assay.
Radiolabeled virus was prepared, and binding to triplicate cell monolayers was measured as described previously (6). For each sample, a virus binding index was calculated as follows: (amount of virus bound for the sample/C-MCF for the sample) × (C-MCF for human DAF/amount of virus bound for human DAF).
RESULTS
Virus attachment to DAF requires human SCR2.
To identify human DAF sequences specifically required for virus attachment and mouse sequences that inhibit attachment, we constructed chimeric DAF molecules in which human sequences were replaced by murine sequences or in which mouse sequences were replaced by human sequences. In Fig. 2, human SCR domains (SCRs) are indicated by H and murine domains are indicated by m; for example, the chimera labeled HHHH includes human SCRs 1 to 4, whereas the chimera labeled HHmm includes human SCRs 1 and 2 and murine SCRs 3 and 4. We measured the attachment of radiolabeled CVB3-RD, the prototype DAF-binding CVB3 isolate (13), to CHO cells expressing each of the chimeric DAF molecules and assessed the surface expression of each chimera by flow cytometry. For each chimera, we computed a virus binding index which took into account both binding and expression, and which was normalized to the results obtained with human DAF.
FIG 2.
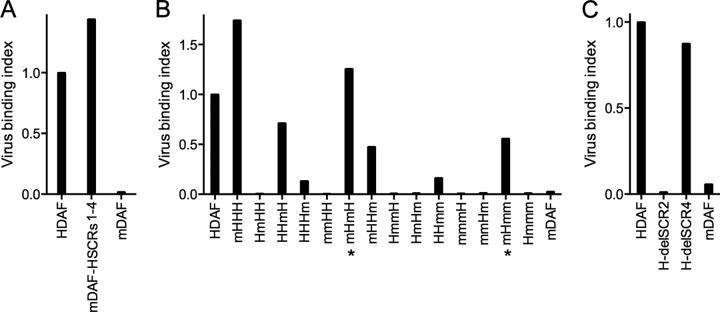
Human SCR2 is essential for virus attachment. (A) Binding of radiolabeled CVB3-RD to CHO cells expressing human DAF (HDAF), murine DAF (mDAF), or murine DAF in which SCRs 1 to 4 have been replaced by human sequences (mouse DAF-human SCRs 1 to 4 [mDAF-HCSRs1-4]). (B) Virus binding to murine-human SCR chimeras. Chimeras are labeled according to the configuration of the human (H) and murine (m) SCR domains: for example, chimera mHHH includes murine SCR1 and human SCRs 2 to 4. Chimeras marked by an asterisk were constructed in a mouse DAF cDNA background and include the mouse DAF ST-rich domain. (C) Virus binding to human DAF from which SCR 2 or 4 was deleted (H-delSCR2 and H-delSCR4, respectively).
Based on previous observations (4, 5, 14), we suspected that the specificity of virus binding would reside within the SCR domains rather than within the membrane-proximal serine-threonine domain. To confirm this, we introduced human SCRs 1 to 4 in place of the murine SCRs in a cDNA encoding murine DAF (Fig. 2A). As expected, virus bound efficiently to this chimeric molecule, despite the presence of the murine serine-threonine domain. We then examined virus attachment to chimeras with each of the possible combination of human and murine SCR domains (Fig. 2B). In every case, replacement of human SCR2 with murine SCR2 (for example, construct HmHH) ablated virus binding to human DAF, and replacement of mouse SCR2 with the human domain (for example, construct mHmm) allowed virus to bind to murine DAF, although with variable efficiency. These results indicate an important role for human SCR2 in virus attachment, consistent with earlier observations (4, 5).
Replacement of human SCR4 with mouse SCR4 appeared to reduce virus binding (for example, compare virus binding indices for HHHH and HHHm), although in no case did the presence of human SCR4 permit binding to a chimera that did not also contain human SCR2. This result was surprising, as we had previously found that deletion of SCR4 had no effect on virus binding to human DAF (5). We therefore generated new constructs in which SCR2 or SCR4 was deleted from human DAF; consistent with our previous observations, we found that deletion of SCR2 ablated binding but deletion of SCR4 had little, if any, effect (Fig. 2C). These results confirm that human SCR4 is not required for CVB3 attachment and suggest further that it does not contribute directly to attachment. We believe that it is more likely that murine SCR4 obstructs virus access to its binding site elsewhere within the DAF molecule, probably within SCR2.
Within human SCR2, serine 104 (S104) is essential but not sufficient for CVB3 attachment.
To identify specific human SCR2 sequences required for virus attachment, we produced human DAF chimeras in which 10-residue segments within SCR2 were replaced by murine sequences (Fig. 3A). Each of these chimeras bound virus less efficiently than wild-type human DAF did, but replacement of the residues between residues 94 and 123—and, particularly, between residues 104 and 113—had the most dramatic effect. Smaller exchanges between residues 104 and 113 revealed that replacement of residues 109 to 113 had little effect, whereas replacement of residues 104 to 108 ablated binding (Fig. 3B). Within this region, replacement of a single amino acid, S104, with the corresponding murine residue was sufficient to prevent virus attachment.
FIG 3.
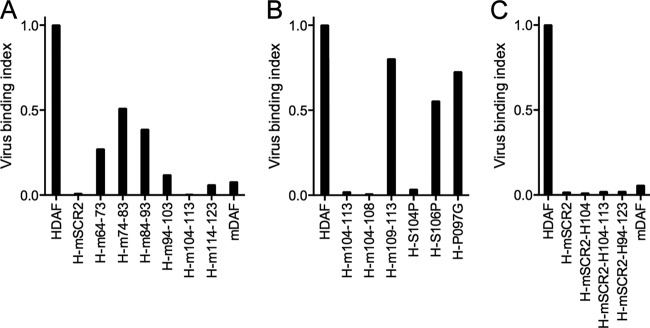
Human DAF residue S104 is essential, but not sufficient, for virus attachment. (A) Virus binding to CHO cells expressing human DAF in which SCR2 was replaced with murine sequences (H-mSCR2) or in which specific residues within SCR2 were replaced; for example, H-m64-73 indicates human DAF with murine residues 64 to 73. (B) Virus binding to CHO cells expressing human DAF in which one or more residues between residues 104 and 113 were replaced by murine residues. (C) Introduction of human residues 94 to 123 into mouse SCR2 does not restore virus binding; specific human residues (indicated by H) were introduced into a chimera consisting of human DAF with murine SCR2.
Introduction of the human residue serine 104 into murine SCR2, even in a chimera otherwise composed of human DAF sequences (H-mSCR2-H104; Fig. 3C), did not permit virus attachment, indicating that this human residue is necessary, but not sufficient, for virus binding to SCR2. Similarly, replacement of larger segments of murine SCR2 with human sequences—including the segment comprising residues 94 to 123—was not sufficient for binding (Fig. 3C).
VP1 T271 is important for CVB3-RD attachment to human DAF but not to CAR.
The available pseudoatomic model of human DAF bound to CVB3-RD (4) suggests that S104 is in close contact with a viral capsid residue, VP1 T271 (Fig. 4A). We suspected that mutation of this capsid residue might specifically inhibit the interaction with DAF. We introduced specific mutations, replacing serine with alanine (A), leucine (L), tryptophan (W), glutamate (E), or arginine (R), into a cDNA clone encoding CVB3-RD and produced mutant viruses. Sequencing of the recovered viruses indicated that T271E rapidly reverted to the wild type, suggesting that this mutation was not tolerated by the virus. However, viruses with T271A, T271L, T271W, and T271R mutations were stable and replicated to wild-type titers in HeLa cells (Fig. 4B). Radiolabeled wild-type virus bound efficiently to human DAF expressed on CHO cells, as did virus with T271A. In contrast, viruses with the T271L, T271W, and T271R mutations all bound poorly to DAF (Fig. 4C), although they bound to human CAR as efficiently as the wild-type virus did. These results indicate that replacement of T271 with bulkier amino acids interferes specifically with virus attachment to human DAF.
FIG 4.

VP1 T271 of CVB3 is critical for binding to human DAF. (A) Interaction of human DAF S104 (orange) with CVB3-RD VP1 T271 (dark blue); the partial ribbon structure of CVB3 is in blue, and that of DAF is in yellow. (B) Replication of wild-type and mutant viruses in HeLa cells (virus input, 2 PFU per cell). (C) Attachment of 35S-labeled wild-type and mutant viruses to CHO cells stably transfected with human DAF or to control CHO cells transfected with the pcDNA 3.1 vector alone. (D) Attachment to CHO cells stably transfected with human CAR or to control CHO cells transfected with the pcDNA 3.1 vector alone. Results in panels C and D are shown as the mean amount of virus bound to triplicate monolayers ± standard deviation.
DISCUSSION
In the work reported here, we used murine-human chimeras to identify human DAF sequences specifically required for interaction with CVB3-RD. The results point to a critical role for human SCR2. Within SCR2, mutation of a specific serine residue ablated virus attachment to human DAF, although introduction of this serine into murine DAF was not sufficient to restore binding.
These results are consistent with previous evidence indicating the importance of SCR2 for virus attachment (5); further, they support a recently refined cryo-EM model of the CVB3-DAF interaction (4), in which SCR2 residues 102 to 104, 110, 113, 114, 116, and 119 (Fig. 1, gray highlights)—but not residues elsewhere in SCR2—appear in close contact with the virus capsid. We previously found that capsid residues VP3 Q234 and VP2 D138 are important for virus attachment to DAF (6); on the basis of the cryo-EM model, these make contact with SCR2 residues K116 and Q113, respectively, neither of which is conserved in murine DAF. In this work, we specifically confirmed the importance of DAF residue S104, which was also implicated by the cryo-EM structure, in which it is seen to interact with the viral capsid residue VP1 T271. As predicted by the current model—but not by an earlier cryo-EM study (4)—we found that mutation of VP1 T271 specifically interfered with CVB3-RD attachment to DAF. The result thus provides support for the current model.
The available cryo-EM structure includes DAF SCRs 1 to 4, but not the membrane-proximal ST-rich domain. We found that replacement of the human ST domain with the murine domain had no inhibitory effect and may have even enhanced virus binding. The ST domain, which is heavily O-glycosylated, is believed to act as a spacer facilitating exposure of the SCRs; sequence differences within this domain may affect the overall flexibility of the molecule and the accessibility of SCR2 to virus. Although the pseudoatomic model predicts close contacts between the virus and SCR3, we found that replacement of human SCR3 with murine SCR3 had only a limited effect on virus attachment, consistent with our earlier observation that complete deletion of human SCR3 has little or no effect (5). Replacement of human SCR4 inhibited (but did not abolish) virus attachment, although contact with SCR4 accounts for only a minimal portion of the total virus-DAF interface and deletion of SCR4 entirely has a minimal effect on virus binding (5) (Fig. 2C). These results suggest that human SCR4 is not directly required for virus binding but that its replacement by murine SCR4 inhibits virus interactions with other parts of the DAF molecule, presumably, SCR2. Given that DAF binds CVB3 with low affinity (15), small structural changes—in surface chemistry or in the angles between individual SCR domains as DAF drapes across the virus surface—may be sufficient to disrupt virus attachment.
ACKNOWLEDGMENTS
We thank Douglas Lublin for human DAF cDNA and Wen-Chao Song for murine DAF cDNA.
This work was supported by grants from the National Institutes of Health (AI052281 to J.M.B. and AI07927 to S.H.), by the Max Lang Junior Faculty Scholar Award (to S.H.), by the Pennsylvania Department of Health using Tobacco CURE funds (to S.H.), and by the Plotkin Endowed Chair in Infectious Diseases at The Children's Hospital of Philadelphia (to J.M.B).
REFERENCES
- 1.Bergelson JM. 2010. Receptors, p 73–86 InEhrenfeld E, Domingo E, Roos RP (ed), The picornaviruses. ASM Press, Washington, DC. [Google Scholar]
- 2.Shieh JTC, Bergelson JM. 2002. Interaction with decay-accelerating factor facilitates coxsackievirus B infection of polarized epithelial cells. J Virol 76:9474–9480. doi: 10.1128/JVI.76.18.9474-9480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coyne CB, Bergelson JM. 2006. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 124:119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 4.Yoder JD, Cifuente JO, Pan J, Bergelson JM, Hafenstein S. 2012. The crystal structure of a coxsackievirus B3-RD variant and a refined 9-angstrom cryo-electron microscopy reconstruction of the virus complexed with decay-accelerating factor (DAF) provide a new footprint of DAF on the virus surface. J Virol 86:12571–12581. doi: 10.1128/JVI.01592-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergelson JM, Mohanty JG, Crowell RL, St John NF, Lublin DM, Finberg RW. 1995. Coxsackievirus B3 adapted to growth in RD cells binds to decay-accelerating factor (CD55). J Virol 69:1903–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan J, Narayanan B, Shah S, Yoder JD, Cifuente JO, Hafenstein S, Bergelson JM. 2011. Single amino acid changes in the virus capsid permit coxsackievirus B3 to bind decay-accelerating factor. J Virol 85:7436–7443. doi: 10.1128/JVI.00503-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spiller OB, Goodefellow IG, Evans DJ, Almond JW, Morgan BB. 2000. Echoviruses and coxsackie B viruses that use human decay-accelerating factor (DAF) as a receptor do not bind the rodent analogues of DAF. J Infect Dis 181:340–343. doi: 10.1086/315210. [DOI] [PubMed] [Google Scholar]
- 8.Yon J, Fried M. 1989. Precise gene fusion by PCR. Nucleic Acids Res 17:4895. doi: 10.1093/nar/17.12.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medof ME, Lublin DM, Holers VM, Ayers DJ, Getty RR, Leykam JF, Atkinson JP, Tykocinski ML. 1987. Cloning and characterization of cDNAs encoding the complete sequence of decay-accelerating factor of human complement. Proc Natl Acad Sci U S A 84:2007–2011. doi: 10.1073/pnas.84.7.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spicer AP, Seldin MF, Gendler SJ. 1995. Molecular cloning and chromosomal localization of the mouse decay-accelerating factor genes: duplicated genes encode glycosylphosphatidylinositol-anchored and transmembrane forms. J Immunol 155:3079–3091. [PubMed] [Google Scholar]
- 11.Song WC, Deng C, Raszmann K, Moore R, Newbold R, McLachlan JA, Negishi M. 1996. Mouse decay-accelerating factor: selective and tissue-specific induction by estrogen of the gene encoding the glycosylphosphatidylinositol-anchored form. J Immunol 157:4166–4172. [PubMed] [Google Scholar]
- 12.Durocher Y, Perret S, Kamen A. 2002. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res 30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reagan KJ, Goldberg B, Crowell RL. 1984. Altered receptor specificity of coxsackie B3 after growth in rhabdomyosarcoma cells. J Virol 49:635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hafenstein S, Bowman VD, Chipman PR, Bator Kelly CM, Lin F, Medof ME, Rossmann MG. 2007. Interaction of decay-accelerating factor with coxsackievirus B3. J Virol 81:12927–12935. doi: 10.1128/JVI.00931-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodfellow IG, Evans DJ, Blom AM, Kerrigan D, Miners JS, Morgan BP, Spiller OB. 2005. Inhibition of coxsackie B virus infection by soluble forms of its receptors: binding affinities, altered particle formation, and competition with cellular receptors. J Virol 79:12016–12024. doi: 10.1128/JVI.79.18.12016-12024.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coyne KE, Hall SE, Thompson ES, Arce MA, Kinoshita T, Fujita T, Anstee DJ, Rosse W, Lublin DM. 1992. Mapping of epitopes, glycosylation sites, and complement regulatory domains in human decay accelerating factor. J Immunol 149:2906–2913. [PubMed] [Google Scholar]
- 17.Moran P, Raab H, Kohr WJ, Caras IW. 1991. Glycophospholipid membrane anchor attachment. Molecular analysis of the cleavage/attachment site. J Biol Chem 266:1250–1257. [PubMed] [Google Scholar]