

ABSTRACT
Marek's disease virus (MDV), an alphaherpesvirus, is the causative agent of a lethal disease in chickens characterized by generalized nerve inflammation and rapid lymphoma development. The extensive colinearity of the MDV genome with those of related herpesviruses has eased functional characterization of many MDV genes. However, MDV carries a number of unique open reading frames (ORFs) that have not yet been investigated regarding their coding potentials and the functions of their products. Among these unique ORFs are two putative ORFs, ORF011 and ORF012, which are found at the extreme left end of the MDV unique long region. Using reverse transcriptase PCR, we showed that ORF011 and ORF012 are not individual genes but form a single gene through mRNA splicing of a small intron, resulting in the novel ORF012. We generated an ORF012-null virus using an infectious clone of MDV strain RB-1B. The deletion virus had a marked growth defect in vitro and could not be passaged in cultured cells, suggesting an essential role for the ORF012 product in virus replication. Further studies revealed that protein 012 (p012) localized to the nucleus in transfected and infected cells, and we identified by site-directed mutagenesis and green fluorescent protein (GFP) reporter fusion assays a nuclear localization signal (NLS) that was mapped to a 23-amino-acid sequence at the protein's C terminus. Nuclear export was blocked using leptomycin B, suggesting a potential role for p012 as a nuclear/cytoplasmic shuttling protein. Finally, p012 is phosphorylated at multiple residues, a modification that could possibly regulate its subcellular distribution.
IMPORTANCE Marek's disease virus (MDV) causes a devastating oncogenic disease in chickens with high morbidity and mortality. The costs for disease prevention reach several billion dollars annually. The functional investigation of MDV genes is necessary to understand its complex replication cycle, which eventually could help us to interfere with MDV and herpesviral pathogenesis. We have identified a previously unidentified phosphoprotein encoded by MDV ORF012. We were able to show experimentally that predicted splicing of the gene based on bioinformatics data does indeed occur during replication. The newly identified p012 is essential for MDV replication and localizes to the nucleus due to the presence of a transferable nuclear localization signal at its C terminus. Our results also imply that p012 could constitute a nucleocytoplasmic shuttle protein, a feature that could prove interesting and important.
INTRODUCTION
Marek's disease (MD) is a viral disease of chickens that is characterized by a general inflammation of peripheral nerves (polyneuritis) and development of solid tumors in multiple organs that originate from transformed T lymphocytes. The lymphomas associated with MD form with reliable kinetics within weeks after infection and can seed in virtually every organ and tissue of the chicken (1). However, in its most acute form, which includes additional symptoms, like torticollis and ataxia due to brain edema, MD causes the death of an infected chicken within days (2). Therefore, and despite the availability of excellent vaccines, MD morbidity and mortality, as well as prophylactic vaccination, represent a massive burden to chicken husbandry worldwide (3).
Within the subfamily Alphaherpesvirinae, MD virus (MDV) represents the prototype strain of the genus Mardivirus (Marek's disease-like viruses) (4, 5). Following the current nomenclature, the genus encompasses three distinct species: pathogenic MDV, also referred to as gallid herpesvirus type 2 (GaHV-2, or MDV; formerly MDV-1); apathogenic gallid herpesvirus type 3 (GaHV-3, formerly MDV-2); and meleagrid herpesvirus type 1 (MeHV-1, or HVT; formerly MDV-3), for which the turkey is the natural host. In cell culture, MDV is highly cell associated and shows very slow replication kinetics, with plaques typically appearing only after several days. In vivo, comparable to human Epstein-Barr virus (EBV), MDV is highly lymphotropic and infects B and T cells (1). Apart from its tropism for lymphocytes, our knowledge of the exact sequence of events of MDV infection, starting with the uptake of the pathogen from the environment to final shedding of cell-free virus from feather follicle epithelia, still has considerable gaps. The current model of MDV replication, referred to as the “Cornell model,” proposes that the infectious cycle starts with virus gaining access to the lung of the chicken by inhalation of contaminated dust and dander. Here, antigen-presenting cells (APC), like macrophages or dendritic cells, are supposedly the first to be infected by MDV. While entering primary lymphoid tissues, the virus infects B cells, the first target cell for massive lytic replication and production of viral progeny. Subsequently, CD4+ T cells become infected and act as a reservoir in which the viral latency program is activated, and transformation of individual T cells leads to the formation of solid lymphomas, the hallmark of MDV infection, and ultimately death of the host (6).
The genes and gene products that execute the complicated viral replication cycle are encoded in a 180-kbp double-stranded DNA genome. It represents a classical E-type genome consisting of a unique long (UL) and a unique short (US) segment, each bracketed by inverted terminal (TRL and TRS) and internal (IRL and IRS) repeats (Fig. 1) (7). MDV contains more than 100 genes or open reading frames (ORFs), the vast majority of which have orthologues within the UL and US regions of herpes simplex virus 1 (HSV-1) and are annotated as such in the MDV gene nomenclature. Notwithstanding the extensive homology to other alphaherpesviruses, some regions of the MDV genome are truly unique and contain genes that are not found in any other herpesvirus described so far. Among these specific genes are genes encoding the multifunctional Meq (8) and a virokine called viral interleukin 8 (vIL-8) (9, 10), which are found in the TRL and IRL, respectively. Both Meq and vIL-8 have been the subjects of extensive functional studies in the past and were shown to play a substantial role in tumor formation (Meq) and replication (vIL-8) (11–13).
FIG 1.

p012 is generated from a spliced transcript. Shown are the position of MDV ORF012 and the splicing of its mRNA. The structure of the MDV genome is outlined, and the positions of the hypothetical ORF011* and ORF012* genes (black) in relation to other genes are indicated. Sizes are given in base pairs (bp) for DNA or bases (b) for RNA. The 82-bp intron in the former ORF011* is indicated in gray. The predicted splicing results in a frameshift and absence of the predicted ORF011* stop codon. Splicing results in fusion of the remaining sequence with the formerly predicted short intergenic region and the 5′ end of the former ORF012*, thus creating the novel ORF012 transcript. The splice donor (GT) and acceptor (AG) sites are indicated in the intron. The primary sequence of p012 with predicted phosphorylation sites (underlined) and a predicted NLS (boxed) is shown. Initiator methionines of p012 and the former p012*, respectively, are marked in boldface. The amino acid corresponding to the exon-exon border (arginine; R) is marked in boldface and italics.
Despite the wealth of information on the roles of some MDV genes, other genome regions contain distinctive genes whose functions remain to be elucidated. In particular, the left terminus of the MDV UL region, positioned upstream of the UL-1 gene (a homologue of the HSV-1 gene encoding the envelope protein gL), is poorly characterized. Remarkably, this region contains several potential ORFs that seem to be restricted to avian alphaherpesviruses, suggesting the genes may govern the host specificity of the bird viruses (14, 15). Within this relatively unexplored region, only ORF010 was characterized in some detail. ORF010 encodes a lipase-like protein (called vLIP) that lacks catalytic activity but nevertheless is required for efficient replication of the virus in vivo (16). Downstream of ORF10, two predicted ORFs were originally annotated as ORF011 and ORF012 and predicted to express two distinct proteins (7). Here, we refer to these ORFs as ORF011* and ORF012*. Since the original annotation of the MDV reference sequence for the Md5 strain in 2000, the most recent annotation postulated splicing within the ORF011* and ORF012* region, leading to a single ORF called MDV ORF012 (Refseq NC_002229) (7). ORF011* was consequently excluded from the annotation, leaving a gap between ORF010 and the predicted novel and spliced ORF012. However, these predictions were solely based on bioinformatic and comparative analyses rather than experimental approaches.
In this work, we addressed the expression, or lack thereof, of the hypothetical ORFs ORF011* and ORF012* and the recently proposed MDV ORF012 splice product. We determined that in fact, both ORF011* and ORF012* are not autonomous units but form a single gene produced from a spliced mRNA. Whereas ORF011* and ORF012* do not result in the production of detectable individual proteins during infection, the product of the spliced ORF012 is essential for viral growth in vitro. In addition, p012 is phosphorylated and capable of entering the nucleus in infected and transfected cells using a nuclear localization signal (NLS) in the C-terminal region of the protein. NLSs are amino acid motifs that function as recognition and docking domains for nuclear transport carriers and thus control the import of proteins exerting functions within this cellular compartment. Experiments using the nuclear export inhibitor leptomycin B (LMB) also point to active export from the nucleus back to the cytoplasm, indicating p012 may act as a nucleocytoplasmic shuttling protein during replication.
MATERIALS AND METHODS
Bioinformatic predictions.
For comparison of p012-related proteins in different avian herpesviruses, amino acid sequences were aligned using Clustal Omega software (http://www.ebi.ac.uk/Tools/msa/clustalo). Splicing of the ORF012 mRNA message was predicted with the help of the NetGene2 server (http://www.cbs.dtu.dk/services/NetGene2/) (17, 18). In order to predict the NLS, the amino acid sequence of p012 was analyzed with the prediction tool NLStradamus (http://www.moseslab.csb.utoronto.ca/NLStradamus/) (19), as well as the tool NucPred (http://www.sbc.su.se/∼maccallr/nucpred/) (20). Phosphorylation was predicted with the NetPhos 2.0 server (http://www.cbs.dtu.dk/services/NetPhos) (21).
Cell culture and viruses.
Primary chicken embryo cells (CEC) were maintained in minimal essential medium (MEM) (Biochrom) supplemented with 1 to 10% fetal bovine serum (FBS) (Biochrom) and 1% penicillin-streptomycin (Applichem). CEC were grown at 37°C under a 5% CO2 atmosphere. The spontaneously immortalized chicken embryonic fibroblast cell line DF-1 (ATCC CRL-12203; kindly provided by L. Martin, MPI Berlin), was maintained in Dulbecco's modified essential medium (DMEM) (Biochrom) supplemented with 10% FBS, 1% penicillin-streptomycin, 5% glutamine (Biochrom), and 2 mM sodium pyruvate (Biochrom). DF-1 cells were grown at 39°C under a 5% CO2 atmosphere and passaged twice a week. For reconstitution of viruses, primary CEC were transfected with purified bacterial artificial chromosome (BAC) DNA using the CaPO4 method described previously (22). The pathogenic MDV strain RB-1B (vRb; GenBank accession no. EF523390.1) represents a very virulent (vv) and clinically relevant virus that is available as an infectious BAC clone (23). Strain 584Ap80C (cloned as BAC20) represents a cell culture-adapted, avirulent strain that was obtained by serial passage of the very virulent plus (vv+) strain 584 (24) and can be grown to high titers in vitro.
Generation of recombinant viruses.
All recombinant viruses were generated with a two-step Red-mediated mutagenesis technique, as previously described (25). Briefly, the aphAI–I-SceI cassette containing a kanamycin resistance marker and a unique intron-encoded SceI (I-SceI) restriction site was amplified from the vector pEPkanS1 using PCR with primers (Table 1) containing the specific mutation to be generated, as well as homologous sequences that allowed the desired recombination events. The PCR products were purified and introduced by electroporation into the Escherichia coli strain GS1783 harboring the specific BAC to be mutated. Kanamycin-resistant clones were analyzed by restriction fragment length polymorphism (RFLP) analysis with multiple restriction enzymes. Following the second recombination step, kanamycin-sensitive clones were analyzed by RFLP to ensure integrity of the genome and by PCR and DNA sequencing to confirm the presence of the desired mutation. An ORF012 start codon mutant virus (vRbΔMet012, where Met is methionine), as well as the respective revertant virus (vRbΔMet012R), were based on vRB-1B. Viruses encoding p012 with C- or N-terminal Flag epitope tags (v20_012Flag or v20_Flag012), as well as a mutant with a deletion in the 3′ end of 012, containing the NLS (bp 1036 to 1467; v20_012ΔNLSFlag), and a mutant containing an alanine substitution for the short NLS (v20_012mutshortNLSFlag), were based on the cell culture-adapted BAC20 (v20) strain.
TABLE 1.
Primers used in this study
| Primer | Sequence (5′→3′)a |
|---|---|
| TS1 | ATGACTAGCGAGAGAGCTCTTACTCT |
| TS2 | TGTACGCCAAATTTTACAACGATTAT |
| TS3 | CTATTCATCATCTGAACTCGACATCC |
| chGAPDH for | ATGGTGAAAGTCGGAGTCAACG |
| chGAPDH rev | TCACTCCTTGGATGCCATGTG |
| vRΔ012 for | AACGAGAGGTTGGTAACAAACAGCTTTTGAAAATAAACTAGCGAGAGAGCTAGGGATAACAGGGTAATCGATTT |
| vRΔ012 rev | TACCAGGCGCGAGAGTAAGAGCTCTCTCGCTAGTTTATTTTCAAAAGCTGGCCAGTGTTACAACCAATTAACC |
| vRΔ012R for | AACGAGAGGTTGGTAACAAACAGCTTTTGAAAAATGACTAGCGAGAGAGCTAGGGATAACAGGGTAATCGATTT |
| vRΔ012R rev | TACCAGGCGCGAGAGTAAGAGCTCTCTCGCTAGTCATTTTTCAAAAGCTGGCCAGTGTTACAACCAATTAACC |
| v20_012Flag for | AGATCTTGTGGTTCTTGGGATGTCGAGTTCAGATGATGAAGACTACAAAGACGATGACGACAAGTAGCATTTGCCAGTGTTACAACCAATTAACC |
| v20_012Flag rev | ACAGTGGATTTGCAATCACACAACATATACACAAATGCTACTTGTCGTCATCGTCTTTGTAGTCTTCATCATTAGGGATAACAGGGTAATCGATTT |
| v20_012ΔNLSFlag for | CTTGGATACCGTTGTCGTTCGAGATCACCCAGTAACACATGACTACAAAGACGATGACGAGCCAGTGTTACAACCAATTAACC |
| v20_012ΔNLSFlag rev | ATATACACAAATGCTACTTGTCGTCATCGTCTTTGTAGTCATGTGTTACTGGGTGATCTCTAGGGATAACAGGGTAATCGATTT |
| v20_012mutshortNLSFlag for | ATAACAGTGAAGATCCAAACCGTAGTCGGAGCCGGAGTCGATCTAGGGAGGCAGCGGCAGCAGCCGCAGCAGTTAGGCCTGCCAGTGTTACAACCAATTAACC |
| V20_012mutshortNLSFlag rev | CCACAAGATCTCGTATAGTTGTAGCCGTACTCCTACGCCCAGGCCTAACTGCTGCGGCTGCTGCCGCTGCCTCCCTAGATTAGGGATAACAGGGTAATCGATTT |
| 012*Flag for | ACTCGAGCGGCCGCGCCACCATGTTTACCGGAGGAGGAACTATTG |
| 012*Flag rev | GAGCTCGGATCCTTACTTGTCGTCATCGTCTTTGTAGTCTTCATCATCTGAACTCGACATCCC |
| 012ΔintFlag for | CTCGAGCGGCCGCGCCACCACCATGACTAGCGAGAGAGCTCTTACTCTCGCGCCTGGTAAAGTTTCGACGGCAGATATTTATGAAGCCGATTTCAGTTTCCGTCGTGAATTTGTACGCCAAATTTTACAACGATTATTCCCAAGGACCTT |
| 012ΔintFlag rev | AACTTAAGCTTCTACTTGTCGTCATCGTCTTTGTAGTCTTCATCATCTGAACTCGACATCCCA |
| GFPcterm for | CAGATCTCGAGTAGTTCGAGATCACCCAGTAACACATCG |
| GFPcterm rev | AATTCGAAGCTTTTATTCATCATCTGAACTCGACATCCC |
| GFP_GSlinker for | CAGATCTCGAGCTCAAGGAGGCAGTGGTGGAGG |
| GFPlongNLS template | AAGGAGGCAGTGGTGGAGGCAGTGGTCGTAGTCGGAGCCGGAGTCGATCTAGGGAGCGTAGGCGAAGACGGCCACGAGTTAGGCCTGGGCGTAGGTAA |
| GFPlongNLS rev | TCGACTGCAGAATTCTTACCTACGCCCAGGCCTAAC |
| GFPshortNLS template | AAGGAGGCAGTGGTGGAGGCAGTGGTCGTAGGCGAAGACGGCCACGATAA |
| GFPshortNLS rev | GTCGACTGCAGAATTCTTATCGTGGCCGTCTTCGC |
| GFP_RSrepeat template | GAGCTCAAGGAGGCAGTGGTGGAGGCAGTGGTCGTAGTCGGAGCCGGAGTCGATCTAGGGAGTAAGAATTC |
| GFP_RSrepeat rev | TCGACTGCAGAATTCTTACTCCCTAGATCGACTCCGG |
| 012mutshortNLS for | TCGGAGCCGGAGTCGATCTAGGGAGGCTGCGGCAGCAGCGGCAGCAGTTAGGCCTGGGCGTAGGAGTACG |
| 012mutshortNLS rev | CGTACTCCTACGCCCAGGCCTAACTGCTGCCGCTGCTGCCGCAGCCTCCCTAGATCGACTCCGGCTCCGA |
| 012mutRSrepeat for | ACAGTGAAGATCCAAACGCCGCAGCGGCTGCAGCAGCTGCCAGGGAGCGTAGGCGAAGACGG |
| 012mutRSrepeat rev | CCGTCTTCGCCTACGCTCCCTGGCAGCTGCTGCAGCCGCTGCGGCGTTTGGATCTTCACTGT |
| 012mutStoA for | GTGAAGATCCAAACCGTGCTCGGGCCCGGGCTCGAGCTAGGGAGCGTAGGCGA |
| 012mutStoA rev | TCGCCTACGCTCCCTAGCTCGAGCCCGGGCCCGAGCACGGTTTGGATCTTCAC |
Regions of interest (restriction sites, mutated sequences, epitope tags, and sequences representing the exon-exon border of ORF012 [primer TS2]) are underlined.
Plaque size assays.
BAC DNA was purified (Qiagen Midi Kit) following the protocol provided by the manufacturer. One microgram of recombinant RB-1B (rRB-1B) BAC DNA (rRb), an rRbΔMet012 mutant, or an rRbΔMet012R revertant was transfected into 1 × 106 CEC by the CaPO4 method, as described previously (22). Six days after transfection, the cells were fixed with 90% ice-cold acetone, air dried, blocked with 10% FBS in phosphate-buffered saline (PBS), and stained with polyclonal anti-MDV chicken sera (26) diluted 1:5,000 in 1% bovine serum albumin (BSA) (Applichem) in PBS. Following three washing steps with PBS, the cells were stained with secondary rabbit anti-chicken Alexa 488 antibody (Invitrogen) diluted 1:1,000. Using an Axio-Observer Z1 fluorescence microscope (ZeissJena), images of at least 50 plaques from each virus group were recorded at ×100 magnification in three independent experiments. Corresponding plaque areas were measured using the NIH Image J 1.410 software (27) and mathematically transformed into plaque diameter values. Graphs were produced with GraphPad Prism 5 (GraphPad Software, Inc.), and the diameters were expressed relative to those of the parental vRB-1B. For statistical analysis, the values were first tested for normality and subsequently analyzed for significance by one-way analysis of variance (ANOVA).
Cloning of expression plasmids and site-directed mutagenesis.
The expression plasmids pc_011*Flag, pc_012*Flag, pc_012Flag, and pc_012ΔintFlag (all based on pcDNA3.1 and containing a C-terminal Flag tag) were generated by PCR cloning. pc_012ΔintFlag was generated by fusion PCR and is devoid of the intron in the 5′ region of the gene. The respective inserts were amplified by standard PCR from rRb BAC DNA with Phusion polymerase (NEB) and primers containing restriction sites for directional cloning and the Flag epitope tag (Table 1). Both vector and inserts were cut with restriction enzymes NotI and BamHI (NEB), gel purified, ligated with T4 DNA ligase (NEB), and transformed into Top10 competent cells (Invitrogen). pc_012ΔintFlag was cloned via NotI/HindIII sites. Positive colonies were selected on ampicillin agar plates and analyzed by restriction digests and Sanger sequencing (LGC Genomics). For site-directed mutagenesis, the QuickChange mutagenesis kit (Agilent Technologies) was used according to the protocol provided by the manufacturer. Primers were designed with the corresponding software available at the Agilent home page (Table 1). The green fluorescent protein (GFP) fusion constructs pGFP-012cterm, pGFP-longNLS, pGFP-shortNLS, and pGFP-RSrepeat were based on the pEGFP-C1 expression vector (Clonetech). Briefly, fragments to be fused to the C terminus of GFP were amplified as described above with primers and templates listed in Table 1 and cloned via SacI/EcoRI restriction sites. pGFP-012cterm was cloned via AvaI/HindIII sites. All forward primers, except those for cloning of pGFP-012cterm, also contained a double glycine-serine (GS) linker that served as a spacer to add flexibility to the fused sequences (28). Positive colonies were selected on kanamycin LB agar plates and analyzed by restriction digestion and agarose gel electrophoresis, as well as Sanger sequencing (LGC Genomics).
RNA extraction and RT-PCR analysis.
To investigate putative mRNA splicing of ORF012, 1 × 106 CEC were infected with MDV vRb or mock infected. Additionally, DF-1 cells were transfected with pc_012Δint as a positive control. Five days postinfection (p.i.) or 24 h posttransfection (p.t.), total RNA was extracted from cells using the RNAeasy Kit (Qiagen), following the manufacturer's protocol. Genomic DNA was removed with gEliminator columns (Qiagen), as well as an additional on-column DNase digest (Qiagen). Eluted RNA was quantified using a spectrophotometer (Nanodrop). Reverse transcriptase (RT) PCR was performed with the indicated primers (Table 1 and Fig. 2) in a two-step reaction. First, cDNA was synthesized from 500 ng of total RNA using the Omniscript RT Kit (Qiagen) in a 20-μl reaction mixture. Half a microliter of the reaction mixtures was used in PCR (95°C for 5 min; 30 cycles of 95°C for 30 s, 62°C for 30 s, and 72°C for 2.5 min; and 72°C for 10 min), and the amplicons were separated on 1% agarose gels. In addition, the amplicons were subjected to Sanger sequencing (LGC Genomics). Reaction mixtures to which no RT was added served as a control for genomic-DNA contamination, and amplification of chicken glyceraldehyde-3-phospate dehydrogenase (GAPDH) served as an internal control.
FIG 2.

Analysis of ORF012 splicing in MDV infection by RT-PCR. (A) Positions of primer binding regions on ORF012 cDNA. Two sets of primers specific for the 5′ and 3′ coding regions of the ORF012 transcript (TS1 and TS3) or the exon-exon border (TS2) were used. The primer sequences are listed in Table 1. (B) Primers were tested using genomic RB-1B DNA. Note that no product should be obtained with the primer combination TS2 and TS3 due to the absence of the exon-exon border in genomic DNA. (C) Total RNA of MDV-infected or mock-infected CEC was extracted 5 days p.i. The RNA was reverse transcribed into cDNA with the indicated primers. Amplification products obtained by subsequent PCR were analyzed by agarose gel electrophoresis. cDNA prepared from DF-1 cells transfected with an expression plasmid encoding an intronless ORF012 construct (termed pc_012Δint) served as a positive control and size marker (lanes +ctrl). The amplicons were subjected to Sanger sequencing and showed 100% identity with the predicted mRNA. cDNA of chicken GAPDH mRNA served as an internal control (bottom), and RT-negative control reactions excluded genomic DNA contamination (middle). (D) To show that splicing of ORF012 is independent of other viral factors, total RNA of DF-1 cells transfected with the pc_012Δint, pc012, or pcDNA3.1 vector (negative control) was extracted 24 h after transfection. Samples were subsequently treated as described for panel C. Sequencing revealed 100% identity with the predicted ORF012 mRNA sequence.
Western blot analysis.
CEC (1 × 106) were infected with the same number of PFU of MDV v20, a mutant virus encoding a C-terminally (v20_012Flag) or N-terminally (v20_Flag012) Flag-tagged p012. Infected cells were harvested 5 days p.i. and lysed in RIPA buffer (20 mM Tris-HCl, 150 mM NaCl, 1% [vol/vol] Nonidet P-40, 0.5% [wt/vol] sodium deoxycholate, 0.1% [wt/vol] SDS) supplemented with Complete Mini protease inhibitor (Roche) and phosphatase inhibitor cocktail (Sigma). The lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the proteins were transferred onto polyvinylidene difluoride membranes (Carl Roth) using the Bio-Rad wet-blot system. Subsequently, the membranes were blocked with 3% BSA in PBS and incubated overnight at 4°C with polyclonal rabbit anti-Flag antibody (Sigma) or rabbit polyclonal anti-actin antibody (Cell Signaling), both diluted 1:1,000 in blocking buffer. Following washing with PBS containing 0.1% Tween 20 (Carl Roth), the membranes were incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (Cell Signaling) diluted 1:10,000. Finally, the membranes were incubated with enhanced chemiluminescence (ECL) Plus Western blot detection reagent (GE Healthcare), and the signal was recorded using a Chemi-Smart 5100 detection system (PeqLab). To remove bound antibodies, the membranes were incubated twice with stripping buffer (50 mM glycine, 2% [vol/vol] SDS) at room temperature on an orbital shaker, washed twice with PBS, blocked with blocking buffer, and reprobed with antibodies. For dephosphorylation experiments, DF-1 cells transfected with pc_012Flag were lysed in RIPA buffer 24 h p.t. Prior to Western blotting, some lysates were treated with λ protein phosphatase (LPP) (NEB) or mock treated for 30 min according to the manufacturer's protocol to analyze the phosphorylated state of proteins.
Phos-tag Western blotting to determine phosphorylation of p012.
In order to validate the phosphorylation of p012, we used the Phos-tag reagent (Wako Chemicals) as described in the manual provided by the supplier. Phos-tag binds specifically to phosphorylated proteins in the presence of manganese ions (MnCl2) and decreases the migration of phosphoproteins in SDS-PAGE (29, 30). Briefly, 25 μM Phos-tag solution and 1 mM MnCl2 solution were added to the gel mixture prior to casting. Subsequent Western blotting was performed as described above.
Transfection of expression plasmids, indirect immunofluorescence microscopy, and quantification of cellular localization.
DF-1 cells (1 × 104) seeded on glass coverslips in a 24-well plate were transfected with 1 μg of plasmid DNA using 3 μl Fugene HD transfection reagent (Promega). The cells were washed with PBS at 24 h p.t., fixed with 3% paraformaldehyde in PBS for 10 min at room temperature, and permeabilized with 0.1% Triton X-100 in PBS for 10 min at room temperature. Following a blocking step with 5% FBS in PBS, the cells were stained with polyclonal rabbit anti-Flag antibody (Sigma) diluted 1:1,000 in 1% BSA-PBS for 1 h at room temperature, washed, and incubated with goat anti-rabbit Alexa 568 antibody (diluted 1:2,000; Life Technologies) for 1 h. Finally, the cells were stained with Hoechst 33342 (Life Technologies) to visualize nuclei, and coverslips were mounted with PermFluor mounting medium (Thermo Scientific). For pEGFP-C1 fusion constructs, cells were fixed 24 h p.t. and stained with Hoechst 33342 (Life Technologies). To quantify the intracellular distribution of p012, NLS deletion proteins, and the GFP fusion constructs, we used a semiquantitative transfection assay based on expression plasmids as described previously by Brock et al. (31). Images of at least 200 fluorescence-positive cells for each transfected construct were captured with an Axiovision microscope (×400 magnification) in a randomized fashion. In replicate experiments, the cellular distribution of the fluorescence signal within each cell was classified by an individual blinded to the experimental groups into one of three categories: (i) predominantly nuclear localization, (ii) mixed nuclear/cytoplasmic localization, or (iii) predominantly cytoplasmic localization.
Leptomycin B treatment.
In order to test the effects of the nuclear export inhibitor leptomycin B (Sigma-Aldrich) on p012 localization, DF-1 cells (1 × 104) plated on glass coverslips in a 24-well plate were transfected with 1 μg of plasmid DNA using Fugene HD. At 6 h p.t., cells in individual wells were incubated with fresh medium containing 2 μM leptomycin B or were mock treated with diluent. The cells were further incubated for 9 h, fixed with 3% paraformaldehyde, and analyzed for subcellular localization as described in the previous paragraph. In a different experiment, cells were incubated with 20 μM leptomycin B at 10 h p.t. and then fixed after 5 h of treatment. Differences between absolute cell numbers were tested for significance by the χ2 test.
RESULTS
Location of ORF012 in the MDV genome.
Despite its extensive colinearity with varicella-zoster virus (VZV) and HSV-1, the MDV genome contains a sequence stretch within the UL region that seems to be exclusively present in avian alphaherpesviruses (Fig. 1). A potential role in the host range has, therefore, been proposed for the genes in this area (14). Nevertheless, the region is poorly characterized regarding its coding capacity. The annotation of MDV ORFs that do not have comparable homologues in other herpesviruses is not a trivial task and usually relies heavily on bioinformatic predictions. Early attempts at genome-wide annotations for several MDV strains deposited in GenBank resulted in the provisional prediction of two potential ORFs, named ORF011 and ORF012, in the 5′ region of the UL segment. We refer to these ORFs as ORF011* and ORF012* in the present work in order to clearly differentiate them from the newly identified ORF012.
Based on early predictions, the region downstream of the MDV lipase-like protein vLIP (ORF010) contains a putative ORF011* with a length of 258 bp (GenBank AAG14191.1), followed by ORF012* with a predicted size of 1,155 bp (GenBank AAG14192.1) (Fig. 1). The two predicted ORFs are separated by a short intergenic sequence of 139 bp. However, a more recent annotation predicted splicing within the ORF011*-ORF012* region (GenBank NC_002229.3). We confirmed these predictions by bioinformatic analysis using the NetGene2 server (17, 18). The analysis revealed putative splicing of a small intron of 82 bp within ORF011*, because the sequence matched the consensus sequence for classical splice acceptor and donor sites with high scores (Fig. 1). The splicing would lead to the fusion of the two putative exons of ORF011* and, as a consequence, result in a frameshift mutation and readthrough into ORF012*. The predicted spliced transcript, therefore, represents an mRNA in which ORF011* and ORF012*, as well as the former intergenic region, are joined together to form a single transcript termed ORF012. The predicted protein encoded by ORF012 is a 489-amino-acid protein with a calculated molecular mass of 55 kDa. Interestingly, ORF012 was already annotated as a putatively spliced gene following an update of the Md5 strain reference sequence in 2007, and consequently, ORF011* was excluded from this new annotation (NC_002229.3). However, to our knowledge, no experimental evidence has been provided for this splicing event, nor has any functional characterization been performed.
Splicing of MDV ORF012 during infection of chicken cells.
In order to determine the predicted splicing of ORF012 by RT-PCR, we generated a synthetic ORF012 construct that was devoid of the predicted intron using fusion PCR and cloned the fragment into the pcDNA3.1 expression vector. The construct was termed pc_012Δint to differentiate it from the original ORF012 sequence that still contains the predicted intron. Additionally, we designed forward and reverse primers specific for the 5′ and 3′ coding regions of the ORF012 transcript (TS1 and TS3), as well as a forward primer spanning the predicted exon-exon border within its 3′-terminal portion (TS2) (Fig. 2A). First, we controlled both primer sets in PCRs using rRb BAC DNA. The combination of primers TS1 and TS3 yielded an expected DNA fragment of 1,552 kb that contained the intron sequence. Due to the nonexistent exon-exon border in genomic DNA, the combination of primers TS2 and TS3 did not yield any product (Fig. 2B). In order to demonstrate splicing, we produced cDNA from total RNA of vRb or mock-infected CEC at 5 days p.i. Using both primer sets, a single fragment from the cDNA of infected cells was amplified that corresponded in size to control cDNA generated from chicken cells transfected with the intronless pc_012Δint (Fig. 2C). In addition, the band was absent in mock-infected cells (Fig. 2C). The PCR products were gel purified and subjected to Sanger sequencing and revealed perfect sequence identity with the predicted spliced mRNA. PCRs performed on RT-negative (−RT) samples served as a control for possible DNA contamination (Fig. 2C, middle). In addition, DNA contamination of RNA was excluded using primer TS2, which is capable of priming only the exon-exon border within the spliced mRNA. Chicken GAPDH levels served as an internal control (Fig. 2C, bottom).
To further validate our results, we repeated the RT-PCR analysis with cDNA generated from chicken DF-1 cells transfected with pc_012Δint (positive control), pc_012, or pcDNA3.1. Again, we were able to amplify a single band of the expected size whose sequence was identical to that of the predicted ORF012 mRNA (Fig. 2D). We therefore concluded that ORF011* and ORF012* do not represent independent genes but one single unit that is comprised of two exons that are separated by an 82-bp intron close to the 5′ region of the novel ORF012. The intron is spliced during both transfection of the expression construct and virus replication, suggesting it is spliced independently of viral factors.
p012, but not p012* by itself, is produced during MDV infection.
Despite the clear indication of a splicing event, we considered the possibility that ORF012 represents only a splice variant and that an individually expressed ORF012* may be produced. That is, p012* could be translated from the predicted in-frame start codon of ORF012* within the ORF012 mRNA (compare Fig. 1). In order to investigate the protein-coding capacity of ORF012 mRNA, we analyzed protein translation from its transcript. We individually cloned ORF012* and ORF012 into the pcDNA3.1 expression vector with C-terminal Flag tags. Furthermore, we used the cell culture-adapted, apathogenic MDV strain v20 (24) to generate FLAG epitope-tagged versions of p012 (v20_012Flag). We hypothesized that due to the significant differences in the predicted molecular masses of p012* and p012 (44 kDa versus 55 kDa, respectively), the v20_012Flag virus would allow the differentiation of each protein by Western blot analysis. We infected CEC with 200 PFU of v20 or v20_012Flag for 5 days and then subjected the cell lysates to Western blot analysis. Lysates of cells transfected with the pc_012*Flag or pc_012Flag expression plasmid served as controls and internal size markers. As shown in Fig. 3, we could detect the presence of a specific band of ∼55 to 60 kDa in lysates of cells transfected with pc_012*Flag (lane 1), as well as a band of 75 to 80 kDa in lysates of cells transfected with pc_012Flag (lane 2), indicating that both proteins can be produced from expression plasmids in DF-1 cells. Most importantly, only a single band corresponding to the size of p012Flag, but not p012*Flag, was present in lysates of v20_012Flag-infected DF-1 cells (lane 4). As expected, no p012Flag-specific band could be detected in the negative control, v20-infected cells (lane 3), using the Flag antibody. Interestingly, both p012* and p012 appeared to have higher molecular masses than predicted. Although some deviation of apparent molecular masses after SDS-PAGE from those predicted based on the amino acid sequence is common (32), the differences observed were much greater and thus could be due to posttranslational modification.
FIG 3.
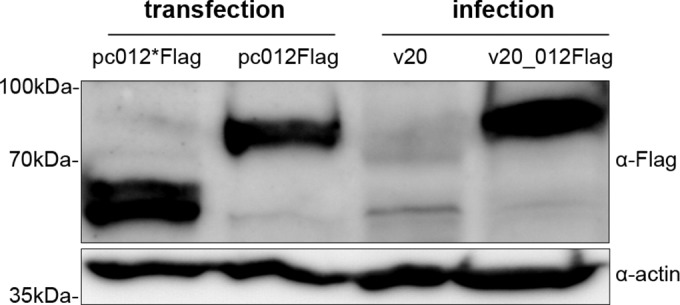
Detection of p012, but not p012*, in MDV-infected cells by Western blot analysis. CEC infected with 200 PFU of MDV v20 or v20_012Flag were collected 5 days p.i. Lysates were separated by SDS-7.5% PAGE, followed by immunoblotting. Lysates of DF-1 cells transfected with pc012*Flag or pc012Flag were used as controls and internal size markers, respectively. Membranes were incubated with polyclonal rabbit anti-Flag (α-Flag) antibody, washed, and incubated with secondary goat anti-rabbit HRP antibody. For the detection of actin as a loading control, the blots were stripped, blocked, and reprobed with rabbit anti-actin antibody. Note the absence of a band corresponding to p012 in lysates of virus-infected cells. The positions of mass marker bands are indicated on the left.
Contrary to expression of p012* alone, we considered production of a protein from ORF011* as highly unlikely due to the efficiency of the splicing process. Mechanisms that suppress splicing of ORF011* (e.g., intron skipping) and that would retain the stop codon would have to be active in order to generate a functional protein. We were unable to detect any such protein (predicted molecular mass of the theoretical protein, 10 kDa) in pc_011*Flag-transfected DF-1 cells when applying indirect immunofluorescence microscopy or Western blotting (data not shown). Nevertheless, to corroborate our results, we introduced a Flag tag immediately downstream of the initiation codon of the putative ORF011* (v20_Flag011). Again, we were unable to detect the presence of a low-molecular-weight protein following Western blotting of infected CEC cells (data not shown). In summary, we concluded that only p012 from a spliced mRNA is synthesized in virus-infected cells.
ORF012 is essential for viral replication in vitro.
Next, we determined whether ORF012 was dispensable for viral replication in vitro. Using two-step Red-mediated mutagenesis, we replaced the start codon of ORF012 in the pathogenic RB-1B strain with a stop codon (rRbΔMet012) to prevent translation of the protein. The resulting mutant virus, vRbΔMet012, was severely replication impaired following reconstitution in CEC. The numbers, as well as the sizes, of plaques were significantly smaller than those of the parental vRb (Fig. 4). ORF012-null virus plaques also displayed a different phenotype, as they appeared less “dense” and reminiscent of a cluster of single infected cells with many interspersed uninfected cells rather than the characteristic dense clusters of infected cells normally seen for the parental MDV. More importantly, we were unable to expand vRbΔMet012 by serial passaging of infected cells despite multiple attempts (n = 6). Even in very early passages following reconstitution, scant signs of cytopathic effects could be observed, indicating a severe impact on viral replication in the absence of p012. Consequently, classical single- or multistep growth kinetics could not be performed.
FIG 4.

p012 is essential for viral replication in vitro. Quantification of viral replication by plaque size assay is shown. Cells were transfected with rRb DNA, a mutant BAC in which the start codon of ORF012 was replaced with a stop codon (rRbΔMet012), and a revertant construct in which the start codon was repaired (rRbΔMet012R). At 5 days after transfection, plaques were stained by indirect immunofluorescence using an MDV-specific polyclonal antiserum, and the diameters of at least 50 plaques in three independent experiments were determined. Whisker plots of plaque diameter distributions relative to wild-type virus are shown (whiskers, minimum to maximum; line, median; box, interquartile range). Representative images of plaques are shown at the top. The results were tested for normality and subsequently analyzed for significance by 1-way ANOVA (***, P < 0.01).
To exclude the possibility of secondary site mutations introduced during BAC mutagenesis, we generated a revertant virus (vRB-1BΔMet012R) in which the start codon was restored. In three independent experiments, the sizes of at least 50 plaques for each virus were determined and compared based on calculated diameter values. As demonstrated in Fig. 4, vRB-1BΔMet012 induced significantly smaller plaques and reached only approximately 30% of the mean diameters determined for wild-type and revertant virus, which were not significantly different from each other. The computed diameters were tested for normality of distribution and for significance by 1-way ANOVA (P < 0.01). Given the dramatically reduced size of the vRB-1BΔMet012 plaques and, more importantly, our inability to expand the virus by serial passaging, we concluded that ORF012 is important for replication of the RB-1B wild-type virus.
p012 localizes predominantly to the nuclei of transfected and infected cells.
Next, we focused on elucidating the potential role p012 may play during MDV replication. First, we determined the protein's subcellular localization in DF-1 cells. p012 exhibited a predominantly nuclear localization in transfected DF-1 cells (Fig. 5A) that were analyzed by indirect immunofluorescence. The remaining fraction of positive cells showed either a mixed nuclear-cytoplasmic or a predominantly cytoplasmic distribution of p012. The same localization pattern was apparent in cells transfected with the pc012Δint control plasmid that expresses the synthetic intronless version of p012 (Fig. 5A). This was not unexpected, since the construct is devoid of the intron but otherwise leads to the production of an identical protein.
FIG 5.

Nuclear/cytoplasmic localization of MDV p012 in transfected cells. (A) DF-1 cells on coverslips were transfected with pc012Flag. The expression plasmid pc012ΔintFlag served as a control. At 24 h after transfection (48 h in the case of p012), the cells were fixed, permeabilized, and stained with polyclonal rabbit anti-Flag antibody (red). The transfected cells were costained with Hoechst 33342 to visualize the nuclei (blue). (B) Localization was quantified by indirect immunofluorescence microscopy as described in Materials and Methods. The results shown are from two independent experiments. nuc, nuclear; cyto, cytoplasmic.
In order to quantify the subcellular localization, we categorized the distribution of p012 in a blinded approach in more than 200 cells per construct by indirect immunofluorescence microscopy. Although our approach is of a semiquantitative nature, this method has already been used to quantify the nuclear localization of viral factors (31). The distribution of p012 was categorized into three classes: (i) predominantly nuclear, (ii) mixed nuclear-cytoplasmic, or (iii) predominantly cytoplasmic. Figure 5B shows the combined results of two independent experiments. The number of cells in each category in relation to all analyzed cells is displayed as percentage values. In approximately 55 to 65% of the analyzed cells, we found an entirely nuclear distribution of the target protein 24 h posttransfection (black). In the remaining cells, 30 to 35% were predominately cytoplasmic (white), while 5 to 10% contained p012 in both the nucleus and cytoplasm (gray). A similar distribution was apparent in cells transfected with the pc_012Δint control plasmid (middle bar). The distribution of p012 was also comparable at 48 h posttransfection (right bar).
Since the synthesis of viral proteins from expression vectors does not necessarily reflect the situation during infection, we validated our results in CEC infected with v20_012Flag. In agreement with our transfection experiments, p012 localized mainly to the nucleus in MDV-infected cells, while a smaller fraction of cells showed predominantly cytoplasmic or mixed (nuclear and cytoplasmic) localization (see below).
p012 contains a functional nuclear localization signal in its C-terminal domain.
The predominant nuclear localization of p012 prompted a bioinformatic search for potential NLSs. Two different NLS analysis tools predicted a potential monopartite NLS in the C-terminal portion of p012. NucPred (20) predicted an NLS comprised of six basic arginines and one proline ranging from amino acids 457 to 463 (457RRRRRPR463) with high probability. We provisionally termed the sequence stretch “short NLS.” In addition, analysis of the protein sequence with the tool NLStradamus (19) identified an NLS with the sequence 447RSRSRSRSRERRRRRPRVRPGRR469, which overlapped with the short NLS but was considerably longer and was thus termed “long NLS” (Fig. 6A).
FIG 6.

Prediction and mapping of an NLS in p012. (A) Schematic representation of the C-terminal 50 amino acids (aa) of p012. The position of two putative overlapping nuclear localization signals is indicated (boxed and grey/underlined). (B) Mutational disruption of the p012 NLS in MDV v20 inhibits nuclear accumulation. CEFs were infected with v20_012Flag, a C-terminal deletion mutant (v20_012ΔNLSFlag), or an alanine substitution mutant corresponding to the predicted short NLS (v20_012mutshortNLS) (bottom). At 5 days p.i., the cells were fixed, permeabilized, and stained with polyclonal rabbit anti-Flag antibody. Arrows indicate cells with representative localization of p012. (C) Mutational mapping of essential NLS regions in p012. Alanine substitutions in the NLS are indicated in gray. DF-1 cells were transfected with pc012Flag, pc012FlagmutRS, or pc012FlagmutshortNLS. Images of representative results, as well as the quantification of three independent experiments, are shown.
In order to determine the importance of the NLS sequence in p012, we deleted the 3′ region of the ORF012 gene within the viral genome of vRB-1B. The deletion removed about 1/3 of the protein, encompassing both potential NLSs. In another mutant virus, we replaced the basic amino acids with alanine residues within the provisionally termed short NLS. Interestingly, both mutant viruses were replication incompetent; however, when these mutations were replaced with wild-type sequences, the respective revertant viruses were rendered fully replication competent again (data not shown).
These results were reminiscent of the growth defect induced by the ORF012-null virus vRbΔMet012 (Fig. 4) and already pointed to the functional importance of the NLS in viral replication. At the same time, they presented us with the problem of being unable to further investigate the localization of p012 in the context of viral infection. We therefore turned to the avirulent and cell culture-adapted v20 MDV strain to further investigate the expression of p012 during replication. As with vRB-1B, we deleted the region containing the predicted NLS in the v20 virus carrying a C-terminal Flag tag, a virus that we had previously used to detect p012 by Western blotting (Fig. 3). Again, the deletion massively impaired the replication of the mutant compared to the parental virus but still allowed visualization of plaques. As seen in Fig. 6B, p012 nuclear localization was virtually absent in cells infected with the deletion mutant v20_012ΔNLSFlag. In contrast, nuclear localization was again observed with the parental virus v20_012Flag. To validate our observations, we investigated the localization of p012 in a virus mutant containing an alanine substitution for the short NLS. The virus, termed v20_012mutshortNLS, exhibited the same phenotype as the deletion mutant (Fig. 6B, bottom). We concluded from our results that the predicted NLS has an essential function in nuclear import of p012 and that not only the presence of p012, but also its nuclear localization during infection, is important for viral growth. It is not known why the effects of the NLS mutations are more severe in the vRB-1B than in the v20 background, but it most likely is the result of the cell culture adaptation of v20, which has led to numerous deletions, insertions, and point mutations, affording it a greater capacity to replicate in vitro (33).
Mutational mapping of the p012 NLS.
Our previous results with a mutant virus indicated the importance of the predicted NLS for nuclear import of p012. Given the fact that two different, but overlapping, NLSs were predicted, we wished to determine the bona fide NLS sequence by substitution mutagenesis and reporter-based mapping approaches. To do this, we employed DF-1-based transfection assays, as described previously. First, the short NLS with the sequence 457RRRRRPR463 was replaced with alanine residues by site-directed mutagenesis in the pc_012Flag expression vector, resulting in plasmid pc_012mutshortNLS (Fig. 6C). Accordingly, we deleted a region encoding an arginine-serine-rich dipeptide repeat motif (447RSRSRSRSR455), which represents approximately the first half of the predicted long NLS but is separated from the short NLS by one glutamic acid residue (Fig. 6C). We termed the plasmid pc_012mutRSrepeat. Mutation of the short NLS sequence almost completely abolished nuclear localization, as seen using immunofluorescence microscopy in transfected cells. Surprisingly, the mutation of the RS repeat sequence had a comparable effect on nuclear localization, reducing the percentage of cells in this category to 7% (Fig. 6C). Nevertheless, we found a small percentage of cells with mixed cytoplasmic-nuclear localization in both cases. We concluded that not only the short NLS region, but also the preceding RS-rich motif is necessary for efficient nuclear import of p012.
The p012 NLS is transferable and can shuttle GFP to the nucleus.
The previously described NLS mutation experiments suggested the involvement of the long NLS in nuclear localization of p012. Nevertheless, deletion experiments alone are not adequate to determine whether a specific sequence within a nuclear protein is sufficient for localization. Therefore, we cloned different p012 NLS-GFP fusion constructs based on the pEGFP-C1 expression vector (Fig. 7A and B). Again, we quantified GFP localization in transfection assays. Figure 7C shows the combined results of two independent experiments. A baseline level of nuclear-cytoplasmic localization in a small percentage of cells transfected with the GFP control vector was apparent and reached about 8%. This effect can probably be attributed to a nonspecific accumulation of GFP and has been documented previously (34). Complementing our previous results of the NLS mutagenesis experiments, GFPs fused with either the C-terminal 150 amino acids of p012 or the long NLS were equally efficient at shuttling GFP to the nucleus. Nuclear localization was found in about 70% of transfected cells in both cases. Importantly, neither a GFP fused to the RS repeat nor a GFP fused to the sequence encompassing the short NLS was able to enter the nucleus above control levels. We concluded from these experiments that the long NLS (447RSRSRSRSRERRRRRPRVRPGRR469) is necessary and sufficient for NLS function and nuclear import of p012. Most likely, the sequence 457RRRRRPR463 constitutes only part of the functional signal.
FIG 7.
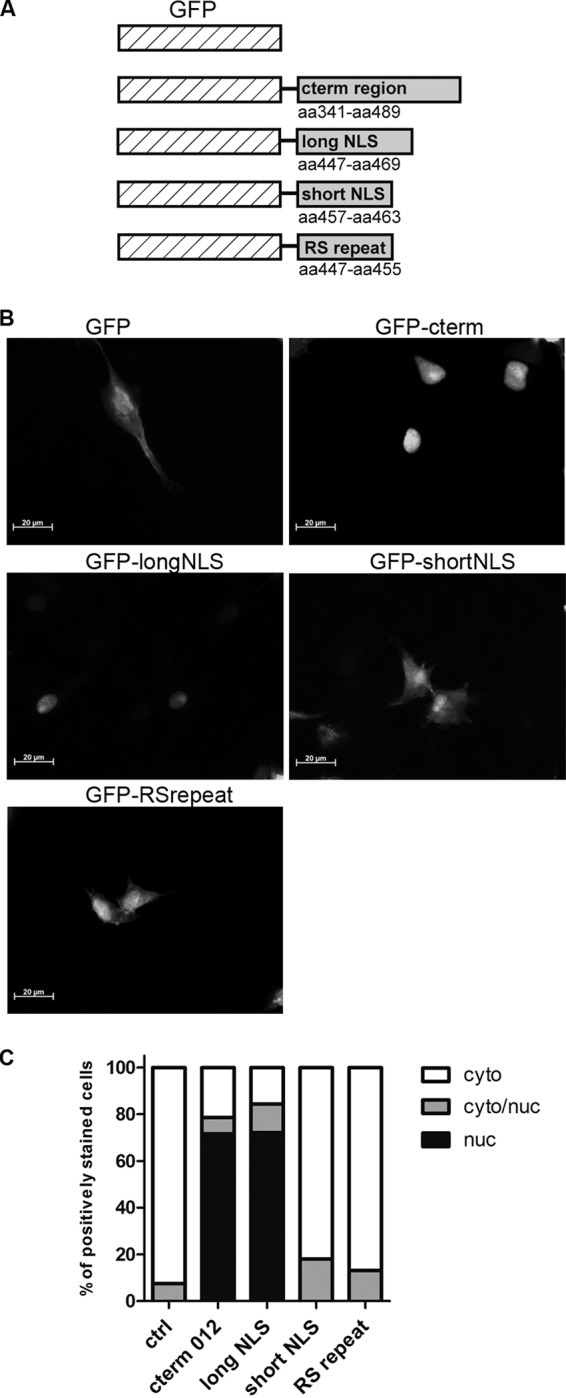
The p012 NLS is transferable and sufficient for nuclear import of GFP. (A) Schematic representation of cloned GFP-NLS fusion constructs. cterm, C-terminal. (B) DF-1 cells were transfected with either pEGFPc-1_cterm, GFP_longNLS, GFP_shortNLS, GFP_RSrepeat, or the empty vector. (C) Subcellular localization of GFP was quantified as described in Materials and Methods. The results of two combined experiments are shown.
Nuclear export of p012 can be inhibited with leptomycin B.
As is evident in Fig. 6 and 7, p012 showed a clear nuclear localization in transfected, as well as infected, chicken cells. When we quantified the distribution of the viral protein in transfected cells, about 70% of the cells showed an exclusively nuclear localization, whereas the remaining 30% of the cells were categorized as predominantly cytoplasmic. This distribution raised the question of whether p012, apart from its NLS-driven nuclear import, could also be actively exported from the nucleus. We tested this hypothesis by quantifying the subcellular distribution of p012 at different times posttransfection over a period of 48 h. Figure 8A shows the results of two combined experiments. Within the first 6 h posttransfection, almost all transfected cells showed nuclear localization of p012 (first bar from left). However, from this time point onward, the protein was found to localize predominantly to the cytoplasm and reached equilibrium around 36 h posttransfection with little change at later time points (Fig. 8A). These results suggested active export of p012 from the nucleus to the cytoplasm, at least in transfected cells. In a second experiment, we applied LMB, a potent inhibitor of nuclear export to transfected cells. LMB acts by binding to the karyopherin export protein CRM-1 and prevents its interaction with proteins harboring leucine-rich nuclear export signals (NES) (35). If p012 were actively exported from the nucleus by CRM-1, LMB treatment would lead to increased nuclear accumulation of the protein. Figure 8B shows the summarized results quantifying the cytoplasmic localization of p012 in three independent experiments under LMB treatment. In one experiment, cells were incubated at 6 h posttransfection with 2 μM LMB for 9 h, an inhibitor concentration and time period that did not induce any visible cytotoxicity (data not shown). We noticed a mean reduction of cells, with cytoplasmic localization of around 18% compared to mock-treated cells (Fig. 8B). In a second experiment, we used 20 μM LMB, the highest concentration recommended by the supplier, for a shorter period. Again, we were able to detect a difference of about 10% versus nontreated cells. The effect of LMB on cells appeared rather mild, but differences in the combined absolute numbers of 5 independent experiments were highly significant, as determined with a χ2 test (P < 0.001). This result indicates that p012 is indeed actively exported from the nucleus. Initial bioinformatic predictions yielded no clear candidates for a classical NES (36, 37), and the potential existence of export signals remains to be established. However, p012 contains 36 leucine residues, amounting to approximately 8% of its 489 total amino acids. Together with arginine, which is a major component of the NLS, leucine is among the most prevalent amino acids in the p012 sequence. Thus, there may potentially be an unknown NES within p012 that bioinformatic analysis cannot predict at this time.
FIG 8.
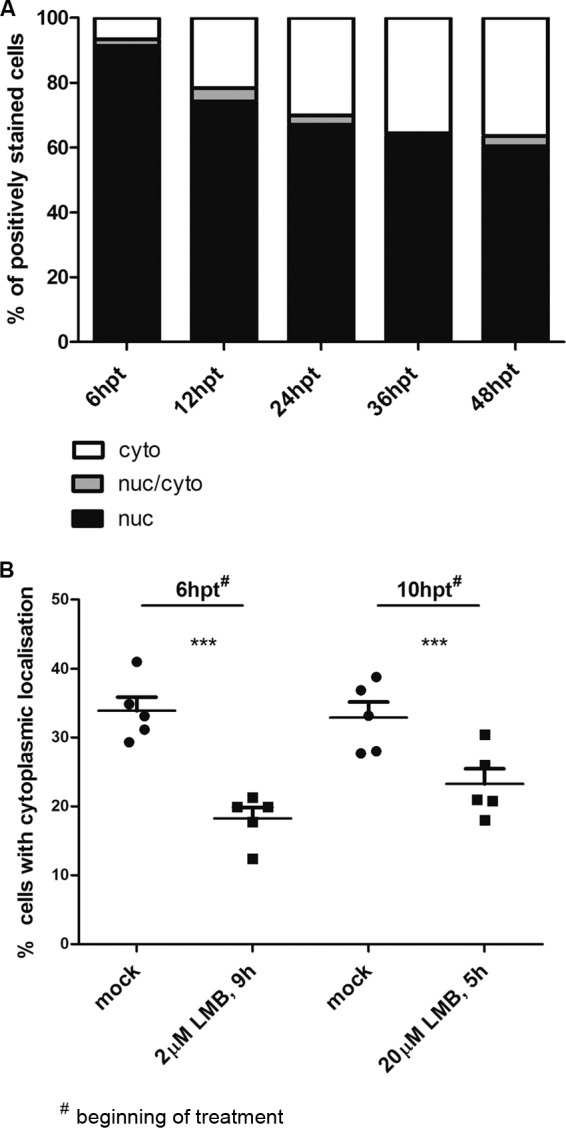
Leptomycin B inhibits nuclear export of p012. (A) DF-1 cells were transfected with pc012Flag. At the indicated time points, cells were fixed and stained. The results of two independent experiments are shown. (B) DF-1 cells were transfected with pc012Flag. At 6 h or 10 h after transfection, the cells were incubated with 2 μM or 20 μM LMB and treated for 9 h or 5 h, respectively, before fixation and staining. The combined results of 5 independent experiments are shown as percentages of cells with predominantly cytoplasmic localization. Lines and error bars represent the mean values and the standard error of the mean (SEM), respectively. Note that the axis is scaled to 50%. Differences were tested for significance by χ2 tests (***, P < 0.01).
Phosphorylation of p012.
The C-terminal portion of p012 near the NLS also contains a number of serine and tyrosine residues. Both amino acids can serve as targets for phosphorylation. This is of particular interest, since phosphorylation of amino acids proximal to an NLS can modulate its activity and influence subcellular protein localization (38). When we performed Western blot analysis of transfected and infected cells (Fig. 3), we noted that p012 migrated as multiple bands that often appeared as a smear, suggesting posttranslational modification of p012. In order to assess whether p012 is a potential target for phosphorylation, we treated lysates of DF-1 cells transfected with pc_012Flag with LPP prior to Western blotting. As shown in Fig. 9A, treatment with LPP clearly changed the migration properties of p012 compared to mock-treated lysates. In particular, the observed band notably decreased in size, an observation that strongly suggests p012 phosphorylation at multiple residues.
FIG 9.
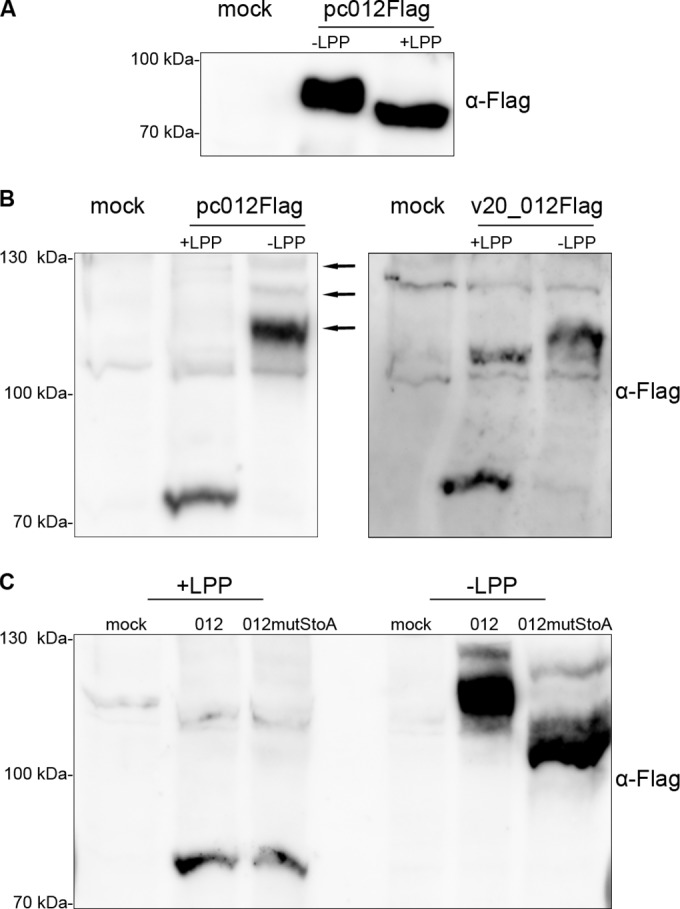
p012 is a phosphorylated protein. (A) Cells transfected with pc012Flag (or mock transfected) were lysed in RIPA buffer. Samples were subsequently treated with LPP or mock treated for 30 min prior to SDS-PAGE and Western blotting. Mock-transfected cell lysates served as a control. (B) Lysates of transfected or infected cells were treated as described for panel A and separated in SDS-PAGE gels containing 25 μM Phos-tag reagent and 1 mM MnCl2. Note that Phos-tag decreases the mobility of phosphorylated proteins due to specific interaction (see Materials and Methods). The arrows indicate the positions of differentially phosphorylated p012. (C) p012mutStoAFlag shows increased mobility compared to p012Flag in the presence of Phos-tag, indicating decreased phosphorylation. The positions of mass marker bands are indicated on the left.
To further confirm our results, we employed a phosphate-binding tag called Phos-tag in combination with Western blotting. The reagent is capable of binding phosphorylated proteins in the presence of manganese ions, thereby inducing slower migration in SDS-PAGE of phosphoproteins than of their unaffected dephosphorylated counterparts (29, 30). However, it has to be noted that the addition of Phos-tag and MnCl2 to acrylamide gels has bystander effects on the migration of proteins within complex cellular lysates, and effects like tailing or waving of bands have been described previously (29).
As seen in Fig. 9B, we could detect a dramatic mobility shift of the phosphorylated form compared to LPP-treated p012, indicating phosphorylation at potentially multiple residues. In addition, we could detect several bands of phosphorylated p012 (Fig. 9B, left, arrows), an effect that could reflect different phosphorylation states of the protein (30). A comparable migration after SDS-PAGE of p012 was detected in lysates of v20_012Flag-infected CEC (Fig. 9B, right). As depicted in Fig. 1, p012 contains several potential targets for phosphorylation that we predicted with high probability. However, the phosphorylation of serines within the RS repeat that we described is of particular interest, since it could influence the functions of the NLS and the protein, respectively (38). Therefore, we generated an expression plasmid in which the four serine residues of the RS repeat were replaced with alanines by site-directed mutagenesis. When we compared the migration of p012 to that of p012mutStoA on Phos-tag Western blots, we could indeed detect faster migration of the mutated form. This result could point to a less phosphorylated state of the modified protein due to the absence of four serine residues. When we investigated the localization of p012mutStoA following transfection, we observed a reduced number of cells with nuclear localization, which, however, was less pronounced than that of p012mutRSrepeat (data not shown).
Avian alphaherpesvirus proteins with similarity to MDV p012.
As previously mentioned, genes that show similarity to ORF012 are encoded in different avian alphaherpesviruses, including duck enteritis virus (DEV), HVT, GaHV-3, and infectious laryngotracheitis virus (ILTV), as well as the recently sequenced falconid herpesvirus 1 (FaHV-1) (15). Table 2 shows an identity percentage matrix based on protein sequence alignment. As expected, the p012 sequences of MDV and apathogenic GaHV-3 share the highest degree of similarity; however, the two proteins deviate by 50% in their compositions. Given the fact that the sequence similarity of proteins of the two closely related viruses usually ranges from 50% to 80% (2), the value is on the lower end of the spectrum. Nevertheless, related proteins in other viruses deviate even more compared to MDV. ILTV UL0 and UL-1, which form a cluster due to a presumed gene duplication event (39), showed the lowest overall identity to MDV p012. Table 3 summarizes mRNA splicing and the occurrence of NLS sequences, as well as phosphorylation, for the different candidate proteins, based on either experimental evidence or bioinformatic predictions. Whereas all of the proteins share similar structural properties, their functional relatedness remains to be established.
TABLE 2.
Identity matrix of proteins with similarity to MDV p012
| Protein | % Identity witha: |
||||||
|---|---|---|---|---|---|---|---|
| HVT Lorf2 | GaHV-3 Lorf2 | MDV ORF012 | DEV Lorf3 | FaHV Lorf3 | ILTV UL0 | ILTV UL-1 | |
| HVT Lorf2 | 100 | 36 | 41 | 23 | 26 | 17 | 15 |
| GaHV-3 Lorf2 | 36 | 100 | 50 | 26 | 27 | 17 | 16 |
| MDV ORF012 | 41 | 50 | 100 | 28 | 26 | 17 | 16 |
| DEV Lorf3 | 23 | 26 | 28 | 100 | 27 | 16 | 17 |
| FaHV Lorf3 | 26 | 27 | 26 | 27 | 100 | 15 | 15 |
| ILTV UL0 | 17 | 17 | 17 | 16 | 15 | 100 | 25 |
| ILTV UL-1 | 15 | 16 | 16 | 17 | 15 | 25 | 100 |
Percent identity based on amino acid alignment of candidate proteins using the Clustal Omega server.
TABLE 3.
Properties of MDV p012 and similar proteins
| Protein name | Splicing | NLS | Phosphorylation |
|---|---|---|---|
| MDV ORF012 | Yes (exptl evidence) | Yes; arginine-rich; “highly basic”; RS repeat | Yes (exptl evidence) |
| GaHV3 Lorf2 | Predicteda | Predicted; arginine-rich; “highly basic”; RS repeat | Predicted; mainly serine |
| HVT Lorf2 | Predicteda | Predicted; arginine-rich; RS repeat | Predicted; mainly serine |
| ILTV UL0 | Yes (exptl evidence)b | Predicted; arginine rich | Predicted; mainly serine |
| ILTV UL-1 | Yes (exptl evidence)b | Not predicted; RS repeat | Predicted; mainly serine |
| FaHV Lorf3 | Predicted potential splice site upstream of genec | Predicted; “highly basic”; arginine rich | Predicted; mainly serine |
| DEV Lorf3 | Not predictedc | Predicted; arginine rich | Predicted; mainly serine |
DISCUSSION
As the majority of MDV genes share homology to their HSV-1 or VZV counterparts, a number of MDV gene products have already been functionally analyzed in detail. However, unique and potentially unidentified genes exist in MDV, which could serve important functions in its complex replication cycle. In this work, we identified a novel MDV nuclear phosphoprotein that is translated from a spliced mRNA of the ORF012 gene. The annotation of this region in MDV has been ambiguous, with different names given to genes and ORFs, including ORF012. The majority of MDV genomes deposited in GenBank still define ORF011* and ORF012* as independent, hypothetical genes. In contrast, other annotations omit ORF011* completely, placing only ORF012* in the region downstream of ORF010 (viral lipase; Lorf2) and upstream of ORF013 (glycoprotein L; UL1) and refer to it as Lorf3. Some of the newer annotations acknowledge the predicted splicing but retain the ORF012 nomenclature. To add even more confusion, the “Lorf terminology,” starting with the first ORF that has its promoter in the UL region, is handled incoherently for different MDV strains. In particular, the inclusion or omission of Lorf1, a potential gene of unknown function, has led to different designations of all the subsequent genes in the UL region. Therefore, depending on the MDV sequence under scrutiny, Lorf2 stands for either viral lipase (16) or the spliced gene that we describe here (GenBank NC_002229). Therefore, here, we propose the term MDV ORF012 when referring to the gene identified in this report and hereafter. In general, our work underlines the fact that bioinformatic predictions, in particular those used for genome-wide annotations, are an excellent tool for the determination of potential ORFs, although, naturally, they cannot replace experimental evidence to prove or refute their implications.
mRNA splicing is a common principle of eukaryotic transcription. Despite its prevalence in eukaryotic cells, the mechanism was first identified in adenovirus-infected cells (40, 41). Apart from adenoviruses, splicing has also been found in herpesviruses. MDV is known to make extensive use of alternative splicing to generate diverse sets of transcripts from single genes, particularly in the repeat long regions. The mechanism serves to maximize the coding capacity of compact viral genomes, usually with strict size limitations. Here, we showed that MDV ORF012 is produced through mRNA splicing, and this splicing occurs independently of other viral factors, since the spliced transcript was also detected in cells transfected with an expression plasmid harboring the target gene. The resulting splice product removed a small, 82-bp intron within the 5′ region of the immature message (Fig. 1). Interestingly, the viral lipase-like vLIP encoded by ORF010 and located immediately upstream of ORF012 is also translated from a spliced transcript. Just as is the case for ORF012, the ORF010 intron is small and in the 5′ region of the gene (16). In order to ensure that ORF012 did not simply represent an alternative splice variant of an individually expressed ORF012*, we used epitope tagging of (putative) viral proteins to analyze the coding capacity of the entire region. We clearly showed that the hypothetical p012* was not produced during infection. Regarding the expression of p012, we were able to show its existence in virus-infected cells, corresponding in size to the control protein expressed in DF-1 cells. Therefore, we concluded that only p012 is produced during viral infection.
One of the first questions that arise with newly identified gene products of viruses is whether the protein is dispensable for viral replication. Using an MDV ORF012-null mutant based on the strain RB-1B, it became evident that p012 is important for viral growth in vitro (Fig. 4). Although small plaques were produced upon reconstitution of infectious DNA in CEC, we were unable to expand the virus by passaging in multiple trials. A recent study published by Hildebrandt et al. identified de novo mutations following extensive passaging of the vv Md5 strain in vitro. Among the mutations, two independent single-nucleotide polymorphisms (SNPs) associated with ORF012, one in the putative promoter region and one within the 82-bp intron, led to reduced virulence in chickens (42). However, in that report, the authors did not investigate whether and to what extent either of the single point mutations may have affected p012 expression or function.
In order to approximate the role of p012 in MDV replication, we used epitope-tagged expression constructs, as well as expression of tagged protein versions from recombinant viruses, to analyze the subcellular localization of p012. Interestingly, p012 showed a predominantly nuclear distribution in transfected cells and an even stronger nuclear accumulation in virus-infected cells (Fig. 5 and 6). The localization was reminiscent of the products of the duplicated UL0 and UL-1 genes of the distantly related ILTV, both of which were shown to be spliced. The UL0 and UL-1 proteins also accumulate in the nuclei of infected cells, but to date, no specific function or NLS has been assigned to either protein (39). In line with our results, it has also been reported that both proteins showed molecular weights after separation by SDS-PAGE and Western blotting considerably higher than the calculated values deduced from their primary sequences. The actual size deviation of p012 from its calculated value, however, can be only partially explained by phosphorylation, since LPP-treated p012 still migrated more slowly than expected. Other posttranslational modifications, therefore, may be responsible for the observation and warrant more extensive investigation in the future.
With the help of bioinformatic prediction tools, we were able to identify an NLS that mapped to a 23-amino-acid stretch in the C-terminal region of p012. The first experimental evidence that the sequence can indeed control nuclear import was provided by our infection experiments using a set of mutant viruses carrying mutations in the NLS. Compared to the parental virus, nuclear localization was completely abrogated in cells infected with these mutants (Fig. 6). In order to map the exact position of the NLS, we utilized expression assays and quantified the effects of targeted alanine substitutions within predicted NLS sequences. In accordance with the infection experiments, deletion of either the basic arginine core or the preceding serine-arginine-rich repeat motif abolished nuclear import (Fig. 6). Furthermore, we used synthetic NLS-GFP fusion constructs to directly test portions of the NLS that were required for nuclear import. Only constructs encompassing the sequence stretch 447RSRSRSRSRERRRRRPRVRPGRR469 accumulated in the nucleus, indicating that the motif can act as a transferable bona fide NLS. However, fine-mapping approaches, like alanine scanning or single amino acid deletions, will be necessary to identify the minimal core sequence. Whereas consensus sequences for classical NLS motifs, either mono- or bipartite, are well established (43–45), it has become evident that many nuclear proteins contain nonclassical signals that differ considerably in sequence (46, 47). Concerning the primary sequence of the p012 NLS, its categorization is not entirely obvious. The signal does not match the structure of a classical bipartite NLS (48), but rather, presents a stretch of basic arginines that is reminiscent of a monopartite simian virus 40 (SV40)-type NLS (49). Boulikas further subdivided classical monopartite NLSs, depending on their compositions (50, 51). In this regard, the core sequence would represent a “highly basic NLS” that usually contains 5 or 6 (K/R) residues (45). Nevertheless, we were able to show that the basic motif (called the short NLS here) is not sufficient for nuclear translocation. Only in combination with the preceding RS repeat were we able to detect cells with a clear nuclear accumulation of GFP. This shows that the p012 NLS constitutes a rather large peptide.
A question that still remains open is whether the entire NLS of p012 represents a docking site for nuclear importins or whether the two motifs fulfill different but complementary functions. In this regard, the phosphorylation state of p012 could play an important role in nuclear transport. It is known that phosphorylation of residues within or near the NLS can up- or downregulate activity (38). The mechanisms behind the modulations can be of various types but are often related to increased (or decreased) affinity for the import factor. The classical SV40 NLS itself is embedded in a sequence of residues that can be phosphorylated by protein kinase CK2, a modification that massively enhances nuclear import (52). The fact that substitution of the phosphorylation-accessible serines within the RS repeat of p012 decreased the extent of phosphorylation (Fig. 9C) and partially inhibited nuclear import could point to a functional involvement of phosphorylation. However, it is also conceivable that predominant localization to either the nucleus or the cytoplasm may influence protein phosphorylation. We will investigate a potential link between localization, the NLS, and the phosphorylation state of p012 in future experiments.
Interestingly, despite the presence of an NLS, the distribution of p012 was not entirely nuclear. A rather constant percentage of cells displayed a predominantly cytoplasmic or mixed distribution in transfected or infected cells. This fraction could be increased significantly by treatment with LMB, which inhibits leucine-rich export signals. Although analysis of the protein sequence did not yield clear candidates for an NES, the high prevalence of leucine residues in p012, in addition to increased nuclear localization during LMB treatment, suggests these sequences may play a part in nuclear export.
Although we have not yet performed a full functional characterization, it is tempting to speculate about potential actions of p012 as a nuclear/cytoplasmic shuttling viral protein. Eukaryotic cells contain a class of proteins that have a characteristic arginine-serine-rich motif in their C termini. These so-called “SR proteins” are capable of nucleocytoplasmic shuttling, can be heavily phosphorylated, and perform various functions ranging from RNA transport to control of mRNA splicing (53). Only recently, however, have strict refinements of the properties defining a SR protein been made (54). The protein must contain one or two N-terminally located RNA binding domains (called RRM boxes) followed by an RS domain, which should contain at least 50 amino acids with an arginine-serine content of more than 40%. Only 12 proteins in the human genome actually fulfill these requirements (54). Given the lack of an obvious RNA binding domain, as well as its short RS domain, p012 does not qualify as an SR protein per se. However, reports show that SR-like proteins that do not fully match all the requirements exist and still carry out functions involving RNA. Herpesviruses encode proteins that are known to interact with cellular SR proteins (55). Among the most intensively studied herpesviral factors is HSV-1 ICP27. ICP27 is a multifunctional regulatory protein that mediates the export of viral RNAs and is capable of inhibiting splicing of viral, as well as cellular, mRNAs. In this regard, the protein performs the function of a host shutoff protein (56). Interestingly, ICP27 is able to interact with cellular SR proteins, modulating their distribution inside the nucleus, as well as their phosphorylation (56). MDV also contains a homologue of ICP27 (57), and the protein was shown to interact with SR proteins and to inhibit splicing (58). Therefore, the hypothetical role of p012 in splicing and/or mRNA export remains to be addressed.
In summary, we have identified a novel nuclear phosphoprotein in MDV that is important for replication and that actively shuttles between the nucleus and the cytoplasm. In support of this, we showed that MDV p012 (i) is expressed as a spliced mRNA product, (ii) is important for replication in vitro, (iii) is primarily located within the nucleus that is dependent on a C-terminally located NLS, and (iv) most likely shuttles between the nucleus and the cytoplasm during replication. Further studies will be directed at addressing its role in shuttling and potential targets for its exact role in MDV replication.
ACKNOWLEDGMENTS
We thank Michaela Zeitlow for excellent technical assistance.
Timo Schippers was supported in part by the ZIBI Graduate School (IMPRS-IDI) Berlin and the Dahlem Research School (DRS) of Freie Universität Berlin. The work was supported in part by DFG grant 143/4-1 to N.O.
REFERENCES
- 1.Calnek BW. 2001. Pathogenesis of Marek's disease virus infection. Curr Top Microbiol Immunol 255:25–55. doi: 10.1007/978-3-642-56863-3_2. [DOI] [PubMed] [Google Scholar]
- 2.Osterrieder N, Kamil JP, Schumacher D, Tischer BK, Trapp S. 2006. Marek's disease virus: from miasma to model. Nat Rev Microbiol 4:283–294. doi: 10.1038/nrmicro1382. [DOI] [PubMed] [Google Scholar]
- 3.Baigent SJ, Smith LP, Nair VK, Currie RJW. 2006. Vaccinal control of Marek's disease: current challenges, and future strategies to maximize protection. Vet Immunol Immunopathol 112:78–86. doi: 10.1016/j.vetimm.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Davison AJ. 2002. Evolution of the herpesviruses. Vet Microbiol 86:69–88. doi: 10.1016/S0378-1135(01)00492-8. [DOI] [PubMed] [Google Scholar]
- 5.Davison AJ. 2010. Herpesvirus systematics. Vet Microbiol 143:52–69. doi: 10.1016/j.vetmic.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarosinski KW, Tischer BK, Trapp S, Osterrieder N. 2006. Marek's disease virus: lytic replication, oncogenesis and control. Expert Rev Vaccines 5:761–772. doi: 10.1586/14760584.5.6.761. [DOI] [PubMed] [Google Scholar]
- 7.Tulman ER, Afonso CL, Lu Z, Zsak L, Rock DL, Kutish GF. 2000. The genome of a very virulent Marek's disease virus. J Virol 74:7980–7988. doi: 10.1128/JVI.74.17.7980-7988.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones D, Lee L, Liu JL, Kung HJ, Tillotson JK. 1992. Marek disease virus encodes a basic-leucine zipper gene resembling the fos/jun oncogenes that is highly expressed in lymphoblastoid tumors. Proc Natl Acad Sci U S A 89:4042–4046. doi: 10.1073/pnas.89.9.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parcells MS, Lin SF, Dienglewicz RL, Majerciak V, Robinson DR, Chen HC, Wu Z, Dubyak GR, Brunovskis P, Hunt HD, Lee LF, Kung HJ. 2001. Marek's disease virus (MDV) encodes an interleukin-8 homolog (vIL-8): characterization of the vIL-8 protein and a vIL-8 deletion mutant MDV. J Virol 75:5159–5173. doi: 10.1128/JVI.75.11.5159-5173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui X, Lee LF, Reed WM, Kung H-J, Reddy SM. 2004. Marek's disease virus-encoded vIL-8 gene is involved in early cytolytic infection but dispensable for establishment of latency. J Virol 78:4753–4760. doi: 10.1128/JVI.78.9.4753-4760.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown AC, Baigent SJ, Smith LP, Chattoo JP, Petherbridge LJ, Hawes P, Allday MJ, Nair V. 2006. Interaction of MEQ protein and C-terminal-binding protein is critical for induction of lymphomas by Marek's disease virus. Proc Natl Acad Sci U S A 103:1687–1692. doi: 10.1073/pnas.0507595103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupiani B, Lee LF, Cui X, Gimeno I, Anderson A, Morgan RW, Silva RF, Witter RL, Kung H-J, Reddy SM. 2004. Marek's disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proc Natl Acad Sci U S A 101:11815–11820. doi: 10.1073/pnas.0404508101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel AT, Selvaraj RK, Kamil JP, Osterrieder N, Kaufer BB. 2012. Marek's disease viral interleukin-8 promotes lymphoma formation through targeted recruitment of B cells and CD4+ CD25+ T cells. J Virol 86:8536–8545. doi: 10.1128/JVI.00556-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Huang B, Ma X, Wu J, Li F, Ai W, Song M, Yang H. 2009. Molecular characterization of the genome of duck enteritis virus. Virology 391:151–161. doi: 10.1016/j.virol.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 15.Spatz SJ, Volkening JD, Ross TA. 2014. Molecular characterization of the complete genome of falconid herpesvirus strain S-18. Virus Res 188:109–121. doi: 10.1016/j.virusres.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 16.Kamil JP, Tischer BK, Trapp S, Nair VK, Osterrieder N, Kung H-J. 2005. vLIP, a viral lipase homologue, is a virulence factor of Marek's disease virus. J Virol 79:6984–6996. doi: 10.1128/JVI.79.11.6984-6996.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunak S, Engelbrecht J, Knudsen S. 1991. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol 220:49–65. doi: 10.1016/0022-2836(91)90380-O. [DOI] [PubMed] [Google Scholar]
- 18.Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouzé P, Brunak S. 1996. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res 24:3439–3452. doi: 10.1093/nar/24.17.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen Ba AN, Pogoutse A, Provart N, Moses AM. 2009. NLStradamus: a simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinformatics 10:202. doi: 10.1186/1471-2105-10-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brameier M, Krings A, MacCallum RM. 2007. NucPred—predicting nuclear localization of proteins. Bioinformatics 23:1159–1160. doi: 10.1093/bioinformatics/btm066. [DOI] [PubMed] [Google Scholar]
- 21.Blom N, Gammeltoft S, Brunak S. 1999. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 22.Sambrook J, Russell WD. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 23.Petherbridge L, Brown AC, Baigent SJ, Howes K, Sacco MA, Osterrieder N, Nair VK. 2004. Oncogenicity of virulent Marek's disease virus cloned as bacterial artificial chromosomes. J Virol 78:13376–13380. doi: 10.1128/JVI.78.23.13376-13380.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schumacher D, Tischer BK, Fuchs W, Osterrieder N. 2000. Reconstitution of Marek's disease virus serotype 1 (MDV-1) from DNA cloned as a bacterial artificial chromosome and characterization of a glycoprotein B-negative MDV-1 mutant. J Virol 74:11088–11098. doi: 10.1128/JVI.74.23.11088-11098.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 26.Jarosinski KW, Osterrieder N, Nair VK, Schat KA. 2005. Attenuation of Marek's disease virus by deletion of open reading frame RLORF4 but not RLORF5a. J Virol 79:11647–11659. doi: 10.1128/JVI.79.18.11647-11659.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abramoff M, Magelhaes P, Ram S. 2004. Image processing with ImageJ. Biophotonics Int 11:36–42. [Google Scholar]
- 28.Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotný J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R. 1988. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci U S A 85:5879–5883. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5:749–757. doi: 10.1074/mcp.t500024-mcp200. [DOI] [PubMed] [Google Scholar]
- 30.Kinoshita E, Kinoshita-Kikuta E, Koike T. 2009. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat Protoc 4:1513–1521. doi: 10.1038/nprot.2009.154. [DOI] [PubMed] [Google Scholar]
- 31.Brock I, Krüger M, Mertens T, von Einem J. 2013. Nuclear targeting of human cytomegalovirus large tegument protein pUL48 is essential for viral growth. J Virol 87:6005–6019. doi: 10.1128/JVI.03558-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Y, Mowery RA, Ashley J, Hentz M, Ramirez AJ, Bilgicer B, Slunt-Brown H, Borchelt DR, Shaw BF. 2012. Abnormal SDS-PAGE migration of cytosolic proteins can identify domains and mechanisms that control surfactant binding. Protein Sci 21:1197–1209. doi: 10.1002/pro.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spatz SJ, Rue C, Schumacher D, Osterrieder N. 2008. Clustering of mutations within the inverted repeat regions of a serially passaged attenuated gallid herpesvirus type 2 strain. Virus Genes 37:69–80. doi: 10.1007/s11262-008-0242-0. [DOI] [PubMed] [Google Scholar]
- 34.Seibel NM, Eljouni J, Nalaskowski MM, Hampe W. 2007. Nuclear localization of enhanced green fluorescent protein homomultimers. Anal Biochem 368:95–99. doi: 10.1016/j.ab.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 35.Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. 1999. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A 96:9112–9117. doi: 10.1073/pnas.96.16.9112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fischer U, Huber J, Boelens WC, Mattaj IW, Lührmann R. 1995. The HIV-1 Rev activation domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 82:475–483. doi: 10.1016/0092-8674(95)90436-0. [DOI] [PubMed] [Google Scholar]
- 37.Wen W, Meinkoth JL, Tsien RY, Taylor SS. 1995. Identification of a signal for rapid export of proteins from the nucleus. Cell 82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- 38.Nardozzi JD, Lott K, Cingolani G. 2010. Phosphorylation meets nuclear import: a review. Cell Commun Signal 8:32. doi: 10.1186/1478-811X-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ziemann K, Mettenleiter TC, Fuchs W. 1998. Infectious laryngotracheitis herpesvirus expresses a related pair of unique nuclear proteins which are encoded by split genes located at the right end of the UL genome region. J Virol 72:6867–6874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chow LT, Gelinas RE, Broker TR, Roberts RJ. 1977. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12:1–8. doi: 10.1016/0092-8674(77)90180-5. [DOI] [PubMed] [Google Scholar]
- 41.Berget SM, Moore C, Sharp PA. 1977. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci U S A 74:3171–3175. doi: 10.1073/pnas.74.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hildebrandt E, Dunn JR, Perumbakkam S, Niikura M, Cheng HH. 2014. Characterizing the molecular basis of attenuation of Marek's disease virus via in vitro serial passage identifies de novo mutations in the helicase-primase subunit gene UL5 and other candidates associated with reduced virulence. J Virol 88:6232–6242. doi: 10.1128/JVI.03869-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Görlich D, Kutay U. 1999. Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol 15:607–660. doi: 10.1146/annurev.cellbio.15.1.607. [DOI] [PubMed] [Google Scholar]
- 44.Chelsky D, Ralph R, Jonak G. 1989. Sequence requirements for synthetic peptide-mediated translocation to the nucleus. Mol Cell Biol 9:2487–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LaCasse EC, Lefebvre YA. 1995. Nuclear localization signals overlap DNA- or RNA-binding domains in nucleic acid-binding proteins. Nucleic Acids Res 23:1647–1656. doi: 10.1093/nar/23.10.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nigg EA. 1997. Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature 386:779–787. doi: 10.1038/386779a0. [DOI] [PubMed] [Google Scholar]
- 47.Pouton CW, Wagstaff KM, Roth DM, Moseley GW, Jans DA. 2007. Targeted delivery to the nucleus. Adv Drug Deliv Rev 59:698–717. doi: 10.1016/j.addr.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 48.Robbins J, Dilworth SM, Laskey RA, Dingwall C. 1991. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell 64:615–623. doi: 10.1016/0092-8674(91)90245-T. [DOI] [PubMed] [Google Scholar]
- 49.Kalderon D, Roberts BL, Richardson WD, Smith AE. 1984. A short amino acid sequence able to specify nuclear location. Cell 39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- 50.Boulikas T. 1994. A compilation and classification of DNA binding sites for protein transcription factors from vertebrates. Crit Rev Eukaryot Gene Expr 4:117–321. doi: 10.1615/CritRevEukarGeneExpr.v4.i2-3.10. [DOI] [PubMed] [Google Scholar]
- 51.Boulikas T. 1994. Putative nuclear localization signals (NLS) in protein transcription factors. J Cell Biochem 55:32–58. doi: 10.1002/jcb.240550106. [DOI] [PubMed] [Google Scholar]
- 52.Xiao CY, Hübner S, Jans DA. 1997. SV40 large tumor antigen nuclear import is regulated by the double-stranded DNA-dependent protein kinase site (serine 120) flanking the nuclear localization sequence. J Biol Chem 272:22191–22198. doi: 10.1074/jbc.272.35.22191. [DOI] [PubMed] [Google Scholar]
- 53.Boucher L, Ouzounis CA, Enright AJ, Blencowe BJ. 2001. A genome-wide survey of RS domain proteins. RNA 7:1693–1701. [PMC free article] [PubMed] [Google Scholar]
- 54.Manley JL, Krainer AR. 2010. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev 24:1073–1074. doi: 10.1101/gad.1934910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ote I, Lebrun M, Vandevenne P, Bontems S, Medina-Palazon C, Manet E, Piette J, Sadzot-Delvaux C. 2009. Varicella-zoster virus IE4 protein interacts with SR proteins and exports mRNAs through the TAP/NXF1 pathway. PLoS One 4:e7882. doi: 10.1371/journal.pone.0007882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sandri-Goldin RM. 2008. The many roles of the regulatory protein ICP27 during herpes simplex virus infection. Front Biosci 13:5241–5256. doi: 10.2741/3078. [DOI] [PubMed] [Google Scholar]
- 57.Ren D, Lee LF, Coussens PM. 1994. Identification and characterization of Marek's disease virus genes homologous to ICP27 and glycoprotein K of herpes simplex virus-1. Virology 204:242–250. doi: 10.1006/viro.1994.1528. [DOI] [PubMed] [Google Scholar]
- 58.Amor S, Strassheim S, Dambrine G, Remy S, Rasschaert D, Laurent S. 2011. ICP27 protein of Marek's disease virus interacts with SR proteins and inhibits the splicing of cellular telomerase chTERT and viral vIL8 transcripts. J Gen Virol 92:1273–1278. doi: 10.1099/vir.0.028969-0. [DOI] [PubMed] [Google Scholar]