

ABSTRACT
Enveloped viruses utilize membrane fusion for entry into, and release from, host cells. For entry, members of the Herpesviridae require at least three envelope glycoproteins: the homotrimeric gB and a heterodimer of gH and gL. The crystal structures of three gH homologues, including pseudorabies virus (PrV) gH, revealed four conserved domains. Domain II contains a planar β-sheet (“fence”) and a syntaxin-like bundle of three α-helices (SLB), similar to those found in eukaryotic fusion proteins, potentially executing an important role in gH function. To test this hypothesis, we introduced targeted mutations into the PrV gH gene, which either disrupt the helices of the SLB by introduction of proline residues or covalently join them by artificial intramolecular disulfide bonds between themselves, to the adjacent fence region, or to domain III. Disruption of either of the three α-helices of the SLB (A250P, V275P, V298P) severely affected gH function in in vitro fusion assays and replication of corresponding PrV mutants. Considerable defects in fusion activity of gH, as well as in penetration kinetics and cell-to-cell spread of PrV mutants, were also observed after disulfide linkage of two α-helices within the SLB (A284C-S291C) or between SLB and domain III (H251C-L432C), as well as by insertions of additional cysteine pairs linking fence, SLB, and domain III. In vitro fusion activity of mutated gH could be partly restored by reduction of the artificial disulfide bonds. Our results indicate that the structure and flexibility of the SLB are relevant for the function of PrV gH in membrane fusion.
IMPORTANCE Mutational analysis based on crystal structures of proteins is a powerful tool to understand protein function. Here, we continued our study of pseudorabies virus gH, a part of the core fusion machinery of herpesviruses. We previously showed that the “flap” region in domain IV of PrV gH is important for its function. We now demonstrate that mutations within domain II that interfere with integrity or flexibility of a syntaxin-like three-helix bundle also significantly impair gH function during fusion. These studies provide important insights into the structural requirements of gH for function in fusion.
INTRODUCTION
Membrane fusion, a crucial process in pro- and eukaryotic organisms, occurs, e.g., during cell division, autophagy, endocytosis, and exocytosis. Fusion events are also indispensable for enveloped viruses and are required for entry into host cells, egress, cell-to-cell spread, and syncytium formation. During entry, viral fusion proteins mediate the merger of the viral envelope with the plasma membrane or with endosomes of the target cell. In contrast to many other viruses (e.g., orthomyxo- or rhabdoviruses), which need only one protein for attachment and entry, herpesviruses require several envelope proteins for the subsequent steps of attachment and penetration. Three of these proteins, designated glycoprotein B (gB), gH, and gL, are conserved and considered essential for this process. Additional, less conserved herpesvirus glycoproteins are required for the first steps of attachment to specific host cell receptors (reviewed in references 1, 2, and 3).
Most herpesviruses initially bind to heparan sulfate chains of cell surface proteoglycans. In alphaherpesviruses like pseudorabies virus (PrV, Suid herpesvirus 1), or herpes simplex viruses 1 and 2 (HSV-1/2, Human herpesvirus 1/2), this step is mediated mainly by glycoprotein C (gC). gC is not essential for replication but improves the efficiency of virus entry (4, 5). In a subsequent step, glycoprotein D (gD) binds to specific cellular receptors like nectin-1 and nectin-2 (6). HSV-1 gD was further shown to trigger membrane fusion, presumably by interacting with gB and gH (7). Although gD is usually essential for virus replication, replication-competent gD deletion mutants of PrV and Bovine herpesvirus 1 (BoHV-1) that possess compensatory mutations in gB and gH could be isolated in vitro (8, 9). Moreover, gD is absent in the alphaherpesvirus varicella-zoster virus (VZV; Human herpesvirus 3) and in the other herpesvirus subfamilies.
In contrast, gB is the most conserved envelope glycoprotein of herpesviruses. HSV-1 gB interacts with heparan sulfate and other cellular receptors and may possess an important accessory function during attachment to specific cell types (1). However, the main role of gB is during membrane fusion. Crystal structures of the HSV-1 and Epstein-Barr virus (EBV, Human herpesvirus 4) gB homologues revealed homotrimers with strong similarities to fusion proteins of other virus families, in particular to vesicular stomatitis virus G protein (10, 11). Conserved “fusion loops” within gB homologues are essential for fusion activity by interacting with the host cell plasma membrane (10, 12). This interaction might induce a conformational change of gB that leads to formation of the fusion pore (1).
Another essential component of the herpesvirus fusion machinery is the heterodimer formed by glycoproteins gH and gL. Although it has long been known that gH/gL is required for herpesvirus penetration (13, 14), the detailed functions of the two proteins are still unknown. In most cases, the type I integral membrane protein gH has to interact with gL, which lacks a membrane anchor, for correct processing and virion incorporation of both proteins (15, 16). However, the PrV, Murid herpesvirus 4 (MuHV-4), and Bovine herpesvirus 4 (BoHV-4) gH homologues are integrated into virions also in the absence of gL (17–19). Moreover, after serial passage, gL-deleted PrV mutants regained infectivity due to compensatory mutations within the gH gene (20). The gH/gL homologues of HSV-1 and EBV interact with cellular receptors like integrins, which might be relevant for attachment and/or fusion (21, 22). In a recent study, HSV-1 gB-expressing cells were exposed to soluble forms of gD and gH/gL, resulting in a low fusion activity (23). This indicated that gH/gL, as well as gD, may be required only for activation of the fusogenic properties of gB. However, in similar studies, soluble EBV gH/gL was not sufficient to induce gB-mediated fusion, but the complex had to be membrane anchored to support fusion (24). Besides their role during virus entry, the gH/gL and gB homologues of HSV-1 may also participate in nuclear egress of nascent virions (25), which, however, does not apply for the PrV homologues (26).
The recently determined crystal structures of soluble gH/gL heterodimers of HSV-2 (27) and EBV (28) and of a core fragment of PrV gH in complex with a monoclonal antibody (29) revealed no homologies to any known viral fusion protein. Furthermore, the structural analyses demonstrated that a previously identified lipid-interacting and fusogenic peptide sequence (30, 31) is not exposed at the surface of gH. However, despite limited sequence homology, the analyses showed markedly similar domain structures of the three gH homologues. The N-terminal domain I of HSV-2 and that of EBV gH exhibit the lowest homology but are both tightly bound to gL (27, 28). The construct used for crystallization of the core fragment of PrV gH did not include domain I (29) (Fig. 1A). The structurally more conserved domain II of all three gH homologues contains a sheet of antiparallel β-chains (fence), which separates the gL binding domain I from the downstream gH domains. Furthermore, gH domain II contains a highly conserved bundle of three α-helices, as well as an integrin-binding site identified in HSV-2 and EBV gH (27–29). Since the arrangement of the three α-helices exhibits structural similarities to the N-terminal domain of syntaxin 1A, they were designated “syntaxin-like-bundle” (SLB) (29) (Fig. 1A). Syntaxins belong to the family of eukaryotic SNARE proteins, which are involved in vesicle fusion processes and, for that purpose, have to undergo conformational changes (32). This structural similarity attracted our attention to possible conformational changes of the SLB during gH-mediated membrane fusion. Domain III of gH is less conserved but consists of eight consecutive α-helices in all analyzed proteins (Fig. 1A). The membrane-proximal domain IV represents the most highly conserved part of gH, implicating an important role for gH function. Domain IV consists of two four-stranded β-sheets, which are connected by an elongated basic polypeptide chain designated the “flap” (29) (Fig. 1A). It has been speculated that receptor binding of gH might induce a conformational change leading to movement of the flap. This would uncover a patch of hydrophobic amino acid residues, which then might interact with the viral envelope and alter membrane curvature to enable fusion (29). This hypothesis has been supported by targeted point mutations within domain IV of PrV gH, leading, e.g., to the loss of conserved disulfide bonds required for positioning of the flap or to introduction of artificial disulfide bonds, which should prevent flap movement (33). In contrast, studies on VZV gH indicated that structural stability rather than flexibility of domain IV might be relevant for function (34).
FIG 1.
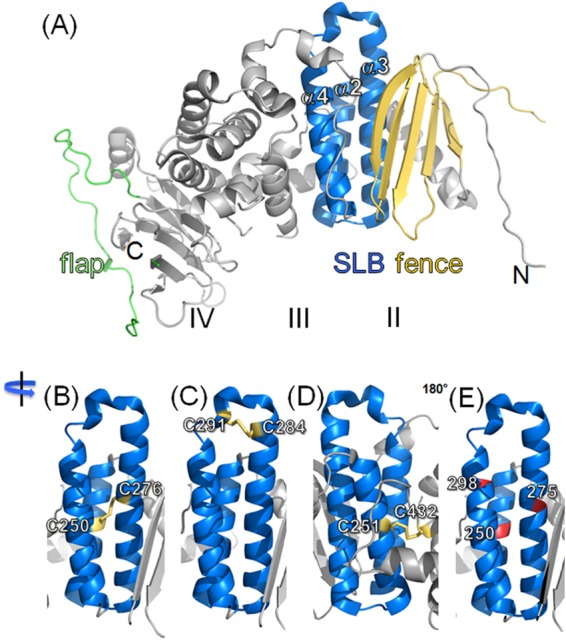
Structure of the PrV gH core fragment and design of the cysteine and proline mutants. (A) The structure of the crystallized core PrV gH is shown. Fence, SLB, and flap are highlighted in yellow, blue, and green, respectively. Domains are marked with roman numbers. Locations of the N and C termini are indicated. The SLB is magnified to illustrate the locations of engineered cysteine pairs and disulfide bonds, colored in yellow (B to D) in a selection of mutants showing interesting phenotypes in this study. (E) The positions of SLB residues mutated to proline are shown in red. The PrV gH core fragment structure is described in reference 29 and is available in the PDB under accession number 2XQY.
In the present study, structure-based mutational analyses were performed in domain II of PrV gH to elucidate its relevance for gH function. By site-directed mutagenesis of the cloned gH gene, spatially adjacent amino acids were changed into cysteines to allow disulfide bond formation, which should immobilize the three α-helices and prevent conformational changes within the SLB, or between fence, SLB, and domain III during the fusion process. To exclude that the observed phenotypic effects were caused by alteration of the amino acid sequence per se, the corresponding single-cysteine mutations and double mutations including one cysteine and one serine were also analyzed. Furthermore, proline residues were introduced to disrupt any of the three α-helices of the SLB. Expression plasmids containing the mutated gH genes were tested in combination with plasmids encoding gB, gD, and gL by in vitro fusion assays in cotransfected rabbit kidney cells. The influence of the disulfide-reducing agent dithiothreitol (DTT) on the fusogenic activity of gH mutants containing artificial cysteine pairs was also investigated. Furthermore, the altered gH genes were inserted into the PrV genome by mutagenesis of an infectious bacterial artificial chromosome (BAC) in Escherichia coli. The replication properties of the obtained virus mutants, including growth and penetration kinetics, as well as cell-to-cell spread, were analyzed.
MATERIALS AND METHODS
Viruses and cells.
All virus mutants described here were derived from the infectious BAC clone pPrV-ΔgGG (Fig. 2A) (33), which had been obtained by insertion of a mini-F plasmid vector, and an expression cassette for enhanced green fluorescent protein (EGFP) at the nonessential gG gene locus of PrV strain Kaplan (PrV-Ka) (35). Viruses were propagated in RK13 cells or in the gH- and gL-expressing cell line RK13-gH/gL (26). Cells were grown at 37°C in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) (Life Technologies).
FIG 2.

Construction of expression plasmids and virus mutants. (A) The PrV genome contains unique (UL, US) and inverted repeat (IR, TR) sequences. BamHI fragments and the positions of the gH and gG genes are indicated. The BAC pPrV-ΔgGG (33) contains an EGFP expression cassette and a mini-F plasmid at the gG locus. (B) Enlargement of the gH gene region with generated plasmids. Viral and vector sequences are drawn as bold or thin lines, respectively, and vertical lines indicate repetitive sequences. The ORFs encoding gH, viral thymidine kinase (TK), and kanamycin resistance (KanR), promoters (PgH, PHCMV), polyadenylation signals (A+), a viral replication origin (OriL), and FLP recombinase target sites (FRT), as well as relevant restriction sites are also shown. Sites reconstituted after ligation of noncompatible fragment ends (*) and generated (in parentheses) or deleted (crossed out) by targeted mutations are labeled. (C) The gH deletion mutants pPrV-ΔgHABKF and pPrV-ΔgHABF, as well as virus revertants expressing wild-type (pPrV-gHK) or mutated gH, were generated by BAC mutagenesis in E. coli (see the text).
Mutagenesis of the cloned PrV gH gene.
For subsequent insertion into the cloned PrV genome, expression plasmid pcDNA-gH (36) containing the gH gene (UL22) of PrV-Ka had to be modified. To this end, the genomic 311-bp BamHI fragment 16 of PrV-Ka containing the 3′-end of the gH gene was cloned into pUC19 (New England BioLabs). A kanamycin resistance (here referred to as KanR) gene was amplified by PCR (Pfx DNA polymerase; Life Technologies) from pACYC177 (New England BioLabs) using primers KanR-F and KanR-R (Table 1). After digestion with EcoRI and BamHI, the PCR product was inserted into a unique BsmFI site downstream of the polyadenylation signal (A+) of the gH mRNA, resulting in plasmids pUC-B16KA and pUC-B16KB (Fig. 2B). After double digestion of pcDNA-gH with HindIII and EcoRI followed by religation, a fragment of the resulting plasmid, pcDNA-gHEH (Fig. 2B), was removed by digestion with BamHI and BclI and replaced by the BamHI insert of pUC-B16KA, resulting in pcDNA-gHK. To obtain the final expression plasmid pcDNA-gHKDE (Fig. 2B), an artificial DrdI site was introduced (Quick-Change II XL site-directed mutagenesis kit; Agilent Technologies) using primers PGHK-DRDF and PGHK-DRDR (Table 1), and a unique EcoRI site was removed by digestion, Klenow treatment, and religation. Klenow treatment was also applied prior to ligation of noncompatible fragment ends in other cloning experiments.
TABLE 1.
Oligonucleotide primersa
| Primer use and name | DNA sequence, 5′ → 3′ | Nucleotide positions |
|---|---|---|
| Site-directed mutagenesisb | ||
| PGHV174C-F | CTCGGCCTGCGCTGCGCGCTGCGCTC | 61,117–61,142 |
| PGHA270C-F | CTACTTTTACCGCGCATGCGTGCGCCTCGGCGTGGC | 61,401–61,436 |
| PGHA252C-F | CGGCGGCGCACTGTGCCGCCCTGGCC | 61,352–61,377 |
| PGHA419C-F | GAGCTCACGCGGGCATGCCTCTCGCCGACGG | 61,849–61,879 |
| PGHH251C-F | GCTGTCGGCGGCATGCGCGGCCGCCC | 61,347–61,372 |
| PGHH251S-F | GCTGTCGGCGGCTAGCGCGGCCGCCC | 61,347–61,372 |
| PGHL432C-F | CGAGCCCTTCAGCTGCCTCGACGTCCTCTCG | 61,890–61920 |
| PGHA250C-F | GCCCAGCTGTCGGCATGCCACGCGGCCGCC | 61,342–61,371 |
| PGHA250S-F | CCAGCTGTCGGCTAGCCACGCGGCCGC | 61,344–61,370 |
| PGHA276C-F | CGCCTCGGCGTGTGCGCATTCGTCTTCTCCG | 61,423–61,453 |
| PGHA253C-F | CGGCGCACGCGTGCGCCCTGGCCGC | 61,355–61,379 |
| PGHA269C-F | CGTACTACTTTTACCGCTGCGCAGTGCGCCTCGGCG | 61,397–61,432 |
| PGHA284C-F | CGTCTTCTCCGAGGCATGCCGCCGCGACCGGC | 61,443–61,474 |
| PGHA284S-F | CTTCTCCGAGGCTAGCCGCCGCGACCG | 61,446–61,472 |
| PGHS291C-F | GACCGGCGCGCCTGTGCACCGGCGCTCC | 61,468–61,495 |
| PGHA250P-F | CAGCTGTCGGCGCCGCACGCGGCC | 61,345–61,368 |
| PGHV298P-F | CGGCGCCGGCGCTCCTGAGGCCTGAGAGCGACGCGCGCC | 61,481–61,519 |
| PGHV275P-F | GTGCGCCTCGGCCCGGCCGCCTTCG | 61,420–61,444 |
| PGHK-DRDF | GGGAGACCCAAGCGTCTTCAAAGTTTGCCGTGCCC | (pcDNA3) |
| PCR amplification and sequencing | ||
| PGH-PSF | TTCACGTCGGAGATGGGG | 60,451–60,468 |
| PGH-PSF2 | GGAAGCCCTTCGACCAG | 61,715–61,731 |
| PGH-PSR | AGTTATGTCATCCAGCAGCC | 62,887–62,868 (r) |
| PGH-PSR2 | GTCGAGCAGGCTGAAGG | 61,911–61,895 (r) |
| PGH-WH3 | TGCACGAGAGCGACGACTACC | 61,319–61,339 |
| PGHK-PSF | TGACGAGCGTAATGGCTGG | (pACYC177) |
| PGHK-PSR | CGCGAGCCCATTTATACCC | (pACYC177) |
| KanR-F | TCCGGATCCCGATTTATTCAACAAAGCCACG | (pACYC177) |
| KanR-R | TTCGAATTCGCCAGTGTTACAACCAATTAACC | (pACYC177) |
| T7 | TAATACGACTCACTATAGGG | (pcDNA3) |
| SP6 | CTCTAGCATTTAGGTGACACTATAG | (pcDNA3) |
Artificial restriction sites used for cloning or fragment isolation are underlined. Altered nucleotides are printed in bold letters. Nucleotide positions refer to GenBank accession number BK001744.
Forward primer sequences are shown for site-directed mutagenesis. The corresponding reverse primers were exactly complementary.
The site-directed mutagenesis kit and complementary pairs of oligonucleotide primers (Table 1) were further used to introduce point mutations in pcDNA-gHKDE, leading to amino acid substitutions within gH, and to create or delete restriction sites for convenient identification of the desired expression plasmids. The presence of the desired alterations and the absence of unwanted ones was verified by DNA sequencing of the complete gH-coding regions (results not shown).
Generation of gH mutants of PrV.
To facilitate the insertion of mutated gH genes into the PrV genome, a new BAC-based gH deletion mutant lacking a major part of UL22 (codons 51 to end) was generated. For that purpose, a 111-bp BamHI fragment of pUC-B16KB (Fig. 2B) containing PrV genome sequences from downstream of the gH gene was inserted into BamHI and BclI doubly digested pcDNA-gHEH (Fig. 2B). From the obtained plasmid, pcDNA-gHB16B (Fig. 2B), 1,828 bp was deleted by digestion with ApaI and BamHI and replaced by a 1,258-bp BstBI fragment of pKD13 containing a KanR gene flanked by FLP recombination target (FRT) sites (37). The insert fragment of the resulting plasmid, pcDNA-ΔgHABKF (Fig. 2B), was amplified by PCR with the T7 promoter primer (New England BioLabs) and PGH-PSR (33) and used for Red-mediated mutagenesis of pPrV-ΔgGG as described previously (33, 38). The KanR gene was removed from pPrV-ΔgHABKF (Fig. 2C) by FLP-mediated recombination after transformation of the bacteria with expression plasmid pCP20 (39), which enabled the reuse of kanamycin resistance for selection of future recombinants based on pPrV-ΔgHABF (Fig. 2C).
Modified PrV gH genes together with a downstream KanR gene were isolated from agarose gels after DrdI digestion of pcDNA-gHKDE derivates (Fig. 2B) and used for Red-mediated mutagenesis of pPrV-ΔgHABF. The desired BAC recombinants were selected on LB agar plates, supplemented with chloramphenicol (30 μg/ml) and kanamycin (50 μg/ml), by incubation at 37°C for 24 h. BAC DNA from overnight liquid cultures of obtained colonies was obtained by alkaline lysis, phenol-chloroform extraction, isopropanol precipitation, and RNase treatment and used for transfection of RK13 or RK13-gH/gL cells (FuGene HD transfection reagent, Promega). Single plaque isolates of virus progenies were further propagated and characterized by restriction analysis and Southern blot hybridization of viral DNA (results not shown), and the presence of the desired mutations was verified by sequencing of the PCR-amplified gH genes.
Western blot analyses.
RK13 cells were harvested 28 h after transfection using X-tremeGENE HP transfection reagent (Roche) with gH and, where indicated, gL expression plasmids, or 20 h after infection with wild-type or mutated PrV at a multiplicity of infection (MOI) of 5. Virions were purified from culture supernatants by sucrose gradient centrifugation (18). Protein samples (5 μg virion proteins or lysates of approximately 104 cells per lane) were separated by discontinuous sodium dodecyl sulfate (SDS)–10% polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and incubated with antibodies as described previously (40). Monospecific rabbit antisera against PrV gH, gB, and pUL34 (20, 38, 41) were used at a dilution of 1:10,000 (anti-gH) or 1:50,000 (gB and pUL34).
IF analyses.
For indirect immunofluorescence (IF) analyses, RK13 cell monolayers were transfected (X-tremeGENE HP transfection reagent; Roche) with gH expression plasmids and fixed 26 h later with 3% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 30 min. Optionally, the fixed cells were subsequently permeabilized by incubation with 3% PFA containing 0.2% Triton X-100 for 15 min. After washing with PBS, the cells were incubated with 10% FBS in PBS and with a gH-specific rabbit antiserum (20) (1:100 in PBS with 10% FBS) for 1 h each. Bound antibodies were detected by incubation with rabbit-specific Alexa Fluor 488-conjugated secondary antibodies (Life Technologies) (1:1,000 in PBS) for 30 min. After each step, the cells were washed repeatedly with PBS and analyzed by fluorescence microscopy (Eclipse Ti; Nikon).
In vitro fusion assays.
RK13 cells were grown in 12-well plates and cotransfected (X-tremeGene HP transfection reagent; Roche) with equal amounts (200 ng each) of eukaryotic expression plasmids for C-terminally truncated PrV gB (pgB-008) (42), PrV gD (gDgI-CMV) (43), PrV gL (pRc/CMV-gL) (36), native or mutated PrV gH (pcDNA-gHKDE or derivatives), and EGFP (pEGFP-N1; Clontech) to facilitate detection of syncytia. In negative controls, the empty expression vector pcDNA3 (Life Technologies) was used instead of a gH expression plasmid. After 36 h, the cells were fixed with 3% PFA in PBS for 30 min, washed repeatedly with PBS, and analyzed by fluorescence microscopy (Eclipse Ti; Nikon). For each gH expression construct, the areas of 50 randomly selected syncytia containing 3 or more nuclei in random sight fields were measured. If no or only very few syncytia were detectable, areas of single fluorescent cells were added. Mean areas and standard deviations were calculated, and the statistical significance of differences was evaluated by Student's t tests.
In vitro fusion assays under reducing conditions.
RK13 cells grown in 12-well plates were cotransfected with expression plasmids for PrV gB-008, gD, gH, gL, and EGFP. After 18 h, the cells were incubated for 5 min at 37°C with medium containing 24 mM DTT, washed, and further incubated in MEM supplemented with 10% FBS. After an additional 48 h, DTT-treated and untreated control cells were fixed with 3% PFA and analyzed as above.
Plaque assays.
For determination of virus titers and plaque sizes, RK13 or RK13-gH/gL cell monolayers in 6-well plates were infected with serial dilutions of virus supernatants. After 2 h at 37°C, the inoculum was replaced by semisolid MEM containing 5% FBS and 6 g/liter methylcellulose, and incubation was continued for 48 h at 37°C. The areas of 30 plaques or foci of infected cells per virus mutant were measured by fluorescence microscopy, and mean sizes as well as standard deviations were calculated as percentages of the plaque areas induced by the mutant pPrV-gHK expressing wild-type gH. Student's t test was used for estimation of the statistical significance of differences.
Replication kinetics.
Confluent monolayers of RK13 cells in 24-well plates were infected with the PrV mutants at an MOI of 0.1 and consecutively incubated on ice and at 37°C for 1 h each. Subsequently, the inoculum was removed, nonpenetrated virus was inactivated by low-pH treatment (44), and fresh medium was added. Immediately thereafter, and after 8, 12, 16, 24, 36, and 48 h at 37°C, the cells were harvested together with the supernatants and lysed by freeze-thawing (−70°C and 37°C). Progeny virus titers were determined by plaque assays on RK13 cells or RK13-gH/gL cells (proline and quadruple mutants), and mean results from three independent experiments per virus were calculated.
Penetration kinetics.
Confluent monolayers of RK13 cells grown in 6-well plates were infected on ice with approximately 500 PFU of the investigated PrV mutants per well. After 1 h, prewarmed medium was added and incubation was continued at 37°C. Before and 5, 10, 20, or 40 min after the temperature shift, nonpenetrated virus was inactivated by acid treatment, and cells were overlaid with semisolid medium. After 48 h at 37°C, the plaques were counted by fluorescence microscopy and compared to the numbers obtained in wells that had not been acid treated. Mean penetration rates from three independent experiments were determined.
RESULTS
Effects of the mutations in gH domain II on protein maturation and localization.
To study the functional relevance of the SLB during membrane fusion, each of the three SLB helices was disrupted by introduction of a single proline residue at position 250, 275, or 298 (Fig. 1 and Table 2). Furthermore, one or two pairs of amino acids in different helices of the SLB and/or in structurally proximal regions of the fence (domain II) or of domain III were replaced by cysteine residues to induce the formation of artificial disulfide bonds that might inhibit structural changes of the SLB during the fusion process (Fig. 1; Table 2). The distances of the introduced cysteine residues were calculated to be between 3.19 and 4.46 Å. The corresponding single amino acid mutations and serine-cysteine double mutations were used as controls. To investigate expression and processing of the analyzed gH variants, lysates of RK13 cells transfected with gH expression plasmid pcDNA-gHKDE (Fig. 2B) or mutagenized derivatives of it were analyzed by Western blotting. These studies revealed wild-type-like expression levels of all gH mutants. However, processing of the proline mutants (A250P, V275P, V298P), of the single cysteine mutant H251C, and of the double mutants A250C-A276C and H251C-L432C, as well as of the quadruple mutants A284C-S291C/A250C-A276C and V174C-A270C/H251C-L432C was apparently impaired, as indicated by a low or even undetectable level of mature, ca. 90-kDa gH (Table 2). Representative Western blots with selected mutants are shown in Fig. 3A. Although it has been demonstrated that in PrV gL is not essential for maturation and virion incorporation of gH (18), lysates of cells cotransfected with gH and gL expression plasmids were also analyzed (Fig. 3B). The presence of gL enhanced the processing efficiency of wild-type and mutated gH, but the impaired mutants still exhibited decreased amounts (H251C, A250C-A276C) or no detectable amounts (H251C-L432C) of mature gH. These findings correlated with reduced surface localization of the corresponding gH mutants as observed in IF analyses of nonpermeabilized RK13 cells (Fig. 4, upper panels; Table 2), whereas IF analyses of permeabilized cells confirmed similar expression levels of all gH mutants in the cytoplasm (Fig. 4, lower panels). Coexpression with gL did not substantially enhance surface localization of wild-type or mutant gH (not shown). However, confocal double-IF investigations revealed that even the gH mutants exhibiting impaired processing and cell surface expression were at least partly transported to the Golgi apparatus (not shown), which is a prerequisite for incorporation into virions.
TABLE 2.
Summary of resultsa
| gH mutation(s) | Localization |
In vitro studies |
In vivo studies |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Processing (WB) | Surface expression (IF) | Expression (IF) | Fusion activity | Virion incorporation | Virus titers | Plaque sizes | Penetration | ||
| V174C | Fence | + | ++ | +++ | +++ | +++ | +++ | +++ | +++ |
| A270C | SLB | + | ++ | ++ | +++ | +++ | +++ | +++ | +++ |
| V174C-A270C | Fence-SLB | + | ++ | +++ | ++ | +++ | +++ | +++ | +++ |
| A252C | SLB | + | ++ | +++ | +++ | +++ | +++ | +++ | +++ |
| A419C | Domain III | + | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| A252C-A419C | SLB-domain III | + | +++ | +++ | ++ | +++ | +++ | +++ | +++ |
| H251C | SLB | − | + | +++ | ++ | +++ | +++ | +++ | +++ |
| L432C | Domain III | + | +++ | ++ | +++ | +++ | +++ | +++ | +++ |
| H251C-L432C | SLB-domain III | − | + | +++ | + | +/− | +++ | + | + |
| H251S-L432C | SLB-domain III | − | + | +++ | ++ | +++ | +++ | +++ | +++ |
| A250C | SLB | + | ++ | +++ | +++ | +++ | +++ | +++ | +++ |
| A276C | SLB | + | +++ | ++ | ++ | +++ | +++ | +++ | +++ |
| A250C-A276C | SLB-SLB | − | + | ++ | ++ | ++ | +++ | +++ | ++ |
| A250S-A276C | SLB-SLB | − | + | +++ | ++ | +++ | +++ | +++ | +++ |
| A253C | SLB | + | +++ | ++ | +++ | +++ | +++ | +++ | +++ |
| A269C | SLB | + | +++ | +++ | ++++ | +++ | +++ | +++ | +++ |
| A253C-A269C | SLB-SLB | + | +++ | +++ | ++++ | +++ | +++ | ++++ | +++ |
| A284C | SLB | + | +++ | +++ | +++ | +++ | +++ | +++ | +++ |
| S291C | SLB | + | +++ | ++ | +++ | +++ | +++ | +++ | +++ |
| A284C-S291C | SLB-SLB | + | ++ | +++ | + | ++ | +++ | + | + |
| A284S-S291C | SLB-SLB | + | ++ | +++ | ++ | +++ | +++ | +++ | +++ |
| A284C-S291C | SLB-SLB | − | + | +++ | − | NT | ++ | + | NT |
| A250C-A276C | SLB-SLB | ||||||||
| V174C-A270C | Fence-SLB | − | + | +++ | − | NT | + | + | NT |
| H251C-L432C | SLB-domain III | ||||||||
| V174C-A270C | Fence-SLB | + | +++ | ++ | ++ | NT | +++ | ++ | NT |
| A252C-A419C | SLB-domain III | ||||||||
| A284C-S291C | SLB-SLB | + | +++ | ++ | ++ | NT | +++ | ++ | NT |
| A253C-A269C | SLB-SLB | ||||||||
| A250P | SLB α2 | − | + | ++ | − | NT | + | + | NT |
| V275P | SLB α3 | − | + | +++ | − | NT | + | + | NT |
| V298P | SLB α4 | − | + | +++ | − | NT | ++ | + | NT |
| gH-WT, pPrV-gHK | + | +++ | ++ | +++ | ++ | +++ | +++ | +++ | |
| No gH, pPrV-ΔgHABF | − | − | − | − | − | NT | |||
Properties and functions of the mutated gH proteins and of the corresponding PrV recombinants were roughly assessed (+ to ++++). −, impaired gH processing, no expression, no in vitro fusion activity, or no detectable productive replication or cell-to-cell spread of virus mutants. Mutations conferring interesting phenotypes (described in the text) are highlighted in boldface. NT, not tested.
FIG 3.

Western blot analyses. Lysates of RK13 cells transfected with wild-type (WT) or mutated gH expression plasmids (A) or cotransfected with gH and gL expression plasmid (B) were separated by SDS-PAGE. Gels were incubated with a gH-specific rabbit antiserum. The signals of immature (gHim) and mature gH are labeled by arrows, and molecular masses of marker proteins are indicated.
FIG 4.
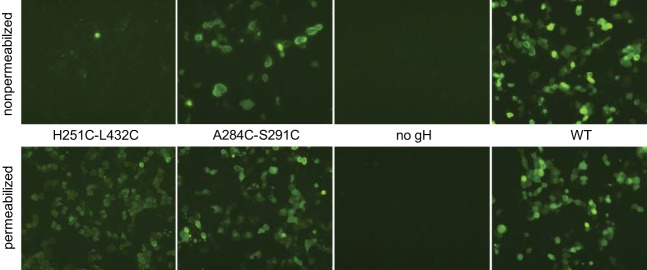
Intracellular localization of gH. RK13 cells were transfected with gH expression plasmids and analyzed by indirect immunofluorescence. Two days after transfection, the cells were fixed with 3% paraformaldehyde (upper panels) and additionally permeabilized with 0.2% Triton X-100 (lower panels). Surface-localized and intracellular gH was detected using a monospecific rabbit antiserum and Alexa Fluor 488-conjugated secondary antibodies.
In vitro fusion activity of mutated gH.
Previous studies demonstrated that transient expression of PrV gB and gH/gL induces the formation of syncytia in plasmid-transfected RK13 cells and that fusion efficiency is enhanced by additional expression of gD (36). Furthermore, it has been shown that C-terminally truncated gB (gB-008) accumulates at the cell surface and exhibits higher in vitro fusion activity than native gB (42). Therefore, all mutated gH expression plasmids were tested in RK13 cells by cotransfection with plasmids encoding wild-type gD, gL, gB-008, and EGFP for convenient detection and evaluation of transfection efficiencies. A plasmid encoding wild-type gH (pcDNA-pGHKDE) and the empty expression vector pcDNA3 were used as positive and negative controls, respectively. Two days after cotransfection, the numbers of syncytia, the numbers of nuclei (not shown), and the areas of polykaryocytes (Fig. 5) were analyzed. Whereas none of the gH mutants containing an artificial proline in one of the SLB α-helices supported cell fusion, most of the single and double cysteine or serine mutations did not impair fusion activity or did so only slightly (Fig. 5 and Table 2). However, the double mutation A250C-A276C moderately affected fusion, and the double mutations H251C-L432C and A284C-S291C led to substantially reduced numbers and sizes of syncytia compared to wild-type gH, as well as to the corresponding single mutations and the serine-cysteine double mutations (Fig. 5), suggesting that gH function was impaired by the formation of the additional disulfide bonds. This was supported by restoration of fusion activity when the cells were briefly exposed to reducing conditions (Fig. 6). Although treatment of cells with DTT slightly reduced fusion activity of wild-type gH, it enhanced fusion activity of gH A250C-A276C, gH H251C-L432C, and to an even greater extent, of gH A284C-S291C but had no effect on the negative control (Fig. 6). As expected, all of the quadruple gH mutants containing one of these cysteine pairs also exhibited severely impaired cell fusion. However, drastic impairment of fusion was also found for a mutant containing two cysteine pairs (V174C-A270C and A252C-A419C), which were rather inconspicuous when introduced separately (Fig. 5 and Table 2). In contrast, the quadruple gH mutant A284C-S291C/A253C-A269C showed slightly less impaired fusion activity than the double mutant A284C-S291C (Fig. 5).
FIG 5.

In vitro fusion assays. RK13 cells were cotransfected with expression plasmids for PrV gB-008, gD, gL, selected gH variants, and EGFP. After 36 h, the areas of induced syncytia were determined by fluorescence microscopy. Mean sizes of 50 syncytia per mutant were calculated, and the percentages of mean wild-type sizes were plotted. Standard deviations (vertical lines) and significant differences from wild-type fusion activity are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
FIG 6.

In vitro fusion assays under reducing conditions. RK13 cells were cotransfected with expression plasmids for PrV gB-008, gD, gL, wild-type (WT) or mutated gH, and EGFP. After 18 h, the indicated samples were incubated with medium containing 24 mM DTT for 5 min. Forty-eight hours later, treated and untreated cells were analyzed by fluorescence microscopy. Areas of 50 syncytia per variant were measured, and the percentages of mean wild-type sizes were calculated. Standard deviations (vertical lines) and significant differences from untreated controls are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Effects of gH mutations on virus replication.
To investigate the influence of the mutations on virus entry into host cells, production of infectious progeny, and direct viral cell-to-cell spread, all mutated gH genes were introduced into the cloned PrV genome by mutagenesis in E. coli. This was facilitated by construction of the new BAC mutant pPrV-ΔgHABF, which lacks almost the complete UL22 open reading frame (ORF) encoding gH (Fig. 2C), and of a novel transfer plasmid, pcDNA-gHKDE, which allows reinsertion of the gH gene together with a downstream kanamycin resistance gene for selection. As expected, the BAC pPrV-ΔgHABF did not replicate in normal RK13 cells (Fig. 7), but infectious virus progeny was obtained after transfection of RK13-gH/gL cells (results not shown). After reinsertion of the wild-type gH gene, plaque sizes and maximum titers of the obtained revertant pPrV-gHK on RK13 cells were identical to those of the parental clone pPrV-ΔgGG (Fig. 7). Thus, integrity of the noncoding, tandem-repeat-containing genome sequences between the polyadenylation site of UL22 and the replication origin ORIL (Fig. 2) is obviously not critical for replication of PrV.
FIG 7.

Final titers and plaque sizes of PrV mutants. (A) RK13 cells were infected with pPrV-gHK, pPrV-ΔgGG, pPrV-ΔgHABF, or virus recombinants containing the indicated gH mutations. Progeny virus titers were determined 48 h after infection at an MOI of 0.1 by plaque assays on RK13 or RK13-gH/gL cells. Shown are the mean results of three experiments and standard deviations. (B) Areas of 30 plaques or foci of infected RK13 cells per virus were determined 48 h after infection by fluorescence microscopy, and mean sizes were plotted as percentages of wild-type (pPrV-gHK) sizes. Standard deviations (vertical lines) and significant size reductions (*, P < 0.05; **, P < 0.01; ***, P < 0.001) are indicated.
Replication studies of most of the PrV mutants containing cysteine mutations within gH revealed only minor differences to the parental pPrV-ΔgGG and wild-type revertant pPrV-gHK, with maximum final titers of 107 to 108 PFU/ml on RK13 cells (Fig. 7A). Remarkably, this also applied to several mutants that exhibited impaired gH processing in plasmid-transfected cells, e.g., pPrV-gH H251C, pPrV-gH H251C-L432C, and pPrV-gH HA250C-A276C. Only the quadruple mutants pPrV-gH V174C-A270C/H251C-L432C and pPrV-gH A284C-S291C/A250C-A276C, as well as the three proline mutants pPrV-gH A250P, pPrV-gH V275P, and pPrV-gH V298P showed significantly reduced virus titers (Fig. 7A) and had to be propagated on complementing RK13-gH/gL cells before and after determination of replication in RK13 cells.
However, unlike the deletion mutant pPrV-ΔgHABF, all gH mutants generated in this study exhibited at least limited replication competence in noncomplementing cells. On the other hand, the proline insertions within the SLB were rather unstable and were reproducibly eliminated or compensated by second-site mutations after several passages on RK13 cells (results not shown).
Effects of gH mutations on cell-to-cell spread.
The influence of the introduced gH mutations on cell-to-cell spread of PrV in RK13 cells was generally congruent with the effects on cell-cell fusion and productive virus replication. In plaque assays, only single or small foci of GFP-expressing cells were observed 2 days after infection with any of the three proline mutants (Fig. 7B). The quadruple cysteine mutants pPrV-gH V174C-A270C/H251C-L432C and pPrV-gH A284C-S291C/A250C-A276C also exhibited drastically reduced plaque sizes compared to any of the corresponding single mutants (Fig. 7B). However, introduction of one of the cysteine pairs present in either of these gH mutants (H251C-L432C or A284C-S291C) also affected cell-to-cell spread of PrV significantly (Fig. 7B), indicating that the intended disulfide bonds between these residues are responsible for impaired gH function. This was supported by the observation that corresponding serine-cysteine double mutations in PrV-gH H251S-L432C or PrV-gH A284S-S291C did not affect plaque formation on RK13 cells, whereas plaque sizes of PrV-gH H251C-L432C or PrV-gH A284C-S291C were reduced to 18% or 30% of wild-type levels, respectively (Fig. 7B). Interestingly, the quadruple mutant pPrV-gH A284C-S291C/A253C-A269C exhibited a slightly less pronounced reduction, to 45% of wild-type plaque sizes, indicating that the defect of pPrV-gH A284C-S291C might have been partly compensated by the additionally introduced cysteine pair (Fig. 7B). Plaque formation of these or any others of the investigated gH mutants on transcomplementing RK13-gH/gL cells was similar to that of wild-type PrV (not shown).
Effects of gH mutations on virus entry.
The time course and efficiency of virus entry were investigated by penetration studies on RK13 cells (Fig. 8). These experiments revealed that none of the single cysteine insertions in gH affected virus internalization significantly, and most of the double mutants also showed wild-type-like penetration kinetics. However, entry of double mutants pPrV-gH A250C-A276C, pPrV-gH H251C-L432C, and pPrV-gH A284C-S291C was significantly delayed (Fig. 8). After 40 min, only 20% of the infectious particles of pPrV-gH H251C-L432C and less than 10% of those of pPrV-gH A284C-S291C were internalized (Fig. 8), and after 3 h the proportions of acid-resistant input virus were still <50% (results not shown). In contrast, the double mutants PrV-gH H251S-L432C and PrV-gH A284S-S291C exhibited wild-type-like entry kinetics (Fig. 8). Penetration of the PrV recombinants containing quadruple cysteine insertions or the single proline mutations was not analyzed since plaque formation of most of these viruses on RK13 cells was severely impaired (Fig. 7B).
FIG 8.
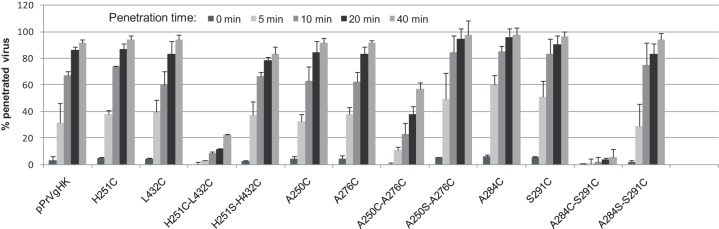
Penetration kinetics of PrV mutants. RK13 cells were infected with approximately 500 PFU pPrV-gHK or virus recombinants containing the indicated gH mutations. After synchronized adsorption at 4°C, the cells were incubated for 0, 5, 10, 20, or 40 min at 37°C prior to acid inactivation of nonpenetrated virus. Numbers of plaques after 2 days were compared to those obtained without inactivation. Mean percentages of results from three independent experiments and standard deviations are shown.
Virion incorporation of mutated gH.
To investigate the influence of the introduced mutations on gH processing and incorporation into virus particles, virions were purified from supernatants of infected RK13 cells and analyzed by Western blotting (Fig. 9; Table 2). Unlike in cells transfected with gH expression plasmids (Fig. 3), only the mature, >90-kDa form of gH was detectable in virus particles of either of the investigated gH mutants, and most of them contained wild-type-like amounts of the protein. However, virions of pPrV-gH H251C-L432C contained considerably less gH, and moderately reduced gH incorporation was also observed for the PrV recombinants containing the double cysteine mutation A250C-A276C (Fig. 9, upper panel; Table 2). In contrast, incorporation of gB was not affected (Fig. 9, middle panel). However, as expected, the amounts of gL, which requires its complex partner gH for virion localization (18), correlated with those of gH (Fig. 9, lower panel). The purity of the virion preparations was confirmed by the absence of the primary envelope protein pUL34 (not shown). Reduced gH/gL incorporation into virions matched the decreased penetration kinetics and cell-to-cell spread of gH H251C-L432C and A250C-A276C mutants, as well as impaired processing after transient gH expression (Fig. 3). However, pPrV-gH A284C-S291C, although exhibiting almost wild-type-like processing and virion incorporation of gH/gL, showed even more pronounced replication defects than the other double-cysteine mutants (Fig. 7B and 8). Because of the inefficient replication of the PrV gH mutants containing proline or quadruple cysteine insertions in RK13 cells (Fig. 7A), virion preparations were not attempted.
FIG 9.
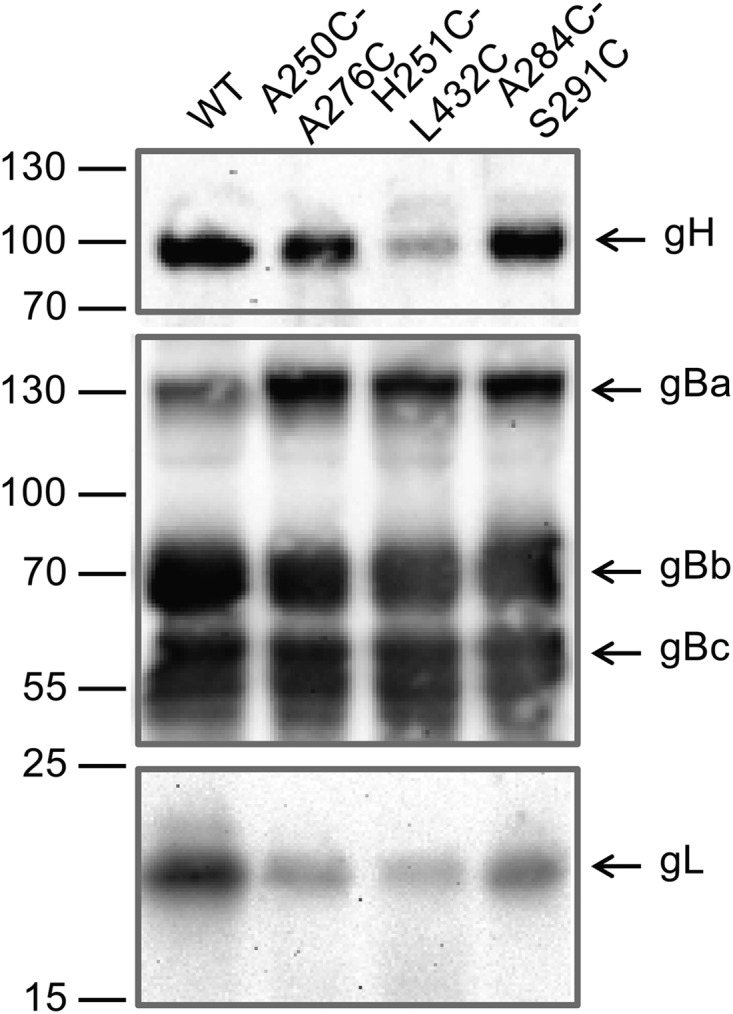
Western blot analyses of purified PrV particles. Virion proteins of pPrV-gHK (WT) and the indicated gH mutants were separated by SDS-PAGE. Blots were incubated with gH-, gB- or gL-specific rabbit antisera. The signals of mature gH, full-length gB (gBa), and furin-cleaved subunits (gBb, gBc) as well as gL are labeled by arrows, and molecular masses of marker proteins are indicated.
DISCUSSION
The crystal structures of HSV-2, EBV, and PrV gH have recently been determined and revealed similar domain architectures (27–29). In previous work, we analyzed the role of domain IV in function of PrV gH during fusion by site-directed mutagenesis (33). The present study focuses on domain II, containing a conserved syntaxin-like bundle of α-helices, which was suggested to be important for the role of gH during membrane fusion (29). To test this hypothesis, we introduced targeted mutations into the gH gene of PrV, which should either disrupt the helices of the SLB by engineered proline residues (Fig. 1E) or covalently join the helices to each other, to the adjacent fence region, or to domain III by intramolecular artificial disulfide bridges. To trigger their formation, pairs of cysteine residues were introduced at structurally proximal positions (Fig. 1B to D). The effects of the expected structural changes in gH or of the loss of flexibility on protein maturation and transport and particularly on function in membrane fusion were investigated using transient expression systems as well as PrV recombinants.
Several of the introduced mutations affected the maturation and transport of transiently expressed gH (Table 2). Proteins containing either of the three proline insertions in the SLB but also mutations including a cysteine at position 251 or a cysteine pair at positions 250 and 276 were barely detectable at the surface of transfected cells, and in Western blot analyses incompletely glycosylated gH was predominantly or exclusively found. However, to some extent all mutated proteins were translocated to the Golgi apparatus as demonstrated by colocalization studies (data not shown). As expected, reduced surface expression of mutated gH correlated with impaired in vitro fusion activity. Several of the gH mutations affecting processing and transport in transfected cells also led to reduced gH/gL incorporation into virions of corresponding PrV recombinants (Table 2). A remarkable exception was the single cysteine mutation H251C, which, unlike double and quadruple mutations including this amino acid substitution, did not detectably reduce the amount of mature gH in purified virus particles. Furthermore, the in vitro fusion activity of gH H251C was much less affected than those of the corresponding double and quadruple mutants, although the glycosylation and transport defects of the transiently expressed proteins appeared to be similar. Similarly, the doubly mutated gH A250C-A276C was barely detectable at the surface of cells transfected with the corresponding expression plasmid but exhibited considerable fusion activity after cotransfection with plasmids encoding gB, gD, and gL. These discrepancies might be explained by contributions of other viral proteins, in particular of the complex partner gL, to processing of PrV gH in infected or cotransfected cells. Although PrV gL is not absolutely required for maturation and virion incorporation of gH (18), essential roles of gL during these steps have been described for other herpesviruses like HSV-1 (15, 16, 46, 47) and VZV (48). A beneficial effect of gL coexpression on processing of PrV gH including mutants like gH A250C-A276C was confirmed in the present study (Fig. 3).
gL has been shown to be closely associated with the N-terminal part of gH (27, 28), which is separated from the SLB by a β-sheet designated “fence” (29). Therefore, it is unlikely that conformational alterations caused, e.g., by disruption of the SLB α-helices by proline insertions or by introduction of artificial disulfide bonds, inhibit interaction with gL. In line with this assumption, our studies revealed that although processing, cell surface localization, and virion incorporation of gH were severely affected by the double mutation H251C-L432C, particles of the corresponding virus mutant still contained the expected amounts of gL. Thus, the correct structure of the SLB per se appears to be relevant for processing, transport, and virion incorporation of gH. Whereas the level of maturation and, consequently, surface exposure of gH is important, it is not the only factor responsible for the fusion activity. This is particularly evident in the comparison between gH H251C and H251C-L432C mutants. Both exhibit maturation defects and strikingly decreased surface localization also when coexpressed with gL, but they demonstrate significantly different fusion activities. On the other hand, the A284C-S291C mutant, which is processed and localized normally, also exhibits strongly reduced fusion activity (Table 2 and Fig. 5). In contrast, neither the corresponding single mutation A284C or S291C nor the double mutation A284S-S291C affected fusion activity of gH significantly, indicating that the desired disulfide bond was indeed formed.
Formation of disulfide bonds was confirmed by transient addition of reducing agents to transfected cell cultures, as has been shown for partial restoration of function of the paramyxovirus virus fusion protein engineered to contain an artificial disulfide bond (49). In a similar approach, we demonstrate that in vitro fusion activity of gH A284C-S291C, but also of gH H251C-L432C and gH A250C-A276C, was partly restored after addition of 24 mM DTT for 5 min. Thus, the function of gH H251C-L432C was apparently affected not only by the processing defect caused by the artificial cysteine in the SLB (H251C) but additionally by an artificial disulfide bond between SLB and domain III (Fig. 1). The beneficial effects of DTT treatment were somewhat surprising, since native disulfide bonds have been shown to be relevant for conformation and function of gH (29, 33) but also of other components of the viral fusion complex like gB (50). Possibly, these disulfide bonds were only partially reduced by the brief DTT treatment. Furthermore, the treatment is able to affect only disulfide bonds in proteins already located at the cell surface, which are eventually replaced by newly synthesized proteins.
Except for A284C-S291C and H251C-L432C, the cysteine pairs engineered in the present study had no or only moderate effects on in vitro fusion activity of gH, and therefore, fusion assays under reducing conditions could not be used to determine whether or not the desired artificial disulfide bonds were formed at all. Possibly, structure determination of the disulfide mutants or comparative mass-spectrometric analyses of proteolytically cleaved, reduced, or nonreduced proteins (51, 52) would be able to answer these questions, but suitable protocols remain to be established for PrV gH.
The generation of infectious, recombinant PrV genomes containing mutated gH genes by BAC mutagenesis in E. coli allowed us to demonstrate that none of the single or multiple amino acid substitutions introduced in the present study completely abolished gH function in the viral context. However, the quadruple cysteine mutations of pPrV-gH V174C-A270C/H251C-L432C and pPrV-gH A284C-S291C/A250C-A276C, as well as either of the three proline insertions in the SLB, inhibited cell-to-cell spread almost completely and reduced infectious progeny virus titers on noncomplementing cells more than 1,000-fold compared to similar PrV recombinants expressing wild-type gH (Fig. 7). The severity of the defects induced by disruption of the α-helices of the SLB was emphasized by reversions of the artificial proline residues to other amino acids or by compensatory mutations at adjacent positions, which occurred after limited cell culture passages of the virus recombinants. Until now, the low titers and spontaneous reversions of the proline mutations hindered elucidation of whether the observed phenotype was due to malfunction or inefficient virion incorporation of gH.
Maximum titers of the other PrV recombinants created in this study were similar to those of wild-type virus, and only their plaque sizes exhibited a clear correlation with the observed in vitro fusion activities of the corresponding gH mutants (Fig. 5 and 7B). Thus, gH double mutations A284C-S291C and H251C-L432C led to inefficient in vitro cell fusion as well as to inefficient cell-to-cell spread of the virus, confirming that the two processes depend on similar mechanisms leading to general or local fusions of adjacent cell membranes (23, 53–55). Furthermore, the observed correlation of results demonstrates that use of the C-terminally truncated gB-008 (44) in transient fusion assays reflects the activity of full-length gB during virus infection.
The role of gH during fusion of viral envelopes with host cell membranes was investigated by analyses of penetration kinetics. In these experiments, the virus recombinants containing artificial disulfide bonds between mutated gH amino acids H251C and L432C or A284C and S291C again exhibited the most pronounced defects, and a less, but still considerably delayed penetration was further observed for the PrV mutant containing gH A250C-A276C, which had been less conspicuous in transient fusion assays (Fig. 5 and 8). Thus, immobilization of the SLB resulted in a delay in penetration, i.e., virus-cell fusion, without necessarily influencing cell-cell fusion. This could be due to alterations in interaction with gD. PrV gD is required for entry but not for plaque formation (56, 57), functionally separating the two membrane fusion events. Impaired interactions with gD as demonstrated for HSV-1 (58) may have a more pronounced effect on PrV penetration than on fusion and direct viral cell-cell spread.
Like in the plaque assays, none of the single cysteine mutations of gH affected virus penetration significantly, confirming that artificial disulfides were most likely responsible for the observed phenotypes. As mentioned above, mutations A250C-A276C as well H251C-L432C led to impaired processing and, consequently, inefficient virion incorporation of gH (Table 2). Therefore, delayed penetration might be partly due to small amounts of gH in the viral envelopes. However, the artificial disulfide bond between mutated SLB residues 284 and 291 did not severely inhibit virion localization of gH (Fig. 9 and Table 2) but caused the most pronounced penetration defects detected in our present studies. Nevertheless, it remains to be elucidated whether steric arrest of the SLB affects the supposed role of gH as a gB activator (46) or, although not inhibiting complex formation, affects functionally relevant interactions between gH and gL (59). Remarkably, as indicated by investigation of long-term penetration kinetics (results not shown), penetration of the three PrV recombinants containing artificial disulfides within the SLB of gH or between SLB and domain III (H251C-L432C) was delayed by several hours but not blocked. However, this delay is apparently compensated by the rapid replication of PrV and is not reflected in final titers even at rather low infection, with an MOI of 0.1. In contrast, all gH proline insertion mutants as well as the quadruple cysteine mutant gH V174C-A270C/H251C-L432C show a strong impairment of viral replication, although titers of viral progeny are noticeably higher than in complete absence of gH. Thus, all gH variants retain a certain level of function. Considering also the rather moderate effects of most mutations introduced into the more highly conserved domain IV of PrV gH (33), it is surprising how robust gH function is maintained even in the face of strategically introduced, as opposed to random, mutations.
In summary, our present work demonstrates the relevance of the syntaxin-like bundle of α-helices within domain II for maturation and virion incorporation of gH, as well as directly for gH function in membrane fusion. The efficiency of these processes obviously depends on the integrity of the three helices, as shown by proline insertions, and on a certain degree of flexibility between them, as indicated by the deleterious effects of artificial disulfide bonds linking amino acid residues 250 and 276 or 284 and 291 (Fig. 1B and C). The effects of an engineered cysteine pair at gH positions 251 and 432 indicate that flexibility between the SLB and the α-helical domain III is also required (Fig. 1D), whereas covalent binding of the SLB to the β-sheet of the fence (V174C-A270C) had only minor effects on gH function. However, it has still not been proven that this disulfide bond is indeed formed, which also applies for several phenotypically innocuous mutants containing cysteine pairs within the SLB or between SLB and domain III (Table 2). In future studies, we will attempt to clarify this and also address the open question as to whether the supposed structural changes of the SLB of gH during membrane fusion are directly required to bring the involved cellular and viral membranes in close proximity or for interaction with other viral or cellular proteins involved in fusion. However, our data clearly demonstrate the importance of the flexibility and structural integrity of the flap region in domain IV (33) as well as the SLB in domain II (this report) for gH function.
ACKNOWLEDGMENTS
These studies were supported by the Deutsche Forschungsgemeinschaft (DFG grant Me 854/11-1).
We thank B. Wanner for providing plasmids for BAC mutagenesis, G. Strebelow for performing sequence analyses, and M. Lenk for help with cell cultures. The technical assistance of C. Ehrlich is greatly appreciated.
REFERENCES
- 1.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spear PG, Longnecker R. 2003. Herpesvirus entry: an update. J Virol 77:10179–10185. doi: 10.1128/JVI.77.19.10179-10185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roizman B KD, Whitney RJ. 2007. Fields virology, 5th ed, p 2501–2601 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Herold BC, WuDunn D, Soltys N, Spear PG. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J Virol 65:1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karger A, Saalmuller A, Tufaro F, Banfield BW, Mettenleiter TC. 1995. Cell surface proteoglycans are not essential for infection by pseudorabies virus. J Virol 69:3482–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 7.Cocchi F, Fusco D, Menotti L, Gianni T, Eisenberg RJ, Cohen GH, Campadelli-Fiume G. 2004. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc Natl Acad Sci U S A 101:7445–7450. doi: 10.1073/pnas.0401883101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt J, Gerdts V, Beyer J, Klupp BG, Mettenleiter TC. 2001. Glycoprotein D-independent infectivity of pseudorabies virus results in an alteration of in vivo host range and correlates with mutations in glycoproteins B and H. J Virol 75:10054–10064. doi: 10.1128/JVI.75.21.10054-10064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schröder C, Keil GM. 1999. Bovine herpesvirus 1 requires glycoprotein H for infectivity and direct spreading and glycoproteins gH(W450) and gB for glycoprotein D-independent cell-to-cell spread. J Gen Virol 80(Part 1):57–61. [DOI] [PubMed] [Google Scholar]
- 10.Backovic M, Jardetzky TS, Longnecker R. 2007. Hydrophobic residues that form putative fusion loops of Epstein-Barr virus glycoprotein B are critical for fusion activity. J Virol 81:9596–9600. doi: 10.1128/JVI.00758-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 12.Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J Virol 83:6825–6836. doi: 10.1128/JVI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Babic N, Klupp BG, Makoschey B, Karger A, Flamand A, Mettenleiter TC. 1996. Glycoprotein gH of pseudorabies virus is essential for penetration and propagation in cell culture and in the nervous system of mice. J Gen Virol 77(Part 9):2277–2285. doi: 10.1099/0022-1317-77-9-2277. [DOI] [PubMed] [Google Scholar]
- 14.Desai PJ, Schaffer PA, Minson AC. 1988. Excretion of non-infectious virus particles lacking glycoprotein H by a temperature-sensitive mutant of herpes simplex virus type 1: evidence that gH is essential for virion infectivity. J Gen Virol 69(Part 6):1147–1156. doi: 10.1099/0022-1317-69-6-1147. [DOI] [PubMed] [Google Scholar]
- 15.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J Virol 66:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roop C, Hutchinson L, Johnson DC. 1993. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J Virol 67:2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gillet L, May JS, Colaco S, Stevenson PG. 2007. Glycoprotein L disruption reveals two functional forms of the murine gammaherpesvirus 68 glycoprotein H. J Virol 81:280–291. doi: 10.1128/JVI.01616-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klupp BG, Fuchs W, Weiland E, Mettenleiter TC. 1997. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J Virol 71:7687–7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lete C, Machiels B, Stevenson PG, Vanderplasschen A, Gillet L. 2012. Bovine herpesvirus type 4 glycoprotein L is nonessential for infectivity but triggers virion endocytosis during entry. J Virol 86:2653–2664. doi: 10.1128/JVI.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klupp BG, Mettenleiter TC. 1999. Glycoprotein gL-independent infectivity of pseudorabies virus is mediated by a gD-gH fusion protein. J Virol 73:3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc Natl Acad Sci U S A 106:20464–20469. doi: 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parry C, Bell S, Minson T, Browne H. 2005. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J Gen Virol 86:7–10. doi: 10.1099/vir.0.80567-0. [DOI] [PubMed] [Google Scholar]
- 23.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirschner AN, Omerovic J, Popov B, Longnecker R, Jardetzky TS. 2006. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J Virol 80:9444–9454. doi: 10.1128/JVI.00572-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104:10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klupp B, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J Virol 82:6299–6309. doi: 10.1128/JVI.00386-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc Natl Acad Sci U S A 107:22635–22640. doi: 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galdiero S, Falanga A, Vitiello M, D'Isanto M, Collins C, Orrei V, Browne H, Pedone C, Galdiero M. 2007. Evidence for a role of the membrane-proximal region of herpes simplex virus Type 1 glycoprotein H in membrane fusion and virus inhibition. Chembiochem 8:885–895. doi: 10.1002/cbic.200700044. [DOI] [PubMed] [Google Scholar]
- 31.Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J Biol Chem 284:17370–17382. doi: 10.1074/jbc.M109.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ungermann C, Langosch D. 2005. Functions of SNAREs in intracellular membrane fusion and lipid bilayer mixing. J Cell Sci 118:3819–3828. doi: 10.1242/jcs.02561. [DOI] [PubMed] [Google Scholar]
- 33.Fuchs W, Backovic M, Klupp BG, Rey FA, Mettenleiter TC. 2012. Structure-based mutational analysis of the highly conserved domain IV of glycoprotein H of pseudorabies virus. J Virol 86:8002–8013. doi: 10.1128/JVI.00690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vleck SE, Oliver SL, Brady JJ, Blau HM, Rajamani J, Sommer MH, Arvin AM. 2011. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc Natl Acad Sci U S A 108:18412–18417. doi: 10.1073/pnas.1111333108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaplan AS, Vatter AE. 1959. A comparison of herpes simplex and pseudorabies viruses. Virology 7:394–407. doi: 10.1016/0042-6822(59)90068-6. [DOI] [PubMed] [Google Scholar]
- 36.Klupp BG, Nixdorf R, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J Virol 74:6760–6768. doi: 10.1128/JVI.74.15.6760-6768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kopp M, Granzow H, Fuchs W, Klupp BG, Mundt E, Karger A, Mettenleiter TC. 2003. The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J Virol 77:5339–5351. doi: 10.1128/JVI.77.9.5339-5351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 40.Pavlova SP, Veits J, Keil GM, Mettenleiter TC, Fuchs W. 2009. Protection of chickens against H5N1 highly pathogenic avian influenza virus infection by live vaccination with infectious laryngotracheitis virus recombinants expressing H5 hemagglutinin and N1 neuraminidase. Vaccine 27:773–785. doi: 10.1016/j.vaccine.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 41.Klupp BG, Granzow H, Mettenleiter TC. 2000. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J Virol 74:10063–10073. doi: 10.1128/JVI.74.21.10063-10073.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nixdorf R, Klupp BG, Karger A, Mettenleiter TC. 2000. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J Virol 74:7137–7145. doi: 10.1128/JVI.74.15.7137-7145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerdts V, Jons A, Mettenleiter TC. 1999. Potency of an experimental DNA vaccine against Aujeszky's disease in pigs. Vet Microbiol 66:1–13. doi: 10.1016/S0378-1135(98)00300-9. [DOI] [PubMed] [Google Scholar]
- 44.Mettenleiter TC. 1989. Glycoprotein gIII deletion mutants of pseudorabies virus are impaired in virus entry. Virology 171:623–625. doi: 10.1016/0042-6822(89)90635-1. [DOI] [PubMed] [Google Scholar]
- 45.Fuchs W, Granzow H, Klupp BG, Kopp M, Mettenleiter TC. 2002. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J Virol 76:6729–6742. doi: 10.1128/JVI.76.13.6729-6742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klyachkin YM, Stoops KD, Geraghty RJ. 2006. Herpes simplex virus type 1 glycoprotein L mutants that fail to promote trafficking of glycoprotein H and fail to function in fusion can induce binding of glycoprotein L-dependent anti-glycoprotein H antibodies. J Gen Virol 87:759–767. doi: 10.1099/vir.0.81563-0. [DOI] [PubMed] [Google Scholar]
- 48.Duus KM, Grose C. 1996. Multiple regulatory effects of varicella-zoster virus (VZV) gL on trafficking patterns and fusogenic properties of VZV gH. J Virol 70:8961–8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zokarkar A, Connolly SA, Jardetzky TS, Lamb RA. 2012. Reversible inhibition of fusion activity of a paramyxovirus fusion protein by an engineered disulfide bond in the membrane-proximal external region. J Virol 86:12397–12401. doi: 10.1128/JVI.02006-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whealy ME, Robbins AK, Enquist LW. 1990. The export pathway of the pseudorabies virus gB homolog gII involves oligomer formation in the endoplasmic reticulum and protease processing in the Golgi apparatus. J Virol 64:1946–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorman JJ, Wallis TP, Pitt JJ. 2002. Protein disulfide bond determination by mass spectrometry. Mass Spectrom Rev 21:183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 52.Goyder MS, Rebeaud F, Pfeifer ME, Kalman F. 2013. Strategies in mass spectrometry for the assignment of Cys-Cys disulfide connectivities in proteins. Expert Rev Proteomics 10:489–501. doi: 10.1586/14789450.2013.837663. [DOI] [PubMed] [Google Scholar]
- 53.Uchida H, Chan J, Shrivastava I, Reinhart B, Grandi P, Glorioso JC, Cohen JB. 2013. Novel mutations in gB and gH circumvent the requirement for known gD receptors in herpes simplex virus 1 entry and cell-to-cell spread. J Virol 87:1430–1442. doi: 10.1128/JVI.02804-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol 6:815–826. doi: 10.1038/nrmicro1972. [DOI] [PubMed] [Google Scholar]
- 55.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84:3825–3834. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rauh I, Mettenleiter TC. 1991. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J Virol 65:5348–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peeters B, de Wind N, Hooisma M, Wagenaar F, Gielkens A, Moormann R. 1992. Pseudorabies virus envelope glycoproteins gp50 and gII are essential for virus penetration, but only gII is involved in membrane fusion. J Virol 66:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fan Q, Longnecker R, Connolly S. 2014. Substitution of herpes simplex virus 1 entry glycoproteins with those of saimiriine herpesvirus 1 reveals a gD-gH/gL functional interaction and a region within the gD profusion domain that is critical for fusion. J Virol 88:6470–6482. doi: 10.1128/JVI.00465-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cairns TM, Friedman LS, Lou H, Whitbeck JC, Shaner MS, Cohen GH, Eisenberg RJ. 2007. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J Virol 81:5102–5111. doi: 10.1128/JVI.00097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]