

Abstract
Introduction: We aimed to determine whether KRAS and BRAF mutant colorectal cancer (CRC) cells exhibit distinct sensitivities to the multi-target angiokinase inhibitor, TKI258 (dovitinib). Materials and methods: We screened 10 CRC cell lines by using receptor tyrosine kinase (RTK) array to identify activated RTKs. MTT assays, anchorage-independent colony-formation assays, and immunoblotting assays were performed to evaluate the in vitro anti-tumor effects of TKI258. In vivo efficacy study followed by pharmacodynamic evaluation was done. Results: Fibroblast Growth Factor Receptor 1 (FGFR1) and FGFR3 were among the most highly activated RTKs in CRC cell lines. In in vitro assays, the BRAF mutant HT-29 cells were more resistant to the TKI258 than the KRAS mutant LoVo cells. However, in xenograft assays, TKI258 equally delayed the growth of tumors induced by both cell lines. TUNEL assays showed that the apoptotic index was unchanged following TKI258 treatment, but staining for Ki-67 and CD31 was substantially reduced in both xenografts, implying an anti-angiogenic effect of the drug. TKI258 treatment was effective in delaying CRC tumor growth in vivo regardless of the KRAS and BRAF mutation status. Conclusions: Our results identify FGFRs as potential targets in CRC treatment and suggest that combined targeting of multiple RTKs with TKI258 might serve as a novel approach to improve outcome of patients with CRC.
Keywords: Colorectal cancer, FGFR, KRAS, BRAF, Dovitinib (TKI258), multi-target angiokinase inhibitor
Introduction
Colorectal cancer (CRC) is the third-most commonly diagnosed cancer in males and the second-most commonly diagnosed cancer in females worldwide [1]. Moreover, CRC incidence is increasing rapidly in several historically low-risk countries such as countries in Eastern Asia and Eastern Europe [2]. Because conventional anti-cancer drugs are not adequate for improving CRC-treatment outcome, we must understand the molecular biology of colon cancer and identify relevant molecular targets to biologically modulate the cancer.
Numerous inhibitors that target various receptor tyrosine kinases (RTKs) have been confirmed to inhibit tumor survival and angiogenesis in preclinical trial models of CRC [3-5]. Bevacizumab, a humanized monoclonal antibody that targets vascular endothelial growth factor A (VEGF-A) was approved for first- or second-line use in metastatic CRC, in combination with conventional chemotherapy [6-8]. Furthermore, certain epidermal growth factor receptor (EGFR)-inhibiting monoclonal antibodies such as cetuximab and panitumumab showed modest efficacy in monotherapy or combination therapy [9-12]. However, the results of several studies in which these RTK inhibitors were used on CRC patients showed limited effect, implying that more effective therapeutic RTK inhibitors are required.
Fibroblast growth factors (FGFs), which promote angiogenesis and tumor growth by binding to tyrosine kinase. FGF receptors (FGFRs), are reported to be overexpressed in CRC patients [13,14]. FGFR genes have been reported to potentially promote tumor growth and invasion in CRC [13,15,16], and FGFR signals were implicated in the intrinsic resistance to EGFR inhibitors [17] in non-small cell lung cancer (NSCLC). Given these results, FGFR inhibitors are considered one of the potential RTK inhibitors that can be used to treat CRC patients; this is not only because FGFR is overexpressed in CRC, but also because this treatment might help overcome the resistance to EGFR inhibitors.
TKI258 is an orally active small molecule that potently inhibits the activity of multiple RTKs including FGFRs, platelet-derived growth factor receptors (PDGFRs), and VEGF receptors (VEGFRs), which participate in tumor growth, survival, angiogenesis, and vascular development [18] through both direct and indirect mechanisms. Inhibiting multiple angiokinases - mainly FGFRs - led to the suppression of their downstream signaling including signaling by RAS-RAF-MAPK molecules and PI3K-AKT related molecules that are mainly involved in cell proliferation, cell survival, and tumor invasion [19].
Mutations in the v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS), which occur in approximately 40% of CRC patients, is considered one of the major negative predictive factors in the treatment response in patients receiving EGFR-directed antibodies [20,21]. In the absence of KRAS mutations, resistance to EGFR inhibitors has been reported to be potentially caused by genetic alterations of molecules related to RAS-RAF-MAPK signaling [19,22]. Furthermore, mutation in v-raf murine sarcoma viral oncogene homolog B1 (BRAF) appears to mediate cetuximab resistance in the absence of KRAS mutation [23,24], and. BRAF mutations have been widely reported to occur only in KRAS-negative colon carcinomas, suggesting that BRAF/KRAS activating mutations might be alternative genetic events in CRC [26-28]. Recently, the mutually exclusive BRAF/KRAS mutation status was suggested to be related to RTK-inhibitor sensitivity in CRC. BIBF 1120, another multi-target angiokinase inhibitor showed efficacy when combined with afatinib against CRC with KRAS mutation in vitro [29]. BIBF 1120 also targets FGFR and PDGFR, but mainly targets VEGFR, and no previous studies reported KRAS/BRAF mutant CRC and FGFR inhibitor sensitivity. We surmised that KRAS or BRAF mutation status might affect FGFR inhibitor - TKI258 - sensitivity in CRC.
In this study, to identify better RTK inhibitors that can improve CRC treatment, we determined whether genetic aberration of a novel target’s downstream signals might affect the efficacy of an RTK inhibitor. First, we hypothesized that inhibition of FGFR will efficiently suppress tumor growth in CRC, and then we hypothesized that the KRAS and BRAF mutant CRC cell lines will exhibit distinct sensitivities to the TKI258, which mainly targets FGFR. The results of this study could lead to the identification of predictive biomarkers and thus facilitate the selection of CRC patients who are likely to benefit from treatment with the FGFR inhibitor. We investigated the anti-tumor activity of TKI258 in CRC cell lines carrying KRAS or BRAF mutations, in vitro and in vivo, to determine whether the sensitivity of these cells to the inhibitor depends on specific gene alteration.
Materials and methods
Cell lines and cell culture
The 6 human CRC cell lines used in this study - KM12SM, KM-12L4, Colo320DM, SNU-C4, SNU-1235, and SW48 - were with no known KRAS or BRAF mutation. And the 10 human CRC cell lines with any of KRAS or BRAF mutation used in this study - DLD-1, HCT-15, COLO205, SW480, HCT-116, LoVo, WiDr, CaCo2, RKO, and HT-29 - were obtained from American Type Culture Collection (Bethesda, MD). All cell lines were cultured in MEM or RPMI-1640 medium supplemented with 10% FBS (GIBCO), 100 U/mL penicillin, and 0.1 mg/mL streptomycin in a humidified 5% CO2 incubator. Among the 10 CRC cell lines used in this study [25,30,31], 5 CRC cell lines had only KRAS mutations (DLD-1, LoVo, SW480, HCT-15, and HCT-116) and 5 CRC cell lines had only BRAFV600E mutations (HT-29, COLO205, RKO, CaCo2, and WiDr). There were no serum starvation or stimulation throughout the experiments.
Receptor tyrosine kinase inhibitor
TKI258 (4-amino-5-fluoro-3-[5-(4-metylpiperazin-1-yl)-1H-benzimidazol-2-yl] quinolin-2(1H)-one, formerly known as CHIR258), which targets FGFRs, PDGFRs, and VEGFRs, was purchased from Eurasian Chemicals Pvt. Ltd. We prepared 10 mM stock solutions of the drug and stored them at -20°C.
Receptor tyrosine kinase array
To screen for the activity of specific RTKs, we used the Human Phospho-RTK Array (R&D Systems) according to the manufacturer’s protocols. Briefly, 300 μg of proteins were added to a blocked membrane and incubated at 4°C. After washing, horseradish peroxidase (HRP)-conjugated anti-phosphotyrosine antibodies (1:2,000) were added to each membrane and incubated. After washing the membranes further, proteins were visualized using an enhanced chemiluminescence (ECL) detection system (Amersham). The activated RTK levels were quantitated using GenePix Pro 4.1 software (Axon Instruments) and normalized using the positive control’s intensity in each membrane. RTK signal intensity was measured as the difference between the mean of each RTK’s intensity and mean of negative signal intensity. Relative RTK intensity was defined as the RTK signal intensity of each sample divided by the positive signal intensity of each membrane.
Immunoblotting analysis
Cells were seeded in 6-well plates and incubated for overnight at 37°C. On the next day, culture media was replaced with serum-free media and the cells were again incubated overnight. Cells were then incubated with the drug (1 μM TKI258 or PBS control) in serum-free media at 37°C for indicated times (0, 0.5, 1, 3, and 6 h). Cells were lysed in NP-40 lysis buffer and proteins were resolved using 6% or 10% SDS polyacrylamide gels and then transferred to PVDF membranes (GE Healthcare) by using electroblotting. The following primary antibodies were used in the experiments: p-FGFR1 (Y653/Y654) (Santa Cruz Biotechnology), p-Akt (S473), AKT, p-ERK1/2 (T202/Y204), ERK1/2, p-PI3K (Y458/Y199), PI3K, p-4EBP1 (Thr37/46), 4EBP1, p-p70S6K (T389), and p70S6 (Cell Signaling). Anti-GAPDH antibody (Abcam) was used to control for equal loading.
Cell-viability assay
Cells were seeded in 96-well plates at a density of 5,000 cells/well and incubated at 37°C. TKI258 was serially diluted using culture media, and various concentrations of the drug were added to each well and incubated for 3 days. MTT (3-(4,5-dimethylthiazol)-2,5-diphenyl tetrazolium) was then added to the medium for 4 h, the medium was removed, the precipitate was dissolved in DMSO, and the absorbance was read at 570 nm. The drug concentrations required to inhibit cell growth by 50% (IC50) were calculated through interpolation of the dose-response curves by using CalcuSyn software (Biosoft).
Anchorage-independent colony formation assay
We added 0.6% Noble agar (Difco) as a basement into each well of culture plates. After the agar solidified, cells (3,000 cells/well) were suspended in 0.4% Noble agar mixed in culture media containing 10% FBS and plated in triplicate on top of the basement. Plates were incubated at 37°C for 3 weeks and colonies featuring a diameter > 200 μm were counted and photographed under an inverted microscope.
Reverse transcription (RT)-PCR analysis
Total RNA was isolated using the Trizol reagent (Invitrogen) and cDNAs were synthesized using Superscript II Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions. RT-PCR reactions were performed using Taq-Gold DNA polymerase (Invitrogen), and the amplified products were separated on ethidium bromide gels containing 1.2% agarose. The house-keeping gene GAPDH was selected as the positive control. The primer set used for RT-PCR amplification of the gene encoding FGFR1 was the following: forward 5’-CAT CCC CAG AAA AGA TGG AA-3’, and reverse 5’-CCT CCC CTG TTC CCA TTA CT-3’.
Xenograft study
Female BALB/c nu/nu mice, 6-8-weeks old, were implanted subcutaneously with LoVo cells (2.0 × 107) or HT-29 cells (1.0 × 107) in the flank. Once the tumors could be measured (mean tumor volume of 200-300 mm3), the mice were assigned to 2 groups, the vehicle only group and the TKI258-treated group (n = 4 per group in the case of LoVo cells; n = 6 per group, HT-29 cells). TKI258 was administered orally at 70 mg/kg per day for 4 weeks. Tumor volumes were measured every other day. After mice were sacrificed, all the organs were thoroughly examined, and formalin-fixed paraffin-embedded tumors were subjected to pathological analysis.
Immunohistochemistry (IHC)
Following deparaffinization and rehydration, tumor sections were treated with an antigen-retrieval solution (DAKO). Endogenous peroxidases were quenched using 3% H2O2. Sections were stained using antibodies against FGFR1 (1:50, Abcam), p-ERK (1:50, Cell Signaling Technology), p-AKT (1:100, Abcam), CD31 (1:100, Abcam), and Ki-67 (1:100, DAKO), which were diluted in an antibody-dilution buffer (DAKO). Staining was visualized using 3,3-diaminobenzidinetrahydro chloride (DAB; DAKO). Lastly, each slide was counterstained with hematoxylin for 5 min, washed with water, mounted, and covered with a coverslip.
TUNEL assay
DNA fragments in tissue sections were detected using a TACS® 2 TdT DAB Kit (Trevigen). Briefly, the enzyme terminal deoxynucleotidyl transferase (TdT) was used to incorporate digoxigenin-conjugated dUTP at the ends of DNA fragments. The signal of TdT-mediated dUTP nick-end labeling (TUNEL) was then detected using an anti-digoxigenin antibody conjugated with peroxidase. TUNEL-positive cells were counted in 5 random high-power fields per section and are reported as a percentage of positive cells in each cellular compartment.
Statistical analysis
The statistical significance of differences between 2 groups of data was assessed using the unpaired t test in SPSS 21.0 software; a P-value of < 0.05 was considered statistically significant.
Results
RTK activity in CRC cell lines
To screen for RTK activity in KRAS or BRAF mutant 10 CRC cell lines, we used a human phospho-RTK array (Figure 1A). In addition to highly activated EGFR, several other druggable targets such as ErbB2, HGF-R, IGF-IR, FGFR1, FGFR3, and VEGFR3 were also highly activated in CRC cell lines. Various RTKs were more activated in the KRAS mutant cell lines than in the BRAF mutant cell lines (Figure 1B). Specifically, EphA7, Mer, TrkA, and FGFR1 appeared to be relatively more activated in KRAS mutant cell lines, whereas EphB2, Insulin R, IGF-IR, and VEGFR3 were relatively more activated in BRAF mutant cell lines (Figure 1C). Because FGFR is considered one of the potential targets in CRC, our results demonstrating that FGFR1 and FGFR3 are highly activated in CRC cell lines provides a strong rationale for treating CRC with an FGFR inhibitor.
Figure 1.
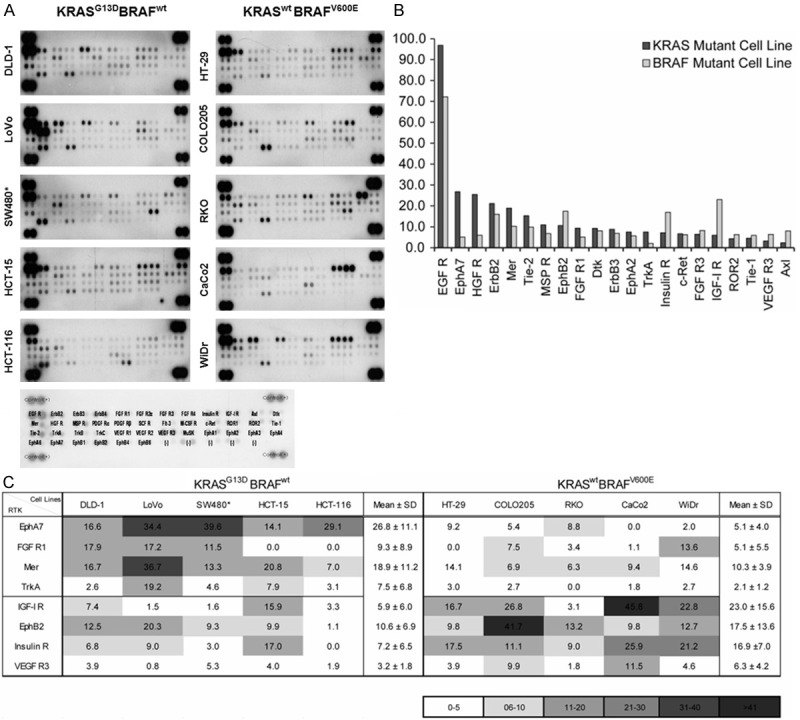
Differently activated RTKs in 10 CRC cell lines carrying KRAS or BRAF mutation. A. Human phospho-RTK array was done among 10 CRC cell lines. Each RTK is spotted in duplicate and positive controls are the pairs of dots in each corner. The activated RTK levels were quantitated using GenePix Pro 4.1 software (Axon Instruments) and relative RTK intensity was calculated comparing with positive control’s intensity in each membrane. B. Among the RTKs tested, the mean values of relative RTK intensity that are the same or > 5.0 are shown on the graph. RTKs appeared to be more highly activated in KRAS mutant cell lines than in BRAF mutant cell lines. C. The 8 RTKs that were differently activated because of KRAS or BRAF mutation in the CRC cell lines are shown in the table and the heat-map. *SW480 carries the KRAS G12V mutation, whereas the other 4 cell lines carry the KRAS G13D mutation.
TKI sensitivity based on genetic variations
Because TKI258 is an ATP-competitive inhibitor belonging to Class III–V RTK inhibitors that are especially sensitive against FGFRs (FGFR1/2/3, with kinase IC50 values of 8-13/21/9-18 nmol/L, respectively [32,33]), we used TKI258 as a novel tyrosine kinase inhibitor (TKI) to inhibit FGFRs in CRC. First, to test whether the KRAS or BRAF mutation status affected cell viability in response to the FGFR inhibitor, we measured the anti-tumor activity of TKI258 against 16 CRC cell lines (6 KRASwt BRAFwt, 5 KRAS mutant cell lines, and 5 BRAF mutant cell lines) (Figure 2). CRC cell lines without any of KRAS or BRAF mutation (KRASwt BRAFwt) were more sensitive to TKI258. We could not tell whether KRAS or BRAF mutation decides sensitivity to TKI258 with this MTT assay result, but there was trend toward KRAS mutant CRC cell lines being more sensitive to TKI258 compared to BRAF mutant (IC50 (nM): KRAS mutant cell lines: 1274.6 ± 1015.9; BRAF mutant cell lines: 1727.6 ± 867.5). Immortalized normal cell lines (CCD-841COTR and CCD-18CO) were more resistant than CRC cell lines (IC50 of 5000 nM, data not shown on figure).
Figure 2.
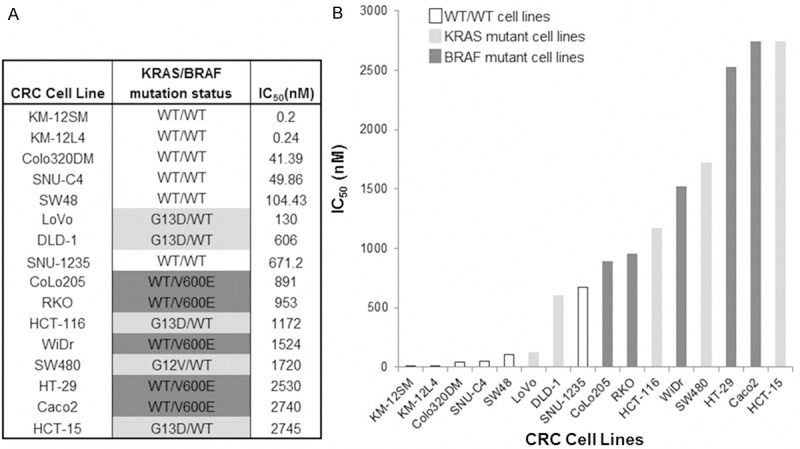
Anti-tumor effect of TKI258 toward CRC cell lines. The growth inhibitor activity of TKI258 toward CRC cell lines was determined by the cell-viability assay (MTT Assay) after 72 hours of continuous drug exposure. IC50 values were calculated (A). CRC cell lines without any of KRAS or BRAF mutation (KRASwt BRAFwt) were more sensitive to TKI258. There was a trend toward KRAS mutant CRC cell lines being more sensitive to TKI258 compared to BRAF mutant (IC50 (nM): KRAS mutant cell lines: 1274.6 ± 1015.9; BRAF mutant cell lines: 1727.6 ± 867.5) (B).
We compared FGFR1 mRNA and protein levels and FGFR1 activity among the CRC cell lines by using RT-PCR and western-blotting analyses; FGFR1 is considered the major target of TKI258. There were no correlation of phosphorylated FGFR1 level and sensitivity to TKI258 measured by MTT assay. The LoVo (KRASG13D BRAFwt) and HT-29 (KRASwt BRAFV600E) cell lines were chosen because phosphorylated FGFR1 was similarly overexpressed in these cells (Figure 3). Assays to determine in vitro anti-tumor effect of TKI258 against LoVo (KRASG13D BRAFwt) and HT-29 (KRASwt BRAFV600E) cell lines are performed. Both cell lines showed a dose-dependent inhibition of cell growth in cell-viability assays (Figure 4A). Whereas LoVo cells were highly sensitive to TKI258 (IC50 of 130 nM), HT-29 cells were relatively more resistant to TKI258 treatment (IC50 of 2,530 nM). When we used the soft agar colony-formation assays to evaluate the anchorage-independent in vitro anti-tumor activity of TKI258 (Figure 4B, 4C), almost no colony was formed in the case of the KRAS mutant LoVo cell line after adding 500 nM TKI258. However, the BRAF mutant HT-29 cells were resistant to TKI258 and showed only 3% reduction in colony formation relative to control.
Figure 3.
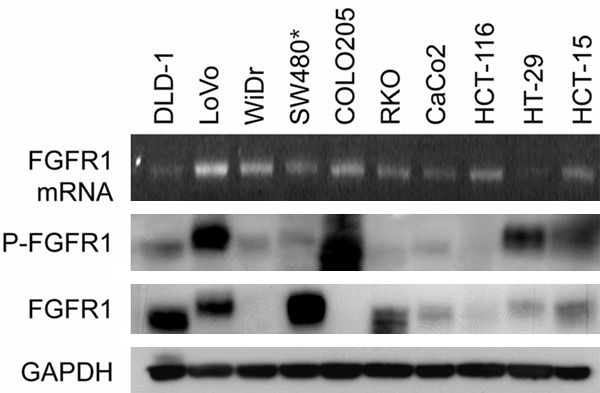
FGFR1 expression among CRC cell lines. We compared FGFR1 mRNA, protein level and its activity among CRC cell lines, via RT PCR and Western blot. FGFR1 was relatively over-expressed among KRAS mutant CRC cell lines (DLD-1, LoVo, SW480, HCT-116, and HCT-15) compared to BRAF mutant (WiDr, COLO205, RKO, CaCo2, and HT-29). P-FGFR1 activities of KRAS mutant LoVo cell line and BRAF mutant HT-29 cell line were similar. *SW480 had KRAS G12V mutation, while other 4 cell lines have KRAS G13D mutation.
Figure 4.
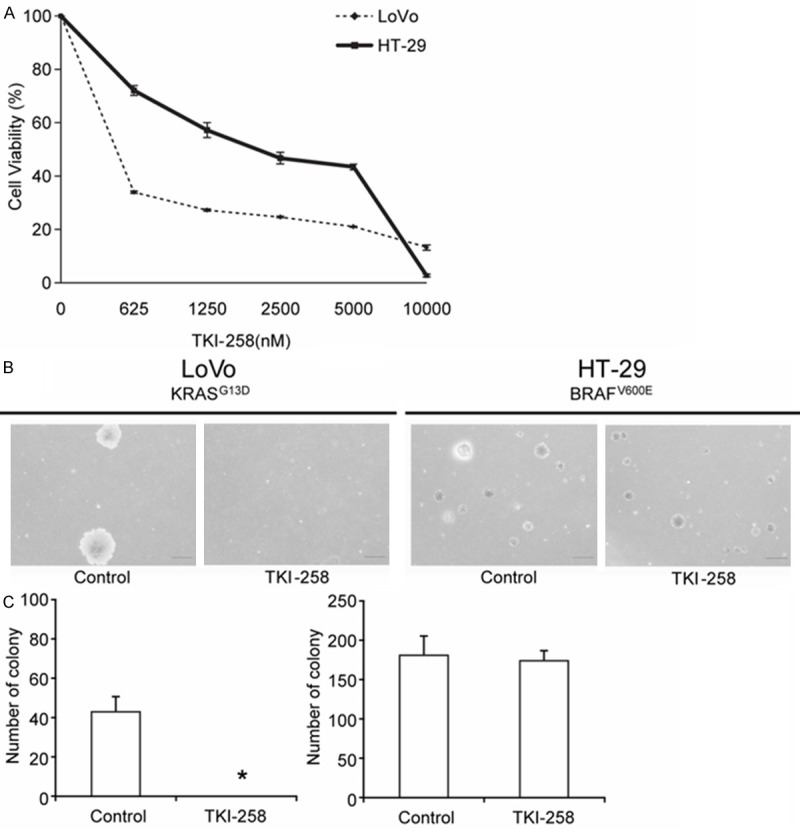
In vitro anti-tumor effect of TKI258 in LoVo and HT-29 cells. A. The GroCell-viability assay (MTT Assay) was performed using the multi-target FGFR inhibitor, TKI258. The KRAS mutant LoVo cells (IC50 = 130 nM) were more sensitive to TKI258 than the BRAF mutant HT-29 cells (IC50 = 2,530 nM). B, C. Anchorage-independent colony-formation assay performed using TKI258 to compare LoVo and HT-29 cell lines. Almost no colonies were formed by the KRAS mutant LoVo cells after adding 500 nM TKI258, and the result was statistically significant; by contrast, the BRAF mutant HT-29 cell line was not affected by the drug treatment. *p < 0.05.
Changes in downstream signaling following TKI258 treatment
To evaluate how downstream signaling molecules are affected after drug treatment in time-course, LoVo and HT-29 cells were treated with 1 μM TKI258 in vitro and then examined using western-blotting analysis (Figure 5). TKI258 showed profound, sustained inhibition of FGFR1 phosphorylation after 30-min treatment in the KRAS mutant LoVo cells, but not in the BRAF mutant HT-29 cells. In HT-29 cells, FGFR1 appeared to show increased and sustained activation after 30 min of TKI258 treatment. Analyzing the PI3K-AKT signaling pathways revealed that TKI258 treatment reduced the levels of phosphorylated PI3K, AKT, and P70S6K without altering the levels of phosphorylated 4EBP1 or the total expression of 4EBP1 in LoVo cells; however, no alteration in downstream signaling molecules was detected in HT-29 cancer cells. By contrast, TKI258 treatment increased the level of activated ERK in both LoVo and HT-29 cells.
Figure 5.
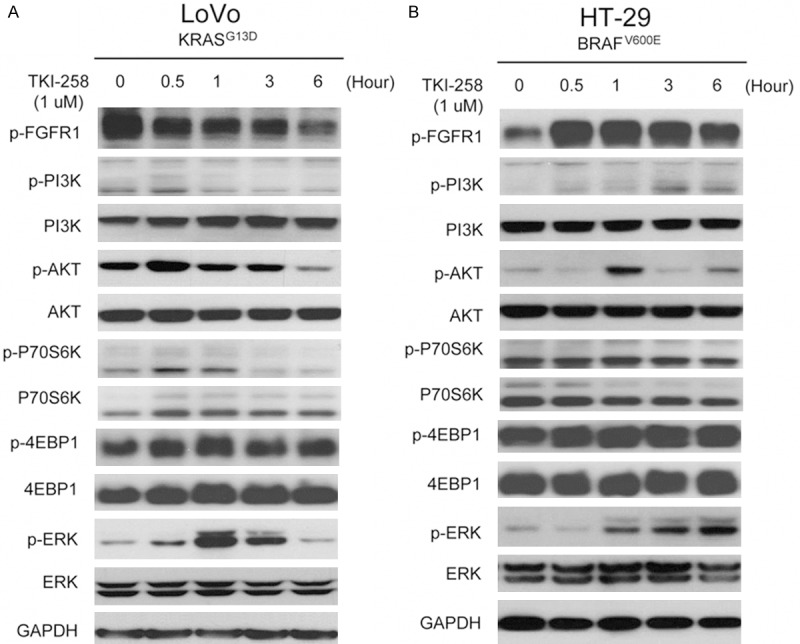
Changes in downstream signaling molecules after TKI258 treatment in LoVo and HT-29 cells. KRAS mutant LoVo and BRAF mutant HT-29 cells were treated with 1 μM TKI258 in vitro to evaluate the time-dependent effect on downstream signaling molecules by using western-blotting analysis. PI3K-AKT and RAS-RAF-ERK pathways appeared to be inhibited by TKI258 in LoVo cells, but the PI3K-AKT pathway was not inhibited in HT-29 cells and, in these cells, the RAS-RAF-ERK pathway appeared to be activated. (24 hour treatment data not shown, since cells were damaged due to long serum free status).
In vivo efficacy and pharmacodynamic marker evaluation using TKI258
To evaluate the anti-tumor effect of TKI258 in vivo, a daily oral dose of TKI258 of 70 mg/kg was administered to mice bearing subcutaneous LoVo or HT-29 human tumor xenografts. Contrasting the in vitro results, no difference in in vivo anti-tumor effect was observed between the 2 xenograft models featuring distinct mutation statuses: TKI258 delayed tumor growth equally in both cell lines compared with the control group (Figure 6). No major toxicity was detected in the treated mice in both groups, and the bodyweights of these mice were not different (data not shown).
Figure 6.
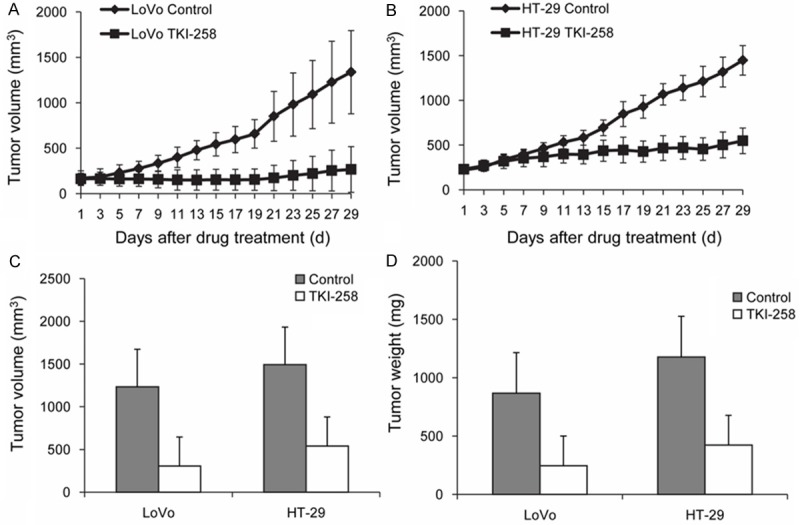
In vivo anti-tumor effect of TKI258 in LoVo and HT-29 xenograft models. (A, B) After treating with the TKI258 (70 mg/kg/d PO for 4 weeks), the in vivo xenograft model showed that TKI258 delayed the growth of tumors induced by both cell lines compared with the control group (shown as Mean ± SE). However, when we precisely measured the differences in tumor volume in the control and drug-treated groups in a time-dependent manner, HT-29 xenografts appeared to be inhibited earlier than LoVo xenografts. Compared with the control groups, in the LoVo and HT-29 cell lines, tumor volume (C) and weight (D) decreased similarly following treatment with TKI258 in vivo.
Next, when we evaluated the pharmacodynamics of target modulation in the tumors (Figure 7), TKI258 treatment was found to substantially reduce the levels of phosphorylated FGFR1 in both xenografts. Cells positive for phosphorylated AKT were not markedly reduced after the treatment, but cells positive for phosphorylated ERK were reduced in the TKI258-treated group. This result differed from the in vitro western-blotting data, which showed that p-ERK staining was not reduced. Large areas of necrosis were also detected in TKI258-treated tumors when compared with control tumors. When CD31 was stained to evaluate the anti-angiogenic activity of TKI258 in vivo, the size and numbers of CD31-positive blood vessels within the tumors were observed to be diminished after the treatment in both xenografts. Furthermore, actively proliferating tumor cells that were identified using Ki-67 staining were markedly reduced in TKI258-treated mice. Moreover, the TUNEL assay used to assess the effect of TKI258 on apoptosis showed no change in the apoptotic index in either xenograft (Figure 8). These results suggest that TKI258 inhibited tumor growth in vivo and that the effect of TKI258 in target-molecule inhibition was similar in KRAS mutant and BRAF mutant cell lines. Taken together, our results suggest that the KRAS and BRAF mutation status does not affect tumor-growth inhibition and changes in target molecules induced by the FGFR inhibitor in vivo.
Figure 7.
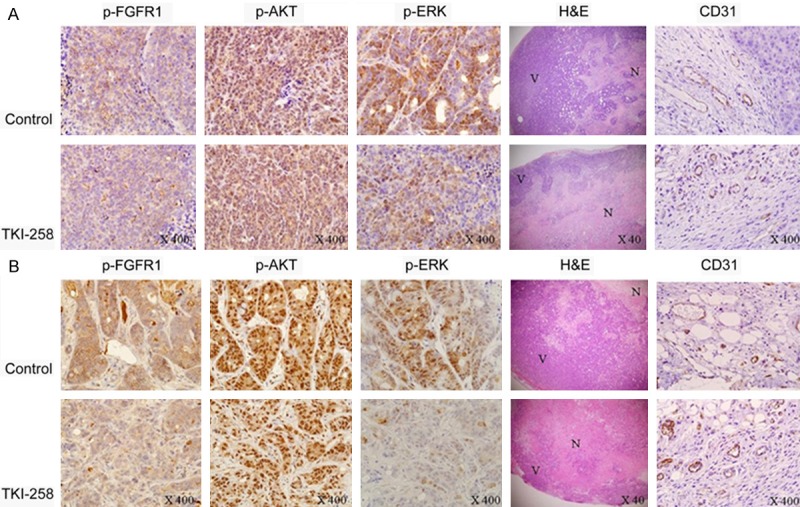
Immunohistochemical staining of human colon tumor xenografts. Tumors were resected and processed and slides were stained using antibodies against p-FGFR1, p-ERK, p-AKT, and CD31. H&E staining was also performed. TKI258 treatment reduced the staining for p-FGFR1, p-ERK, and CD31 in tumor xenografts induced by both cell lines. However, p-AKT stained tumor cells were similar in the control and treated groups. Large areas of necrosis were also detected using H&E staining in the TKI258-treated tumors when compared with the control tumors (A: LoVo; B: HT-29). All magnifications 400×, except H&E magnification (40×); N = massive necrosis; V = viable tumor.
Figure 8.
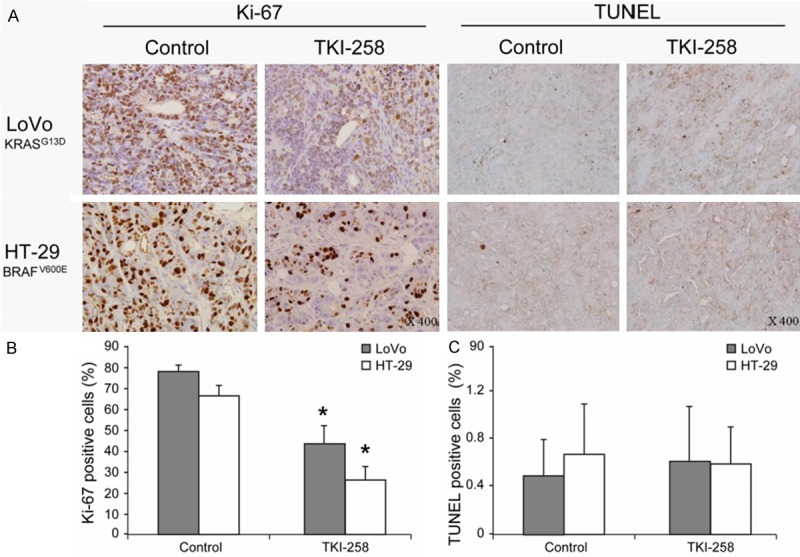
Ki-67 staining and TUNEL assay of xenograft models. Ki-67 staining and TUNEL assay were also performed (A). TKI258-treated mice showed a significant reduction of Ki-67-positive cells in tumors induced by both cell lines (B), but the numbers of TUNEL-positive cells were not statistically different (C). *P < 0.05.
Discussion
Numerous RTK inhibitors are currently available that target various cancers and these inhibitors are continuing to show clinical success in various cancer treatments. To treat metastatic CRC, anti-EGFR monoclonal antibodies such as cetuximab and panitumumab and anti-angiogenic agents such as bevacizumab and regorafenib are becoming widely used. However, because certain disappointing results such as drug resistance have been obtained, more effective RTK targets are sought for CRC treatment.
We screened 10 KRAS or BRAF mutant CRC cell lines using an RTK array to evaluate which RTKs are up-regulated and to check for novel target-RTK candidates. EGFR was the single most activated RTK, and we also detected other activated RTKs such as HGFR, ErbB2, FGFR1, FGFR3, and VEGFR3, which already have inhibitors being used in other types of cancer treatment. Certain RTKs showed varying activity patterns according to KRAS or BRAF mutation status: EphA7, Mer, TrkA, and FGFR1 were highly activated in 5 KRAS mutant cell lines, and EphB2, Insulin R, IGF-IR, and VEGFR3 were highly activated in 5 BRAF mutant cell lines. Among these RTKs, although EphA7 and EphB2 belong to the same ephrin-receptor family, EphA7 was markedly upregulated in KRAS mutant cell lines and EphB2 was upregulated in BRAF mutant cell lines. Ephrin-family proteins are known to affect cytoskeletal and cell-adhesion mechanisms and thus regulate cell position and motility. Interestingly, EphA7, which acts as a tumor suppressor in follicular lymphoma [34], was reported to be down-regulated in CRC relative to normal tissue [35]. The finding for VEGFR3 is also surprising since VEGFR3 is generally associated with lymphangiogenesis. The clinical relevance of these findings in relation to KRAS or BRAF mutation status of CRC remains unknown and further studies are warranted.
FGFR is one of the RTKs that plays key biological roles in cancer, and FGFR has emerged as a potential therapeutic target [36-38]. Recently, FGFR1, FGFR2, or FGFR3 overexpression in CRC was reported to result in increased growth and invasion in preclinical or clinical settings [13,15,16,39]. In this study, 10 CRC cell lines we tested showed overexpressed FGFRs (relative RTK intensity among KRAS-mutant cell lines: FGFR1 > FGFR3 > FGFR2α; and among BRAF-mutant cell lines: FGFR3 > FGFR1 > FGFR2α; almost no FGFR4 was detected in either cell line); this result identifies FGFR as a potential target in CRC treatment. Moreover, considering that tumor progression is mediated not only by the malignant cancer cells themselves but also by surrounding non-malignant stromal cells such as fibroblasts, FGFR is considered an attractive candidate target and several drugs against this RTK family are being developed. Because the initiation and progression of most cancers rarely depend on a single growth factor or a simple genetic alteration, multi-target RTK inhibition might represent an alternative strategy to inhibit complex signaling pathways in cancer cells, even if increased toxicity is occasionally detected. Herein, we have described the anti-tumor activities of TKI258, a multi-target angiokinase inhibitor, by inhibiting Class III, IV, and V RTKs, notably those of the FGFR, VEGFR, and PDGFR subfamilies.
We evaluated 16 CRC cell lines for their sensitivity to TKI258 in relation to the KRAS or BRAF mutation status. Our in vitro data showed that TKI258 exhibited significant anti-tumor activity against CRC cell lines without any of KRAS or BRAF mutation, compared to CRC cell lines with any of KRAS or BRAF mutation. Among KRAS or BRAF mutant CRC cell lines, it was hard to tell which mutation type was more sensitive to TKI258, but there was trend toward KRAS mutant CRC cell lines being more sensitive. In KRAS mutant HT-29 cells, TKI258 inhibited PI3K-AKT activities, whereas these activities in BRAF mutant LoVo cells were not affected by the treatment. ERK appeared to be activated in both cell lines after treatment with TKI258, which suggests that KRAS or BRAF mutation leading to the activation of the RAS-RAF-MAPK pathway might function as a resistance mechanism against the FGFR inhibitor. Studies conducted using EGFR inhibitors also suggested that drug resistance results from the KRAS mutation directly activating RAS-RAF-MAPK downstream signaling [40,41]. Interestingly, ERK activation peaked at 1 hour after treatment and then appeared to diminish in LoVo cells, where p-ERK levels increased in a time-dependent manner in HT-29 cells. TKI258’s ability to inhibit multiple angiokinases might have played a role here. For instance, initial inhibition of FGFR by the drug might have blocked the PI3K-AKT pathway but activated the RAS-RAF-MAPK pathway, and the sequential delayed effects of PDGFR or VEGFR inhibition might have influenced RAS-RAF pathway to inactivate ERK subsequently. Conversely, because KRAS functions not only through the RAS-RAF-MAPK pathway but also through multiple effector-mediated cytoplasmic signaling networks including the PI3K pathway [42], inhibition of the PI3K pathway by the drug might have subsequently affected RAS-RAF-MAPK signaling in KRAS mutant cells. Following TKI258 treatment in the BRAF mutant HT-29 cells, an up-regulation of p-ERK was detected, whereas p-PI3K and p-AKT were repressed. Previously, anchorage-independent growth and tumorigenic growth of BRAF mutation-positive CRC cells were shown to depend on the function of the mutant BRAF and to depend critically on persistent ERK activation [43]. Furthermore, TKI258 might have not altered PI3K pathway in the BRAF mutant HT-29 cells because HT-29 cell lines are known to carry a PI3K-activating E545K mutation, whereas LoVo cells do not exhibit any PI3K mutation.
Because our in vitro data revealed that LoVo cell lines were more sensitive than HT-29 cell lines to TKI258, we expected tumors generated by implanted LoVo cells to have regressed more than tumors generated by the HT-29 cells. However, relative to control, both of the xenografts tested in our study exhibited a significant reduction in tumor growth regardless of the KRAS or BRAF mutation status. Our IHC results were also distinct from the in vitro immunoblotting results after TKI258 treatment: p-ERK-positive cells were reduced but p-AKT-positive cells were similar in both xenografts, whereas the in vitro study showed increased p-ERK in both cell lines but reduced p-AKT in the LoVo cell line considering the crosstalk between the pathways, resulting in drug resistance. Recently, concomitant AKT/mTOR inhibition was suggested to be required for BRAF-mutant CRC [44], and we could talk into account including an mTOR inhibitor as a combination partner in further studies. Treatment with the FGFR inhibitor has been reported to reduce Ki67 staining and increase TUNEL staining in prostate cancer cells and head and neck cancer cells, implying that the inhibitor not only suppressed cell proliferation but also induced apoptosis [45,46]. In our result, TKI258 treatment inhibited cell proliferation and angiogenesis but did not induce apoptosis. This disparity could be explained related to apoptosis induction as the distinct origins of the tumor and the inhibition of different subclasses of FGFRs.
One explanation of the in vitro and in vivo differences might be from the innate difference of two systems including the delayed effect of the drug, because we used a 4-week treatment in the xenograft study. Another possible explanation is the ability of TKI258 to target multiple RTKs and its effect on the tumor microenvironment: TKI258 targets not only FGFRs but also VEGFR and PDGFR. A key factor of the tumor microenvironment is an abnormal but abundant vasculature, which supplies sufficient nutrients and oxygen for sustenance and ensures tumor progression [47]. The VEGFR-inhibiting feature of the multi-target inhibitor TKI258 might play a critical role in equally suppressing proliferation of both KRAS and BRAF mutated tumor cells in vivo; we observed that CD31-stained vessel size and numbers were diminished after treatment with TKI258. In addition, not only VEGFR but also FGFR signaling potently affects the microenvironment, because one of the well-known direct effects of FGF signaling is the promotion of angiogenesis through endothelial cells and other vascular cells [48]. Hence, TKI258 will show anti-angiogenic effect not only by inhibiting VEGFRs, but also by inhibiting FGFRs.
FGF and FGFRs are considered to act synergistically with the VEGF pathway to promote neovascularization, implying one mechanism of adaptive resistance to VEGF inhibitor at least in renal cell carcinoma or pancreatic cancer [49]. Because EGFR and VEGF inhibitors are the only approved RTK inhibitors widely used to treat CRC, TKI258 targeting FGFRs might show promising results among CRC patients who are refractory to EGFR or VEGF inhibitors. Previously, LoVo and HT-29 cells were reported to be resistant to the EGFR inhibitor cetuximab in vivo [50]. However, our results showing that these cell lines are sensitive to TKI258 suggest that the resistance to the EGFR inhibitor might be overcome by using TKI258.
Our results indicate that treatment with the multi-target angiokinase inhibitor TKI258 was effective in KRAS mutant LoVo cells but not in BRAF mutant HT-29 cells in vitro. However, our in vivo studies showed that KRAS and BRAF mutant xenograft tumors were both inhibited by TKI258 treatment, which might have resulted from the angiogenesis-suppressing effect of the multi-target inhibitor TKI258. Moreover, our results revealed the possibility of using TKI258 to overcome resistance to EGFR or VEGF inhibitors in CRC. The results relating to the in vivo effect of the drug would be more compelling if further studies with more cell lines were conducted. In conclusion, FGFR is an effective target for CRC treatment regardless of KRAS or BRAF mutation and our results suggest that combined targeting of multiple RTKs, especially FGFR and VEGFR, might be a novel approach to improve the outcome of CRC patients, possibly among selected patients in whom FGFR is overexpressed or amplified.
Acknowledgements
This research was supported by the Public Welfare & Safety Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning (2010-0020841). Dr. Chung’s work was supported by a Grant from the Korea Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea (0405-BC01-06040002).
Disclosure of conflict of interest
The authors declare no conflict of interes
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Center MM, Jemal A, Ward E. International trends in colorectal cancer incidence rates. Cancer Epidemiol Biomarkers Prev. 2009;18:1688–1694. doi: 10.1158/1055-9965.EPI-09-0090. [DOI] [PubMed] [Google Scholar]
- 3.Shaheen RM, Davis DW, Liu W, Zebrowski BK, Wilson MR, Bucana CD, McConkey DJ, McMahon G, Ellis LM. Antiangiogenic therapy targeting the tyrosine kinase receptor for vascular endothelial growth factor receptor inhibits the growth of colon cancer liver metastasis and induces tumor and endothelial cell apoptosis. Cancer Res. 1999;59:5412–5416. [PubMed] [Google Scholar]
- 4.Shaheen RM, Tseng WW, Davis DW, Liu W, Reinmuth N, Vellagas R, Wieczorek AA, Ogura Y, McConkey DJ, Drazan KE, Bucana CD, McMahon G, Ellis LM. Tyrosine kinase inhibition of multiple angiogenic growth factor receptors improves survival in mice bearing colon cancer liver metastases by inhibition of endothelial cell survival mechanisms. Cancer Res. 2001;61:1464–1468. [PubMed] [Google Scholar]
- 5.Laird AD, Cherrington JM. Small molecule tyrosine kinase inhibitors: clinical development of anticancer agents. Expert Opin Investig Drugs. 2003;12:51–64. doi: 10.1517/13543784.12.1.51. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy M. Antiangiogenesis drug promising for metastatic colorectal cancer. Lancet. 2003;361:1959. doi: 10.1016/S0140-6736(03)13603-3. [DOI] [PubMed] [Google Scholar]
- 7.Ortega J, Vigil CE, Chodkiewicz C. Current progress in targeted therapy for colorectal cancer. Cancer Control. 2010;17:7–15. doi: 10.1177/107327481001700102. [DOI] [PubMed] [Google Scholar]
- 8.Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, Chung DC, Sahani DV, Kalva SP, Kozin SV, Mino M, Cohen KS, Scadden DT, Hartford AC, Fischman AJ, Clark JW, Ryan DP, Zhu AX, Blaszkowsky LS, Chen HX, Shellito PC, Lauwers GY, Jain RK. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–147. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J. Clin. Oncol. 2007;25:1658–1664. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 10.Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 11.Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–2048. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 12.Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, Vega-Villegas ME, Eng C, Steinhauer EU, Prausova J, Lenz HJ, Borg C, Middleton G, Kroning H, Luppi G, Kisker O, Zubel A, Langer C, Kopit J, Burris HA 3rd. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008;26:2311–2319. doi: 10.1200/JCO.2007.13.1193. [DOI] [PubMed] [Google Scholar]
- 13.Sato T, Oshima T, Yoshihara K, Yamamoto N, Yamada R, Nagano Y, Fujii S, Kunisaki C, Shiozawa M, Akaike M, Rino Y, Tanaka K, Masuda M, Imada T. Overexpression of the fibroblast growth factor receptor-1 gene correlates with liver metastasis in colorectal cancer. Oncol Rep. 2009;21:211–216. [PubMed] [Google Scholar]
- 14.Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 15.Sonvilla G, Allerstorfer S, Heinzle C, Stattner S, Karner J, Klimpfinger M, Wrba F, Fischer H, Gauglhofer C, Spiegl-Kreinecker S, Grasl-Kraupp B, Holzmann K, Grusch M, Berger W, Marian B. Fibroblast growth factor receptor 3-IIIc mediates colorectal cancer growth and migration. Br J Cancer. 2010;102:1145–1156. doi: 10.1038/sj.bjc.6605596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henriksson ML, Edin S, Dahlin AM, Oldenborg PA, Oberg A, Van Guelpen B, Rutegard J, Stenling R, Palmqvist R. Colorectal cancer cells activate adjacent fibroblasts resulting in FGF1/FGFR3 signaling and increased invasion. Am J Pathol. 2011;178:1387–1394. doi: 10.1016/j.ajpath.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kono SA, Marshall ME, Ware KE, Heasley LE. The fibroblast growth factor receptor signaling pathway as a mediator of intrinsic resistance to EGFR-specific tyrosine kinase inhibitors in non-small cell lung cancer. Drug Resist Updat. 2009;12:95–102. doi: 10.1016/j.drup.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Renhowe PA, Pecchi S, Shafer CM, Machajewski TD, Jazan EM, Taylor C, Antonios-McCrea W, McBride CM, Frazier K, Wiesmann M, Lapointe GR, Feucht PH, Warne RL, Heise CC, Menezes D, Aardalen K, Ye H, He M, Le V, Vora J, Jansen JM, Wernette-Hammond ME, Harris AL. Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: a novel class of receptor tyrosine kinase inhibitors. J Med Chem. 2009;52:278–292. doi: 10.1021/jm800790t. [DOI] [PubMed] [Google Scholar]
- 19.Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J. Clin. Oncol. 2010;28:1254–1261. doi: 10.1200/JCO.2009.24.6116. [DOI] [PubMed] [Google Scholar]
- 20.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 21.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 22.Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008;26:5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 23.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 24.Lee J, Lee I, Han B, Park JO, Jang J, Park C, Kang WK. Effect of simvastatin on cetuximab resistance in human colorectal cancer with KRAS mutations. J Natl Cancer Inst. 2011;103:674–688. doi: 10.1093/jnci/djr070. [DOI] [PubMed] [Google Scholar]
- 25.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 26.Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, Settleman J, Giovannini M, Jacks T. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–608. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouche O, Landi B, Louvet C, Andre T, Bibeau F, Diebold MD, Rougier P, Ducreux M, Tomasic G, Emile JF, Penault-Llorca F, Laurent-Puig P. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008;26:374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 28.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 29.Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, Heckel A, Rettig WJ. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68:4774–4782. doi: 10.1158/0008-5472.CAN-07-6307. [DOI] [PubMed] [Google Scholar]
- 30.Yeh JJ, Routh ED, Rubinas T, Peacock J, Martin TD, Shen XJ, Sandler RS, Kim HJ, Keku TO, Der CJ. KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther. 2009;8:834–843. doi: 10.1158/1535-7163.MCT-08-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliveira C, Pinto M, Duval A, Brennetot C, Domingo E, Espin E, Armengol M, Yamamoto H, Hamelin R, Seruca R, Schwartz S Jr. BRAF mutations characterize colon but not gastric cancer with mismatch repair deficiency. Oncogene. 2003;22:9192–9196. doi: 10.1038/sj.onc.1207061. [DOI] [PubMed] [Google Scholar]
- 32.Squires M, Ward G, Saxty G, Berdini V, Cleasby A, King P, Angibaud P, Perera T, Fazal L, Ross D, Jones CG, Madin A, Benning RK, Vickerstaffe E, O’Brien A, Frederickson M, Reader M, Hamlett C, Batey MA, Rich S, Carr M, Miller D, Feltell R, Thiru A, Bethell S, Devine LA, Graham BL, Pike A, Cosme J, Lewis EJ, Freyne E, Lyons J, Irving J, Murray C, Newell DR, Thompson NT. Potent, selective inhibitors of fibroblast growth factor receptor define fibroblast growth factor dependence in preclinical cancer models. Mol Cancer Ther. 2011;10:1542–1552. doi: 10.1158/1535-7163.MCT-11-0426. [DOI] [PubMed] [Google Scholar]
- 33.Lee SH, Lopes de Menezes D, Vora J, Harris A, Ye H, Nordahl L, Garrett E, Samara E, Aukerman SL, Gelb AB, Heise C. In vivo target modulation and biological activity of CHIR-258, a multitargeted growth factor receptor kinase inhibitor, in colon cancer models. Clin Cancer Res. 2005;11:3633–3641. doi: 10.1158/1078-0432.CCR-04-2129. [DOI] [PubMed] [Google Scholar]
- 34.Oricchio E, Wendel HG. Functional genomics lead to new therapies in follicular lymphoma. Ann N Y Acad Sci. 2013;1293:18–24. doi: 10.1111/nyas.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herath NI, Spanevello MD, Doecke JD, Smith FM, Pouponnot C, Boyd AW. Complex expression patterns of Eph receptor tyrosine kinases and their ephrin ligands in colorectal carcinogenesis. Eur J Cancer. 2012;48:753–762. doi: 10.1016/j.ejca.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, Ullrich RT, Menon R, Maier S, Soltermann A, Moch H, Wagener P, Fischer F, Heynck S, Koker M, Schottle J, Leenders F, Gabler F, Dabow I, Querings S, Heukamp LC, Balke-Want H, Ansen S, Rauh D, Baessmann I, Altmuller J, Wainer Z, Conron M, Wright G, Russell P, Solomon B, Brambilla E, Brambilla C, Lorimier P, Sollberg S, Brustugun OT, Engel-Riedel W, Ludwig C, Petersen I, Sanger J, Clement J, Groen H, Timens W, Sietsma H, Thunnissen E, Smit E, Heideman D, Cappuzzo F, Ligorio C, Damiani S, Hallek M, Beroukhim R, Pao W, Klebl B, Baumann M, Buettner R, Ernestus K, Stoelben E, Wolf J, Nurnberg P, Perner S, Thomas RK. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Billerey C, Chopin D, Aubriot-Lorton MH, Ricol D, Gil Diez de Medina S, Van Rhijn B, Bralet MP, Lefrere-Belda MA, Lahaye JB, Abbou CC, Bonaventure J, Zafrani ES, van der Kwast T, Thiery JP, Radvanyi F. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol. 2001;158:1955–1959. doi: 10.1016/S0002-9440(10)64665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jang JH, Shin KH, Park JG. Mutations in fibroblast growth factor receptor 2 and fibroblast growth factor receptor 3 genes associated with human gastric and colorectal cancers. Cancer Res. 2001;61:3541–3543. [PubMed] [Google Scholar]
- 39.Matsuda Y, Hagio M, Seya T, Ishiwata T. Fibroblast growth factor receptor 2 IIIc as a therapeutic target for colorectal cancer cells. Mol Cancer Ther. 2012;11:2010–2020. doi: 10.1158/1535-7163.MCT-12-0243. [DOI] [PubMed] [Google Scholar]
- 40.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S, Bardelli A. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67:2643–2648. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 41.Laurent-Puig P, Lievre A, Blons H. Mutations and response to epidermal growth factor receptor inhibitors. Clin Cancer Res. 2009;15:1133–1139. doi: 10.1158/1078-0432.CCR-08-0905. [DOI] [PubMed] [Google Scholar]
- 42.Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14:639–647. doi: 10.1016/j.tcb.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 43.Hao H, Muniz-Medina VM, Mehta H, Thomas NE, Khazak V, Der CJ, Shields JM. Context-dependent roles of mutant B-Raf signaling in melanoma and colorectal carcinoma cell growth. Mol Cancer Ther. 2007;6:2220–2229. doi: 10.1158/1535-7163.MCT-06-0728. [DOI] [PubMed] [Google Scholar]
- 44.Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L, Wang WV, Vecchione L, de Vriendt V, Weinstein BJ, Bronson RT, Tejpar S, Xavier RJ, Engelman JA, Martin ES, Hung KE. Concomitant BRAF and PI3K/mTOR Blockade Is Required for Effective Treatment of BRAFV600E Colorectal Cancer. Clin Cancer Res. 2013;19:2688–2698. doi: 10.1158/1078-0432.CCR-12-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng S, Shao L, Yu W, Gavine P, Ittmann M. Targeting Fibroblast Growth Factor Receptor Signaling Inhibits Prostate Cancer Progression. Clin Cancer Res. 2012;18:3880–8. doi: 10.1158/1078-0432.CCR-11-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sweeny L, Liu Z, Lancaster W, Hart J, Hartman YE, Rosenthal EL. Inhibition of fibroblasts reduced head and neck cancer growth by targeting fibroblast growth factor receptor. Laryngoscope. 2012;122:1539–1544. doi: 10.1002/lary.23266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ellis LM. Angiogenesis and its role in colorectal tumor and metastasis formation. Semin Oncol. 2004;31:3–9. doi: 10.1053/j.seminoncol.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 48.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 49.Saylor PJ, Escudier B, Michaelson MD. Importance of fibroblast growth factor receptor in neovascularization and tumor escape from antiangiogenic therapy. Clin Genitourin Cancer. 2012;10:77–83. doi: 10.1016/j.clgc.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 50.Wild R, Fager K, Flefleh C, Kan D, Inigo I, Castaneda S, Luo F, Camuso A, McGlinchey K, Rose W. Cetuximab preclinical antitumor activity (monotherapy and combination based) is not predicted by relative total or activated epidermal growth factor receptor tumor expression levels. Mol Cancer Ther. 2006;5:104–113. doi: 10.1158/1535-7163.MCT-05-0259. [DOI] [PubMed] [Google Scholar]