

Abstract
Myeloid cell leukemia-1 (Mcl-1) is a highly expressed anti-apoptotic Bcl-2 protein in cancer. Therefore, inhibition of its expression induces apoptosis in cancer cells and enhances sensitivity to cancer treatment. The aims of this study were to evaluate whether Mcl-1 affects the oncogenic behaviors of colorectal cancer cells, and to document the relationship of its expression with various clinicopathological parameters in colorectal cancer. Mcl-1 knockdown induced apoptosis by activating cleaved caspase-3 and -9, and increasing the expression of the pro-apoptotic protein, PUMA. Mcl-1 knockdown induced cell cycle arrest by decreasing cyclin D1, CDK4 and 6, and by increasing p27 expression. Mcl-1 knockdown decreased both endothelial cell invasion and tube formation, and decreased the expression of VEGF. The phosphorylation level of STAT3 was decreased by Mcl-1 knockdown. The mean apoptotic index value of Mcl-1 positive tumors was significantly lower than that of Mcl-1 negative tumors. The mean microvessel density value of Mcl-1 positive tumors was significantly higher than that of negative tumors. Mcl-1 expression was significantly increased in colorectal cancer, also associated with tumor stage, lymph node metastasis, and poor survival. These results indicate Mcl-1 is associated with tumor progression through its inhibition of apoptosis and enhancement of angiogenesis in colorectal cancer.
Keywords: Myeloid cell leukemia-1, apoptosis, angiogenesis, prognosis, colorectal neoplasm
Introduction
Colorectal cancer is a common cause of cancer-related morbidity and mortality worldwide [1]. Despite recent advances in diagnosis and treatment, invasion and metastasis remain the major contributors of cancer-related morbidity and mortality in colorectal cancer [1]. Therefore, it is of great importance to understand the molecular and biological mechanisms of colorectal cancer progression, such as invasion and metastasis, to develop more effective cancer surveillance and therapy strategies.
Cancer progression is a very complex process, including tumor cell transformation, growth, invasion, angiogenesis, dissemination and survival in circulation, and subsequent adhesion and colonization in the distant organ or tissue. Among these events, aberrant cell apoptosis, proliferation, and angiogenesis play crucial roles in the growth and dissemination of tumors, and promote cancer progression [2-4].
The B cell leukemia/lymphoma (Bcl)-2 family proteins are the main regulators of the apoptotic process implicated in the intrinsic mitochondrial pathway and are divided into anti-apoptotic and pro-apoptotic members based on their structural and functional properties. The anti-apoptotic Bcl-2 proteins include Bcl-2, Bcl-xL, Bcl-w, myeloid cell leukemia-1 (Mcl-1) and A1, whereas pro-apoptotic Bcl-2 proteins include Bax and Bak with multiple Bcl-2 homology (BH) domains, and Bad, Bid, Bim, Bik, NOXA, and PUMA with only BH3 domains. The balance between the levels of anti-apoptotic and pro-apoptotic proteins determines the survival or death of the cells [5]. Any mechanism breaking down this balance in normal apoptosis pathways may contribute to a number of pathologic diseases. Increased expression of anti-apoptotic Bcl-2 proteins occurs in significant subsets of human cancers [6-8].
Mcl-1 is an anti-apoptotic Bcl-2 protein with a very short half-life and is highly expressed in a variety of human cancers. Increased expression of Mcl-1 has been shown to contribute to carcinogenesis and result in the inhibition of apoptosis and cell cycle progression, promotion of cancer cell replication, invasion, metastasis and chemoresistance [9-15]. Furthermore, the increased expression of Mcl-1 was found to be an indicator of cancer progression and poor outcomes in several human cancers [16-19].
The aims of current study were to investigate the possible roles of Mcl-1 in human colorectal cancer cell apoptosis, proliferation, and angiogenesis, and to examine its correlation with clinicopathological features including prognosis.
Materials and methods
Patients and tissue samples
This study was approved by the Institutional Review Board of Chonnam National University Hwasun Hospital (Jeonnam, Korea). Fresh colorectal cancer tissues, paired normal tissues, and metastatic or nonmetastatic lymph node tissues from same patients were obtained in colonoscopic biopsy specimens and surgical specimens. The colorectal cancer tissues were obtained from 155 consecutive patients who underwent surgery for colorectal cancer at Chonnam National University Hwasun Hospital between January 2004 and December 2004. None of the patients had received chemotherapy or irradiation before surgery. The tumors were staged at the time of surgery using the American Joint Committee on Cancer’s standard criteria for TNM staging [20]. Survival was measured from the time of surgery until the final follow-up by December 31, 2011.
Cell culture and siRNA transfection
Human colorectal cancer cell lines, Caco2 and DLD1 were obtained from the American Type Culture Collection Line, Inc. and maintained in DMEM (Hyclone, Loan, UT, USA) supplemented with 10% fetal bovine serum (Hyclone). For the tube formation assay, human umbilical vein endothelial cells (HUVECs, LONZA, Walkersville, MD, USA) were maintained in EGM®-2 MV SingleQuots® (LONZA, Walkersville, MD, USA). The Mcl-1 and scramble small interfering RNA (siRNA) duplexes used in this study were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Qiagen (MD, USA), respectively. Mcl-1 siRNA and scramble siRNA were transfected with lipofectamineTM RNAiMAX (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s recommendations.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted using the Trizol reagent (Invitrogen). RT-PCR was performed using MMLV transcription reagents (Invitrogen), according to the manufacturer’s recommendations. PCR amplification of cDNA was performed using gene-specific primers and Go Taq® DNA polymerase (Promega, Madison, WI, USA). The following specific primers were used; Mcl-1 5’-TCCTCTTGCCACTTGCTTTT-3’/ 5’-TGCTGGAGTAGGAGCTGGTT-3’; GAPDH 5’-ACCACAGTCCATGCCATCAC-3’/5’-TCCACCACCCTGTTGCTGTA-3’. Reaction products were analyzed on 1.2% agarose gels, and the bands were visualized by ethidium bromide.
Western blotting
Isolated proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel and transferred to PVDF membrane (Millipore, Billerica, MA, USA). The specific proteins were blotted with a primary antibody. The following antibodies were used: antibodies against Mcl-1, vascular endothelial growth factor (VEGF), hypoxia-inducible factor-1α (HIF-1α) and GAPDH were purchased from Santa Cruz Biotechnology (CA, USA). Antibodies against cleaved caspases (3, 7, 9), Bcl-xL, Bax, Bak, PUMA, cyclin D1, cyclin-dependent kinase 4 (CDK4), CDK6, p27, phospho-signal transducers and activators of transcription-3 (phospho-STAT3), phospho-AKT, phospho-glycogen synthase kinase-3β (phospho-GSK3β), and mitogen-activated protein kinase (MAPK) sampler kit were purchased from Cell Signaling (Danvers, MA, USA). The target proteins were developed using the enhanced chemiluminescent substrate (Millipore) and the luminescent image analyzer LAS-4000 (Fujifilm, Tokyo, Japan).
Proliferation assay
Cell proliferation was measured with the EZ-CyTox (tetrazolium salts, WST-1) cell viability assay kit (Daeil Lab Inc., Seoul, Korea). The transfected cells were plated to a 96-well plate and treated with WST-1 reagent at 37°C. The absorbance intensity was measured using a microplate reader (Infinite M200, Tecan, Austria GmbH, Austria) with Magellan V6 data analysis software (Tecan).
Flow cytometric analysis
For apoptosis detection, the transfected cell suspensions were incubated in APC Annexin V and 7-amino-actinomycin D (7-AAD) (BD Biosciences, San Jose, CA, USA). For cell cycle analysis, cells were incubated in 10 μg/ml ribonuclease A (Sigma-Aldrich, St. Louis, MO, USA) and 50 μg/ml propidium iodide (PI). The BD Cell Quest® version 3.3 (Becton Dickinson, San José, CA, USA) and WinMDI version 2.9 (The Scripps Research Institute, San Diego, CA, USA) were used to analyze the population of Annexin-V positive cells and cell cycle.
Matrigel invasion assay
A Matrigel invasion assay was performed using a Matrigel (1 mg/ml, BD Bioscience) coated Transwell chamber (8 μm-pore, Costar, Cambridge, MA). The HUVECs were inoculated at a density of 1 × 105 cells in the upper Matrigel chamber. The lower chambers were filled with conditioned media (CM) from gene-transfected cells. After incubation for 16 h, the non-invaded cells on the upper membrane surface were removed with a cotton swab and the invaded cells on the bottom surface of the transwell were stained with Diff-Quik soln (Sysmex, Kobe, Japan). The numbers of invaded cells were counted in five random fields per sample. Data are expressed as mean ± standard deviation (SD) of the number of cells/field from 3 independent experiments.
Matrigel in vitro endothelial tube formation assay
Matrigel Basement Membrane Matrix (BD Bioscience) was used to coat culture plates according to the manufacturer’s instructions. About 50 μl of thawed Matrigel in 4°C was applied to each well of 96-multiwell plates and polymerized at 37°C for 1 h. Trypsinized HUVECs (3 × 104 cells/well) were seeded on the Matrigel substratum and incubated with cell-free CM from gene-transfected cells for 18 h in a CO2 incubator. After incubation, tube-like structures were photographed using an inverted microscope. The tube length and area quantification were determined using the WIMtube image analysis platform (WIMASIS GmbH, Munich, Germany) from the ibidi website (www.ibidi.com).
Immunohistochemistry
Paraffin tissue sections from patients were deparaffinized, rehydrated, and retrieved with retrieval buffer. To block the endogenous peroxidase activity, tissues were treated with a purchased peroxidase-blocking solution (Dako, Carpinteria, CA, USA) and were incubated with anti-human Mcl-1, Ki-67 (Dakopatts, Glostrup, Denmark), and CD34 (Novocstra Lab., Newcastle, UK) antibodies in a primary diluent solution (Invitrogen) overnight at 4°C. After washing in TBST, tissues were stained using the Dako RealTM Envision HRP/DAB detection system (Dako) and counterstained with hematoxylin. Stained tissues were scored and photographed using a light microscope.
Evaluation of Mcl-1 expression
Immunohistochemically-stained specimens were evaluated by two dependent observers without knowledge of the clinicopathological data. The degree of intensity of staining cancer cells were graded on a scale of four grades: 0, no staining of cancer cells; 1, weak staining; 2, moderate staining; and 3, strong staining. The percentage of stained cancer cells was also graded on a scale with four grades: 0, none; 1, < 25%; 2, 25-50%; 3, 51-75%; and 4, > 75%. The intensity rating was multiplied by the percent stain rating to obtain an overall score. The mean overall score for 155 tumors analyzed, was 6.0. Therefore, the mean overall score of 6.0 was chosen as the cut-off point to classify the status of Mcl-1 expression. The specimens with a score of more than 6 were regarded as positive for Mcl-1 expression, and those with a score ≤ 6 as a negative expression.
Assessment of tumor cell proliferation and apoptosis in tissue
Tumor proliferative cells were visualized by immunohistochemical staining using an anti-Ki-67 antibody (Dakopatts). A distinct nuclear immunoreactivity for Ki-67 was considered positive. The Ki-67 labeling index (KI) was presented as the number of Ki-67-positive nuclei per 1000 tumor cell nuclei. The determination of KI has been used to estimate the proliferative ability of tumor cells. To detect and quantify apoptosis, the DeadEndTM Colorimetric terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) System (Promega) was used, according to the instructions of the manufacturer. The number of positive staining cells with an apoptotic morphology were counted in five random fields per sample. The apoptotic index (AI) results were expressed as the number of positive nuclei, including apoptotic bodies, among 1000 tumor cell nuclei.
Determination of microvessel density
A detection procedure for microvessels was performed using a human anti-CD34 antibody (Novocstra Lab.). Microvessel quantification was performed according to an international consensus [21]. Any single brown-stained cell or cluster of endothelial cells that was clearly separate from adjacent microvessels, tumor cells, and other connective tissue elements was considered a vessel. The stained sections were screened at 4x magnification to identify the areas of the highest vascular density within the tumor. Vessels were counted in the 5 areas of highest vascular density at 200x magnification. Microvessel density (MVD) was expressed as the mean number of vessels in these areas.
Statistical analysis
In intergroup comparisons, student’s t test was used to determine statistical significance and data were presented as the mean ± SD. To compare Mcl-1 expression with various clinicopathological parameters, statistical analyses were performed using the χ2-test and Fisher exact test. The Kaplan-Meier method was used to survival analysis of patients with positive or negative Mcl-1 expression, and statistical significance was further tested by the log-rank test. The statistical analysis was performed with the Statistical Package for the Social Sciences (SPSS/PC + 15.0, Chicago, IL). P-values of less than 0.05 were considered to indicate statistical significance.
Results
Knockdown of Mcl-1 induces apoptosis in human colorectal cancer cells
To evaluate the impact of Mcl-1 knockdown on oncogenic behaviors of human colorectal cancer cells, all experiments were performed using siRNA-transfected cells. Mcl-1 gene expression in all tested cells showed a specific reduction in both the mRNA and protein levels by transfection of Mcl-1 siRNA (data not shown). To evaluate the impact of Mcl-1 on apoptosis, we performed flow cytometric analyses. The proportion of early apoptotic cells induced by transfection of Mcl-1 siRNA was greater than that induced by transfection of the scramble siRNA in Caco2 and DLD1 cells (16.7 vs. 10.9 and 20.3 vs. 12.2%, respectively) (Figure 1A). We further investigated caspase-specific activities to determine the activation of caspases, key enzymes in apoptosis, during the Mcl-1 knockdown. The cleaved caspase-3 and -9 expressions were up-regulated in the Caco2 and DLD1 cells after Mcl-1 knockdown (Figure 1B). We further examined whether Mcl-1 knockdown-induced apoptosis is associated with the modulation of apoptosis regulatory proteins. As shown in Figure 1C, Mcl-1 knockdown led to an increase in the pro-apoptotic protein, PUMA. However, levels of the anti-apoptotic protein, Bcl-xL and pro-apoptotic proteins, Bax and Bak were not altered in response to Mcl-1 knockdown.
Figure 1.
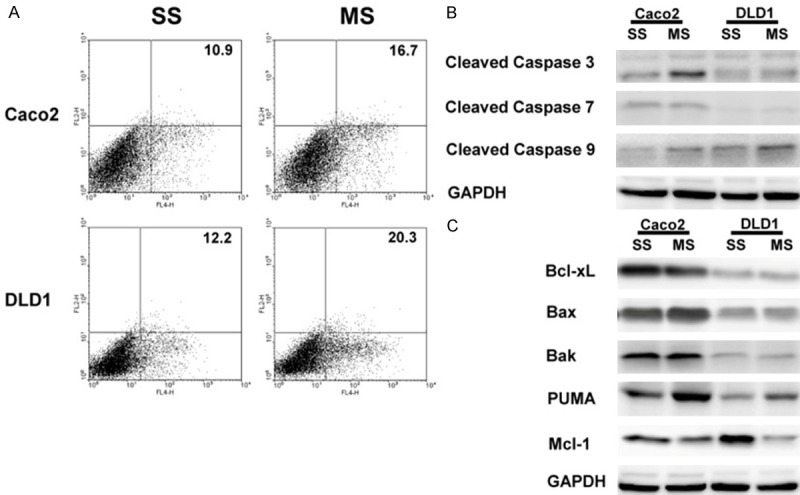
Knockdown of Mcl-1 induces apoptosis in human colorectal cancer cells. A. The proportion of early apoptotic cells induced by transfection of MS was greater than that induced by transfection of SS (16.7 vs. 11.0 and 20.3 vs. 12.2%, respectively) in Caco2 and DLD1 cells. B. Expression of cleaved caspase-3, -7 and -9 proteins. An increase in caspase-3 and -9 expressions was detected in Caco2 and DLD1 cells after Mcl-1 knockdown. C. Expression of apoptosis regulatory proteins. Mcl-1 knockdown led to an increase in the pro-apoptotic protein, PUMA. However, levels of the anti-apoptotic protein, Bcl-xL and pro-apoptotic proteins, Bax and Bak were not altered in response to the Mcl-1 knockdown. One representative experiment of the three independent experiments is shown. SS; scramble siRNA, MS; Mcl-1 siRNA.
Knockdown of Mcl-1 induces cell cycle arrest in human colorectal cancer cells
To detect whether Mcl-1 could change cell cycle distribution, we performed flow cytometric analyses. Mcl-1 knockdown induced cell cycle arrest at the G2/M phase in Caco2 and DLD1 cells (Figure 2A). Next, we evaluated the effects of Mcl-1 on positive regulators including cyclins and CDKs, and negative regulators, including CDK inhibitors (CDKIs), involved in cell cycle progression in human colorectal cancer cells. As shown in Figure 2B, the cyclin D1 and CDK6 protein levels were significantly decreased, and the CDKI, p27 protein level was significantly increased by Mcl-1 knockdown in the Caco2 and DLD1 cells. The CDK4 protein level was significantly decreased by Mcl-1 knockdown in Caco2 cells.
Figure 2.
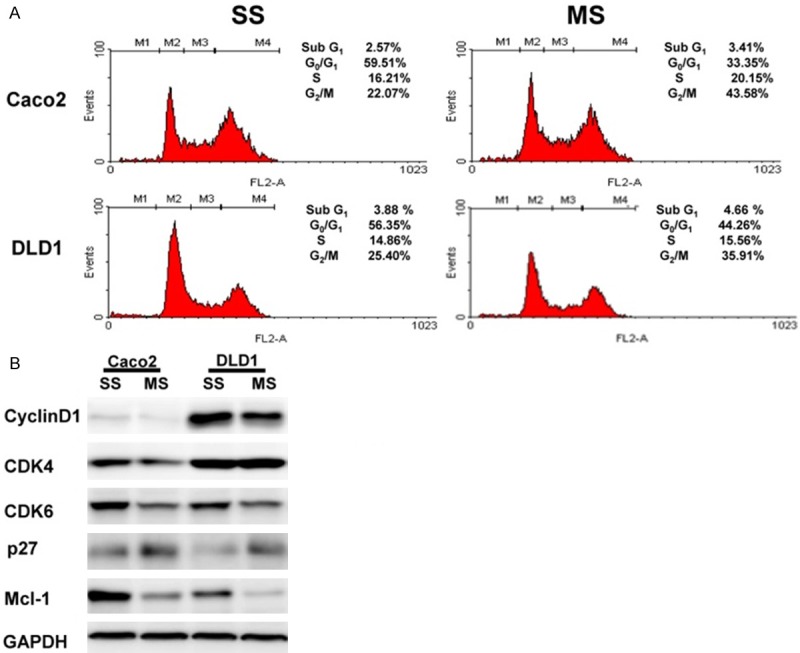
Knockdown of Mcl-1 induces cell cycle arrest in human colorectal cancer cells. A. Cell cycle analysis demonstrated that Mcl-1 knockdown induced cell cycle arrest of the G2/M phase in Caco2 and DLD1 cells. B. Expression of cyclins, CDK, and CDKI proteins. The cyclin D1 and CDK6 protein levels were significantly decreased, and the p27 protein level was significantly increased by Mcl-1 knockdown in Caco2 and DLD1 cells. The CDK4 protein level was significantly decreased by Mcl-1 knockdown in Caco2 cells. SS; scramble siRNA, MS; Mcl-1 siRNA, CDK; cyclin-dependent kinase, CDKI; cyclin-dependent kinase inhibitor.
The impact of Mcl-1 knockdown on proliferation of human colorectal cancer cells
To determine the potential Mcl-1 effects on cell proliferation, a cell proliferation assay was performed 1, 2, and 3 days after a transfection with Mcl-1 siRNA. However, in all tested cells, there was no significant difference of cell proliferation between Mcl-1 siRNA-transfected cells and the scramble siRNA-transfected cells at 1, 2, and 3 days (Figure 3).
Figure 3.
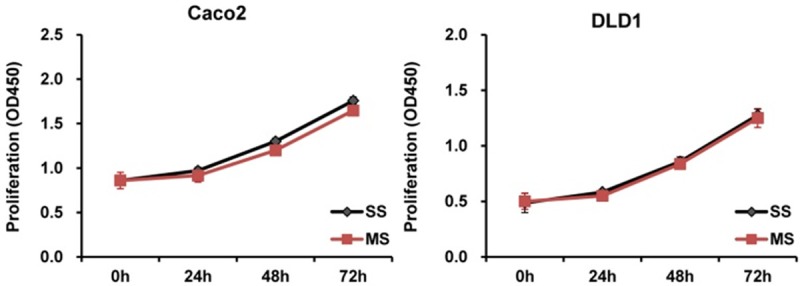
The impact of Mcl-1 knockdown on proliferation of human colorectal cancer cells. In all tested cells, there was no significant difference of cell proliferation between Mcl-1 siRNA-transfected cells and the scramble siRNA-transfected cells at 1, 2, or 3 days. SS; scramble siRNA, MS; Mcl-1 siRNA.
The impact of Mcl-1 knockdown on angiogenesis of human colorectal cancer cells
Solid tumors recruit new blood vessels for growth, maintenance and metastasis. Endothelial cell tube formation and invasiveness are critical in angiogenesis. To evaluate the effects of Mcl-1 to induce angiogenesis in HUVECs, we performed matrigel invasion and tube formation assays using the conditioned media (CM) from Mcl-1 and scramble siRNA-transfected Caco2 and DLD1 cells. The invasion of HUVECs was significantly decreased in the CM of Mcl-1 siRNA-transfected Caco2 and DLD1 cells, compared to the scramble siRNA-transfected cells (p = 0.042 and < 0.001, respectively) (Figure 4A). The results of HUVECs tube formation showed that CM from Mcl-1 siRNA-transfected Caco2 and DLD1 cells inhibited the formation of endothelial tubes, compared to CM from the scramble siRNA-transfected cells (p = 0.045 and 0.020, respectively) (Figure 4B). Mcl-1 knockdown led to a decrease of the angiogenic inducer, VEGF, in all tested cells. But the another angiogenic inducer, HIF-α, was not altered in response to Mcl-1 knockdown in all tested cells (Figure 4C).
Figure 4.
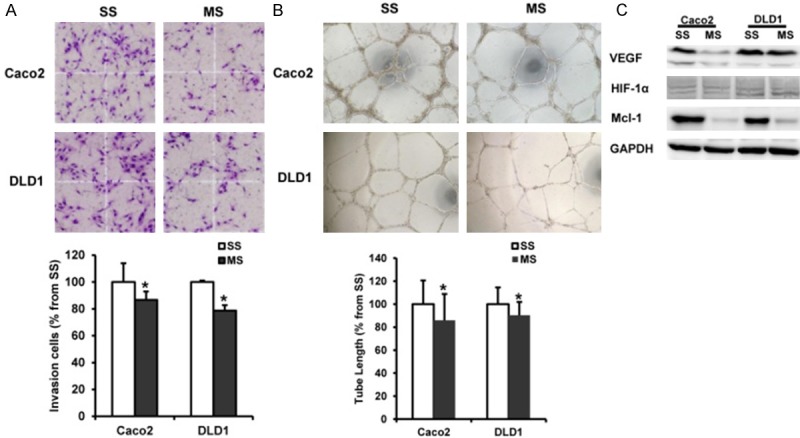
The impact of Mcl-1 knockdown on angiogenesis of human colorectal cancer cells. A. The Invasion of HUVECs was significantly decreased in the conditioned medium (CM) from Mcl-1 siRNA-transfected Caco2 and DLD1 cells compared to the scramble siRNA-transfected cells. B. CM from Mcl-1 siRNA-transfected Caco2 and DLD1 cells inhibited the tube formation of HUVECs compared to CM from the scramble siRNA-transfected cells. C. Mcl-1 knockdown led to a decrease of the angiogenic inducer, VEGF, in Caco2 and DLD1 cells. However, the another angiogenic inducer, HIF-1α was not altered in response to Mcl-1 knockdown in all tested cells. SS; scramble siRNA, MS; Mcl-1 siRNA, VEGF; vascular endothelial growth factor, HIF-1α; Hypoxia inducible factor-1α; *p < 0.05 versus control.
The impact of Mcl-1 knockdown on intracellular signaling pathways involved in transcriptional regulation of Mcl-1 expression in human colorectal cancer cells
To observe the contribution of specific intracellular signaling pathways involved in the regulation of Mcl-1 expression in human colorectal cancer cells, we determined the phosphorylation level of STAT3, AKT, GSK3β, and MAPK cascades signaling proteins using western blotting. The phosphorylation level of STAT3 was decreased by Mcl-1 knockdown Caco2 and DLD1 cells. But the phosphorylation levels of AKT, GSK3β, p38, and ERK1/2 showed no change when tested by the Mcl-1 knockdown (Figure 5).
Figure 5.
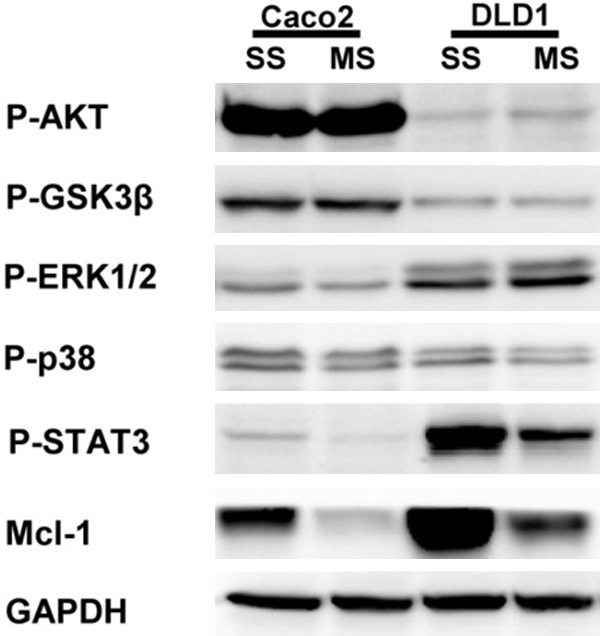
The impact of Mcl-1 knockdown on intracellular signaling pathways involved in regulation of Mcl-1 expression in human colorectal cancer cells. The phosphorylation level of STAT3 was decreased by Mcl-1 knockdown in Caco2 and DLD1 cells. But the phosphorylation levels of AKT, GSK3β, p38, and ERK1/2 showed no change with the Mcl-1 knockdown. SS; scramble siRNA, MS; Mcl-1 siRNA.
Expression of Mcl-1 is increased in human colorectal cancer and metastatic lymph node tissues compared to normal colorectal mucosa and non-metastatic lymph node tissues
To confirm the results of the colorectal cancer cell line study, we evaluated the expression of Mcl-1 at the mRNA and protein levels by RT-PCR and immunohistochemistry in human colorectal cancer tissues, and compared the results with normal mucosa, and metastatic or non-metastatic lymph node tissue of same patient taken by colonoscopic biopsy and surgical specimens. We confirmed the up-regulation of Mcl-1 expression in cancer tissues compared to paired normal mucosa at mRNA level (p = 0.011) (Figure 6A). In the colorectal cancer tissues, the immunostaining of the Mcl-1 protein was predominantly identified in the cytoplasm of cancer cells and not detectable in the tumor stroma. Immunostaining of Mcl-1 protein showed weak to no staining in the normal colorectal mucosa. The immunostaining of Mcl-1 in metastatic lymph node tissues was significantly stronger than that in the non-metastatic lymph node tissues (Figure 6B). The overall score for immunostaining of Mcl-1 in metastatic lymph node tissues was significantly higher than that in the non-metastatic lymph node tissues (p = 0.015) (Figure 6C).
Figure 6.
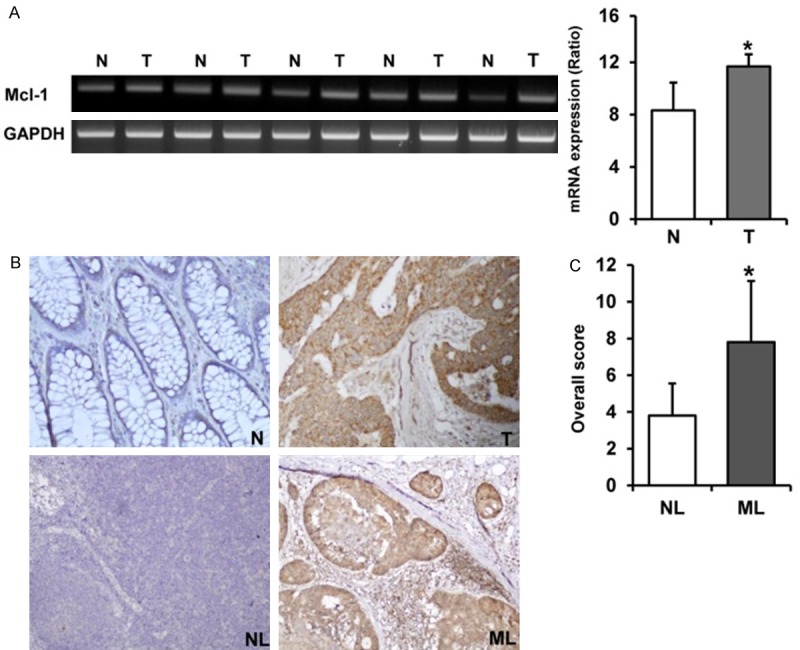
Expression of Mcl-1 is increased in human colorectal cancer and metastatic lymph node tissues compared to normal gastric mucosa and non-metastatic lymph node tissues. A. Expression of Mcl-1 is up-regulated in cancer tissues compared to paired normal mucosa at the mRNA level in fresh colonoscopic biopsy specimens. Each bar represents the mean ± SE of 20 cases. *p < 0.05 vs. normal colorectal mucosa. B. In the colorectal cancer tissues, the immunostaining of Mcl-1 protein was predominantly observed in the cytoplasm of cancer cells and not detectable in the tumor stroma. The Mcl-1 protein showed no or weak immunostaining in the normal colorectal mucosa. The immunostaining of Mcl-1 in metastatic lymph node tissues was significantly stronger than that in non-metastatic lymph node tissues (200x). C. The overall score for immunostaining of Mcl-1 in metastatic lymph node tissues was significantly higher than that in non-metastatic lymph node tissues (*p < 0.05). T; Colorectal cancer tissue, N; Paired normal colorectal mucosa, NL; Non-metastatic lymph node tissue, ML; Metastatic lymph node tissue.
Expression of Mcl-1 is associated with poorer prognosis in human colorectal cancer
To study the prognostic role of Mcl-1 in human colorectal cancer progression, we investigated the expression of the Mcl-1 protein immunohistochemically in formalin-fixed, paraffin-embedded tissue blocks obtained from 155 colorectal cancer patients with clinicopathological data. We analyzed survival rates and the correlation between Mcl-1 immunostaining and clinicopathological parameters. Mcl-1 immunostaining was significantly associated with tumor stage and lymph node metastasis (p < 0.001 and p < 0.001, respectively) (Table 1). Moreover, the overall survival rate for patients with positive Mcl-1 immunostaining was significantly lower than that for patients without (p = 0.029) (Figure 7).
Table 1.
Correlation between Mcl-1 expression and the clinicopathological parameters of human colorectal cancer
| Mcl-1 expression | ||||
|---|---|---|---|---|
|
|
||||
| Parameters | Total (n = 155) | Negative (n = 84) | Positive (n = 71) | P-value |
| Age (years) | 0.242 | |||
| < 66.3 | 69 | 41 | 28 | |
| ≥ 66.3 | 86 | 43 | 43 | |
| Sex | 0.292 | |||
| Male | 90 | 52 | 38 | |
| Female | 65 | 32 | 33 | |
| Tumor size (cm) | 0.245 | |||
| < 5.1 | 95 | 55 | 40 | |
| ≥ 5.1 | 60 | 29 | 31 | |
| Stage | < 0.001 | |||
| I | 19 | 15 | 4 | |
| II | 57 | 40 | 17 | |
| III | 69 | 26 | 43 | |
| IV | 10 | 3 | 7 | |
| Perineuronal invasion | 0.537 | |||
| Negative | 115 | 64 | 51 | |
| Positive | 40 | 20 | 20 | |
| Lymphovascular invasion | 0.187 | |||
| Negative | 96 | 56 | 40 | |
| Positive | 59 | 28 | 31 | |
| Histologic type | 0.280 | |||
| WD | 90 | 45 | 45 | |
| MD | 47 | 25 | 22 | |
| PD | 6 | 5 | 1 | |
| Mucinous | 11 | 8 | 3 | |
| Signet | 1 | 1 | 0 | |
| Depth of invasion (T) | 0.087 | |||
| T1 | 7 | 5 | 2 | |
| T2 | 20 | 11 | 9 | |
| T3 | 120 | 67 | 53 | |
| T4 | 8 | 1 | 7 | |
| Lymph node metastasis (N) | < 0.001 | |||
| N0 | 78 | 55 | 23 | |
| N1-3 | 77 | 29 | 48 | |
| Distant metastasis (M) | 0.112 | |||
| M0 | 145 | 81 | 64 | |
| M1 | 10 | 3 | 7 | |
WD: well differentiated; MD: moderately differentiated; PD: poorly differentiated.
Figure 7.
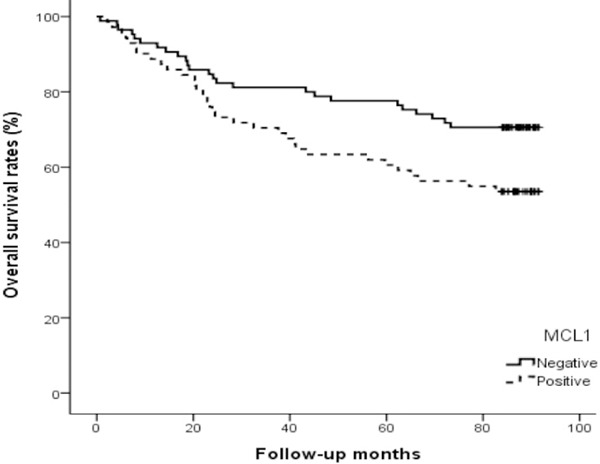
Kaplan-Meier survival curve correlating overall survival with positive expression (solid line) and negative expression (dotted line) of Mcl-1. The overall survival for patients with positive Mcl-1 immunostaining was significantly lower than that for patients without (p = 0.029).
Correlation between the expression of Mcl-1 and tumor cell apoptosis, proliferation or angiogenesis in human colorectal cancers
The standard morphologic criteria for identifying apoptotic cells using the TUNEL assay are the beaded or shrunken chromatin and an apoptotic body with a clear halo (Figure 8A). The AI for the 155 tumors ranged from 1.7 to 30.0 with a mean AI of 8.7 ± 5.7. The mean AI value of Mcl-1 positive tumors was 6.4 ± 2.8 and was significantly lower than the AI of Mcl-1 negative tumors (p = 0.020) (Table 2). Ki-67 immunoreactivity was predominantly found in the nuclei of cancer cells (Figure 8B). The KI for the 155 tumors ranged from 21.9 to 85.4 with a mean KI of 52.4 ± 14.1. There was no significant difference between Mcl-1 expression and KI (p = 0.215) (Table 2). Any single brown-stained cell or cluster of endothelial cells that was clearly separate from adjacent microvessels, tumor cells, and other connective tissue elements was considered a vessel (Figure 8C). The MVD for 155 tumors ranged from 50.0 to 295.0 with a mean MVD of 127.5 ± 50.5. The mean MVD value of Mcl-1 positive tumors was 141.2 ± 55.2 and was significantly higher than the MVD of Mcl-1 negative tumors (p = 0.044) (Table 2).
Figure 8.
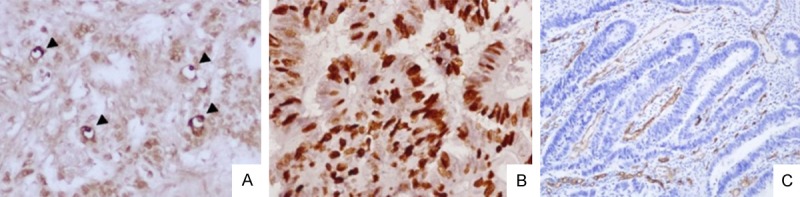
Assessment of tumor cell apoptosis, proliferation or angiogenesis in human colorectal cancers. A. Detection of apoptotic cells by TUNEL staining. Standard morphologic criteria for apoptotic cells using TUNEL assay are the presence of beaded or shrunken chromatin and apoptotic body with a clear halo (arrowhead). B. Immunostaining of Ki-67. Ki-67 immunoreactivity was predominantly found in the nuclei of cancer cells. C. Immunostaining of CD34. Any single brown-stained cell or cluster of endothelial cells that were clearly separate from adjacent microvessels, tumor cells, and other connective tissue elements were considered vessels. TUNEL, terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick end labeling.
Table 2.
Correlation between Mcl-1 expression and tumor cell apoptosis, proliferation, or angiogenesis in human colorectal cancers
| Mcl-1 expression | P-value | |||
|---|---|---|---|---|
|
|
||||
| Parameters | Total (n = 155) | Negative (n = 84) | Positive (n = 71) | |
| AI (Mean ± SD) | 8.7 ± 5.7 | 11.8 ± 7.1 | 6.4 ± 2.8 | 0.020 |
| KI (Mean ± SD) | 52.4 ± 14.1 | 51.8 ± 11.3 | 52.8 ± 16.2 | 0.215 |
| MVD (Mean ± SD) | 127.5 ± 50.5 | 111.0 ± 39.3 | 141.2 ± 55.2 | 0.044 |
AI: Apoptotic index; KI: Ki-67 labeing index; MVD: Microvessel density; SD: Standard deviation.
Discussion
Cancer results from the accumulation of alterations in genes that participate in the regulation of many normal cell processes, including proliferation, adhesion, migration, invasion, apoptosis, cell cycle progression and angiogenesis [2-4].
Bcl-2 family proteins are the key activators of mitochondrial apoptotic processes and include anti-apoptotic and pro-apoptotic members [5-8]. Mcl-1 is an anti-apoptotic member of the Bcl-2 family proteins and is highly amplified in a variety of human cancers, suggesting that its function may contribute to malignant cell growth and evasion of apoptosis [9-15].
We first investigated whether gene silencing of Mcl-1 using siRNA affects tumor cell behaviors in human colorectal cancer cells. Our study showed that Mcl-1 knockdown induces apoptosis and cell cycle arrest, while suppressing angiogenesis in human colorectal cancer cells. These results indicate that Mcl-1 expression is associated with the invasive and oncogenic phenotypes of human colorectal cancer cells.
In the apoptotic process, the expression of cleaved caspase-3 and -9 was up-regulated in human colorectal cancer cells after Mcl-1 knockdown. Mcl-1 knockdown also led to an increase in the pro-apoptotic protein, PUMA. However, levels of the anti-apoptotic protein, Bcl-xL and pro-apoptotic proteins, Bax and Bak were not altered in response to Mcl-1 knockdown. Previously, it was determined that PUMA inhibits the anti-apoptotic proteins of Bcl-2 family including Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and A1, and directly triggers apoptosis mediated by pro-apoptotic proteins Bax and Bak [9-15,22]. Therefore, apoptosis induced by the Mcl-1 knockdown may be achieved either by inhibiting Mcl-1 expression or by enhancing expression of its endogenous inhibitor, PUMA in human colorectal cancer cells. Indeed, the pro-apoptotic BH3-only proteins, including PUMA, compete against the pro-apoptotic proteins Bak and Bax to bind to Mcl-1 [9-15,22].
Cell proliferation involves successive cycles of DNA replication and the cell cycle. Orderly progression of the cell cycle is governed by a family of cyclins and CDKs, which are restrictively counterbalanced by CDKIs. Disturbances in cell cycle regulation are of major importance in carcinogenesis [23]. Our study showed that Mcl-1 knockdown induced cell cycle arrest at the G2/M phase by decreasing cyclin and CDKs, and by increasing CDKI, p27 expression. Deregulation of cyclin, CDKs, and CDKIs is commonly observed and associated with neoplastic formation in many types of human cancers, including colon cancer [24-26]. Also, Mcl-1 has been shown to regulate cell proliferation through its interactions with CDK1 and proliferating cell nuclear antigen [27-29]. These results suggest that Mcl-1 has a potential role in the determination of cell fate by regulating the cell cycle as well as inhibiting apoptosis in human colon cancer.
Angiogenesis has been shown to be a critical aspect of tumor growth and metastasis. The induction of angiogenesis by a tumor is controlled process, influenced by angiogenic and angiostatic factors involving a complex interaction between the tumor and endothelial cells [30]. Our study showed that the Mcl-1 knockdown decreased both endothelial cell invasion and tube formation of HUVECs, key events in angiogenesis and neovascularization. Also, Mcl-1 knockdown led to a decrease of the angiogenic inducer, VEGF, in human colorectal cancer cells. Previously, VEGF has been shown to up-regulate Mcl-1 expression and protect cells against the apoptosis in multiple myeloma and prostate cancer [31,32]. These results showed that Mcl-1 may play an important role in tumor angiogenesis in concert with VEGF in human colorectal cancer.
PI3K/AKT, MAPK, GSK-3β, and STAT signaling pathways play a pivotal role in the tumorigenesis, enhanced cell proliferation, angiogenesis, and inhibition of apoptosis in various human cancers [33-36]. Mcl-1 is tightly regulated at the levels of transcription, translation, and posttranslational modification for its maintenance [9-15]. In our study, the phosphorylation level of STAT3 was decreased by Mcl-1 knockdown in colorectal cancer cells. But the phosphorylation levels of PI3K/AKT, GSK3β, and MAPK showed no change by Mcl-1 knockdown. Previously, it was found that STAT3 is often constitutively activated in human cancer cells and tissues where it promotes the development of human cancer by regulating tumor cell apoptosis, proliferation, and angiogenesis [37-39]. These results suggest that Mcl-1 may be associated with activation of STAT3 important for tumor cell survival and angiogenesis in human colorectal cancer.
Next, we evaluated the expression of Mcl-1 in colorectal cancer tissues and paired normal colorectal mucosa of same patients, and documented the expression of Mcl-1 in a well-defined series of human colorectal cancers, including long-term and complete follow-up, with special reference to patient prognosis. In our study, Mcl-1 expression was increased in human colorectal cancer and metastatic lymph node tissues, compared to normal colorectal mucosa and non-metastatic lymph node tissues. Also, Mcl-1 expression was significantly associated with tumor stage, lymph node metastasis, and poor survival. Previously, one report showed that Mcl-1 expression was correlated with tumor grade and stage, despite an overall decrease of Mcl-1 expression during cancer progression in human colorectal cancer [19]. These results suggest that Mcl-1 expression promotes cancer progression and helps in predicting poor clinical outcomes of human colorectal cancer.
Finally, we evaluated the correlation between Mcl-1 expression and tumor cell apoptosis, proliferation, and angiogenesis in human colorectal cancer tissues to confirm the results of human colorectal cancer cell line studies. In our study, the mean AI value of Mcl-1 positive tumors was significantly lower than that of Mcl-1 negative tumors. However, there was no significant difference between Mcl-1 expression and KI. And the mean MVD value of Mcl-1 positive tumors was significantly higher than that of Mcl-1 negative tumors. These results thus confirm the in vitro inhibition of apoptosis and enhancement of angiogenesis inducing potential of Mcl-1 to in vivo.
Taken together, Mcl-1 is associated with tumor progression via the inhibition of apoptosis and enhancement of angiogenesis in human colorectal cancer.
Acknowledgements
This work was supported by a grant (0720570) from the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea, and partly by research funds from the Research Institute of Clinical Medicine, Chonnam National University Hwasun Hospital in 2014 (HCRI 14028-21), Republic of Korea.
Disclosure of conflict of interest
The authors declare no competing financial interests.
References
- 1.Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490–1502. doi: 10.1016/S0140-6736(13)61649-9. [DOI] [PubMed] [Google Scholar]
- 2.Riethdorf S, Wikman H, Pantel K. Review: Biological relevance of disseminated tumor cells in cancer patients. Int J Cancer. 2008;123:1991–2006. doi: 10.1002/ijc.23825. [DOI] [PubMed] [Google Scholar]
- 3.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 4.Kim ER, Kim YH. Clinical Application of Genetics in Management of Colorectal Cancer. Intest Res. 2014;12:184–193. doi: 10.5217/ir.2014.12.3.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem. 2011;351:41–58. doi: 10.1007/s11010-010-0709-x. [DOI] [PubMed] [Google Scholar]
- 6.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21:12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weyhenmeyer B, Murphy AC, Prehn JH, Murphy BM. Targeting the anti-apoptotic Bcl-2 family members for the treatment of cancer. Exp Oncol. 2012;34:192–199. [PubMed] [Google Scholar]
- 8.Davids MS, Letai A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J. Clin. Oncol. 2012;30:3127–3135. doi: 10.1200/JCO.2011.37.0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584:2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 10.Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66:1326–1336. doi: 10.1007/s00018-008-8637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, Oyesanya RA, Dasgupta S, Dent P, Grant S, Rahmani M, Curiel DT, Dmitriev I, Hedvat M, Wei J, Wu B, Stebbins JL, Reed JC, Pellecchia M, Sarkar D, Fisher PB. Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs. 2011;20:1397–1411. doi: 10.1517/13543784.2011.609167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mandelin AM 2nd, Pope RM. Myeloid cell leukemia-1 as a therapeutic target. Expert Opin Ther Targets. 2007;11:363–373. doi: 10.1517/14728222.11.3.363. [DOI] [PubMed] [Google Scholar]
- 13.Perciavalle RM, Opferman JT. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013;23:22–29. doi: 10.1016/j.tcb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartman ML, Czyz M. Anti-apoptotic proteins on guard of melanoma cell survival. Cancer Lett. 2013;331:24–34. doi: 10.1016/j.canlet.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 15.Bose P, Grant S. Mcl-1 as a Therapeutic Target in Acute Myelogenous Leukemia (AML) Leuk Res Rep. 2013;2:12–14. doi: 10.1016/j.lrr.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang T, Zhao C, Luo L, Zhao H, Cheng J, Xu F. The expression of Mcl-1 in human cervical cancer and its clinical significance. Med Oncol. 2012;29:1985–1991. doi: 10.1007/s12032-011-0005-y. [DOI] [PubMed] [Google Scholar]
- 17.Likui W, Qun L, Wanqing Z, Haifeng S, Fangqiu L, Xiaojun L. Prognostic role of myeloid cell leukemia-1 protein (Mcl-1) expression in human gastric cancer. J Surg Oncol. 2009;100:396–400. doi: 10.1002/jso.21344. [DOI] [PubMed] [Google Scholar]
- 18.Luo L, Zhang T, Liu H, Lv T, Yuan D, Yao Y, Lv Y, Song Y. MiR-101 and Mcl-1 in non-small-cell lung cancer: expression profile and clinical significance. Med Oncol. 2012;29:1681–1686. doi: 10.1007/s12032-011-0085-8. [DOI] [PubMed] [Google Scholar]
- 19.Henderson-Jackson EB, Helm J, Ghayouri M, Hakam A, Nasir A, Leon M, Bui M, Yeatman T, Coppola D. Correlation between Mcl-1 and pAKT protein expression in colorectal cancer. Int J Clin Exp Pathol. 2010;3:768–774. [PMC free article] [PubMed] [Google Scholar]
- 20.Greene FL. AJCC cancer staging manual. Springer; 2002. [Google Scholar]
- 21.Vermeulen PB, Gasparini G, Fox SB, Toi M, Martin L, McCulloch P, Pezzella F, Viale G, Weidner N, Harris AL, Dirix LY. Quantification of angiogenesis in solid human tumours: an international consensus on the methodology and criteria of evaluation. Eur J Cancer. 1996;32A:2474–2484. doi: 10.1016/s0959-8049(96)00379-6. [DOI] [PubMed] [Google Scholar]
- 22.Vavrova J, Rezacova M. Importance of proapoptotic protein PUMA in cell radioresistance. Folia Biol (Praha) 2014;60:53–56. doi: 10.14712/fb2014060020053. [DOI] [PubMed] [Google Scholar]
- 23.Grana X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs) Oncogene. 1995;11:211–219. [PubMed] [Google Scholar]
- 24.Sotak M, Sumova A, Pacha J. Cross-talk between the circadian clock and the cell cycle in cancer. Ann Med. 2014;46:221–232. doi: 10.3109/07853890.2014.892296. [DOI] [PubMed] [Google Scholar]
- 25.Shah MA, Schwartz GK. Cyclin-dependent kinases as targets for cancer therapy. Cancer Chemother Biol Response Modif. 2003;21:145–170. doi: 10.1016/s0921-4410(03)21007-3. [DOI] [PubMed] [Google Scholar]
- 26.Mani S, Wang C, Wu K, Francis R, Pestell R. Cyclin-dependent kinase inhibitors: novel anticancer agents. Expert Opin Investig Drugs. 2000;9:1849–1870. doi: 10.1517/13543784.9.8.1849. [DOI] [PubMed] [Google Scholar]
- 27.Jamil S, Sobouti R, Hojabrpour P, Raj M, Kast J, Duronio V. A proteolytic fragment of Mcl-1 exhibits nuclear localization and regulates cell growth by interaction with Cdk1. Biochem J. 2005;387:659–667. doi: 10.1042/BJ20041596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujise K, Zhang D, Liu J, Yeh ET. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J Biol Chem. 2000;275:39458–39465. doi: 10.1074/jbc.M006626200. [DOI] [PubMed] [Google Scholar]
- 29.Germain M, Duronio V. The N terminus of the anti-apoptotic BCL-2 homologue MCL-1 regulates its localization and function. J Biol Chem. 2007;282:32233–32242. doi: 10.1074/jbc.M706408200. [DOI] [PubMed] [Google Scholar]
- 30.Mittal K, Ebos J, Rini B. Angiogenesis and the tumor microenvironment: vascular endothelial growth factor and beyond. Semin Oncol. 2014;41:235–251. doi: 10.1053/j.seminoncol.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 31.Le Gouill S, Podar K, Amiot M, Hideshima T, Chauhan D, Ishitsuka K, Kumar S, Raje N, Richardson PG, Harousseau JL, Anderson KC. VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood. 2004;104:2886–2892. doi: 10.1182/blood-2004-05-1760. [DOI] [PubMed] [Google Scholar]
- 32.Zhang S, Zhau HE, Osunkoya AO, Iqbal S, Yang X, Fan S, Chen Z, Wang R, Marshall FF, Chung LW, Wu D. Vascular endothelial growth factor regulates myeloid cell leukemia-1 expression through neuropilin-1-dependent activation of c-MET signaling in human prostate cancer cells. Mol Cancer. 2010;9:9. doi: 10.1186/1476-4598-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014;4:64. doi: 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haagenson KK, Wu GS. Mitogen activated protein kinase phosphatases and cancer. Cancer Biol Ther. 2010;9:337–340. doi: 10.4161/cbt.9.5.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Rakus D, Gizak A, Demidenko ZN, Cocco L, Martelli AM, Cervello M. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5:2881–2911. doi: 10.18632/oncotarget.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amoyel M, Anderson AM, Bach EA. JAK/STAT pathway dysregulation in tumors: a Drosophila perspective. Semin Cell Dev Biol. 2014;28:96–103. doi: 10.1016/j.semcdb.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, Dey S, Sung B. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171:59–76. doi: 10.1111/j.1749-6632.2009.04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lassmann S, Schuster I, Walch A, Gobel H, Jutting U, Makowiec F, Hopt U, Werner M. STAT3 mRNA and protein expression in colorectal cancer: effects on STAT3-inducible targets linked to cell survival and proliferation. J Clin Pathol. 2007;60:173–179. doi: 10.1136/jcp.2005.035113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Q, Lai R, Chirieac LR, Li C, Thomazy VA, Grammatikakis I, Rassidakis GZ, Zhang W, Fujio Y, Kunisada K, Hamilton SR, Amin HM. Constitutive activation of JAK3/STAT3 in colon carcinoma tumors and cell lines: inhibition of JAK3/STAT3 signaling induces apoptosis and cell cycle arrest of colon carcinoma cells. Am J Pathol. 2005;167:969–980. doi: 10.1016/S0002-9440(10)61187-X. [DOI] [PMC free article] [PubMed] [Google Scholar]