

Abstract
Objectives: The increased rate of glucose uptake necessary to support the growth of tumor cells is mediated by glucose transporters, and glucose transporter 1 (GLUT1) is overexpressed in several types of cancer in correlation with poor prognosis. And WNT2B overexpression is thought to be involved in tumor progression. Here, we investigated the effects of WNT2B in GLUT1 overexpressing cisplatin resistant head and neck squamous cell carcinoma (HNSCC) in vitro and in vivo. Materials and methods: We generated GLUT1 overexpressing cisplatin resistant CAL27 and SCC25 oral cancer cells. Lentiviral mediated knock-down of WNT2B was performed in CAL27 and SCC25. QRT-PCR and Western blot analysis were used to detect the mRNA and protein expression of GLUT1, WNT2B, Cyclin D1 and β-catenin. Cell viability was assessed by MTT analysis. Colony formation assay was performed by staining with 0.5% crystal violet. The role of WNT2B in HNSCC was examined in vivo through the generation of a CAL27 (or cisplatin resistant CAL27 or cisplatin resistant CAL27 with WNT2B knock-down) nude mice xenograft model of HNSCC. Results: Knock-down of WNT2B in decreased cell viability and colony formation in cisplatin resistant CAL27 and SCC25 in association with the downregulation of GLUT1, cyclin D1 and β-catenin. In a cisplatin resistant CAL27 mouse xenograft model, shRNA mediated silencing of WNT2B increased survival and decreased tumor growth in correlation with the downregulation of GLUT1, cyclin D1 and β-catenin. Conclusion: WNT2B plays a role in tumorigenesis and chemotherapy resistance in oral cancer and provide a potential therapeutic target for the treatment of patients with HNSCC.
Keywords: WNT2B, GLUT1, head and neck squamous cell carcinoma, cisplatin
Introduction
Glucose is an essential substrate for the metabolism of all mammalian cells. Glucose is a hydrophilic compound and as such, it cannot pass through the lipid bilayer by simple diffusion, requiring specific carrier proteins to mediate its transport into the cytosol [1]. Two classes of hexose transporters, the sodium-dependent glucose transporter family (SGLT) and the GLUT family of proteins transport sugars across the cellular membrane. GLUTs are transmembrane proteins of approximately 500 amino acids and 14 members of the GLUT family have been identified in mammals with GLUT1 as the most widely expressed member of the family [2]. Glucose transporters are often overexpressed in tumors, which are characterized by an increased glycolysis rate to meet the requirements of rapidly growing cells. GLUT1 overexpression has been detected in several malignancies including breast cancer, thyroid cancer, head and neck cancer, and lung cancer and its overexpression has been correlated with poor prognosis in several cancers including head and neck cancer [3,4]. In a previous study, we showed that GLUT1 expression in oral cancer cells is correlated with cisplatin resistance and plays an important role in cell survival in head and neck squamous cell carcinoma (HNSCC) via a mechanism involving the activation of the NFκB pathway [5]. We subsequently identified WNT2B as a downregulated gene in GLUT1 knock-down CAL27 oral cancer cells by microarray analysis.
Wnt genes encode small secreted proteins that are present in all animal genomes and the Wnt signaling pathway controls a significant number of diverse biological phenomena [6]. Most mammalian genomes encode approximately 20 Wnt proteins divided into 12 subfamilies [7]. Wnts form high-order receptor complexes routing downstream signaling, and three different pathways are activated upon Wnt-receptor activation, the canonical Wnt/β-catenin cascade, the noncanonical planar cell polarity pathway and the Wnt/Ca2+ pathway [7,8]. Mutations in components of the Wnt pathway have been linked to cancer and other diseases. Human WNT2B was cloned and characterized in 1996 and later shown to function in the canonical Wnt pathway by transducing signals through Frizzled and LRP5/LRP6 receptors to release β-catenin from the AXIN-APC degradation complex, resulting in its stabilization and nuclear translocation for the transcriptional activation of target genes [9-11]. Cyclin D1, a key regulator of the cell cycle that allows the progression from G1 to S phase, was first identified as a transcriptional target of β-catenin in colorectal cancer cell lines [12,13]. WNT2B is expressed in many organs and in several types of human cancer such as basal cell carcinoma, gastric cancer, breast cancer, head and neck squamous cell carcinoma, cervical cancer and leukemia [14,15]. WNT2B is upregulated in certain human cancers although its exact involvement in cancer and inflammation remain unclear.
In the present study, we examined the role of WNT2B and its relation with GLUT1 expression in HNSCC in vitro and in vivo. We showed that WNT2B is upregulated in HNSCC cell lines in association with cisplatin resistance and silencing its expression decreased cell viability, colony formation, and downregulated the expressions of GLUT1, cyclin D1 and β-catenin in vitro and increased survival and decreased tumor growth in vivo. These results shed light on the important role of the Wnt/β-catenin pathway in tumorigenesis and chemotherapy resistance in oral cancer and integrate Wnt and energy signals in the context of increased cell proliferation associated with cancer progression.
Materials and methods
Cell lines and culture
The HNSCC cell lines CAL27 and SCC25 were obtained from the American type culture collection. SCC25 cells were grown in Dulbecco’s minimal essential medium (DMEM)/Ham’s F-12 medium supplemented with 20% fetal bovine serum (FBS, HyClone, Logan, UT, USA), hydrocortisone (40 ng/mL) and sodium pyruvate (1 mM). CAL27 cells were grown in DMEM supplemented with 10% FBS.
Induction of cisplatin resistance
CAL27 and SCC25 cells were cultured in DMEM containing 0.2 μM cisplatin (CDDP) and proliferating cells were repeatedly sub-cultured in DMEM containing increasing concentrations of CDDP over a 6-month period. CAL27 and SCC25 cells that grew in 20 μM CDDP were designated as CAL27-CDDP and SCC25-CDDP and maintained in DMEM containing 3 μM cisplatin.
Cell proliferation assay
Cell viability was assessed using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, Sigma) assay as described previously [16] with minor modifications. Briefly, 7 × 103 cells/100 μL media were seeded in DMEM supplemented with 10% or 20% FBS in 96-well plates and grown for the indicated times. MTT was added to a final concentration of 0.5 mg/mL and incubated for 4 h in a moist chamber at 37°C. The medium was then removed and 0.2 mL DMSO was added. The absorbance (A) of the formazan product was measured at 570 nm with a microtiter plate reader (Bio-Rad Labs, Sunnyvale, CA). IC50 (the concentration required for 50% inhibition of cell proliferation) values were calculated by interpolation from a sigmoidal dose response curve fit of the log transformed survival data, derived using GraphPad Prism version 4.0 for Mac OS X (GraphPad Software Inc., San Diego, USA).
Lentivirus transfection
Cells were transfected with the lentivirus vector pLKO.1 (Sigma) encoding shRNA targeting WNT2B or control shRNA. The shRNA sequences were as follows: control shRNA, 5’-ACCTCGTGTAAGACAGTACAC-3’, WNT2B-shRNA1: 5’-GCACGAGTGATCTGTGACAAT-3’, WNT2B-shRNA2: 5’-CAACTCTCCAGATTACTGT-3’, WNT2B-shRNA3: 5’-CGGTGCAAGGAATGCAGAAAT-3’.
High titer lentiviruses were generated by transfection of 293T cells with lentivirus-shRNA, and CAL27 and SCC25 cell lines were incubated with infectious lentivirus medium.
Quantitative real-time PCR
Total RNA was prepared from cells using the TRIzol reagent (Invitrogen) or the RNeasy mini kit (Qiagen, Valencia, CA), preserved in RNAlater reagent (Qiagen), and 2 μg of each sample was reverse transcribed using the RetroScript kit (Ambion, Austin, TX) according to the manufacturer’s instructions. β-actin was used to normalize the quantity of cDNA used in the PCR reaction. QRT-PCR was performed using the SYBR Green Rox Master Mix (Qiagen) on an ABI 7300 instrument (Applied Biosystems, Foster City, CA). Primer sequences used in qRT-PCR were as follows: GLUT1 forward: 5’-TTGGCTCCGGTATCGTCAAC-3’, reverse: 5’-GCCAGGACCCACTTCAAAGA-3’; WNT2B forward: 5’-CCTGTAGCCAGGGTGAACTG-3’, reverse: 5’-CGGGCATCCTTAAGCCTCTT-3’; β-actin forward: 5’-GTCACCAACTGGGACGACAT-3’, reverse: 5’-GCCAGAGGCGTACAGGGATA-3’. The comparative CT method was used to quantitate relative gene expression [17].
Western blotting
Cell or tumor lysates were separated by SDS-PAGE followed by transfer to polyvinylidene difluoride membranes (Millipore, MA, USA). Membranes were blocked with 5% milk in TBS-T (TBS containing Tween 20) for 30 min to 1 h and incubated in primary antibodies overnight at 4°C followed by incubation in goat anti-rabbit IgG H&L (HRP) at 1/2000 dilution for 1 to 2 h. The primary antibodies (all from Abcam) and dilutions used were as follows: GLUT1 (1/1000), WNT2B (1/2000), cyclin D1 (1/100), β-catenin (1/5000), with β-actin (1/2000) and GAPDH (1/2500) as the loading controls. Proteins were visualized by autoradiography and films were scanned and quantified using a gel image analysis system (Bio-Rad, Hercules, USA).
Colony-formation assay
Cells were plated in 6-well plates at a density of 1 × 103 cells per well. The medium was changed every three days. After 1 week, cells were fixed, stained with 0.5% crystal violet, photographed and counted. All experiments were performed in triplicate and were representative of 3 independent experiments.
HNSCC xenograft tumors
All animal experiments were approved by the Animal Research Committee at Shanghai Tongji University and were carried out in accordance with established International Guiding Principles for Animal Research. Five-week old female athymic nude mice were injected with CAL27, CAL27-CDDP and CAL27-CDDP cells transfected with lentivirus carrying WNT2B-shRNA1 (4 × 106 cells/mouse) in the right flank to form xenograft tumors. Twenty mice were used for each treatment group. Two to four animals were housed in each sterile cage and given free access to sterile water and food. Tumor dimensions were measured with digital calipers every three days and tumor volume was calculated according to the following formula: TV (mm3) = (length × width)2 × 0.5. Tumor xenografts were harvested at 28 days after cell transplantion and lysates were prepared for western blot analysis.
Statistical analysis
All data were expressed as means ± SD from triplicate experiments performed in a parallel manner unless otherwise indicated. For statistical analysis of the data, group means were compared by one-way ANOVA, and Bonferroni’s test was used to identify differences between groups. Statistical differences were considered significant at the P < 0.05 or P < 0.01 level. All the figures shown in this article were obtained from at least three independent experiments.
Results
Upregulation of GLUT1 and WNT2B is associated with cisplatin resistance
The HNSCC cell lines CAL27 and SCC25 were cultured in increasing concentrations of cisplatin to generate the cisplatin resistant cell lines CAL27-CDDP and SCC25-CDDP. Cell viability was determined using the MTT assay in CAL27-CDDP and SCC25-CDDP exposed to increasing concentrations of CDDP for 72 h. The results showed that CAL27-CDDP and SCC25-CDDP cells were more resistant to cisplatin than the parental cell lines, with IC50 values of 22.06 μM and 19.68 μM, respectively, which was 6.8 and 4.5 fold higher than the IC50 values for CAL27 and SCC25 (3.25 μM and 4.39 μM, respectively) (Figure 1A and 1B).
Figure 1.
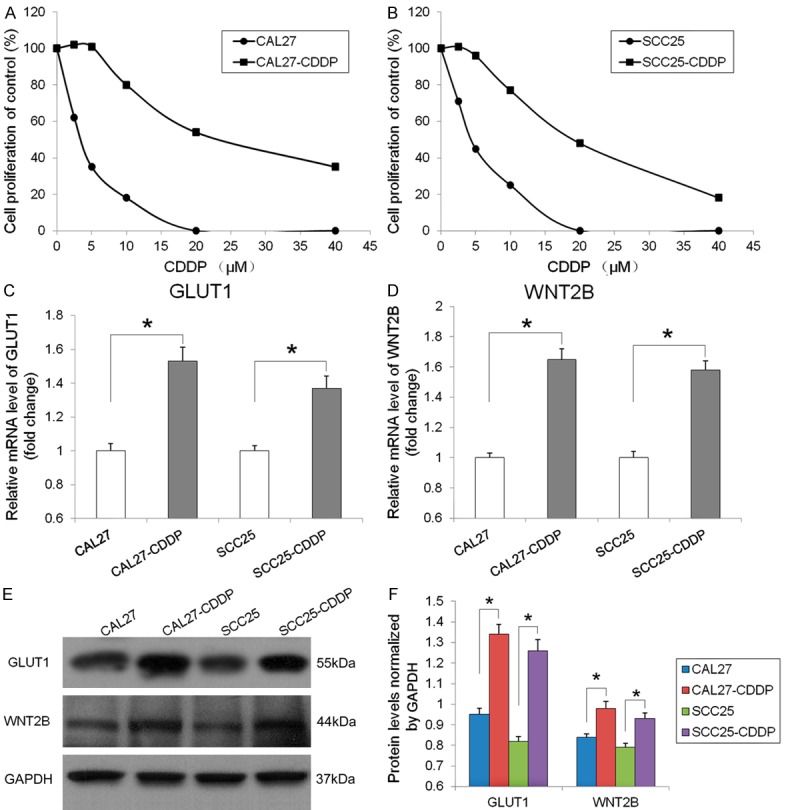
GLUT1 and WNT2B expression in cisplatin resistant HNSCC cells. (A and B) Cell viability was assessed using the MTT assay in CAL27 and SCC25 cells and their cisplatin resistant counterparts (CAL27-CDDP and SCC25-CDDP) after culture for 72 h with different concentrations of cisplatin (CDDP). (C) GLUT1 and (D) WNT2B mRNA levels detected by qRT-PCR and normalized to the levels of β-actin are expressed as fold-change relative to the parental cell lines. *P < 0.05. (E) GLUT1 and WNT2B protein expression detected by western blotting with GAPDH as the loading control. (F) Quantification of GLUT1 and WNT2B protein levels normalized to GAPDH, *P < 0.05.
QRT-PCR detection of GLUT1 and WNT2B mRNA expression and normalization to β-actin showed that GLUT1 levels were 1.53 and 1.37 fold higher and WNT2B levels were 1.65 and 1.58 fold higher in CAL27-CDDP and SCC25-CDDP cells than in the parental cell lines, respectively (P < 0.05) (Figure 1C and 1D). Western blot analysis and quantification of band density relative to the expression of GAPDH showed significantly higher GLUT1 and WNT2B protein levels in CAL27-CDDP and SCC25-CDDP cells than in the parental cell lines (GLUT1: 1.34 vs. 0.95 and 1.26 vs. 0.82, respectively; WNT2B: 0.98 vs. 0.84 and 0.93 vs. 0.79, respectively) (Figure 1D-F).
Silencing of WNT2B decreases cell viability and colony formation in cisplatin resistant oral cancer cells
To examine the correlation between GLUT1 and its target gene WNT2B and their effect on the survival of HNSCC cells, the WNT2B gene was knocked-down by infection of CAL27-CDDP and SCC25-CDDP with lentiviral vectors carrying shRNA targeting WNT2B (WNT2B-shRNA1-3) or control shRNA for 48 h. WNT2B mRNA levels were measured by qRT-PCR and normalized to the level of β-actin, which showed a significant downregulation of WNT2B expression by all three vectors in both cell lines (P < 0.01) (Figure 2A). Assessment of WNT2B protein levels by western blotting showed a marked downregulation in CAL27-CDDP and SCC25-CDDP cells transfected with the three WNT2B-shRNA constructs compared to control shRNA infected cells (Figure 2B). Figure 2C shows the quantification of bands in Figure 2B, indicating a significant downregulation of WNT2B protein expression (P < 0.01). To further determine the role of WNT2B in mediating the effect of GLUT1 in squamous cell carcinoma, the effect of WNT2B silencing on the viability of CAL27-CDDP and SCC25-CDDP cells was assessed at different time points using the MTT assay. The results showed that the viability of CAL27-CDDP and SCC25-CDDP cells transfected with WNT2B-shRNA1 decreased to approximately 50% and 66%, respectively, of the untreated or control shRNA transfected cells at 96 h (P < 0.01 and P < 0.05, respectively) (Figure 3A and 3B). The results of colony formation assays confirmed the effect of WNT2B silencing in CAL27-CDDP and SCC25-CDDP cells, with an approximately 2.1 and 2.4 fold lower number of colonies in WNT2B-shRNA1 transfected cells than in their untreated or control shRNA transfected counterparts (P < 0.01) (Figure 3C and 3D).
Figure 2.
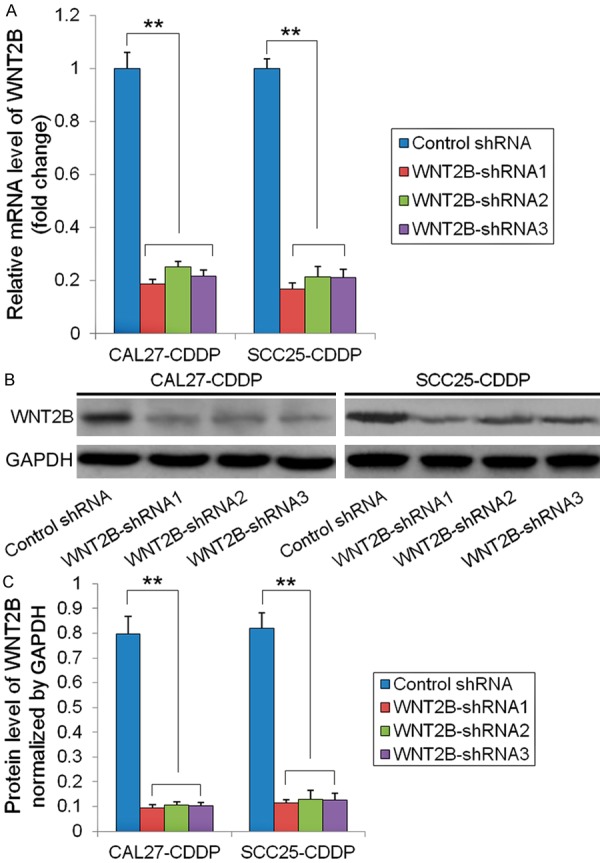
Effectiveness of shRNA mediated silencing of WNT2B. CDDP resistant cells (CAL27-CDDP and SCC25-CDDP) were infected with lentivirus expressing three WNT2B-shRNAs or control shRNA for 48 h. A. WNT2B mRNA expression was detected by qRT-PCR, normalized to β-actin expression and depicted as fold-change relative to the control shRNA group, **P < 0.01. B. WNT2B protein expression detected by western blotting with GAPDH as the loading control. C. Quantification of WNT2B protein level normalized to GAPDH, **P < 0.01.
Figure 3.
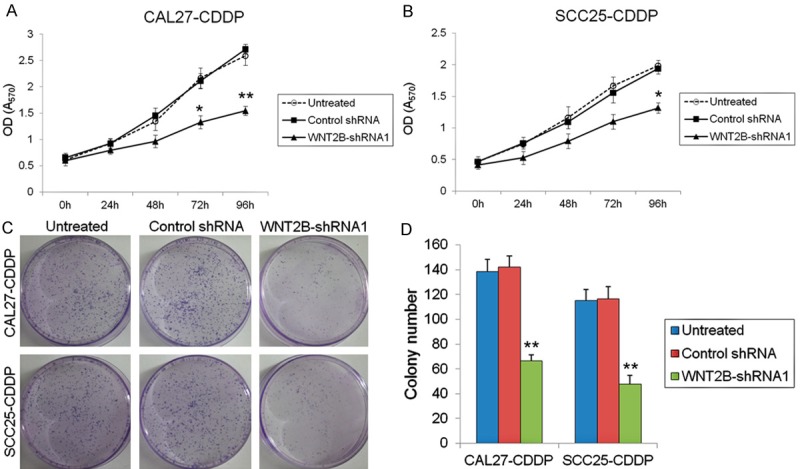
Silencing of WNT2B decreases cell viability and colony formation in cisplatin resistant oral cancer cells. A and B. Cell viability in untreated, WNT2B-shRNA or control shRNA transfected CDDP-resistant cells was determined by the MTT assay, *P < 0.05, **P < 0.01 vs. untreated cells. C. Representative images of clone formation assays. D. Stained colonies larger than 1 mm in diameter were counted, **P < 0.01 vs. untreated cells.
Silencing of WNT2B downregulates cyclin D1 and β-catenin in cisplatin resistant oral cancer cells
To examine the effect of WNT2B knock-down on the downstream effectors and transcriptional targets of the Wnt pathway, the levels of expression of β-catenin and cyclin D1 were determined in CAL27-CDDP and SCC25-CDDP cells transfected with WNT2B-shRNA1 in comparison to untreated or control shRNA transfected cells by western blotting and normalized to the levels of β-actin. ShRNA-mediated silencing of WNT2B significantly decreased the protein levels of GLUT1, cyclin D1 and β-catenin in both cisplatin resistant cell lines. GLUT1 levels were decreased by 1.4 and 1.2 fold (P < 0.05, Figure 4B), cyclin D1 by 2.3 and 2.2 fold (P < 0.05, Figure 4C) and β-catenin by 8.6 and 5.8 fold (P < 0.01, Figure 4D) in CAL27-CDDP and SCC25-CDDP cells transfected with WNT2B-shRNA1, respectively, compared to control shRNA transfected cells.
Figure 4.
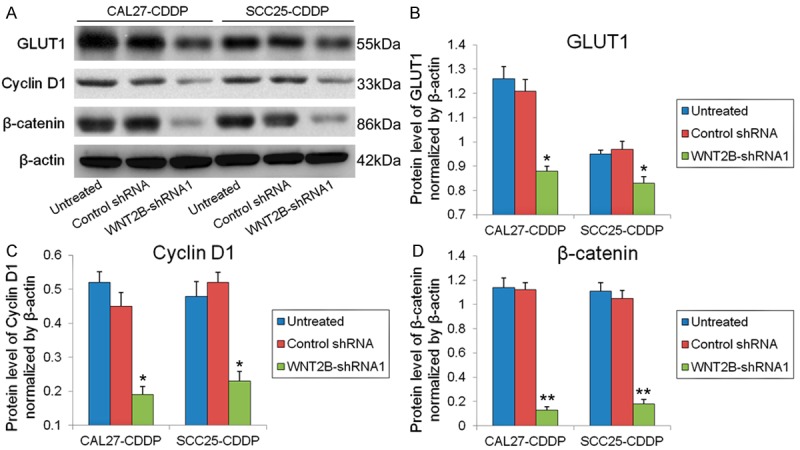
Silencing of WNT2B downregulates cyclin D1 and β-catenin in cisplatin resistant oral cancer cells. (A) GLUT1, cyclin D1 and β-catenin levels in untreated, WNT2B-shRNA or control shRNA transfected CDDP-resistant cells as determined by western blotting with β-actin as the loading control. Quantification of GLUT1 (B), Cyclin D1 (C) and β-catenin (D) protein levels normalized to β-actin, *P < 0.05, **P < 0.01 vs untreated cells.
Silencing of WNT2B increases survival and decreases tumor volume in a xenograft mouse model
To investigate the role of GLUT1 and WNT2B in vivo, a xenograft tumor model was generated by injecting mice with CAL27, CAL27-CDDP and CAL27-CDDP-WNT2B-shRNA cells. The survival rate of mice carrying WNT2B-shRNA tumors was significantly higher than that of mice carrying CAL27 or CAL27-CDDP tumors (70% vs. 50% vs. 30%, respectively, on day 28) (Figure 5A). Tumor dimensions were measured every three days and tumor volume was calculated, which showed that tumor growth was significantly inhibited in mice carrying tumors generated with CAL27-CDDP cells transfected with WNT2B-shRNA compared to those bearing CAL27 or CAL27-CDDP tumors (average tumor volume in mm3 at 28 days: 332.92 ± 46.73 vs. 530.67 ± 54.80 vs. 912.56 ± 40.78, respectively) (Figure 5B). Images of representative tumors excised from xenograft mice treated as indicated are shown in Figure 5C. The expression levels of WNT2B and GLUT1 were examined in protein lysates from xenograft tumors by western blotting, which confirmed that GLUT1 and WNT2B were upregulated in tumors from untreated CAL27-CDDP cells compared to those bearing CAL27 cells and the significant downregulation of WNT2B and GLUT1 in xenograft tumors generated from CAL27-CDDP-WNT2B-shRNA cells compared to those bearing CAL27 or CAL27-CDDP cells (Figure 5D). Figure 5E shows the results of densitometric quantification of WNT2B and GLUT1 protein levels normalized to GAPDH (CAL27-CDDP-WNT2B-shRNA P < 0.05 vs. CAL27 group, P < 0.05 vs. untreated CAL27-CDDP group).
Figure 5.
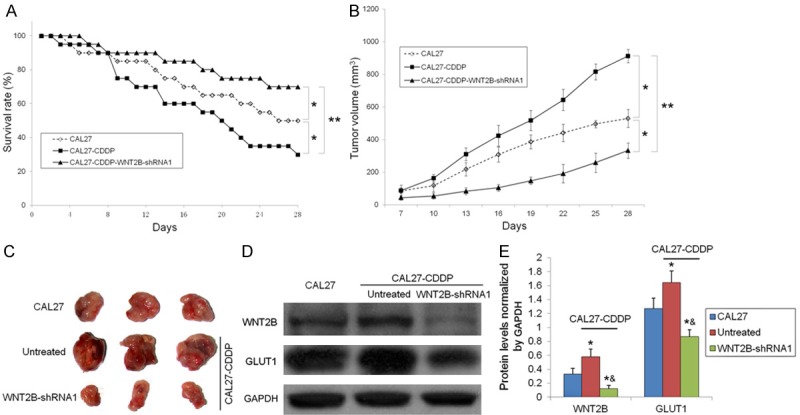
Silencing of WNT2B increases survival and decreases tumor volume in a xenograft mouse model. A. Survival analysis in xenograft mice for 28 days. *P < 0.05, **P < 0.01. B. Tumor volume was determined every three days after day 7. *P < 0.05, **P < 0.01. C. At 28 days post implantation, tumor xenografts were harvested. Representative images of tumors are shown. D. WNT2B and GLUT1 protein expression in tumor tissues was detected by western blotting with GAPDH as the loading control. E. Quantification of WNT2B and GLUT1 protein levels normalized to GAPDH, *P < 0.05 vs. CAL27 mice, &P < 0.05 vs. untreated CAL27-CDDP mice.
Discussion
Cancer cells require increased nutrient uptake to support an enhanced rate of proliferation and the uptake of glucose in malignant cells is mediated by glucose transporters. GLUT1 expression is upregulated in several cancers including head and neck cancer and its expression is associated with the resistance of HNSCC cells to cisplatin [3,4]. We previously showed that silencing of GLUT1 expression inhibited HNSCC cell proliferation and tumorigenesis in vitro and in vivo whereas overexpression of GLUT1 had the opposite effect [5]. In the present study, we investigated the association between the WNT2B protein and GLUT1 and examined the role of a potential signaling axis composed of GLUT1, WNT2B and β-catenin in the resistance of HNSCC cells to cisplatin.
In the present study, we showed that the expression of GLUT1 and WNT2B was upregulated in two cisplatin resistant oral cancer cell lines and that silencing of WNT2B decreased proliferation and colony forming ability, suggesting the involvement of the Wnt pathway in chemotherapy resistance in HNSCC. WNT2B has been involved in tumor progression and metastasis in different cancers and has been suggested to control the metastatic ability and drug resistance of tumor cells [18-20]. In an early study, blocking WNT-2 protein expression by monoclonal antibody or siRNA downregulated β-catenin and induced apoptosis in non-small cell lung cancer through the inactivation of survivin [21]. Silencing of WNT2B inhibits cell viability and colony formation, induces apoptosis, reduces invasiveness and suppresses cisplatin resistance in ovarian cancer cells [18]. In malignant pleural mesothelioma, intratumoral WNT2B expression is correlated with the levels of the Wnt/β-catenin target genes c-Myc and survivin, leading to the acceleration of tumor proliferation [19]. The therapeutic efficacy of WNT2B silencing was suggested in a recent study in which an adenoviral vector expressing a shRNA targeting WNT2B had antitumor effects in a WNT2B overexpressing lung carcinoma xenograft tumor model [22]. These results underscore the importance of WNT2B in cancer and support our results linking GLUT1 and WNT2B to tumorigenesis and chemotherapy resistance in HNSCC.
We showed that knock-down of WNT2B downregulated the expressions of GLUT1, cyclin D1 and β-catenin in cisplatin resistant cell lines and xenograft tumors in association with increased survival and decreased tumor growth. The involvement of the Wnt/β-catenin pathway in the resistance of cancer cells to chemotherapy has been reported previously. In lung adenocarcinoma, cisplatin resistance was shown to be associated with abnormal expression of Wnt/β-catenin signaling proteins [23]. Activation of Wnt/β-catenin signaling and upregulation of survivin associated with inhibition of glycogen synthase kinase-3β (GSK-3β), which targets β-catenin for ubiquitination and proteasomal degradation in the absence of Wnt, was identified as the underlying mechanism of cisplatin resistance in non-small cell lung cancer. Interestingly, GSK-3 regulates glucose transport and metabolism and was shown to negatively regulate GLUT1 expression [24]. Our results suggest that the involvement of WNT2B in the resistance of oral cancer cells to cisplatin may be mediated by another Wnt/β-catenin target gene, cyclin D1. Further investigation is necessary to determine the effect of WNT2B silencing on cell cycle progression and the expression of other Wnt/β-catenin target genes in relation to cisplatin resistance in oral cancer.
In conclusion, we showed that GLUT1 overexpression is associated with the upregulation of WNT2B and lentiviral mediated knock-down of WNT2B decreased cell viability and colony formation in cisplatin resistant HNSCC cells. Silencing of WNT2B downregulated GLUT1, cyclin D1 and β-catenin and was correlated with decreased tumor growth and increased survival in a xenograft tumor model. These results indicate that a regulatory network involving GLUT1 and the Wnt/β-catenin pathway may underlie the resistance of oral cancer cells to chemotherapy and suggest a novel therapeutic target for the treatment of HNSCC.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 81102047), the Fundamental Research Funds for the Central Universities.
Disclosure of conflict of interest
There are no potential conflicts of interest.
References
- 1.Szablewski L. Expression of glucose transporters in cancers. Biochim Biophys Acta. 2013;1835:164–169. doi: 10.1016/j.bbcan.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Thorens B, Mueckler M. Glucose transporters in the 21st Century. Am J Physiol Endocrinol Metab. 2010;298:E141–145. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho H, Lee YS, Kim J, Chung JY, Kim JH. Overexpression of glucose transporter-1 (GLUT-1) predicts poor prognosis in epithelial ovarian cancer. Cancer Invest. 2013;31:607–615. doi: 10.3109/07357907.2013.849722. [DOI] [PubMed] [Google Scholar]
- 4.Wang YD, Li SJ, Liao JX. Inhibition of glucose transporter 1 (GLUT1) chemosensitized head and neck cancer cells to cisplatin. Technol Cancer Res Treat. 2013;12:525–535. doi: 10.7785/tcrt.2012.500343. [DOI] [PubMed] [Google Scholar]
- 5.Li S, Yang X, Wang P, Ran X. The effects of GLUT1 on the survival of head and neck squamous cell carcinoma. Cell Physiol Biochem. 2013;32:624–634. doi: 10.1159/000354466. [DOI] [PubMed] [Google Scholar]
- 6.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 7.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 8.Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- 9.Katoh M, Hirai M, Sugimura T, Terada M. Cloning, expression and chromosomal localization of Wnt-13, a novel member of the Wnt gene family. Oncogene. 1996;13:873–876. [PubMed] [Google Scholar]
- 10.Katoh M, Kirikoshi H, Terasaki H, Shiokawa K. WNT2B2 mRNA, up-regulated in primary gastric cancer, is a positive regulator of the WNT- beta-catenin-TCF signaling pathway. Biochem Biophys Res Commun. 2001;289:1093–1098. doi: 10.1006/bbrc.2001.6076. [DOI] [PubMed] [Google Scholar]
- 11.Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 12.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 14.Katoh M. Differential regulation of WNT2 and WNT2B expression in human cancer. Int J Mol Med. 2001;8:657–660. doi: 10.3892/ijmm.8.6.657. [DOI] [PubMed] [Google Scholar]
- 15.Katoh M. Transcriptional regulation of WNT2B based on the balance of Hedgehog, Notch, BMP and WNT signals. Int J Oncol. 2009;34:1411–1415. [PubMed] [Google Scholar]
- 16.Cui J, Germer K, Wu T, Wang J, Luo J, Wang SC, Wang Q, Zhang X. Cross-talk between HER2 and MED1 regulates tamoxifen resistance of human breast cancer cells. Cancer Res. 2012;72:5625–5634. doi: 10.1158/0008-5472.CAN-12-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Fan L, Xia X, Rao Y, Ma Q, Yang J, Lu Y, Wang C, Ma D, Huang X. Silencing Wnt2B by siRNA interference inhibits metastasis and enhances chemotherapy sensitivity in ovarian cancer. Int J Gynecol Cancer. 2012;22:755–761. doi: 10.1097/IGC.0b013e3182540284. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi M, Huang CL, Sonobe M, Kikuchi R, Ishikawa M, Kitamura J, Miyahara R, Menju T, Iwakiri S, Itoi K, Yasumizu R, Date H. Intratumoral Wnt2B expression affects tumor proliferation and survival in malignant pleural mesothelioma patients. Exp Ther Med. 2012;3:952–958. doi: 10.3892/etm.2012.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li G, Liu Y, Su Z, Ren S, Zhu G, Tian Y, Qiu Y. MicroRNA-324-3p regulates nasopharyngeal carcinoma radioresistance by directly targeting WNT2B. Eur J Cancer. 2013;49:2596–607. doi: 10.1016/j.ejca.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 21.You L, He B, Xu Z, Uematsu K, Mazieres J, Mikami I, Reguart N, Moody TW, Kitajewski J, McCormick F, Jablons DM. Inhibition of Wnt-2-mediated signaling induces programmed cell death in non-small-cell lung cancer cells. Oncogene. 2004;23:6170–6174. doi: 10.1038/sj.onc.1207844. [DOI] [PubMed] [Google Scholar]
- 22.Liu D, Kadota K, Ueno M, Nakashima N, Yokomise H, Huang CL. Adenoviral vector expressing short hairpin RNA targeting Wnt2B has an effective antitumour activity against Wnt2B2-overexpressing tumours. Eur J Cancer. 2012;48:1208–1218. doi: 10.1016/j.ejca.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Gao Y, Liu Z, Zhang X, He J, Pan Y, Hao F, Xie L, Li Q, Qiu X, Wang E. Inhibition of cytoplasmic GSK-3beta increases cisplatin resistance through activation of Wnt/beta-catenin signaling in A549/DDP cells. Cancer Lett. 2013;336:231–239. doi: 10.1016/j.canlet.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 24.Buller CL, Loberg RD, Fan MH, Zhu Q, Park JL, Vesely E, Inoki K, Guan KL, Brosius FC 3rd. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am J Physiol Cell Physiol. 2008;295:C836–843. doi: 10.1152/ajpcell.00554.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]