

Abstract
Benzo(a)pyrene (BaP) is a ubiquitously distributed environmental pollutant. BaP is a known carcinogen and can induce malignant transformation of rodent and human cells. Many evidences suggest that inhibitor of poly(ADP-ribose) glycohydrolase (PARG) is potent anticancer drug candidate. However, the effect of PARG on BaP carcinogenesis remains unclear. We explored this question in a PARG-deficient human bronchial epithelial cell line (shPARG cells) treated with various concentration of BaP for 15 weeks. Soft agar assay was used to examine BaP-induced cell malignancy of human bronchial epithelial cells and shPARG cells. Mechanistic investigations were used by 2D-DIGE and mass spectrometry. Western blot analysis and Double immunofluorescence detection were used to confirm some of the results obtained from DIGE experiments. We found that PARG silencing could dramatically inhibit BaP-induced cell malignancy of human bronchial epithelial cells in soft agar assay. Altered levels of expression induced by BaP were observed within shPARG cells for numerous proteins, including proteins required for cell mobility, stress response, DNA repair and cell proliferation pathways. Among these proteins, TCTP and Cofilin-1 involved in malignancy, were validated by western blot analysis and immunofluorescence assay. PARG inhibition contributed to down-regulation of TCTP and Cofilin-1. This is the first experimental demonstration of a link between PARG silencing and reduced cell migration after BaP exposure. We propose that PARG silencing might down-regulate TCTP and Cofilin-1 associated with metastasis in BaP carcinogenesis.
Keywords: poly(ADP-ribosyl)ation, poly(ADP-ribose) glycohydrolase, benzo(a)pyrene, DNA damage, human bronchial epithelial cells (16HBE cells)
Introduction
Poly(ADP-ribosyl)ation is a post-translational protein modification catalyzed by poly(ADP-ribose) polymerases (PARPs) and involved in various cellular processes such as chromatin structure remodeling, DNA repair, regulation of gene expression, and integration of cellular signaling pathways [1-3]. Poly(ADP-ribose) (PAR) is heterogenic molecule synthesized from NAD by PARPs. Following DNA damage, activated PARPs catalyzes the elongation and branching of PAR attached to target proteins implicated in many important cellular processes [4]. PAR accumulation is transient, which is rapidly degraded to ADP-ribose by poly(ADP-ribose) glycohydrolase (PARG) [5]. PARG has both endoglycosidase and exoglycosidase activities, which is responsible for the hydrolysis of free (non-protein-bound) or protein-bound linear or branched poly-ADP-ribose (PAR) [6]. The local accumulation of PAR acts as a loading platform for the recruitment of chromatin remodeling and DNA repair factor. The dynamics of the system is derived from both the contribution of PAR-synthesizing PARPs and PAR catabolism by PARG [7]. The interplay between PARP and PARG is responsible for the transient and dynamic nature of the PAR. PARG is recruited to DNA damage sites after ionizing irradiation. PARG could be a novel potential therapeutic target for cancer chemotherapy [8,9]. Functional inhibition of PARG leads to sensitization of tumor cells to some chemo- and radiation therapies [10]. PARG inhibition in combination with alkylating agents enhances either apoptosis or necrotic cell death through a defect in DSB repair and S-phase arrest [11]. Taken together, these data underscore PARG plays a role of mediator in a wide spectrum of biological processes.
Benzo(a)pyrene (BaP), one of the most widely studied PAHs, is a well-known carcinogen with cytotoxicity and/or genotoxicity to lung, stomach and skin tissue [12]. Previously investigations have shown that necrosis induced by BaP involved PARP activation, NAD and ATP depletion [13,14]. Although the study reflects that poly(ADP-ribosyl)ation play an important role in the cytotoxicity and/or genotoxicity induced by BaP, it still remains unclear the role of PARG in BaP carcinogenesis.
Thus, the aim of the current study was to investigate the effect of PARG in BaP carcinogenesis. For this purpose, we used a PARG-deficient human bronchial epithelial cell line as an in vitro model and examined the effect of PARG in BaP-induced cell malignancy, then make further investigation on the possible molecular mechanisms of PARG silencing in BaP carcinogenesis by 2D-DIGE and mass spectrometry. We showed that the altered levels of proteins induced by BaP were observed in PARG-deficient cells involved in cell mobility, stress response, DNA repair and cell proliferation pathways. Our major finding is that PARG silencing down-regulated expression of TCTP and Cofilin-1 and up-regulated UCHL1 expression in BaP-induced cell transformation. These data shed new light on molecular mechanisms of PARG silencing in BaP carcinogenesis.
Material and methods
Cell culture and treatments
PARG-deficient human bronchial epithelial cells (shPARG cells) were previously established by our work [14]. Normal human bronchial epithelial cells (16HBE cells) and shPARG Cells were cultured in minimum essential Eagle’s medium (MEM) containing 10% fetal bovine serum (FBS). Penicillin (100 U/ml) and streptomycin (100 µg/ml) were added to the culture media. The cultured cells grew to 80% confluence were treated with BaP (BaP was dissolved in DMSO, and the final concentration of DMSO in the culture was 0.1%). Cells were washed twice with PBS before cell lysis. All media and supplements used for cell cultures were obtained from Gibco®.
Soft agar assay
The 16HBE cells and shPARG cells were treated with BaP in concentrations of 0, 10, 20, 40 μmol/l for 15 weeks. Cells were released from adherent cultures using trypsin, washed with PBS. Soft agar assay was performed as: 2.5 ml of 0.5% agar in basal modified Eagle’s medium (BMEM) supplemented with 10% FBS was layered onto each well of 6-well plates. Approximately 1.0 × 104 cells were mixed with 1 ml of 0.5% agar BMEM supplemented with 10% FBS layered on top of the 0.5% agar layer. The plates were incubated at 37°C in 5% CO2 for more than 2 weeks until visible colonies were formed and then the colonies were photographed and counted.
Sample preparation for DIGE
Cells from different treatment groups were washed once in PBS and collected by scraping into lysis buffer (8 M urea, 4% (w/v) CHAPS, 40 mM Tris base) containing DNase/RNase and protease inhibitors (Roche), and subjected to three rapid (2 min) cycles of freezing in liquid nitrogen/defrosting at 30°C with vigorous vortexing. Samples were centrifuged at 13,000 g for 30 min at 4°C and then precipitated with the 2D-Clean up Kit (GE Healthcare), resuspended in 50 μL of sample buffer (8 M urea, 2% (w/v) CHAPS) and the concentration was determined using Protein assay kit (BioRad). Protein extracts were aliquoted into single-use sample and stored at -80°C until analysis.
DIGE analysis
Each fifty-microgram sample was labelled with 400 pmol of N-hydroxysuccinimidyl-ester derivate of cyanine dyes Cy2, Cy3, and Cy5 according to the manufacturer’s instructions for minimal labelling dyes (GE Healthcare) with the following modifications. Protein samples were labeled with the appropriate CyDye (Cy2, Cy3, or Cy5) according to the DIGE minimal labeling protocol (GE Life Sciences, Sweden). An internal standard, which comprised a mixture of equal amounts of two cell lines, was labelled with Cy2. Samples were applied to 240 mm Immobiline Drystrips IPG pH 3-11 (GE Healthcare). The IEF was run on an Ettan IPGphor II (GE Healthcare) with an active rehydration at 50 V (20°C) for 16 h, followed by a three-step ramping focusing conditions for a total of 56,000 Vh at 20°C and a maximum current setting of 50 mA per strip. Following IEF, IPG strips were incubated in equilibration buffer (6 M urea; 30% (w/v) glycerol; 2% SDS; 50 mM Tris/HCl, pH 8.0) first with 0.5% dithiothreitol and then with 2% iodoacetamide each for 15 min. The strips were loaded onto 10% SDS-PAGE gels (Ettan Dalt Six gel system, GE Healthcare; 1 W/gel for 2 h and 3 W/gel for 12-16 h). Images were acquired with the multifluorescent point laser scanner Typhoon 9410 (GE Healthcare) and analyzed by the image analysis software (DeCyderTM software, GE Healthcare). The Differential In-Gel Analysis (DIA) module was used to detect and quantify the protein spots on the individual gels and the Biological Variation Analysis (BVA) module was used to match the gels relative to an internal standard (Cy2 labeled) and to statistically analyze differences in normalized protein spot abundance between the gels. ANOVA was used to assess differences between matched spots among gels and t-test was subsequently used to analyze and compare individual pairs of matched protein spots. Spots displaying a P-value < 0.05 when compared between treatment groups were regarded as statistically significant. Following fluorescence scanning, DIGE gels were also stained with colloidal Coomassie CBB G-250 to allow the visual detection of differential abundances of spots. Protein spots that showed differential abundance in 16HBE cells and shPARG cells were selected for mass spectrometry.
Identification of proteins by MALDI-TOF/TOF-MS/MS analysis
Bands excised from the gels were rinsed with de-ionized water, and then 100 µL of 25 mM ammonium bicarbonate in 10% acetonitrile was added. Proteins in the gel were reduced by adding 3 µL of 45 mM DTT and incubating for 30 min at 37°C. Alkylation was accomplished by adding 3 µL of 100 mM idoacetamide and incubating the mixture in the dark for 20 min. Then 100 µL of 25 mM ammonium bicarbonate in 10% acetonitrile and 0.1% octyl β- D-glycopyranoside was added. Finally, the samples were incubated at 37°C for 20-24 h with 5 µL of 0.1 mg/ml trypsin. MALDI analysis was performed on the ABI 4700 proteomics analyzer (Applied Biosystems, Framingham, MA). Peptide masses measured by MALDI-TOF/TOF-MS/MS were submitted to PeptIdent for searching against the SwissProt database. Results were evaluated and prioritized based on sequence coverage and matching of the theory to the measured molecular mass and pI.
Gene Ontology (GO) biological process annotations for abnormally expressed proteins were assigned according to those reported in the UniProt Database and confirmed using the Panther Classification System (http://www.pantherdb.org/panther/globalSearch.jsp?).
Cell lysates and western blot analysis
Cells from different treatment groups were washed once in PBS and collected by scraping into 200 μL ice-cold lysis buffer containing 62.5 mM Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, 50 mM dithiothreitol (DTT), 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4, and protease inhibitors. Protein (20 μg/lane) was loaded onto polyacrylamide gels. Proteins were separated by electrophoresis and then transferred onto nitrocellulose membranes. For immunoblotting, membranes were blocked with 5% nonfat dried milk in Trisbuffered saline (TBS) for 90 min, incubated overnight at 4°C with primary antibodies: a mouse monoclonal anti-pADPr 10H (1:400, abcam, Cambridge, UK); a rabbit polyclonal anti-PARG(O-23) (1:1000, Santa Cruz Biotechnology); a mouse monoclonal anti-TCTP antibody (ab58362, Abcam, USA), a rabbit polyclonal anti-Cofilin antibody (ab42824, Abcam, USA), a mouse monoclonal IgG1 anti-GAPDH (6C5, sc-32233, Santa Cruz Biotechnology). Then followed by secondary antibodies at 1:4000 dilution: goat anti-mouse IgG-HRP (sc-2005, Santa Cruz Biotechnology), goat anti-rabbit IgG-HRP (sc-203l, Santa Cruz Biotechnology). Blots were incubated in Supersignal® West Dura Extended Duration Substrate (Thermo scientific) and detected with ECL™ western blotting detection system (ImageQuant™ RT, GE Healthcare).
Double immunofluorescence detection
Cells grown on glass coverslips washed with PBS and fixed 15 min with 4% formaldehyde at room temperature. Incubated cells in the mixture of two primary antibodies: a mouse monoclonal anti-TCTP antibody (ab58362, Abcam, USA) and a rabbit polyclonal anti-Cofilin antibody (ab42824, Abcam, USA) in 1% BSA in PBST in a humidified chamber for 1 hr at room temperature. After washing, cells were incubated with the mixture of two secondary antibodies which are raised in different species (Texas Red-conjugated against rabbit and FITC-conjugated against mouse) for 90 min at room temperature. After washing three times with PBS, 0.1% Tween (v:v), DNA was counterstained with Dapi. Pictures were taken with Confocal Laser Scanning Microscopy (Leica Tcs sp5, Leica Microsystems).
Statistical analysis
Statistical analyses were performed using SPSS 17.0 software. A one-way ANOVA test was used to compare differences between matched spots among gels, and unpaired Student’s t-test was used for single comparison. A value of P < 0.05 was regarded as statistically significant.
Results
Silencing of PARG leads to increase and prolong levels of PAR after BaP treatment
PAR is transient and rapidly degraded to ADP-ribose by PARG. In order to detect the role of poly(ADP-ribosyl)ation, the shPARG cell line as previously reported [14] was used. Then, we investigated level of PARG expression in the shPARG cells and demonstrated that PARG silencing decreased PARG levels approximately 80% (Figure 1A, 1B).
Figure 1.
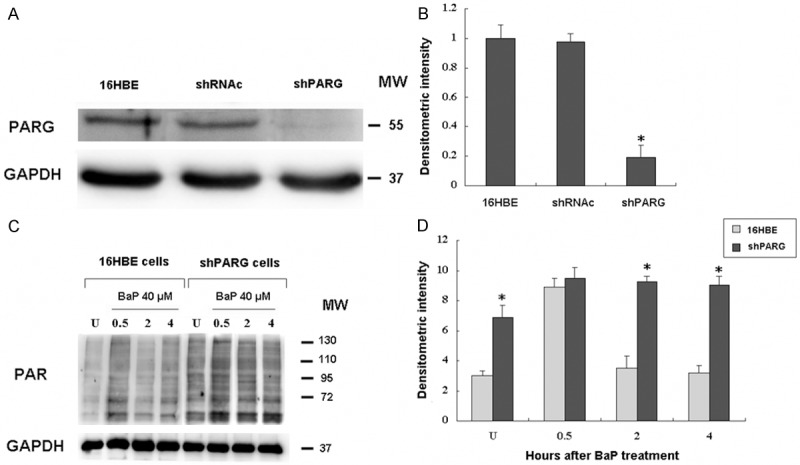
Silencing of poly(ADP-ribose) glycohydrolase (PARG) by shRNA in 16HBE cells. 16HBE cells were transfected with shRNA oligos for PARG as previously reported [14]. A: Western blotting analysis of shRNAc (lane 1) and shPARG (lane 2) cell lysates probed with antibodies against PARG and GAPDH. B: Densitometric quantification of PARG protein bands from A. C: The 16HBE cells and shPARG cells were treated with 40 µmol/L BaP for 20 min, then analyzed for levels of PAR by immunoblot from 0.5-4 h. Equal protein loading per lane was verified by immunoblotting detection of GAPDH. D: Densitometric quantification of PAR levels from C. *indicates a significant difference (P < 0.05) between 16HBE and shPARG cells. Error bars represent the SEM. All experiments were repeated at least 3 times with similar results.
Analysis of PAR levels after BaP treatment in normal cells demonstrated peak levels of PAR at 0.5 h, with a decrease in PAR levels from 2-4 h thereafter (Figure 1C). In the shPARG cells, however, a high level of PAR was observed in untreated cells. At all the time points (0.5-4 h), PAR levels were significantly elevated in shPARG cells as compared to the normal cells (Figure 1D).
These results suggested that there is significant down-regulation of PARG in the shPARG cell and PARG knockdown poses a hurdle to PAR degradation. This allowed us to highlight the role of PARG in BaP carcinogenesis.
PARG silencing inhibits cell malignancy induced by BaP
To investigate the effect of PARG, we treated cells with various concentrations BaP for 15 weeks. To determine the malignant potential of cells in vitro, cells were assessed for colony forming efficiency in soft agar, a measure of anchorage independent growth that is characteristic of transformed cells. As shown in Figure 2, PARG silencing significantly inhibited BaP-induced anchorage-independent colony formation in a dose-dependent manner in 16HBE cells.
Figure 2.
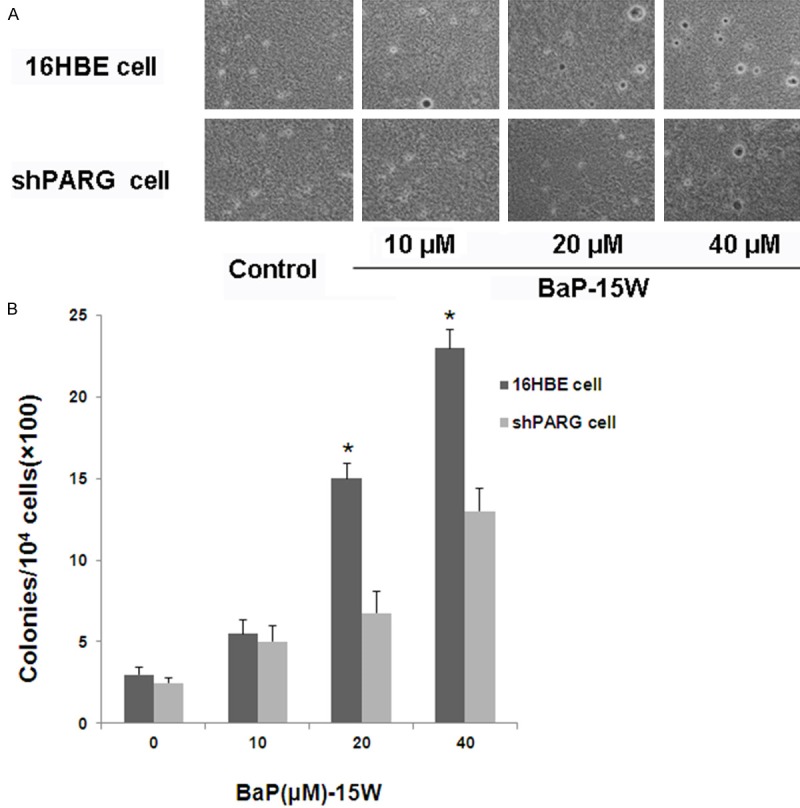
Colony formation in soft agar of BaP induced cells. (A) Representative images of colonies of cells in soft agar. (B) The number of colonies was counted under microscopy in soft agar and the results were presented as colonies per 10,000 cells from three independent experiments. The asterisk (*) indicates a significant different in the 16HBE cell treated with different concentration of BaP compared with the shPARG cell (P < 0.05).
Abnormally expressed proteins of silencing PARG in BaP-induced cell transformation
One of the main goals of this study was to reveal the role of poly(ADP-ribosyl)ation in BaP carcinogenesis and investigate its mechanism in carcinogenic process. We focused our attention on the biological effect of long-term exposure of BaP. Therefore, we treated the shPARG cells and 16HBE cells with BaP (40 μmol/l) for 15 weeks and investigated changes in proteins expression of two different cell lines induced by BaP. To comparatively analyze the 2D-DIGE, a total of 4 large format 2DE gels were obtained. In the image analysis, 45 protein spots showed significant differences when comparing the shPARG cells with 16HBE cells after BaP treatment (Figure 3). With MALDI-TOF-MS/MS analysis, 41 (91.1%) out of the 45 differentially regulated protein spots were successfully identified with high confidence after searching databases (Table 1). Down-regulated proteins identified in the shPARG cells induced by BaP were found to be mainly involved in cell proliferation and migration, DNA replication, tumor protein, and cytoskeleton protein (Table 2). Conversely, proteins involved in antioxidant and deubiquitination were up-regulated in the shPARG cells induced by BaP (Table 3). Protein (Ubiquitin carboxyl-terminal hydrolase isozyme L1) was identified in different localization on 2D-Gel, indicating potentially diverse protein forms, thus suggesting the occurrence of post-translational modification in the shPARG cells.
Figure 3.

DIGE analysis of cells after treated with BaP (PH = 3-11, NL, 240 mm). Overlay of Cy3 and Cy5 derived from a single gel, highlighting differentially expressed protein spots. Information about the proteins corresponding to the spot numbers is listed in Table 1.
Table 1.
Forty-one protein spots were identified by MALDI-TOF-MS/MS
| No. | Accession No. | Protein name | Mascot Score | Peptidesf | Protein MW | pI-Value |
|---|---|---|---|---|---|---|
| 1 | P13639 | Elongation factor 2 | 383 | 10 | 96246 | 6.4 |
| 2 | P21980 | Protein-glutamine gamma-glutamyltransferase 2 | 593 | 19 | 78420 | 5 |
| 3 | Q12931 | Heat shock protein 75 kDa, mitochondrial | 540 | 21 | 80345 | 9 |
| 4 | P41250 | Glycyl-tRNA synthetase | 186 | 5 | 83854 | 6.7 |
| 5 | No identification | |||||
| 6 | P31040 | Succinate dehydrogenase (ubiquinone) flavoprotein subunit, mitochondrial | 78 | 5 | 73672 | 7.3 |
| 7 | Q92945 | Far upstream element-binding protein 2 | 93 | 10 | 73355 | 7 |
| 8 | P02768 | Serum albumin | 112 | 9 | 71317 | 5.9 |
| 9 | P02768 | Serum albumin | 58 | 2 | 71317 | 5.9 |
| 10 | P08107 | Heat shock 70 kDa protein 1A/1B | 49 | 6 | 70294 | 5.4 |
| 11 | P02768 | Serum albumin | 118 | 9 | 71317 | 5.9 |
| 12 | P13797 | Plastin-3 | 274 | 8 | 71279 | 5.3 |
| 13 | P02768 | Serum albumin | 106 | 11 | 71317 | 5.9 |
| 14 | P11142 | Heat shock cognate 71 kDa protein | 386 | 15 | 71082 | 5.2 |
| 15 | P10809 | 60 kDa heat shock protein, mitochondrial | 432 | 15 | 61187 | 5.6 |
| 16 | P47895 | Aldehyde dehydrogenase family 1 member A3 | 191 | 13 | 56871 | 7.7 |
| 17 | P31943 | Heterogeneous nuclear ribonucleoprotein H | 88 | 10 | 49484 | 5.9 |
| 18 | P04264 | Keratin, type II cytoskeletal 1 | 123 | 9 | 66170 | 8.8 |
| 19 | P25705 | ATP synthase subunit alpha, mitochondrial | 129 | 4 | 59828 | 9.6 |
| 20 | No identification | |||||
| 21 | Q04695 | Keratin, type I cytoskeletal 17 | 559 | 20 | 48361 | 4.8 |
| 22 | P06733 | Alpha-enolase | 323 | 19 | 47481 | 7.7 |
| 23 | P05787 | Keratin, type II cytoskeletal 8 | 145 | 13 | 53671 | 5.4 |
| 24 | O75874 | Isocitrate dehydrogenase [NADP] cytoplasmic | 367 | 27 | 46915 | 6.6 |
| 25 | P30740 | Leukocyte elastase inhibitor | 642 | 21 | 42829 | 5.9 |
| 26 | P04264 | Keratin, type II cytoskeletal 1 | 195 | 6 | 66170 | 8.8 |
| 27 | Q9UJZ1 | Stomatin-like protein 2, mitochondrial | 349 | 19 | 38624 | 7.7 |
| 28 | P04264 | Keratin, type II cytoskeletal 1 | 140 | 6 | 66170 | 8.8 |
| 29 | P09651 | Heterogeneous nuclear ribonucleoprotein A1 | 206 | 26 | 38837 | 9.6 |
| 30 | P62937 | Peptidyl-prolyl cis-trans isomerase A | 340 | 36 | 18229 | 9 |
| 31 | P09211 | Glutathione S-transferase P | 314 | 23 | 23569 | 5.3 |
| 32 | Q9HB71 | Calcyclin-binding protein | 235 | 21 | 26308 | 9 |
| 33 | P07737 | Profilin-1 | 438 | 43 | 15216 | 9.4 |
| 34 | P13693 | Translationally-controlled tumor protein | 280 | 37 | 21626 | 5.2 |
| 35 | Q06323 | Proteasome activator complex subunit 1 | 234 | 14 | 28876 | 5.7 |
| 36 | P08729 | Keratin, type II cytoskeletal 7 | 151 | 14 | 51430 | 5.3 |
| 37 | P50395 | Rab GDP dissociation inhibitor beta | 53 | 8 | 51087 | 6.1 |
| 38 | No identification | |||||
| 39 | Q04695 | Keratin, type I cytoskeletal 17 | 78 | 14 | 48361 | 4.8 |
| 40 | Q99439 | Calponin-2 | 67 | 13 | 34074 | 7.7 |
| 41 | P09936 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 | 80 | 17 | 25151 | 5.2 |
| 42 | P09936 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 | 106 | 25 | 25151 | 5.2 |
| 43 | P23528 | Cofilin-1 | 74 | 16 | 18719 | 9.1 |
| 44 | P49411 | Elongation factor Tu, mitochondrial | 91 | 13 | 49852 | 7.9 |
| 45 | No identification |
Number of unique peptides according to proteins were identified by mass spectrometry.
Table 2.
Down-regulated proteins of the shPARG cells induced by BaP identified in the proteomic study
| No.a | Accession No. | Protein identify | Mascot score | Protein MW | pI-Value | Ratiob/Probc | Ratiob/Probc | Peptidesf |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| P-C vs B-Cd | P-T vs B-Te | |||||||
| Protein biosynthesisg | ||||||||
| 1 | P13639 | Elongation factor 2 | 383 | 96246 | 6.4 | NDg | -1.67/0.036 | 10 |
| Protein chaperones and stress proteing | ||||||||
| 3 | Q12931 | Heat shock protein 75 kDa, mitochondrial | 540 | 80345 | 9 | NDg | -1.72/0.030 | 21 |
| 10 | P08107 | Heat shock 70 kDa protein 1A/1B | 49 | 70294 | 5.4 | NDg | -1.30/0.041 | 6 |
| 14 | P11142 | Heat shock cognate 71 kDa protein | 386 | 71082 | 5.2 | NDg | -2.49/0.009 | 15 |
| RNA processing and protein bindingg | ||||||||
| 29 | P09651 | Heterogeneous nuclear ribonucleoprotein A1 | 206 | 38837 | 9.6 | -1.13/0.043 | -1.27/0.002 | 26 |
| Cell proliferation and tumor proteing | ||||||||
| 32 | Q9HB71 | Calcyclin-binding protein | 235 | 26308 | 9.0 | NDg | -1.24/0.026 | 21 |
| 33 | P13693 | Translationally-controlled tumor protein | 280 | 21626 | 5.2 | NDg | -1.32/0.038 | 37 |
| Cell migration and DNA replicationg | ||||||||
| 16 | P47895 | Aldehyde dehydrogenase family 1 member A3 | 191 | 56871 | 7.7 | NDg | -1.64/0.028 | 13 |
| 27 | Q9UJZ1 | Stomatin-like protein 2, mitochondrial | 349 | 38624 | 7.7 | -1.16/0.029 | -1.26/0.001 | 19 |
| Calcium binding proteinsg | ||||||||
| 12 | P13797 | Plastin-3 | 274 | 71279 | 5.3 | NDg | -1.93/0.008 | 8 |
| Cytoskeleton proteing | ||||||||
| 33 | P07737 | Profilin-1 | 438 | 15216 | 9.4 | -1.17/0.012 | -1.38/0.009 | 43 |
| 43 | P09936 | Cofilin-1 | 74 | 18719 | 9.1 | NDg | -1.49/0.001 | 16 |
Spot No.- The numbers correspond to the specific spots as indicated in Figure 3.
Average ratios of spot abundance of control or BaP-treated samples relative to the cell line, represent data from three separate experiments.
Student’s t test p-value are given as a measure of confidence for the ratio of each spot measured.
P-C, the control group of shPARG cells; B-C, the control group of 16HBE cells.
P-T, the BaP-treated group of shPARG cells; B-T, the BaP-treated group of 16HBE cells.
Number of unique peptides according to proteins were identified by mass spectrometry.
Functional categories according to Gene ontology and panther biological process annotations.
Table 3.
Up-regulated proteins of the shPARG cells induced by BaP identified in the proteomic study
| No.a | Accession No. | Protein identify | Mascot score | Protein MW | pI-Value | Ratiob/Probc | Ratiob/Probc | Peptidesf |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| P-C vs B-Cd | P-T vs B-Te | |||||||
| Protein chaperones and stress proteing | ||||||||
| 15 | P10809 | Heat shock protein 60, mitochondrial | 432 | 61187 | 5.6 | NDg | 1.29/0.048 | 15 |
| Antioxidant/detoxificationg | ||||||||
| 24 | O75874 | Cytosolic NADP-isocitrate dehydrogenase | 367 | 46915 | 6.6 | NDg | 2.28/0.002 | 27 |
| Protein biosynthesis/foldingg | ||||||||
| 30 | P62937 | Peptidyl-prolyl cis-trans isomerase A | 340 | 18229 | 9 | 1.42/0.002 | 1.61/0.004 | 36 |
| 44 | P49411 | Elongation factor Tu, mitochondrial | 91 | 49852 | 7.9 | NDg | 1.33/0.003 | 13 |
| Protein deubiquitinationg | ||||||||
| 41 | P09936 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 | 80 | 25151 | 5.2 | NDg | 1.23/0.028 | 17 |
| 42 | P09936 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 | 106 | 25151 | 5.2 | NDg | 1.26/0.019 | 25 |
Spot No.- The numbers correspond to the specific spots as indicated in Figure 3.
Average ratios of spot abundance of control or BaP-treated samples relative to the cell line, represent data from three separate experiments.
Student’s t test p-value are given as a measure of confidence for the ratio of each spot measured.
P-C, the control group of shPARG cells; B-C, the control group of 16HBE cells.
P-T, the BaP-treated group of shPARG cells; B-T, the BaP-treated group of 16HBE cells.
Number of unique peptides according to proteins were identified by mass spectrometry.
Functional categories according to Gene ontology and panther biological process annotations.
Expression of TCTP and Cofilin-1 are confirmed by western blot
To confirm some of the results obtained from DIGE experiments, Translationally-controlled tumor protein (TCTP) and Cofilin-1 were selected for verification of protein-expression changes between the shPARG cells and 16HBE cells treated with various concentrations of BaP for 15 weeks. GAPDH was used as internal control. We confirmed that BaP-induced increase in abundance of TCTP and Cofilin-1 in 16HBE cells which were unchanged in the shPARG cells (Figure 4). These data corroborated the results from our proteomic analysis.
Figure 4.
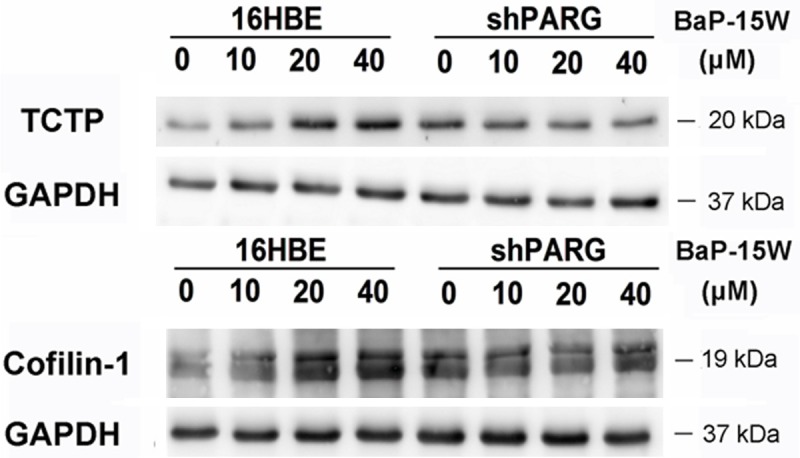
Validation of proteomic data. Expression of TCTP and cofilin-1 in two different cell lines treated with 0, 10, 20, 40 μM BaP for 15 weeks. GAPDH was used as a loading control. 16HBE - 16HBE cells, shPARG - shPARG cells.
Up-regulation and co-localization of TCTP and Cofilin-1 are required for BaP-induced cell transformation
Immunocytochemistry was further performed to determine whether TCTP effects on the expression of Cofilin-1 after BaP treatment. As assessed by immunocytochemical analysis (Figure 5), BaP treatment caused up-regulation of TCTP and Cofilin-1 in 16HBE cells. According to photos, we found that TCTP and cofilin-1 co-localized in cytoplasm and exhibited strong signal with low background. However, there was no significant difference of TCTP and Cofilin-1 expression in BaP-treated shPARG cells compared with the untreated control. Overall, these data demonstrate that up-regulation and co-localization of TCTP and Cofilin-1 play a key role in BaP-induced cell transformation.
Figure 5.
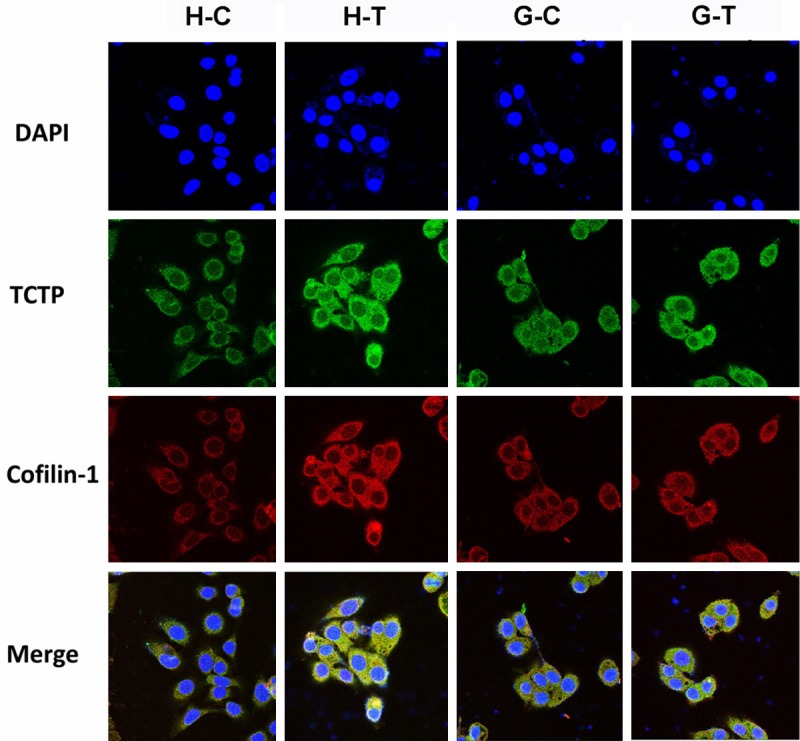
Expressions of TCTP and Cofilin-1 analyze by confocal imaging. BaP-induced TCTP and Cofilin-1 up-regulation in 16HBE cells were detected by immunocytochemistry. H-C, 16HBE cell treated with 0 μM BaP for 15 weeks; H-T, 16HBE cell treated with 40 μM BaP for 15 weeks; G-C, shPARG cell treated with 0 μM BaP for 15 weeks; G-T, shPARG cell treated with 40 μM BaP for 15 weeks. Scale bar: 20 μm.
Discussion
Carcinogenesis is multistage and may involve not only gene mutations but also abnormal dynamics of chromosomal organization, possibly also caused by non-genotoxic factors. Research on poly(ADP-ribosyl)ation has progressed rapidly, the recent study reveals that poly(ADP-ribosyl)ation is dynamic and important for the regulation of critical cell functions, including mechanisms suppressing carcinogenesis [15-17]. Possible applications in the therapy and prevention of cancer are also discussed [18,19].
BaP belongs to the polycyclic aromatic hydrocarbon (PAH) family of environmental carcinogens that are present in cigarette smoke, automobile exhaust and barbecued food. BaP is tumorigenic in laboratory animals and a suspected human carcinogen [20-22]. In the present study, we found that 16HBE cells have a greatly increased proliferation rate, and form colonies in soft agar after BaP exposure. However, PARG silencing significantly inhibited BaP-induced colony formation. Previously investigations had shown that PARP-1 played an important role in early repair of DNA damage caused by BaP [13]. This conclusion was further supported by our observation of PARG silencing reduced BaP-induced genetic toxicity.
To reveal the possible molecular mechanisms of PARG in BaP carcinogenesis, a comparative proteomic approach was carried out between normal and PARG silencing human bronchial epithelial cells in response to long-term BaP stimulation. In our study, soluble proteomes of cells were separated with a high-resolution 2D-DIGE, with a mean number of 1129 ± 72 spots. A total of 41 differentially expressed protein spots were identified between normal and PARG-deficient cells induced by BaP. Most are involved cell mobility, stress response, DNA repair and cell proliferation pathways.
Among these proteins, we focused on TCTP because of its involvement in malignancy. In our study, BaP treatment caused protein TCTP up-regulation in 16HBE cells, but it was found in relatively low level in PARG-deficient cells. TCTP is a highly conserved multifunctional protein [23], which has been implicated in many cellular processes, such as cell growth, cell cycle progression, apoptosis, malignant transformation, and the regulation of pluripotency. Overexpression of TCTP was detected in many types of tumors [24]. TCTP was initially identified as a growth-related protein on the basis of its translationally dependent regulation of expression in mouse ascetic tumor and erythroleukemic cells [25,26].
In this study, we also found that TCTP and cytoskeleton proteins in the normal cells were up-regulated in a dose-dependent manner. They are consistent with recent findings identifying TCTP reveals homology to the actin-binding region of Cofilin and TCTP interacts with actin cytoskeleton in cancer cells [27,28]. Cofilin is implicated in chemotaxis [29,30] and cell growth [31] and is believed to promote tumor metastatis by enhancing actin dynamics at the leading cell edge [32]. We showed in the current paper that TCTP and Cofilin-1 down-regulated synchronously in the PARG-deficient cell. Our data suggest that TCTP may involve in BaP carcinogenesis and PARG silencing may reduce cell migration to prevent tumor metastases.
Our proteomic analysis revealed that expression of Ubiquitin C-terminal hydrolase L1 (UCHL1) protein in PARG-deficient cell was significantly higher than the normal 16HBE cells after BaP exposure. UCHL1 was first identified as a deubiquitinating enzyme that hydrolyses the peptide bond at the C terminus of ubiquitin [33]. Protein ubiquitination plays a critical role in various biological processes including cell proliferation, cell cycle, apoptosis, signal transduction, while its deregulation contributes to tumor initiation and progression [34,35]. UCHL1 is a dual-regulator of the ubiquitin proteasome pathway, controls intracellular protein stability by transferring ubiquitin directly to protein substrates and releasing ubiquitin from tandemly conjugated ubiquitin monomers [36,37]. It is a tumor-suppressor gene and may inactivate by promoter methylation or gene deletion in several types of human cancers [38]. Here, PARG silencing up-regulate UCHL1 expression, which perhaps result in regulation of the ubiquitin pathway, inducing apoptosis and maintaining genomic stability. These data also provide a new evidence that poly(ADP-ribosyl)ation may has a close interaction with ubiquitination.
In summary, this study provides the first evidence that: (i) PARG silencing reduced cell migration after BaP exposure, (ii) PARG silencing down-regulated TCTP and Cofilin-1 to prevent tumor metastases, (iii) poly(ADP-ribosyl)ation has a close interaction with ubiquitination. Based on the results of the present study we propose PARG silencing might down-regulate TCTP and Cofilin-1 associated with metastasis in BaP carcinogenesis, which is depicted in Figure 6.
Figure 6.
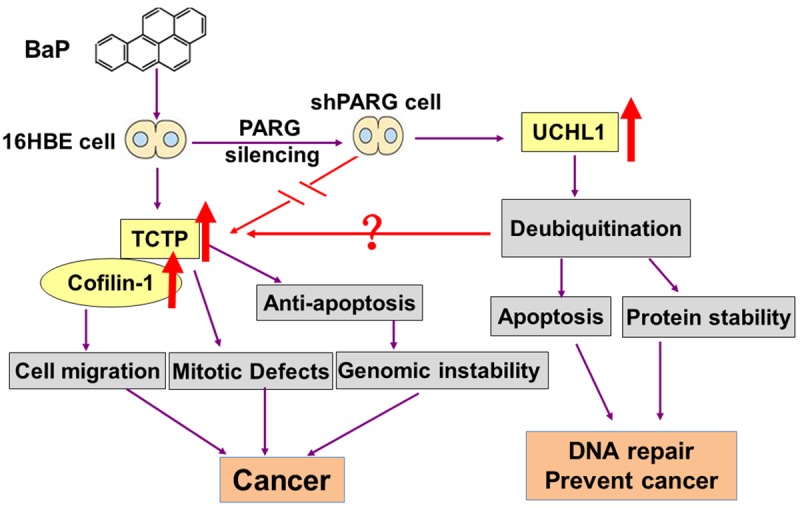
Role of PARG silencing and the possible molecular mechanism in BaP carcinogenesis.
Further analysis of TCTP interactions both with UCHL1 and PARG should give us more insights into the role of PARG in the regulation of BaP carcinogenesis. This is the first experimental demonstration of a link between PARG silencing and reduced cell migration after BaP exposure. We propose that PARG silencing might down-regulate TCTP and Cofilin-1 associated with metastasis in BaP carcinogenesis.
Acknowledgements
This study was supported by NSFC (Project No: 81001261, 81370080, 81273127 and 81260434), the Science and Technology Program of Shenzhen (Project No: 201302238), the Future Industry Special Project of Shenzhen (No. ZDSYS20140509101335476) and the Shenzhen Science and Technology Development Fund Project (Project No: JCYJ20130329103949642).
Disclosure of conflict of interest
None.
References
- 1.Drel’ VR, Shymans’kyĭ IO, Sybirna NO, Velykyĭ MM. Role of PARP and protein poly-ADP-ribosylation process in regulation of cell functions. Ukr Biokhim Zh. 2011;6:5–34. [PubMed] [Google Scholar]
- 2.Miwa M, Masutani M. PolyADP-ribosylation and cancer. Cancer Sci. 2007;10:1528–1535. doi: 10.1111/j.1349-7006.2007.00567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masutani M, Nakagama H, Sugimura T. Poly(ADP-ribosyl)ation in relation to cancer and Figure 6. Role of PARG silencing and the possible molecular mechanism in BaP carcinogenesis. autoimmune disease. Cell Mol Life Sci. 2005;62:769–783. doi: 10.1007/s00018-004-4509-x. [DOI] [PubMed] [Google Scholar]
- 4.Pears CJ, Couto CA, Wang HY, Borer C, Kiely R, Lakin ND. The role of ADP-ribosylation in regulating DNA double-strand break repair. Cell Cycle. 2012;11:48–56. doi: 10.4161/cc.11.1.18793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, Ahel I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature. 2011;7366:616–620. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-Ribosylation Reactions in Mammalian Cells: Where Are We Today and Where Are We Going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Min W, Wang ZQ. Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front Biosci (Landmark Ed) 2009;14:1619–1626. doi: 10.2741/3329. [DOI] [PubMed] [Google Scholar]
- 9.Amé JC, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, Schreiber V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J Cell Sci. 2009;122:1990–2002. doi: 10.1242/jcs.039115. [DOI] [PubMed] [Google Scholar]
- 10.Fujihara H, Ogino H, Maeda D, Shirai H, Nozaki T, Kamada N, Jishage K, Tanuma S, Takato T, Ochiya T, Sugimura T, Masutani M. Poly(ADP-ribose) Glycohydrolase deficiency sensitizes mouse ES cells to DNA damaging agents. Curr Cancer Drug Targets. 2009;9:953–962. doi: 10.2174/156800909790192419. [DOI] [PubMed] [Google Scholar]
- 11.Shirai H, Poetsch AR, Gunji A, Maeda D, Fujimori H, Fujihara H, Yoshida T, Ogino H, Masutani M. PARG dysfunction enhances DNA double strand break formation in S-phase after alkylation DNA damage and augments different cell death pathways. Cell Death Dis. 2013;4:e656. doi: 10.1038/cddis.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Augusto S, Pereira MJ, Máguas C, Soares A, Branquinho C. Assessing human exposure to polycyclic aromatic hydrocarbons (PAH) in a petrochemical region utilizing data from environmental biomonitors. J Toxicol Environ Health A. 2012;75:819–830. doi: 10.1080/15287394.2012.690685. [DOI] [PubMed] [Google Scholar]
- 13.Tao GH, Yang LQ, Gong CM, Huang HY, Liu JD, Liu JJ, Yuan JH, Chen W, Zhuang ZX. Effect of PARP-1 deficiency on DNA damage and repair in human bronchial epithelial cells exposed to Benzo(a)pyrene. Mol Biol Rep. 2009;8:2413–2422. doi: 10.1007/s11033-009-9472-z. [DOI] [PubMed] [Google Scholar]
- 14.Huang HY, Cai JF, Liu QC, Hu GH, Xia B, Mao JY, Wu DS, Liu JJ, Zhuang ZX. Role of poly(ADP-ribose) glycohydrolase in the regulation of cell fate in response to benzo(a)pyrene. Exp Cell Res. 2012;5:682–690. doi: 10.1016/j.yexcr.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Bieche I, de Murcia G, Lidereau R. Poly(ADP-ribose) polymerase gene expression status and genomic instability in human breast cancer. Clin Cancer Res. 1996;2:1163–1167. [PubMed] [Google Scholar]
- 16.Raval-Fernandes S, Kickhoefer VA, Kitchen C, Rome LH. Increased susceptibility of vault poly(ADP-ribose) polymerase-deficient mice to carcinogen induced tumorigenesis. Cancer Res. 2005;65:8846–8852. doi: 10.1158/0008-5472.CAN-05-0770. [DOI] [PubMed] [Google Scholar]
- 17.Shiokawa M, Masutani M, Fujihara H, Ueki K, Nishikawa R, Sugimura T, Kubo H, Nakagama H. Genetic alteration of poly(ADPribose) polymerase-1 in human germ cell tumors. Jpn J Clin Oncol. 2005;35:97–102. doi: 10.1093/jjco/hyi028. [DOI] [PubMed] [Google Scholar]
- 18.Calabrese CR, Almassy R, Barton S, Batey MA, Calvert AH, Canan-Koch S, Durkacz BW, Hostomsky Z, Kumpf RA, Kyle S, Li J, Maegley K, Newell DR, Notarianni E, Stratford IJ, Skalitzky D, Thomas HD, Wang LZ, Webber SE, Williams KJ, Curtin NJ. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96:56–67. doi: 10.1093/jnci/djh005. [DOI] [PubMed] [Google Scholar]
- 19.Gallmeier E, Kern SE. Absence of specific cell killing of the BRCA2-deficient human cancer cell line CAPAN1 by poly(ADP-ribose) polymerase inhibition. Cancer Biol Ther. 2005;4:703–706. doi: 10.4161/cbt.4.7.1909. [DOI] [PubMed] [Google Scholar]
- 20.Xiao H, Rawal M, Hahm ER, Singh SV. Benzo[a] pyrene-7,8-diol-9,10-epoxide causes caspase-mediated apoptosis in H460 human lung cancer cell line. Cell Cycle. 2007;22:2826–2834. doi: 10.4161/cc.6.22.4891. [DOI] [PubMed] [Google Scholar]
- 21.International Agency for Research on Cancer . Polynuclear Aromatic Compounds, Part 1. Chemical, Environmental, and Experimental Data. Vol. 32. Lyon, France: International Agency for Research on Cancer; 1983. Monographs on the evaluation of the Carcinogenic risk of Chemicals to Humans. [PubMed] [Google Scholar]
- 22.Hu X, Benson PJ, Srivastava SK, Xia H, Bleicher RJ, Zaren HA, Awasthi S, Awasthi YC, Singh SV. Induction of glutathione S-transferase π as a bioassay for the evaluation of potency of inhibitors of benzo(a)pyrene-induced cancer in a murine model. Int J Cancer. 1997;73:897–902. doi: 10.1002/(sici)1097-0215(19971210)73:6<897::aid-ijc23>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 23.Bazile F, Pascal A, Arnal I, Le Clainche C, Chesnel F, Kubiak JZ. Complex relationship between TCTP, microtubules and actin microfilaments regulates cell shape in normal and cancer cells. Carcinogenesis. 2009;30:555–65. doi: 10.1093/carcin/bgp022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan TH, Chen L, Guan XY. Role of Translationally Controlled Tumor Protein in Cancer Progression. Biochem Res Int. 2012;2012:369384. doi: 10.1155/2012/369384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yenofsky R, Cereghini S, Krowczynska A, Brawerman G. Regulation of mRNA utilization in mouse erythroleukemia cells induced to differentiate by exposure to dimethyl sulfoxide. Mol Cell Biol. 1983;3:1197–1203. doi: 10.1128/mcb.3.7.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chitpatima ST, Makrides S, Bandyopadhyay R, Brawerman G. Nucleotide sequence of a major messenger RNA for a 21 kilodalton polypeptide that is under translational control in mouse tumor cells. Nucleic Acids Res. 1988;16:2350. doi: 10.1093/nar/16.5.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsarova K, Yarmola EG, Bubb MR. Identification of a cofilin-like actin-binding site on translationally controlled tumor protein (TCTP) FEBS Lett. 2010;584:4756–4760. doi: 10.1016/j.febslet.2010.10.054. [DOI] [PubMed] [Google Scholar]
- 28.Bazile F, Pascal A, Arnal I, Le Clainche C, Chesnel F, Kubiak JZ. Complex relationship between TCTP, microtubules and actin microfilaments regulates cell shape in normal and cancer cells. Carcinogenesis. 2009;4:555–565. doi: 10.1093/carcin/bgp022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishita M, Tomizawa C, Yamamoto M, Horita Y, Ohashi K, Mizuno K. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J Cell Biol. 2005;171:349–359. doi: 10.1083/jcb.200504029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mouneimne G, DesMarais V, Sidani M, Scemes E, Wang W, Song X, Eddy R, Condeelis J. Spatial and temporal control of cofilin activity is required for directional sensing during chemotaxis. Curr Biol. 2006;16:2193–2205. doi: 10.1016/j.cub.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 31.Tsai CH, Chiu SJ, Liu CC, Sheu TJ, Hsieh CH, Keng PC, Lee YJ. Regulated expression of cofilin and the consequent regulation of p27(kip1) are essential for G(1) phase progression. Cell Cycle. 2009;8:2365–2374. doi: 10.4161/cc.8.15.9072. [DOI] [PubMed] [Google Scholar]
- 32.van Rheenen J, Condeelis J, Glogauer M. A common cofilin activity cycle in invasive tumor cells and inflammatory cells. J Cell Sci. 2009;122:305–311. doi: 10.1242/jcs.031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chu KY, Li H, Wada K, Johnson JD. Ubiquitin C-terminal hydrolase L1 is required for pancreatic beta cell survival and function in lipotoxic conditions. Diabetologia. 2012;55:128–140. doi: 10.1007/s00125-011-2323-1. [DOI] [PubMed] [Google Scholar]
- 34.Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol. 2005;23:4776–4789. doi: 10.1200/JCO.2005.05.081. [DOI] [PubMed] [Google Scholar]
- 35.Orlowski RZ, Dees EC. The role of the ubiquitination-proteasome pathway in breast cancer: applying drugs that affect the ubiquitin-proteasome pathway to the therapy of breast cancer. Breast Cancer Res. 2003;5:1–7. doi: 10.1186/bcr460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT Jr. The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell. 2002;111:209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 37.Osaka H, Wang YL, Takada K, Takizawa S, Setsuie R, Li H, Sato Y, Nishikawa K, Sun YJ, Sakurai M, Harada T, Hara Y, Kimura I, Chiba S, Namikawa K, Kiyama H, Noda M, Aoki S, Wada K. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet. 2003;12:1945–1958. doi: 10.1093/hmg/ddg211. [DOI] [PubMed] [Google Scholar]
- 38.Kato N, Yamamoto H, Adachi Y, Ohashi H, Taniguchi H, Suzuki H, Nakazawa M, Kaneto H, Sasaki S, Imai K, Shinomura Y. Cancer detection by ubiquitin carboxyl-terminal esterase L1 methylation in pancreatobiliary fluids. World J Gastroenterol. 2013;19:1718–1727. doi: 10.3748/wjg.v19.i11.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]