

ABSTRACT
The Kaposi's sarcoma-associated herpesvirus (KSHV) ORF57 gene product is essential for lytic KSHV replication and virion production. Recombinant ORF57-null mutants fail to accumulate several lytic cycle mRNAs at wild-type levels, leading to decreased production of lytic proteins necessary for efficient replication. Several mechanisms by which ORF57 may enhance expression of lytic KSHV mRNAs have been proposed, including mRNA stabilization, mRNA nuclear export, increased polyadenylation, and transcriptional activation. ORF57 activity is also gene specific, with some genes being highly dependent on ORF57, whereas others are relatively independent. Most experiments have utilized transfection models for ORF57 and have not systematically examined the gene specificity and potential mechanisms of action of ORF57 in the context of KSHV-infected cells. In this study, the KSHV genes that are most highly upregulated by ORF57 during KSHV lytic replication were identified by a combination of high-throughput deep RNA sequencing, quantitative PCR, Northern blotting, and rapid amplification of cDNA ends methods. Comparison of gene expression from a ΔORF57 KSHV recombinant, a rescued ΔORF57 KSHV recombinant, and wild-type KSHV revealed that two clusters of lytic genes are most highly dependent on ORF57 for efficient expression. Despite contiguous location in the genome and shared polyadenylation of several of the ORF57-dependent genes, ORF57 regulation was promoter and polyadenylation signal independent, suggesting that the mRNAs are stabilized by ORF57. The eight genes identified to critically require ORF57 belong to both early and late lytic temporal classes, and seven are involved in DNA replication, virion assembly, or viral infectivity, explaining the essential role of ORF57 in infectious KSHV production.
IMPORTANCE Kaposi's sarcoma-associated herpesvirus (KSHV) is a human herpesvirus involved in the causation of several human cancers. The KSHV ORF57 protein is required for KSHV to replicate and produce infectious virus. We have identified several KSHV genes whose expression is highly dependent on ORF57 and shown that ORF57 increases expression of these genes specifically. These genes code for proteins that are required for the virus to replicate its DNA and to infect other cells. Identifying the targets and mechanism of action of ORF57 provides further approaches to discover antiviral therapy.
INTRODUCTION
Kaposi's sarcoma associated herpesvirus (KSHV; human herpesvirus 8) is a human gammaherpesvirus associated with the development of Kaposi's sarcoma and several pathogenic lymphoproliferative syndromes (for a review, see reference 1). Lytic KSHV replication plays a role in pathogenesis by maintaining the infected cell reservoir, and lytic-phase KSHV proteins contribute to cell proliferation, inflammation, and immune evasion (2, 3). Understanding the mechanism of action of proteins required for lytic herpesvirus replication is therefore critical for further understanding of oncogenic mechanisms and the development of new antiviral agents.
The KSHV ORF57 gene product is essential for lytic replication and virion production (4, 5). Recombinant ORF57-null mutants fail to accumulate many lytic mRNAs at wild-type (WT) levels, leading to decreased production of the lytic proteins necessary for efficient replication (4, 6–8). Several mechanisms by which ORF57 may enhance transcriptional and posttranscriptional expression of lytic KSHV mRNAs have been proposed, and these have received various degrees of experimental support. These mechanisms include mRNA stabilization, mRNA nuclear export, increased polyadenylation, and cooperation with the KSHV ORF50/Rta transcriptional activator (4, 6, 7, 9–17). In addition, ORF57 activity appears to be gene specific, with some genes being highly dependent on ORF57, whereas others are relatively independent of ORF57 (4, 5, 8, 18, 19). Most experiments have utilized transfection models for ORF57 and have not systematically examined the gene specificity and potential mechanisms of action of ORF57 in the context of KSHV-infected cells. Because ORF57 belongs to a family of essential proteins homologous with proteins in each human herpesvirus and because these proteins do not have orthologs in human cells, these proteins represent attractive therapeutic targets (4, 20–24). Further clarification of the mechanism of action and target gene specificity of these proteins is therefore an important goal.
In this study, we set out to ask which of the KSHV genes are the most directly and highly upregulated by ORF57 during KSHV reactivation and lytic replication.
We previously showed that KSHV ORF57 is essential for infectious virion production using a BAC36 bacmid to generate a ΔORF57 recombinant (4). Recent sequencing data have shown that the BAC36 clone has a duplication of a 9-kb unique DNA fragment (25). Therefore, we generated a new ΔORF57 recombinant using another recently constructed infectious clone of the full-length KSHV genome derived from JSC1 cells, BAC16 (26). Using the ΔORF57 BAC16 recombinant, we compared gene expression during lytic reactivation of ΔORF57 and WT KSHV. Using a combination of high-throughput deep RNA sequencing, quantitative PCR (qPCR), Northern blotting, and rapid amplification of cDNA ends (RACE) methods, we delineated the effects of ORF57 on the KSHV transcriptome, established which KSHV RNAs are the most highly upregulated by ORF57, and explored the potential mechanisms by which ORF57 exerts its effects in the context of the entire virus during KSHV replication. The data indicate that ORF57 has its most important effects on two specific clusters of KSHV genes that are critical at both early and late stages of lytic replication.
MATERIALS AND METHODS
Cells, plasmids, and DNA transfections.
SLK cells containing a doxycycline-inducible ORF50 transactivator gene (iSLK cells) were cultured at 37°C in Dulbecco's modified Eagle's medium (DMEM; Cellgro) supplemented with 10% charcoal-stripped fetal calf serum and 1% GlutaMAX supplement (Life Technologies) (27). iSLK cells were infected with either WT or ΔORF57 KSHV bacmid-derived KSHV and maintained in 1,200 μg/ml hygromycin, 1.0 μg/ml puromycin, and 250 μg/ml active G418. 293T and HeLa cells were maintained in DMEM and 10% fetal calf serum. HeLa cell transfections were performed with the Lipofectamine Plus reagent with 1.0 μg of total DNA in 6-well plates according to the manufacturer's protocols (Invitrogen). 293T cells were transfected with the TransIT293 transfection reagent (Mirus) per the manufacturer's protocol.
The ORF57, GPCR, and ORF59 coding regions were PCR amplified and cloned into the mammalian expression vector pcDNA3 (Invitrogen) as previously described (4, 18, 19). The ORF4, ORF6, and ORF11 cDNAs were made by PCR amplification of 1.653 kb, 3.399 kb, and 1.224 kb, respectively, from iSLK cells and cloned into the mammalian expression vector pCDNA3. ORF4-4pA was made by PCR amplification of a 2,653-nucleotide (nt) DNA fragment containing the ORF4 coding domain sequence plus 1.0 kb of the 3′ untranslated region sequence and cloned into pCDNA3 after removal of the bovine growth hormone (βGH) polyadenylation signal from pCDNA3. To generate the ORF4 gene with an ORF11 poly(A) signal, the ORF11 poly(A) signal was PCR amplified and cloned downstream of the ORF4 coding sequence in place of the βGH poly(A) sequence in pCDNA3. All constructs were verified by DNA sequencing.
Generation of ΔORF57 KSHV in BAC16.
ΔORF57 KSHV was generated from the BAC16 cosmid parent as previously described (28). The entire ORF57 coding sequence (ATG-TAA) was removed from the KSHV genome using the λ Red recombinase system, resulting in a markerless deletion. The structure of ΔORF57 recombinant bacmids was verified by PCR and pulsed-field gel electrophoresis (PFGE). ΔORF57 bacmid DNA was isolated from bacteria using a Large-Construct kit (Qiagen) according to the manufacturer's recommendations.
Generation of iSLK cells carrying ΔORF57 KSHV BAC16.
293T cells were transfected with ΔORF57 bacmid DNA, and transfected cells were selected with 1.0 μg/ml puromycin for 1 week. Infectious ΔORF57 KSHV preparations were made from ΔORF57 KSHV-infected 293T cells 5 days after induction of viral replication by transfection with ORF50 and ORF57 plasmid DNAs. Cleared supernatants were filtered through 0.8-μm-pore-size cellulose acetate filters and were used to infect iSLK cells by the spin inoculation method. Infected iSLK cells were cultured in DMEM containing 1.0 μg/ml puromycin, 1,200 μg/ml hygromycin, and 250 μg/ml active G418 for 3 weeks. Individual clones were selected and screened for virus yield after transfection with ORF57 plasmid DNA and treatment with doxycycline. WT KSHV BAC16 was also passaged in iSLK cells in a similar fashion. The presence or absence of ORF57 in the ΔORF57 KSHV or WT KSHV clones passaged in iSLK cells was verified by Western blotting of cell lysates, made after addition of doxycycline to induce lytic gene expression, for ORF57.
RNA isolation and Northern blotting.
Total RNA was purified from pellets of iSLK and HeLa cells by standard protocols using QIAzol and miRNeasy columns according to the instructions in the Qiagen kit. RNA was quantitated by spectrophotometry, 3 to 5 μg of total RNA was resolved on 1% denaturing agarose gels, and Northern blotting was performed as previously described (29). Gene-specific 32P-labeled DNA probes were generated using a random oligonucleotide labeling kit (Amersham), and membranes were hybridized with probes overnight at 65°C. The blots were washed, and the radioactive signal was visualized by phosphorimaging and autoradiography.
Western blotting.
Protein cell lysates were made from iSLK cells stably infected with WT or ΔORF57 KSHV 48 h after viral replication was induced by doxycycline treatment. Samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane. Blots were probed with anti-ORF57 polyclonal antibody using horseradish peroxidase-conjugated secondary antibody, and the signal was visualized by chemiluminescence (Bio-Rad), as previously described (8).
Measurement of viral DNA replication and infectious viral particle titers.
KSHV lytic replication in iSLK cells infected with either WT or ΔORF57 KSHV was induced by treating the cells with 1.0 μg/ml doxycycline. Cell pellets were harvested 3 days and 5 days after doxycycline treatment, and DNA was purified using a Qiagen blood DNeasy kit. Fifty nanograms of total cell DNA was analyzed by qPCR using ORF59-specific primers to quantitate the viral genome copy number by comparison to the cellular GAPDH (glyceraldehyde-3-phosphate dehydrogenase) DNA copy number. Infectious viral particles were measured in cell supernatants at 5 days after induction of KSHV replication by 1.0 μg/ml doxycycline and 2 mM sodium butyrate. Cleared supernatants were filtered through 0.8-μm-pore-size cellulose acetate filters, and dilutions of supernatants were used to infect 293T cells by spin inoculation. Cells were centrifuged at 2,000 × g for 1.0 h at room temperature with virus-containing supernatant and incubated at 37°C. The numbers of green fluorescent protein (GFP)-positive 293T cells representing infectious KSHV particles were easily visualized under a fluorescence microscope after 2 days postinfection, and virus-infected cells were quantitated by flow cytometry as previously described (30, 31).
qPCR.
qPCR was performed with a SYBR green PCR master mix (ABI) according to the manufacturer's protocol using an Applied Biosystems StepOne Plus real-time PCR thermocycler. Poly(A) RNA was selected from total RNA using Oligotex mRNA kits (Qiagen). cDNAs were prepared with a high-capacity cDNA reverse transcription kit (Applied Biosystems) using oligo(dT) primers. Controls with no template were included in each analysis. Each PCR amplification was performed in triplicate with gene-specific primers. Cellular GAPDH was used as the endogenous control for all reactions. Selected KSHV transcripts were quantitated by qPCR using the primer pairs shown in Table 1.
TABLE 1.
Primer pairs used for qPCR
| Gene | Sequence |
|
|---|---|---|
| 5′ primer | 3′ primer | |
| ORF6 | CTGCCATAGGAGGGATGTTTG | CCATGAGCATTGCTCTGGCT |
| ORF8 | CCCGACGTAGATCGCAGG | GTTTTTGATTTCCTCCCGTGTT |
| ORF9 | TAGGCGCTTCGTGCTGG | CCGGATTGCTGCACTCGTA |
| ORF10 | GGGCGTGGCAATGGC | AAGCTGTATGGTGCCTGGCT |
| ORF11 | CGGAATGGCGCCCAA | GACGGGATGATCACTCGTGTT |
| ORF56 | CACAGATTCCCGTCAATACAAA | GTATCTTCAGTAGGCGGCAGAG |
| ORF58 | GCCAAAGGCAGGAGAACAAA | TGTCATGCGTGGGCGTAT |
| ORF59 | CTCCCTCGGCAGACACAGAT | GCGTGGTGCACACCGACGCCC |
RNA sequencing and data analysis.
Total RNAs were isolated from iSLK cells using the Qiagen miRNeasy kit. Unfractionated RNA (1.5 μg) was used for RNA sequencing at the Microarray and Genomic Analysis Core Facility, Huntsman Cancer Institute, University of Utah. cDNA libraries were prepared from poly(A)-selected RNA using Illumina TrueSeq kits and verified using an Agilent bioanalyzer. cDNA libraries were sequenced on a HiSeq2000 instrument with 50-cycle single end reads. Sequence reads were mapped to the KSHV genome, and gene expression analysis was done as described previously (32). Briefly, the reads were aligned to the transcriptome reference index described above using the Novoalign (v2.08.01) program, allowing up to 50 alignments for each read. USeq's SamTranscriptomeParser application was used to select the best alignment for each read and convert the coordinates of reads aligning to splices back to genomic space. Differential gene expression was measured using USeq's Defined Region Differential Seq application. Briefly, the numbers of reads aligned to each gene annotation were calculated. The counts were then used in the DESeq package, which normalizes the signal and determines differential expression (33). Fold change values of KSHV gene expression were not variance stabilized. KSHV transcript boundaries that were established by RACE, Northern blotting, and comparison with recently published data that differed from previously published NCBI annotations were used to map sequence reads as follows. The NCBI annotations were updated to reflect the predicted transcript boundaries. Human cDNA sequences (ensemble release 77) were combined with updated KSHV sequences. The Trinity toolkit (34) was used to estimate transcript abundance and determine differential expression. Trinity was set to use the RSEM software package for abundance estimation and the DESeq package for differential expression detection.
KSHV qPCR array.
KSHV replication in iSLK cells infected with ΔORF57 or WT KSHV bacmids was induced by doxycycline. RNA was isolated at 48 h after viral replication, and RNA was purified using Qiagen miRNeasy kits. Purified RNA was poly(A) selected using Oligotex mRNA kits, and cDNA was made as described above. cDNA samples were analyzed at the Vironomics Core Lab at the University of North Carolina as previously described (35).
RACE.
3′ RACE was performed by standard protocols as described previously (36). Briefly, cDNAs were made from 5.0 μg of total RNA using oligo(dT) adapter primers with SuperScript II reverse transcriptase, followed by RNase treatment (Invitrogen). cDNA pools were diluted to 500 μl with TE (Tris-EDTA) buffer, and PCR was performed using AccuPrime Pfx DNA polymerase (Invitrogen), gene-specific 5′ primers, universal 3′ primers complementary to the adapter sequence (TAGACGCGTGCTAGCTCGAGATCTT), and the following cycling conditions: 95°C for 3 min, 55°C for 2 min, and 68°C for 40 min, followed by 40 cycles at 95°C for 15 s, 55°C for 30 s, and 68°C for 3 min (31). The gene-specific primers used are listed in Table 2. PCR products were separated on agarose gels, purified, and analyzed by DNA sequencing. Sequences were mapped to the BAC16 KSHV genome (GenBank accession number GQ994935.1). 5′ RACE for the ORF9 gene was performed as follows. Briefly, cDNAs were synthesized from 5.0 μg of total RNA using gene-specific 3′ primers (CAAAGGATAGGATTGTGTATGG) and SuperScript II reverse transcriptase (Invitrogen), followed by RNase treatment. cDNA was purified using Qiagen PCR purification kits. Next, either dGTP or dCTP tailing was performed in a separate tube using terminal transferase (New England BioLabs) according to standard protocols with optimization of the reaction times. Second-strand synthesis was performed using dGTP or dCTP tailed DNA template with either d(C) adapter primer AAAAGATCTGTCGACCCCCCCCCCCCCCC or d(G) adapter primer GGCCACGCGTCGACTAGTACGGGIIGGGIIGGGIIG, respectively, and ORF9-3′ specific primers with Taq DNA polymerase (TaKaRa, Japan). The PCR products were separated by agarose gel electrophoresis, and the fragments were purified. The purified products were either sent directly for DNA sequencing or sequenced after cloning into the pBluescript plasmid.
TABLE 2.
Primers used for 3′ RACE analysis
| Gene | 5′ primer sequence |
|---|---|
| ORF6 | GGCCCTTATGCCGTACTTGG |
| ORF8 | GCTGGTACACGCGGTAGC |
| ORF10 | CAATAGTGGGCGTGGCAAT |
| ORF11 | CACGAGTGATCATCCCGTC |
| ORF58 | GTCATGCGTGGGCGTATC |
| ORF59 | GTAAACCGATCTGTGTCTGC |
| ORF60 | CAGTCACGCAGAGGTCCAG |
| ORF61 | GCGCCGTTTGTAGACCAG |
RESULTS
Construction and characterization of a BAC16 ΔORF57 KSHV bacmid.
Our previous work showed that KSHV ORF57 is essential for infectious virion release using the BAC36 KSHV bacmid to generate a ΔORF57 recombinant (4). Recent sequencing data have demonstrated that the BAC36 clone has a duplication of a 9-kb unique DNA fragment within the terminal repeat region (25), rendering it less desirable for studying global KSHV gene regulation by ORF57. We therefore employed another recently described infectious bacmid clone of the full-length KSHV genome derived from JSC1 cells named BAC16, which has been fully sequenced and shown to be free of sequence duplications or deletions (26). In order to study the magnitude and specificity of the effects of ORF57 on KSHV lytic cycle gene expression, we constructed a ΔORF57 KSHV clone in the BAC16 clone, removing the entire ORF57 coding region. The ORF57 coding sequence (ATG-TAA) was removed from KSHV BAC16 in a modified version of the protocol used to create markerless ORF57 deletions within BAC16 (28). Because the ORF57 deletion removes an NheI site in the KSHV genome, the resulting recombinant bacmid clones were screened and verified to contain the ORF57 deletion by NheI digestion followed by PFGE (Fig. 1). Restriction digest analysis of WT and ΔORF57 bacmid clones showed that several clones (clones 2, 3, 4, 6, 7, 8, and 11 which appear in the lanes with the corresponding numbers in Fig. 1B) had the correct deletion and had a highly visible 33-kb band that contained the terminal repeats. These clones were further verified by PCR using ORF57-specific primers, as shown in Fig. 1C. A 1.5-kb ORF57 amplicon was present only in WT bacmid and was absent in all ΔORF57 bacmid clones, as shown in Fig. 1D.
FIG 1.

Generation of ΔORF57 KSHV from BAC16 bacmid. (A) Schematic diagram of the KSHV genome with the NheI restriction map. The ORF57 gene in the KSHV genome is shown in a red box. (B) Ethidium bromide-stained gel of WT and ΔORF57 KSHV recombinant clones after NheI digestion and PFGE. A diagnostic 20-kb band present in the WT bacmid is absent in mutant ORF57 due to the loss of an NheI site in the deleted region. Clones 2, 3, 4, 6, 7, 8, and 11, which appear in the lanes with the corresponding numbers, have strong 33-kb terminal repeat bands and a loss of the 20-kb fragment. (C) Schematic diagram of the ORF57 gene structure and primers used for ΔORF57 verification. Amplification of the WT and ΔORF57 KSHV clones is predicted to yield a 1,556-bp fragment and an 80-bp fragment, respectively. (D) PCR analysis of the ORF57 deletion in ΔORF7 and WT KSHV clones. The amplification products obtained using the primers shown in panel C were analyzed by agarose gel electrophoresis. Recombinant clone numbers are shown above each lane.
Generation of iSLK cells carrying the ΔORF57 KSHV bacmid.
In order to study the role of ORF57 in KSHV lytic-phase gene regulation, we used an epithelial cell line known as iSLK, which inducibly expresses the Rta (ORF50) protein only in the presence of doxycycline (37). iSLK cells exhibit robust KSHV lytic replication and infectious virion release when treated with doxycycline. To produce stable ΔORF57 KSHV-infected iSLK cells, 293T cells were first transfected with the ΔORF57 bacmid and selected with hygromycin. These cells were then transfected with ORF50 and ORF57 to rescue KSHV virion production. Virus-containing supernatant was collected 5 days after transfection and used to infect iSLK cells, which were then selected with hygromycin. A WT KSHV strain was also similarly passaged in iSLK cells. Individual clones were selected and screened for ORF57-dependent virus production by transfection with either empty vector or ORF57 plasmid DNA and treatment with doxycycline. ΔORF57 virus-infected clones released infectious virus only with ORF57 rescue (data not shown). As expected, ΔORF57 KSHV did not express the ORF57 protein (Fig. 2A), but WT KSHV did produce the ORF57 protein during lytic replication. The absence of ORF57 in ΔORF57 KSHV-infected cells was further verified by Southern blotting of DNA isolated from ΔORF57 and WT KSHV-infected cells. Digestion with two separate restriction endonucleases (Fig. 2B) and hybridization with an ORF57-specific probe demonstrated the presence of the ORF57 sequence in WT KSHV-infected cells but not in ΔORF57 KSHV-infected cells. The ORF57 deletion was further confirmed by DNA sequencing.
FIG 2.

ORF57 is required for KSHV lytic replication and infectious virion release. iSLK cells stably infected with ΔORF57 or WT bacmids were mock treated (−D) or treated with doxycycline (+D) to induce ORF50-dependent lytic replication. (A) Western blot analysis to confirm the absence of ORF57 in ΔORF57 (Δ57) KSHV during replication in iSLK cells. The positive-control lysate from ORF57-transfected cells is shown in the lane labeled 57. (B) Southern blot analysis to confirm deletion of ORF57 from the BAC16 genome was performed by probing BamHI-digested DNA (left) and NheI-digested DNA (right) with an ORF57-specific probe. (C, D) Quantification of lytic DNA replication in ΔORF57 KSHV-infected cells (C) or WT KSHV-infected cells (D) by qPCR. Total cellular DNA isolated at 3 and 5 days after viral replication was induced with doxycycline, and KSHV genomes were quantitated by qPCR. (E) Quantification of infectious virion production. Virus replication in ΔORF57 and WT KSHV-infected iSLK cells was induced with doxycycline and sodium butyrate, and serial dilutions of the virus-containing supernatant were used to infect 293T cells. The number of GFP-positive 293T cells, representing infectious KSHV particles, was measured by flow cytometry. The virus titer (in number of GFP-inducing units/ml) is shown above each bar. U, uninduced; I, induced.
ORF57 is required for release of infectious virus from iSLK cells.
We have previously demonstrated that ORF57 is essential for KSHV lytic DNA replication and virion release in the supernatant (4, 30). In order to confirm that KSHV replication was ORF57 dependent in our newly generated BAC16 ΔORF57 virus-infected iSLK cells, we compared lytic KSHV replication in iSLK cells infected with ΔORF57 and WT KSHV. KSHV genome amplification from total cellular DNA isolated at 3 days and 5 days after induction of lytic replication was measured by qPCR. There were approximately 2-fold and 7-fold increases in intracellular KSHV DNA levels at 3 and 5 days after induction of replication in WT KSHV-infected iSLK cells, respectively (Fig. 2D). However, no significant increase in DNA copy number was observed in ΔORF57 KSHV-infected cells after induction (Fig. 2C), verifying the requirement for ORF57 in KSHV replication in iSLK cells. The release of infectious virus particles in supernatant harvested 5 days after induction of virus replication was also quantitated. Filtered supernatants from either ΔORF57 or WT KSHV-infected cells were used to passage released virus (which expresses GFP) in 293T cells, and infected cells were visualized by fluorescence microscopy. Virtually no virus was detectable in the supernatant from ΔORF57 KSHV-infected cells, whereas abundant virus release from WT-infected cells was visible. The virus yields were quantitated by flow cytometry of 293T cells infected by passaged KSHV. As shown in Fig. 2E, approximately 1 × 106 GFP-inducing units/ml were released from WT KSHV-infected iSLK cells. However, no virus release over the background was detectable in ΔORF57 KSHV-derived supernatants. These data confirm that ORF57 is essential for lytic DNA replication and infectious virion release and that the system provides a means to study gene regulation by ORF57 in the context of physiologic replication of a fully sequenced and intact KSHV genome.
Identification of ORF57-dependent gene regulation during KSHV lytic replication.
Our aim was to determine why ORF57 is essential for lytic cycle replication. In order to find out which genes are most highly ORF57 dependent during KSHV replication, we compared KSHV gene expression from ΔORF57 KSHV-infected cells that were transfected with either an empty vector or ORF57 and then induced to enter lytic replication. When gene expression was measured by qPCR arrays spanning the KSHV genome, several clusters of RNAs were differentially upregulated in the presence of ORF57 (Fig. 3). The KSHV array data, displayed as a waterfall plot in Fig. 4A, show that expression of most lytic genes was increased to various degrees in the presence of ORF57. The genes that were the most highly upregulated (log2 ratio, >2) in the presence of ORF57 are shown in Fig. 4B. In order to obtain a more quantitative and higher-resolution map of the effects of ORF57 on the lytic KSHV transcriptome, deep RNA sequencing was employed to compare uninduced and induced transcription between ΔORF57 and WT KSHV. As shown in Fig. 5A, robust induction of lytic transcription was evident in both WT and ΔORF57 KSHV-infected cells. However, upon lytic replication, there was relatively little induction of gene transcription in two regions of the ΔORF57 KSHV genome compared to that in the WT KSHV genome. When these most highly differentially expressed transcripts were mapped to the KSHV genome, it was apparent that nine KSHV genes (ORFs 4, 6, 7, 8, 9, 58, 59, 60, and 61) were the most highly dependent on ORF57, being expressed from 4- to 24-fold more highly by the WT KSHV genome than by the ΔORF57 KSHV genome. To confirm the ORF57 dependence of this group of genes, we rescued ORF57 expression by transfecting either an empty vector or a plasmid carrying ORF57 into ΔORF57 KSHV-infected iSLK cells and compared the KSHV transcript abundance in RNA from (i) uninduced, (ii) replication-induced (doxycycline-treated), and (iii) ORF57-rescued and replication-induced samples. The net effects of ORF57 on gene expression were again clearly seen at the two regions of the KSHV genome spanning ORF4 to ORF9 and ORF58 to ORF60 (Fig. 5C and D). These data correlate almost perfectly with the PCR array data presented in Fig. 4. Upregulation of ORF71 (vFLIP) and ORF72 (v-cyc) was observed only when ORF57 was transfected to rescue the ORF57 deletion, possibly reflecting the effects of constitutive ORF57 expression from a cytomegalovirus (CMV) promoter as opposed to expression from its native promoter in the KSHV genome.
FIG 3.

KSHV gene expression profile of ΔORF57 KSHV during lytic replication. KSHV qPCR array data from ΔORF57 KSHV-infected iSLK cells transfected with empty plasmid vector DNA (mock), induced with doxycycline (induced), or induced with doxycycline and rescued by transfection with ORF57 (induced 57 rescue). Total cellular RNAs were isolated at 48 h after induction of lytic replication, and polyadenylated RNA was selected with oligo(dT) and amplified with gene-specific primers to compare KSHV gene expression in the absence and presence of ORF57. The gene expression profiles obtained without ORF50 (left), with ORF50 (middle), and with ORF50 and ORF57 (right) are shown. Primer locations are shown at the left, and several representative upregulated genes are shown at the right. Blue, unchanged gene expression; brown, upregulated gene expression.
FIG 4.
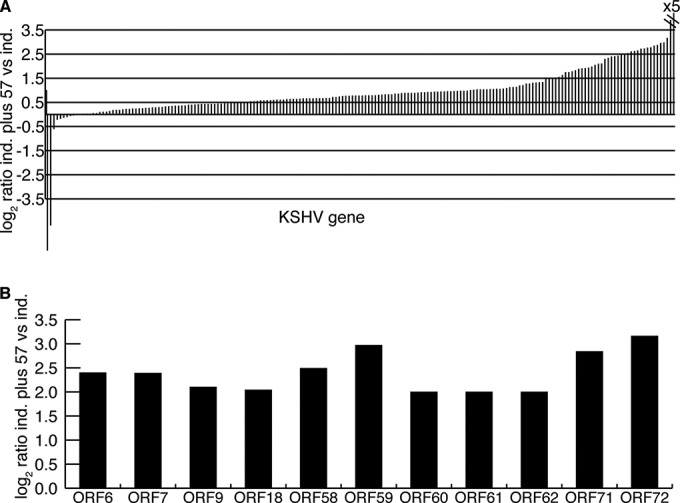
KSHV ORF57-dependent gene regulation. (A) Relative change in KSHV gene expression at 48 h in cells induced to replicate and rescued with ORF57 (ind. plus 57) from that in cells induced to replicate but not rescued (ind.). (B) KSHV genes whose expression was increased (log2 ratio, >2.0) in the presence of ORF57 during replication.
FIG 5.

Comparison of KSHV RNA expression profiles in the presence or absence of ORF57. (A) Comparison of transcriptomes ΔORF57 KSHV-infected cells and WT KSHV-infected cells. ΔORF57 KSHV-infected and WT KSHV-infected iSLK cells were either mock treated (−D) or treated with doxycycline (+D) to induce lytic replication. RNA was collected at 48 h after induction of viral replication and analyzed by high-throughput RNA sequencing. Sequenced reads mapped to the KSHV genome on the x axis are shown as normalized read counts on the y axis. Red bars denote two clusters of genes whose expression was increased in WT KSHV-infected iSLK cells compared to that in ΔORF57 KSHV-infected iSLK cells. The differential expression of ORF57 in WT KSHV is also shown. (B) ORF57-dependent gene expression is shown as the ratio of RNA abundance in WT KSHV-infected iSLK cells versus that in ΔORF57 KSHV-infected iSLK cells. Genes most highly upregulated by ORF57 are marked in red. (C) Transcriptome comparison of ΔORF57 KSHV-infected iSLK cells and ORF57-rescued ΔORF57 KSHV-infected iSLK cells. Sequenced reads mapped to the KSHV genome are shown as normalized read counts in samples from uninduced ΔORF57 KSHV-infected iSLK cells transfected with empty vector (Uninduced), ΔORF57 KSHV-infected iSLK cells induced to replicate and transfected with empty vector (Induced), or ΔORF57 KSHV-infected iSLK cells induced to replicate and transfected with ORF57 (57 rescue). Clusters of genes whose expression was increased in ORF57-rescued cells compared to their level of expression in induced ΔORF57 KSHV-infected iSLK cells are marked with red bars. ORF57 expression in rescued cells is also shown (arrow). (D) ORF57-dependent gene expression is shown as the ratio of RNA abundance in replication-induced ORF57-rescued ΔORF57 KSHV-infected iSLK cells versus that in replication-induced ΔORF57 KSHV-infected iSLK cells. Genes most highly upregulated by ORF57 are marked in red.
We further validated the transcriptome sequencing (RNA-seq) data by performing quantitative reverse transcription-PCR (qRT-PCR) on polyadenylated RNA for selected ORF57-dependent and -independent KSHV target genes (Fig. 6). A significant increase in ORF 6, 8, 9, 58, and 59 RNA levels in WT KSHV compared to those in ΔORF57 KSHV was observed, a finding which correlates well with the RNA-seq data. Conversely, expression of ORF10, ORF11, viral interleukin-6 (vIL-6; K2), and ORF56, which were relatively unaffected by ORF57 in the RNA-seq experiments, was not significantly different between WT KSHV and ΔORF57 KSHV. Polyadenylated nuclear (PAN) RNA expression was increased approximately 1.5-fold.
FIG 6.
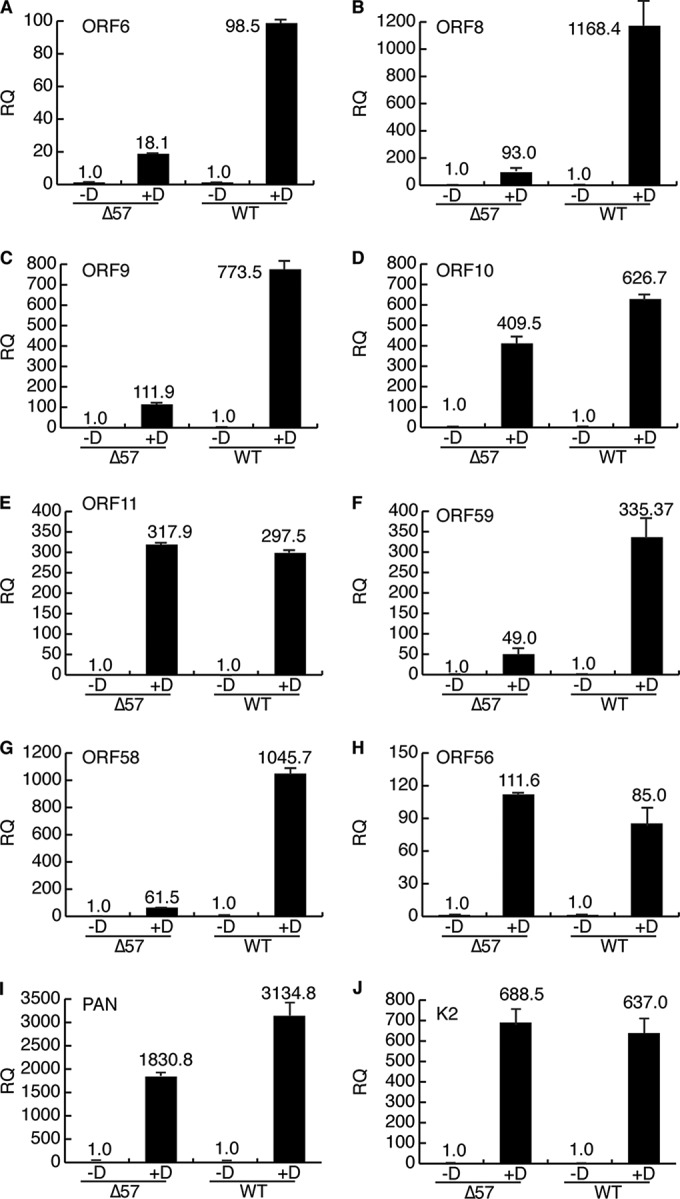
Quantification of ORF57 effects on KSHV genes by qPCR. Polyadenylated RNA was isolated from WT KSHV- and ΔORF57 KSHV-infected iSLK cells, and the abundance of specific mRNAs was measured by qRT-PCR. Analysis was performed with RNA harvested 48 h after mock treatment (−D) or induction of lytic replication with doxycycline (+D). Each bar represents the average of the results from three independently obtained RNA samples. The fold increase in mRNA abundance after induction is shown above each bar. RQ, relative quantity (RNA units).
Characterization of transcription and polyadenylation of the ORF4 to ORF11 gene cluster and effect of ORF57.
ORF57 has been suggested to act partly by enhancing the polyadenylation of target transcripts (12, 13, 38). The clustering of ORF57-dependent transcripts in two discrete regions of the genome and the previous annotation of these genes as belonging to polycistronic units that undergo coterminal polyadenylation at a single canonical polyadenylation site suggested that ORF57 might be affecting polyadenylation of the ORF4, 6, 7, 8, and 9 and the ORF58 to ORF62 gene clusters. To investigate whether these RNAs were polyadenylated at different sites or more efficiently in the presence of ORF57, we first scanned the genome sequence for the presence of canonical or noncanonical poly(A) signals in these regions and found several potential canonical and noncanonical polyadenylation sites located downstream of each open reading frame (ORF). We correlated these potential polyadenylation sites with the appearance of the RNA-seq profiles to identify sites where the read density decreased abruptly, indicating the possible presence of transcriptional termination. In addition, we searched the RNA-seq data for the presence of five or more A residues which did not map to the KSHV genome, in order to identify potential polyadenylation sites, as previously described (39). We also used Northern blot analysis to map the size of each transcript in RNA isolated from cells infected with ΔORF57 or WT KSHV and performed 3′ RACE to map the polyadenylation and cleavage sites of the mRNAs. By combining these data with published information from prior studies (31, 40–43), we generated comprehensive maps of the transcription and posttranscriptional processing of the mRNAs in question.
Analysis of the ORF4 to ORF11 cluster of genes by Northern blotting of oligo(dT)-selected RNAs from ΔORF57 KSHV- and WT KSHV-infected cells suggested that, on the basis of their size, contrary to previously published annotations (44), ORF4, ORF6, ORF7, and ORF9 mRNAs have unique 3′ cleavage and polyadenylation sites that yield monocistronic mRNAs (Fig. 7 and 8). However, the size of the ORF8 mRNA indicated that it may be part of a larger transcript, possibly extending downstream of ORF10 and ORF11. Nevertheless, consistent with our RNA-seq and qPCR data, Northern blot analysis confirmed that ORF4, ORF6, ORF7, and ORF9, as well as ORF8, are all highly ORF57 dependent for expression and are virtually undetectable in ΔORF57 KSHV-infected cells unless the cells are rescued with ORF57. Further, ORF10 and ORF11 were robustly expressed in the absence of ORF57, comparable to their expression in WT KSHV-infected cells, confirming that they are ORF57 independent. These data therefore indicated that the basis of ORF57 dependence was not likely due to the common utilization of an ORF57-activated polyadenylation site.
FIG 7.
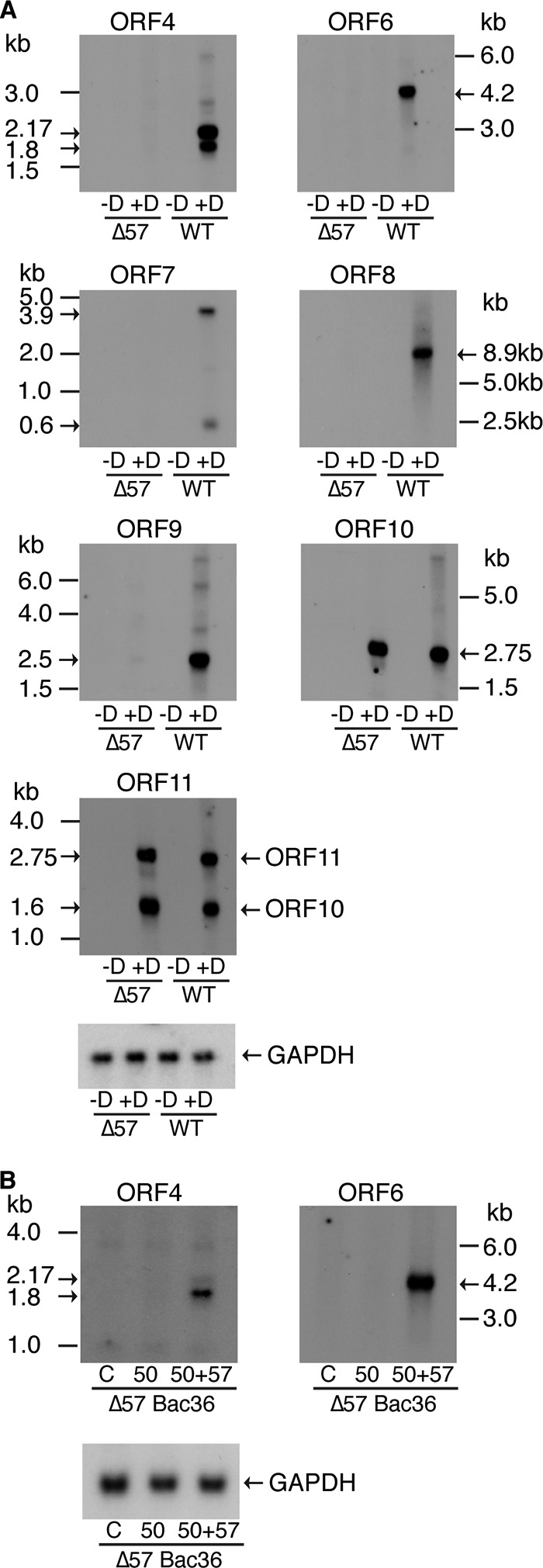
Analysis of ORF57-dependent and -independent gene expression in the ORF4 to ORF11 gene cluster by Northern blotting. (A) RNA was isolated from iSLK cells carrying either ΔORF57 or WT KSHV treated with doxycycline to induce replication (+D) or mock treated (−D) and analyzed by Northern blotting using probes specific for ORFs 4, 6, 7, 8, 9, 10, and 11, as shown above each panel. RNA molecular size markers are shown at the side. The calculated size of each mRNA is shown with an arrow. Blots were stripped and probed with GAPDH probes as a loading control. (B) The effects of ORF57 on KSHV RNA expression in 293T cells infected with ΔORF57 BAC36 were also analyzed by Northern blotting. Lytic KSHV replication in 293T cells was induced by transfection with a plasmid expressing ORF50 and cotransfected either with an empty vector (lanes 50) or with a plasmid expressing ORF57 (lanes 50+57). Cells were also transfected with empty vector alone as an uninduced control (lanes C). The gene probe used is shown above each panel, and RNA molecular size markers and mRNA sizes are shown at the side, as described in the legend to panel A. Blots were stripped and probed with GAPDH probes as a loading control.
In order to further define the complex transcriptional and posttranscriptional processing of this cluster of genes, we combined the data from 3′ RACE and the sequencing profile to generate the map shown in Fig. 8, which confirms the utilization of unique polyadenylation signals by ORFs 4, 6, and 9 and a common coterminal polyadenylation signal by ORFs 8, 10, and 11. Although the predicted ORF9 is 3,039 nucleotides in length, careful measurement of the size of the ORF9 mRNA detected on Northern blots indicated a size closer to 2.5 kb. This suggested either the existence of a novel transcriptional start site or premature termination. 5′ RACE and sequencing of RT-PCR amplicons revealed that transcription actually begins at nucleotide 11826, downstream of the first methionine in the ORF, indicating that termination occurs shortly after the stop codon at nt 14382. The KSHV DNA polymerase in KSHV from JSC-1 cells is therefore likely to be much smaller than previously thought, initiating translation from the third methionine in the originally predicted ORF and yielding a protein of 801 amino acids (aa) rather than one of 1,013 aa.
FIG 8.
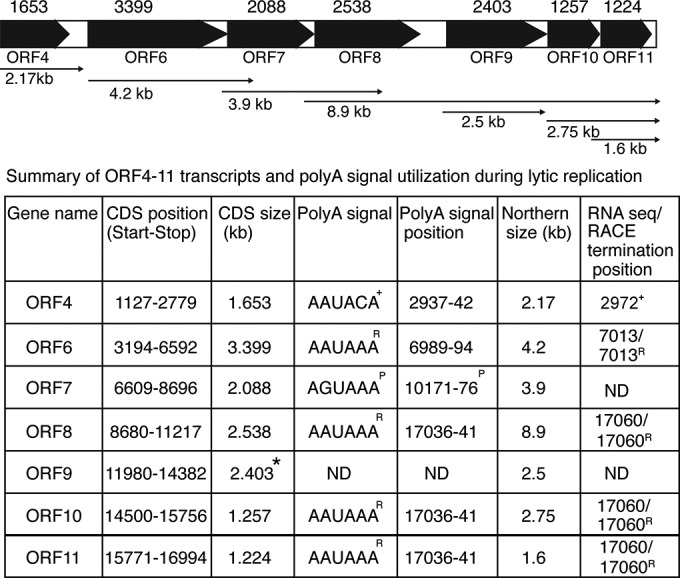
Structure of the ORF 4 to ORF11 gene cluster. The coding domain of each gene is shown as a black bar with an arrowhead. Each transcript is denoted by arrows below the bars, with the predicted sizes (in kb) being indicated below the arrows. Transcript sizes are based on a combination of the sizes predicted by Northern blotting, RACE analysis, and inspection of the read abundance profile from deep sequencing, as shown in the table. The location of the putative canonical or noncanonical cleavage and polyadenylation signal based on the transcript size or RACE analysis for each transcript is shown, except where no such signal could be identified in the KSHV genome sequence (ORF9). Abbreviations and symbols: R, confirmed by RACE in this study; +, RACE result from other studies; P, putative closest polyadenylation signal by Northern mapping; *, mapping by Northern blotting and 5′ RACE; ND, not determined; CDS, coding domain sequence.
We performed a similar analysis on the ORF58 to ORF62 gene cluster utilizing a combination of RACE and Northern blotting (Fig. 9 and 10). The results in this case clearly indicate that all five transcripts originate from unique promoters but terminate at a common site downstream of two overlapping canonical polyadenylation signals, consistent with previously published data (41). Although all five mRNAs coterminate and their accumulation is clearly ORF57 enhanced, the relative levels of the mRNAs are quite different, suggesting that their promoters are not equally active or that the degree of posttranscriptional upregulation by ORF57 varies among the different mRNAs.
FIG 9.

Analysis of ORF57 enhanced gene expression in the ORF58 to ORF62 gene cluster. (A) RNA was isolated from iSLK cells carrying either ΔORF57 or WT KSHV, treated with doxycycline to induce replication (+D) or mock treated (−D), and analyzed by Northern blotting using probes specific for ORFs 58 to 62. RNA molecular size markers are shown at the side. The calculated size of each mRNA is shown with an arrow. (B) The effects of ORF57 on KSHV RNA expression in 293T cells infected with ΔORF57 BAC36 were also analyzed by Northern blotting. Lytic KSHV replication in 293T cells was induced by transfection with a plasmid expressing ORF50 and cotransfected with either an empty vector (lanes 50) or a plasmid expressing ORF57 (lanes 50+7). Cells were also transfected with empty vector alone as an uninduced control (lanes C). The gene probe used is shown above each panel, and RNA molecular size markers are shown at the side. Blots were stripped and probed with GAPDH probes as a loading control.
FIG 10.

Structure of the ORF58 to ORF62 gene cluster. The coding domain of each gene is shown as a black bar with an arrowhead. Each transcript is denoted by arrows below the bars, with the predicted sizes (in kb) being indicated below the arrows. Transcript sizes are based on a combination of the sizes predicted by Northern blotting and RACE analysis, as shown in the table. The location of the putative canonical cleavage and polyadenylation signal based on transcript size or RACE analysis for each transcript is shown in the table. Abbreviations: pA, polyadenylation; R, confirmed by RACE in this study; P, putative closest polyadenylation signal by Northern mapping.
These complex mRNA processing patterns result in overlapping transcripts for ORF8, ORF10, and ORF11. Similarly, ORFs 58 to 62 are processed to yield a ladder of overlapping transcripts. This could therefore result in the overrepresentation of downstream transcripts in the RNA-seq analysis because transcripts originating from upstream promoters would include downstream ORFs. For example, reads mapped to ORF58 would include those from overlapping ORF 59, 60, and 61 transcripts. Indeed, examination of the Northern blot in Fig. 9 suggests that ORF59 is much more enhanced by ORF57 than ORF58 is. The qPCR measurements would be potentially susceptible to the same inflation of downstream transcript abundance. We therefore reexamined the RNA-seq data using the Trinity platform after reannotating the KSHV genome to correct the transcription termination sites on the basis of our mapping, as shown in Fig. 8 and 10. The software uses an expectation maximization algorithm to estimate the abundance of isoforms and fractionally assigns reads to the various isoforms. The results of such an analysis are shown in Fig. 11. While the estimates of differential expression in the presence or absence of ORF57 differ somewhat in magnitude from the previous direct read assignments, the most highly ORF-enhanced transcripts remained essentially the same, consisting of the ORF4 to ORF9 and ORF59 to ORF61 clusters. However, ORF58 was not highly overrepresented in the presence of ORF57, and combined with the Northern blot data, it appears that while ORF58 may be slightly upregulated in the presence of ORF57, it does not belong to the most highly expressed subset of ORF57-dependent genes.
FIG 11.

Comparison of KSHV RNA expression profiles in the presence or absence of ORF57 analyzed with an expectation maximization algorithm. ORF57-dependent gene expression is shown as the ratio of RNA abundance in WT KSHV-infected iSLK cells versus that in ΔORF57 KSHV-infected iSLK cells on the y axis. Genes most highly upregulated by ORF57 are marked in red. The fold changes for genes whose expression was greater than the maximum are shown numerically above individual bars. Read assignment and differential expression analysis were performed using the Trinity platform and the DESeq program.
Both clusters of genes (ORF4 to ORF9 and ORF59 to ORF61) consist of both early and late lytic replicative-phase genes, as well as encode essential proteins that carry out important structural and enzymatic functions, as shown in Table 3. Of these eight genes, six are involved in DNA replication and packaging and one is a glycoprotein important for virion assembly and membrane fusion upon infection, indicating that the essentiality of ORF57 in lytic replication is due to its direct role in upregulating the expression of several critical genes that are required for both virion production and infectivity.
TABLE 3.
Most highly ORF57-dependent genes
| Gene | Function | Biological role | Temporal class |
|---|---|---|---|
| ORF4 | Complement regulatory protein | Immune evasion | Early |
| ORF6 | Single-stranded DNA binding protein | DNA replication | Early |
| ORF7 | Terminase subunit | DNA packaging | Late |
| ORF8 | Glycoprotein B | Membrane fusion, virion assembly | Late |
| ORF9 | DNA polymerase | DNA replication | Early |
| ORF59 | Processivity factor | DNA replication | Early |
| ORF60 | Ribonucleotide reductase subunit | DNA replication | Early |
| ORF61 | Ribonucleotide reductase subunit | DNA replication | Early |
The coding sequence of target mRNAs is important for ORF57 target gene regulation.
The fact that most of the ORFs in the cluster of ORF57-dependent genes located near oriLyt L, i.e., ORFs 4, 6, 7, and 9, utilized unique polyadenylation signals suggested that ORF57 acted on their individual mRNA coding sequences rather than their polyadenylation signals. However, ORFs 58 to 62 did share a polyadenylation signal, leaving open the possibility that the effects of ORF57 may be dependent on polyadenylation to some degree. In order to determine whether the ORF57 dependence of these ORFs could be demonstrated without the promoters and polyadenylation signals found in the KSHV genome, we cloned several ORF57-dependent and -nondependent genes into the pcDNA3 expression vector, which contains a CMV promoter and a bovine growth hormone (βGH) polyadenylation signal. We next investigated the effect of ORF57 on the expression of each of these KSHV genes in a cotransfection assay. Each expression vector was transfected into HeLa cells with either an ORF57 expression plasmid or an empty vector, and RNAs were analyzed by Northern blotting. As shown in Fig. 12, ORF 4, 6, and 59 RNA was barely detectable in the absence of ORF57 but highly expressed in the presence of ORF57. However, ORF11 and KSHV G protein-coupled receptor (GPCR) RNAs were not ORF57 dependent; i.e., they were expressed well without ORF57 and their expression was only slightly enhanced by ORF57, similar to their pattern of expression from the viral genome in infected cells. These experiments therefore indicate that while ORF57 may affect polyadenylation, it is not likely to be the main reason that certain ORFs require ORF57 for efficient expression. Rather, specific sequences present in the coding regions of ORFs 4 to 9 and ORFs 59 to 61 can confer ORF57 dependence, regardless of their promoter or polyadenylation signal.
FIG 12.

Effect of ORF57 on expression of the coding region RNAs of ORF57-independent versus ORF57-dependent genes. HeLa cells were transfected with a plasmid expressing ORF4, ORF6, or ORF59 (highly ORF57 dependent) or plasmid expressing ORF11 or viral GPCR (ORF57 independent) in combination with either an ORF57 expression plasmid (lanes 57) or an empty vector (lanes C). RNAs were analyzed by Northern blotting using gene-specific probes, as shown above each panel. Blots were stripped and reprobed with GAPDH as a loading control, as shown to the right of each blot.
Providing a strong poly(A) sequence does not rescue ORF57-dependent gene expression.
It remained possible that poor expression of ORF57-dependent genes in the absence of ORF57 might be due to the presence of a weak but ORF57-responsive polyadenylation signal. To test this hypothesis, we cloned the ORF4 coding domain into a CMV promoter-driven expression vector with either its own poly(A) signal, the βGH polyadenylation signal, or the ORF11 polyadenylation signal. These plasmids were transfected into HeLa cells with and without the ORF57 expression vector, and RNAs were analyzed by Northern blotting. As shown in Fig. 13, ORF4 RNA was barely detectable in the absence of ORF57 when cloned with its own polyadenylation signal or the βGH polyadenylation signal. In the presence of the ORF11 polyadenylation sequence, expression was increased, indicating that it is a stronger polyadenylation signal than the ORF4 or βGH polyadenylation signal. Again, however, ORF4 expression was highly upregulated in the presence of ORF57. These results indicate that ORF4 RNA is not expressed well, regardless of the strength of the polyadenylation signal, unless ORF57 is present, suggesting that the need for ORF57 is based on inherent properties of the ORF4 coding sequence RNA.
FIG 13.

ORF57-enhanced gene expression is independent of the polyadenylation signal. ORF4 was cloned downstream of the CMV promoter in expression vector pCDNA3 with either the ORF4 poly(A) signal (ORF4-4pA), the ORF11 poly(A) signal (ORF4-11pA), or the bovine growth hormone poly(A) signal (ORF4-βGHpA). Each construct was transfected into HeLa cells with either an empty vector (lanes C) or ORF57 (lanes 57). RNA was isolated 48 h after transfection and analyzed by Northern blotting with an ORF4-specific probe. Blots were stripped and reprobed with GAPDH as a loading control, as shown to the right of each blot.
DISCUSSION
The exact reason that ORF57 is essential for KSHV lytic replication and virion production has not been conclusively established. While it has been demonstrated that RNAs of a few individual KSHV genes accumulate more efficiently when ORF57 is provided in trans to ΔORF57 KSHV, the global effect of ORF57 on the KSHV transcriptome has not been determined. The mechanisms by which ORF57 acts to enhance KSHV gene expression also remain a matter of some controversy. Most experiments have been performed using individual KSHV target gene reporter constructs in transfection assays and have suggested that ORF57 may enhance RNA stability, nuclear export, polyadenylation, and transcription. Binding of ORF57 to particular regions of KSHV transcripts has been demonstrated on the basis of RNA UV cross-linking and immunoprecipitation studies, but the relevance of these findings to physiological ORF57 targets has likewise not been established, except in an individual case (45). In this study, we compared the expression of all KSHV transcripts during lytic replication in the presence or absence of ORF57 by a combination of high-throughput RNA sequencing, qRT-PCR, Northern blotting, and RACE analysis. The results demonstrate that while the majority of KSHV lytic cycle transcripts are expressed more abundantly in the presence of ORF57, eight transcripts are particularly ORF57 dependent. While members of this group of eight genes are expressed during early or late lytic replication, almost all perform functions that are predicted to be critical for production of infectious virions. Thus, five of the proteins encoded by these genes are involved in DNA replication, one is involved in DNA packaging, and one is a glycoprotein important for virion assembly and membrane fusion (46). Only one, ORF4, is involved in an immune-modulatory function, acting as a complement regulatory protein (47).
Interestingly, these genes are clustered together in two loci of the KSHV genome, initially raising the possibility that their ORF57 dependence was based on a mechanism of coordinate control. This hypothesis was also initially supported by the fact that the published genome annotation of KSHV available in the GenBank database (GenBank accession number NC_009333.1 [http://www.ncbi.nlm.nih.gov/nuccore/139472801?report=GenBank]) indicated that both sets of genes were coterminally polyadenylated, using a polyadenylation signal 3′ to the last gene in the cluster. It has also been suggested that ORF57 may enhance polyadenylation (12, 13, 48, 49). We have mapped the 3′ termini of the heretofore uncharacterized transcripts, and combined with additional recently published data (31, 40), it is clear that while the ORF 58 to 62 genes are, in fact, transcribed as polycistronic messages, the transcriptional pattern of ORFs 4 to 11 is much more complex. Thus, while ORF8, ORF10, and ORF11 do share a cleavage and polyadenylation site, the other ORFs, including ORF9, the DNA polymerase, which is located between ORF8 and ORF10, possess their own unique polyadenylation signals and are processed to yield monocistronic mRNAs (Fig. 8 and 10). In the case of ORF4 and ORF7, the evidence indicates utilization of a noncanonical polyadenylation signal. Despite extensive attempts, the precise location of the polyadenylation signal of ORF9 could not be determined by RACE, and there is no canonical or typical noncanonical signal present at the predicted location, based on the transcript size from Northern blotting and the sequence of the coding domain that was amplified by RT-PCR and 5′ RACE.
Despite the unusual nature of the polyadenylation pattern in ORFs 4 to 11 and the coterminal polyadenylation seen in ORFs 58 to 62, the evidence does not support the hypothesis that ORF57 acts to enhance the expression of these genes by enhancing polyadenylation. First, ORF57-dependent genes continue to display a requirement for ORF57 even when removed from the context of the KSHV genome and expressed with heterologous promoters and polyadenylation signals (Fig. 12). Second, while expression of these genes could be increased by provision of a stronger polyadenylation signal, they still require ORF57 for robust RNA accumulation. Finally, exchanging the polyadenylation signal of an ORF57-independent gene did not confer ORF57 independence on ORF4 or ORF59. While we cannot rule out the possibility that ORF57 acts to enhance polyadenylation in the intact virus, ORFs of ORF57-dependent genes remain highly ORF57 dependent and responsive even when provided with a strong heterologous polyadenylation signal. Conversely, ORF57-independent genes remain so even when expressed without their cognate polyadenylation signals. Further, despite the fact that ORFs 58 to 62 share a coterminal polyadenylation signal, ORF58 is not highly upregulated, nor are the upstream transcripts of the cluster, particularly ORF59, suggesting that polyadenylation effects are not the basis of the ORF57 mechanism.
While direct binding of ORF57 to nucleotide sequences and stabilization of target transcripts are the most obvious explanations for the specificity of ORF57 activity, other mechanistic possibilities exist. First, a response element for ORF57 may be a structure rather than a specific nucleotide sequence, leading to a lack of an identifiable consensus ORF57 response element. We have examined the ORF57 transcripts identified in this study for the presence of sequences similar to those previously implicated as ORF57 response elements in both vIL-6 and PAN (12, 38, 45) and did not find any potential consensus sites. Alternatively, ORF57 may act to relieve a transcriptional elongation block in some transcripts. Such a mechanism has not been directly ruled out for ORF57 transcripts in the context of cells actively undergoing lytic replication. A role for ORF57 in transcriptional activation has also been suggested (9, 50). While ORF57 responsiveness is not promoter dependent in transfection studies, this does not exclude the possibility that ORF57 may be required for the optimal activity of specific viral promoters in the KSHV genome, which is chromatinized in latency. Finally, it is also possible that ORF57 may prevent destabilization and degradation of specific transcripts by indirect means involving interactions with proteins that target particular unstable transcripts.
Previous work has indicated that expression of both vIL-6 and PAN can be increased by ORF57. In the case of vIL-6, it was shown, using another bacmid lacking ORF57, that ORF57 could enhance vIL-6 expression from a replicating virus. Neither PAN nor vIL-6 was particularly ORF57 responsive in our study, as measured by both RNA-seq and qPCR. PAN expression was increased approximately 1.5-fold, and vIL-6 expression was not detectably increased (Fig. 6I and J). It should be noted that PAN expression is extremely robust in the absence of ORF57 (Fig. 5, peak at approximately nt 29,000). In fact, PAN is the most highly expressed lytic transcript, even in the absence of ORF57. Thus, while PAN expression may be increased by ORF57, it is by no means dependent on ORF57 for expression during lytic replication.
In conclusion, we have determined the physiological targets of ORF57 during KSHV replication. These genes are clustered together, but the ORF57 dependence of each transcript resides in its coding sequence rather than in a shared regulatory element. The identification of this group of genes explains why ORF57 is essential for KSHV lytic replication and infectious virion production, as a lack of ORF57 would compromise DNA replication and virion assembly, production, and entry due to inefficient synthesis of several key proteins involved in these processes. The teleological reason for the evolution of such specific control points as targets of posttranscriptional regulation remains speculative, but one possibility is that ORF57 and related herpesvirus proteins act as precise checkpoints in the temporal progression of lytic replication, preventing frequent and abortive initiation of the lytic replicative cycle.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants CA-81133 (to S.S.), CA88763 (to R.R.), and RC2CA148407 (to R.R.) from the National Cancer Institute and by the National Center for Research Resources of the National Institutes of Health under award number 1S10RR026802-01. Viral transcription profiling was conducted by the UNC Vironomics Core (http://www.med.unc.edu/vironomics) using custom arrays designed with support from grants AI107810 and CA019014.
We thank Peter Turner for critical readings of the manuscript, Eun A. Kim and Jacob Thompson for technical assistance, and Tim Mosbruger for bioinformatic analyses.
REFERENCES
- 1.Ganem D. 2007. Kaposi's sarcoma-associated herpesvirus, p 2847–2888 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Staskus KA, Sun R, Miller G, Racz P, Jaslowski A, Metroka C, Brett-Smith H, Haase AT. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. J Virol 73:4181–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N Engl J Med 340:1063–1070. [DOI] [PubMed] [Google Scholar]
- 4.Han Z, Swaminathan S. 2006. Kaposi's sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J Virol 80:5251–5260. doi: 10.1128/JVI.02570-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majerciak V, Pripuzova N, McCoy JP, Gao SJ, Zheng ZM. 2007. Targeted disruption of Kaposi's sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, K8alpha, and K8.1 and the production of infectious virus. J Virol 81:1062–1071. doi: 10.1128/JVI.01558-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majerciak V, Uranishi H, Kruhlak M, Pilkington GR, Massimelli MJ, Bear J, Pavlakis GN, Felber BK, Zheng ZM. 2011. Kaposi's sarcoma-associated herpesvirus ORF57 interacts with cellular RNA export cofactors RBM15 and OTT3 to promote expression of viral ORF59. J Virol 85:1528–1540. doi: 10.1128/JVI.01709-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Majerciak V, Yamanegi K, Allemand E, Kruhlak M, Krainer AR, Zheng ZM. 2008. Kaposi's sarcoma-associated herpesvirus ORF57 functions as a viral splicing factor and promotes expression of intron-containing viral lytic genes in spliceosome-mediated RNA splicing. J Virol 82:2792–2801. doi: 10.1128/JVI.01856-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nekorchuk M, Han Z, Hsieh TT, Swaminathan S. 2007. Kaposi's sarcoma-associated herpesvirus ORF57 protein enhances mRNA accumulation independently of effects on nuclear RNA export. J Virol 81:9990–9998. doi: 10.1128/JVI.00896-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunter OV, Sei E, Richardson RB, Conrad NK. 2013. Chromatin immunoprecipitation and microarray analysis suggest functional cooperation between Kaposi's sarcoma-associated herpesvirus ORF57 and K-bZIP. J Virol 87:4005–4016. doi: 10.1128/JVI.03459-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malik P, Blackbourn DJ, Cheng MF, Hayward GS, Clements JB. 2004. Functional co-operation between the Kaposi's sarcoma-associated herpesvirus ORF57 and ORF50 regulatory proteins. J Gen Virol 85:2155–2166. doi: 10.1099/vir.0.79784-0. [DOI] [PubMed] [Google Scholar]
- 11.Malik P, Blackbourn DJ, Clements JB. 2004. The evolutionarily conserved Kaposi's sarcoma-associated herpesvirus ORF57 protein interacts with REF protein and acts as an RNA export factor. J Biol Chem 279:33001–33011. doi: 10.1074/jbc.M313008200. [DOI] [PubMed] [Google Scholar]
- 12.Sahin BB, Patel D, Conrad NK. 2010. Kaposi's sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog 6:e1000799. doi: 10.1371/journal.ppat.1000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stubbs SH, Hunter OV, Hoover A, Conrad NK. 2012. Viral factors reveal a role for REF/Aly in nuclear RNA stability. Mol Cell Biol 32:1260–1270. doi: 10.1128/MCB.06420-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirshner JR, Lukac DM, Chang J, Ganem D. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 57 encodes a posttranscriptional regulator with multiple distinct activities. J Virol 74:3586–3597. doi: 10.1128/JVI.74.8.3586-3597.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyne JR, Colgan KJ, Whitehouse A. 2008. Recruitment of the complete hTREX complex is required for Kaposi's sarcoma-associated herpesvirus intronless mRNA nuclear export and virus replication. PLoS Pathog 4:e1000194. doi: 10.1371/journal.ppat.1000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyne JR, Jackson BR, Taylor A, Macnab SA, Whitehouse A. 2010. Kaposi's sarcoma-associated herpesvirus ORF57 protein interacts with PYM to enhance translation of viral intronless mRNAs. EMBO J 29:1851–1864. doi: 10.1038/emboj.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson BR, Boyne JR, Noerenberg M, Taylor A, Hautbergue GM, Walsh MJ, Wheat R, Blackbourn DJ, Wilson SA, Whitehouse A. 2011. An interaction between KSHV ORF57 and UIF provides mRNA-adaptor redundancy in herpesvirus intronless mRNA export. PLoS Pathog 7:e1002138. doi: 10.1371/journal.ppat.1002138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verma D, Kim EA, Swaminathan S. 2013. Cell-based screening assay for antiviral compounds targeting the ability of herpesvirus posttranscriptional regulatory proteins to stabilize viral mRNAs. J Virol 87:10742–10751. doi: 10.1128/JVI.01644-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta AK, Ruvolo V, Patterson C, Swaminathan S. 2000. The human herpesvirus 8 homolog of Epstein-Barr virus SM protein (KS-SM) is a posttranscriptional activator of gene expression. J Virol 74:1038–1044. doi: 10.1128/JVI.74.2.1038-1044.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gruffat H, Batisse J, Pich D, Neuhierl B, Manet E, Hammerschmidt W, Sergeant A. 2002. Epstein-Barr virus mRNA export factor EB2 is essential for production of infectious virus. J Virol 76:9635–9644. doi: 10.1128/JVI.76.19.9635-9644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sacks WR, Greene CC, Aschman DP, Schaffer PA. 1985. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J Virol 55:796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen JI, Krogmann T, Ross JP, Pesnicak L, Prikhod'ko EA. 2005. Varicella-zoster virus ORF4 latency-associated protein is important for establishment of latency. J Virol 79:6969–6975. doi: 10.1128/JVI.79.11.6969-6975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashi ML, Blankenship C, Shenk T. 2000. Human cytomegalovirus UL69 protein is required for efficient accumulation of infected cells in the G1 phase of the cell cycle. Proc Natl Acad Sci U S A 97:2692–2696. doi: 10.1073/pnas.050587597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sato B, Sommer M, Ito H, Arvin AM. 2003. Requirement of varicella-zoster virus immediate-early 4 protein for viral replication. J Virol 77:12369–12372. doi: 10.1128/JVI.77.22.12369-12372.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yakushko Y, Hackmann C, Gunther T, Ruckert J, Henke M, Koste L, Alkharsah K, Bohne J, Grundhoff A, Schulz TF, Henke-Gendo C. 2011. Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome contains a duplication of a long unique-region fragment within the terminal repeat region. J Virol 85:4612–4617. doi: 10.1128/JVI.02068-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. doi: 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 29.Ruvolo V, Wang E, Boyle S, Swaminathan S. 1998. The Epstein-Barr virus nuclear protein SM is both a post-transcriptional inhibitor and activator of gene expression. Proc Natl Acad Sci U S A 95:8852–8857. doi: 10.1073/pnas.95.15.8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li DJ, Verma D, Swaminathan S. 2012. Binding of cellular export factor REF/Aly by Kaposi's sarcoma-associated herpesvirus (KSHV) ORF57 protein is not required for efficient KSHV lytic replication. J Virol 86:9866–9874. doi: 10.1128/JVI.01190-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai Z, Huang Y, Li W, Zhu Y, Jung JU, Lu C, Gao SJ. 2014. Genomewide mapping and screening of Kaposi's sarcoma-associated herpesvirus (KSHV) 3′ untranslated regions identify bicistronic and polycistronic viral transcripts as frequent targets of KSHV microRNAs. J Virol 88:377–392. doi: 10.1128/JVI.02689-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li DJ, Verma D, Mosbruger T, Swaminathan S. 2014. CTCF and Rad21 act as host cell restriction factors for Kaposi's sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog 10:e1003880. doi: 10.1371/journal.ppat.1003880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, Macmanes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han Z, Verma D, Hilscher C, Dittmer DP, Swaminathan S. 2009. General and target-specific RNA binding properties of Epstein-Barr virus SM posttranscriptional regulatory protein. J Virol 83:11635–11644. doi: 10.1128/JVI.01483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scotto-Lavino E, Du G, Frohman MA. 2006. 3′ end cDNA amplification using classic RACE. Nat Protoc 1:2742–2745. doi: 10.1038/nprot.2006.481. [DOI] [PubMed] [Google Scholar]
- 37.Myoung J, Ganem D. 2011. Infection of lymphoblastoid cell lines by Kaposi's sarcoma-associated herpesvirus: critical role of cell-associated virus. J Virol 85:9767–9777. doi: 10.1128/JVI.05136-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sei E, Conrad NK. 2011. Delineation of a core RNA element required for Kaposi's sarcoma-associated herpesvirus ORF57 binding and activity. Virology 419:107–116. doi: 10.1016/j.virol.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Grady T, Cao S, Strong MJ, Concha M, Wang X, Splinter Bondurant S, Adams M, Baddoo M, Srivastav SK, Lin Z, Fewell C, Yin Q, Flemington EK. 2014. Global bidirectional transcription of the Epstein-Barr virus genome during reactivation. J Virol 88:1604–1616. doi: 10.1128/JVI.02989-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, Holdorf M, Weissman JS, Ganem D. 2014. KSHV 2.0: a comprehensive annotation of the Kaposi's sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog 10:e1003847. doi: 10.1371/journal.ppat.1003847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Majerciak V, Yamanegi K, Zheng ZM. 2006. Gene structure and expression of Kaposi's sarcoma-associated herpesvirus ORF56, ORF57, ORF58, and ORF59. J Virol 80:11968–11981. doi: 10.1128/JVI.01394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majerciak V, Ni T, Yang W, Meng B, Zhu J, Zheng ZM. 2013. A viral genome landscape of RNA polyadenylation from KSHV latent to lytic infection. PLoS Pathog 9:e1003749. doi: 10.1371/journal.ppat.1003749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McClure LV, Kincaid RP, Burke JM, Grundhoff A, Sullivan CS. 2013. Comprehensive mapping and analysis of Kaposi's sarcoma-associated herpesvirus 3′ UTRs identify differential posttranscriptional control of gene expression in lytic versus latent infection. J Virol 87:12838–12849. doi: 10.1128/JVI.02374-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A 93:14862–14867. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang JG, Pripuzova N, Majerciak V, Kruhlak M, Le SY, Zheng ZM. 2011. Kaposi's sarcoma-associated herpesvirus ORF57 promotes escape of viral and human interleukin-6 from microRNA-mediated suppression. J Virol 85:2620–2630. doi: 10.1128/JVI.02144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davison A. 2007. Comparative analysis of the genomes. InArvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 47.Spiller OB, Robinson M, O'Donnell E, Milligan S, Morgan BP, Davison AJ, Blackbourn DJ. 2003. Complement regulation by Kaposi's sarcoma-associated herpesvirus ORF4 protein. J Virol 77:592–599. doi: 10.1128/JVI.77.1.592-599.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Conrad NK, Mili S, Marshall EL, Shu MD, Steitz JA. 2006. Identification of a rapid mammalian deadenylation-dependent decay pathway and its inhibition by a viral RNA element. Mol Cell 24:943–953. doi: 10.1016/j.molcel.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 49.Massimelli MJ, Kang JG, Majerciak V, Le SY, Liewehr DJ, Steinberg SM, Zheng ZM. 2011. Stability of a long noncoding viral RNA depends on a 9-nt core element at the RNA 5′ end to interact with viral ORF57 and cellular PABPC1. Int J Biol Sci 7:1145–1160. doi: 10.7150/ijbs.7.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmeri D, Spadavecchia S, Carroll KD, Lukac DM. 2007. Promoter- and cell-specific transcriptional transactivation by the Kaposi's sarcoma-associated herpesvirus ORF57/Mta protein. J Virol 81:13299–13314. doi: 10.1128/JVI.00732-07. [DOI] [PMC free article] [PubMed] [Google Scholar]