

ABSTRACT
Nonmuscle myosin heavy chain IIA (NMHC-IIA) has been reported to function as a herpes simplex virus 1 (HSV-1) entry coreceptor by interacting with viral envelope glycoprotein B (gB). Vertebrates have three genetically distinct isoforms of the NMHC-II, designated NMHC-IIA, NMHC-IIB, and NMHC-IIC. COS cells, which are readily infected by HSV-1, do not express NMHC-IIA but do express NMHC-IIB. This observation prompted us to investigate whether NMHC-IIB might associate with HSV-1 gB and be involved in an HSV-1 entry like NMHC-IIA. In these studies, we show that (i) NMHC-IIB coprecipitated with gB in COS-1 cells upon HSV-1 entry; (ii) a specific inhibitor of myosin light chain kinase inhibited cell surface expression of NMHC-IIB in COS-1 cells upon HSV-1 entry as well as HSV-1 infection, as reported with NMHC-IIA; (iii) overexpression of mouse NMHC-IIB in IC21 cells significantly increased their susceptibility to HSV-1 infection; and (iv) knockdown of NMHC-IIB in COS-1 cells inhibited HSV-1 infection as well as cell-cell fusion mediated by HSV-1 envelope glycoproteins. These results supported the hypothesis that, like NMHC-IIA, NMHC-IIB associated with HSV-1 gB and mediated HSV-1 entry.
IMPORTANCE Herpes simplex virus 1 (HSV-1) was reported to utilize nonmuscle myosin heavy chain IIA (NMHC-IIA) as an entry coreceptor associating with gB. Vertebrates have three genetically distinct isoforms of NMHC-II. In these isoforms, NMHC-IIB is of special interest since it highly expresses in neuronal tissue, one of the most important cellular targets of HSV-1 in vivo. In this study, we demonstrated that the ability to mediate HSV-1 entry appeared to be conserved in NMHC-II isoforms. These results may provide an insight into the mechanism by which HSV-1 infects a wide variety of cell types in vivo.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) infects a wide variety of cultured cells in vitro, and two of the most important cellular targets for HSV-1 in vivo are epithelial cells at the initial site of infection and neurons for the establishment of latent infection (1). For HSV-1 entry into a cell, the initial interaction of HSV-1 with the cell is binding of virion envelope glycoprotein C (gC) and gB to cell surface glycosaminoglycans, preferentially heparan sulfate, which mediates virus attachment to the cell (2, 3). Although not essential for entry, this attachment provides a stable interaction between the virion and cell that facilitates the next entry steps (4). Subsequent viral penetration requires fusion between the virion envelope and host cell membrane and depends on gB, the heterodimer gH/gL, gD, and a gD receptor (5–7), which are thought to act in a cascade resulting in nucleocapsid entry into the cell (8–10). The gD receptors for HSV-1 reported to date fall into three classes (7): (i) HVEM (herpesvirus entry mediator), a member of the tumor necrosis factor (TNF) receptor family (11); (ii) nectin-1 and nectin-2, members of the immunoglobulin (Ig) superfamily (12, 13); and (iii) specific sites on heparan sulfate (3-O-S-HS) generated by certain 3-O-sulfotransferases (3-O-ST) (14). Studies using mice with HVEM and/or nectin-1 knocked out have elucidated the roles of gD receptors in viral replication and disease in vivo (15).
Accumulating evidence supports the hypothesis that, in addition to the interaction of gD with a gD receptor, gB binding to a cellular receptor other than heparan sulfate is required for HSV-1 entry. These data include the following: (i) a soluble form of gB binds to heparan sulfate-deficient cells and blocks HSV-1 infection in some cell lines (16); (ii) paired Ig-like type 2 receptor α (PILRα), a paired receptor expressed mainly in immune cells (17–19), associates with gB and functions as an HSV-1 entry coreceptor (20); and (iii) HSV-1 infection of primary monocytes expressing both HVEM and PILRα is blocked by either an anti-HVEM or an anti-PILRα antibody (20). PILRα appears to play a significant role in viral replication and pathogenesis in vivo, based on the observation that mutation(s) in gB Thr-53 and/or Thr-480, residues required to have sialylated O-glycosylation for binding of gB to PILRα (21, 22), abrogates PILRα-dependent viral entry without affecting viral replication efficiency in PILRα-negative cells and reduces viral replication, pathogenesis, and neuroinvasiveness in mice (22). Furthermore, myelin-associated glycoprotein (MAG), a member of the sialic acid-binding Ig-like lectin family, which has homology with PILRα, can also act as an HSV-1 entry coreceptor and associate with gB when expressed exogenously (23).
We previously identified nonmuscle myosin IIA (NM-IIA) as a novel HSV-1 entry coreceptor that associates with gB (24). NM-IIA is an actin-binding motor protein and plays critical roles in the control of cell adhesion, cell migration, and tissue architecture (25). NM-IIA consists of three pairs of subunits: two heavy chains (NMHC-IIAs), two regulatory light chains (RLCs), and two essential light chains (ELCs), with a fraction of NMHC-IIA expressed on the cell surface (26–28). gB associates with NMHC-IIA on the cell surface, and this interaction mediates membrane fusion and HSV-1 entry (24). It has been shown that subcellular localization of NM-IIA is regulated in part by phosphorylation of its RLCs at residues Thr-18 and Ser-19, and one of the protein kinases responsible for NM-IIA RLC phosphorylation at these sites is myosin light chain kinase (MLCK) (29). Cell surface expression of NMHC-IIA appears to be regulated by MLCK-mediated signaling events and to be required for efficient HSV-1 infection, based on the observations that (i) NM-IIA RLC phosphorylation as well as cell surface expression of NMHC-IIA upon HSV-1 entry is inhibited by ML-7, a specific inhibitor of MLCK, and (ii) ML-7 inhibits HSV-1 infection in vitro and in vivo (24). Recently, NMHC-IIA was also reported to serve as an entry receptor for severe fever with thrombocytopenia syndrome virus (SFTSV) (30). Like HSV-1 entry, cell surface expression of NMHC-IIA was induced upon SFTSV infection (30). Moreover, NMHC-II activity for cellular protrusions such as filopodia, retraction fibers, and microvilli has been reported to be required for entry of several viruses, including Kaposi's sarcoma-associated herpesvirus (KSHV), papillomavirus, vaccinia virus, and murine leukemia virus (MLV) (31–34). Thus, it appears that NMHC-IIA might be involved in entry of viruses other than HSV-1.
Vertebrates have three genetically distinct isoforms of NMHC-II (designated NMHC-IIA, NMHC-IIB, and NMHC-IIC), with the NMHC-II isoform determining the NM-II isoform (designated NM-IIA, NM-IIB, and NM-IIC, respectively) (25). The three NMHC-II isoforms are highly conserved, with 80% identity and 89% similarity between the amino acid sequences of NMHC-IIA and NMHC-IIB, and 64% identity and ∼80% similarity between NMHC-IIC and both NMHC-IIA and NMHC-IIB (35). The three isoforms also have both overlapping and unique properties (25). Most human tissues express different ratios of the NM-II isoforms (35, 36). In particular, NM-IIB predominates in neuronal tissue, one of the most important cellular targets of HSV-1 in vivo. COS cells are also readily infected by HSV-1 and express NM-IIB but not NM-IIA. These observations suggested that NM-II isoforms other than NM-IIA might also be involved in HSV-1 entry and associate with gB. In this study, we focused on NM-IIB and examined whether this NM-II isoform associated with HSV-1 gB and mediated HSV-1 entry.
MATERIALS AND METHODS
Cells and viruses.
Vero, COS-1, and IC21 cells were described previously (37, 38). Wild-type HSV-1(F) and influenza virus (A/WSN/33; H1N1) were described previously (24). Recombinant HSV-1 YK333 carried an enhanced green fluorescent protein (EGFP) expression cassette in the UL3-UL4 intergenic region under the control of the Egr-1 promoter (39). YK333 has been shown to be phenotypically the same as wild-type HSV-1(F) in cell cultures, and cells infected with YK333 efficiently express EGFP (39). Recombinant HSV-1 YK711 expresses gB tagged with an MEF tag consisting of Myc and Flag epitopes and a tobacco etch virus (TEV) protease cleavage site at the amino-terminal domain (MEF-gB), as described previously (24).
Mutagenesis of viral genomes and generation of recombinant HSV-1.
Recombinant virus YK716, which expresses gH tagged with the MEF tag at the carboxyl terminus (gH-MEF), was constructed by the two-step Red-mediated mutagenesis procedure using Escherichia coli GS1783 containing pYEbac102, a full-length infectious HSV-1(F) clone (38), as described previously (40), except for the use of primers 5′-GGTTCTCCGGACAAGTGTCCCGTTTTTTTGGAGACGCGAAATGGAGCAAAAGCTCATTTC-3′ and 5′-TCGGTCGGGCGGATAAACGGCCGAAGCCACGCCCCCTTTATTAATCTTTGTCATCGTCGTC-3′.
Plasmids.
Plasmid pSSSP-NMHC-IIB, used to generate a stable cell line expressing short hairpin RNA (shRNA) against human NMHC-IIB, was constructed as follows. Oligonucleotides 5′-TTTGGATTCCATCAGAACGCCATGGCTTCCTGTCACCATGGCGTTCTGATGGAATCCTTTTTTG-3′ and 5′-AATTCAAAAAAGGATTCCATCAGAACGCCATGGTGACAGGAAGCCATGGCGTTCTGATGGAATC-3′ were annealed and cloned into the BbsI and EcoRI sites of pmU6 (41). The BamHI-EcoRI fragment of the resultant plasmid, containing the U6 promoter and the sequence containing shRNA against human NMHC-IIB, was cloned into the BamHI and EcoRI sites of pSSSP (41), which is a derivative of retrovirus vector pMX containing a puromycin resistance gene, to produce pSSSP-NMHC-IIB. Plasmid pSSSP-Cre containing shRNA against Cre recombinase was described previously (41). Plasmids pPEP98-gB, pPEP99-gD, pPEP101-gL, and pPEP100-gH were used for expression of HSV-1 gB, gD, gL, and gH, respectively, as described previously (6). Plasmids pCAGT7, encoding T7 RNA polymerase, and pT7EMCLuc, carrying the firefly luciferase gene under the control of the T7 promoter, were used to determine fusion efficiency as described previously (42).
For generating a fusion protein of glutathione S-transferase (GST) and the rod domain of human NMHC-IIB (GST–NMHC-IIB), a plasmid (pGEX-NMHC-IIB) was constructed by amplifying the sequence containing NMHC-IIB codons 1672 to 1976 by PCR from pEGFP-BRF305 (43) (a generous gift from M. Takahashi) and cloning the DNA fragment into pGEX-4T3 (GE Healthcare) in frame with GST. Plasmid pGEX-NMHC-IIA, encoding a fusion protein of GST and the rod domain of human NMHC-IIA, was described previously (24) and used to generate GST-NMHC-IIA. Plasmid pME-Fc-gB, used to generate an extracellular domain of gB (codons 31 to 730) fused to a mutant Fc fragment of human IgG1 with low binding affinity for cellular Fc receptors (gB-Fc), and plasmid pME-Fc-gD, used to generate an extracellular domain of gD (codons 31 to 336) fused to the mutant Fc fragment of human IgG1 (gD-Fc), were constructed as described earlier (24). pME-Fc-CD200 (20) was used to generate control Fc fusion protein. The plasmids NMHC-IIB in pCAG-Flag-IRES-puro (a generous gift from R. Nishinakamura), pMXs-NMHC-IIA, pMXs-nectin-1, and pMxs-puro, which were used for generating IC21 cells overexpressing NMHC-IIB, NMHC-IIA, and nectin-1 and control IC21 cells, respectively, were described previously (22, 24, 44).
Antibodies.
Mouse monoclonal antibodies to Flag (M2; Sigma), α-tubulin (DM1A; Sigma), pan-cadherin (CH-19; Sigma), gB (1105; Goodwin Institute), gE (9H3; Abcam), and nectin-1 (CK8; Zymed) were used in this study. Rabbit polyclonal antibodies to the C-terminal peptide of NMHC-IIA and to the C-terminal peptide of NMHC-IIB were purchased from Sigma. Anti-influenza virus antibody R309 was described previously (24).
Immunoprecipitation.
COS-1 cells were incubated with wild-type HSV-1(F), YK711 expressing MEF-gB, or YK716 expressing gH-MEF at a multiplicity of infection (MOI) of 50 at 4°C for 2 h to allow viral adsorption. The cells were then transferred to 37°C for 2 min (a permissive temperature for viral penetration), harvested, washed with phosphate-buffered saline (PBS), and lysed in TNE buffer (1% NP-40, 150 mM NaCl, 10 mM Tris-HCl [pH 7.8], and 1 mM EDTA) containing a proteinase inhibitor cocktail. After centrifugation, the supernatants were precleared by incubation with protein A-Sepharose beads for 30 min at 4°C. After a brief centrifugation, the supernatants were reacted with an anti-Flag, anti-gE, or anti-gB antibody for 2 h at 4°C. Protein A-Sepharose beads were then added and allowed to react, with rotation, for an additional 1 h at 4°C. The immunoprecipitates were collected by a brief centrifugation, washed extensively with TNE buffer, and analyzed by immunoblotting with anti-NMHC-IIB antibody.
GST pulldown.
GST-NMHC-IIA or GST-NMHC-IIB was expressed in bacteria, lysed with HEPES buffer (1% Triton X-100, 250 mM NaCl, 50 mM HEPES [pH 7.4], and 10 mM MgCl2), purified on glutathione-Sepharose beads, and eluted with 10 mM glutathione. gB-Fc, gD-Fc, or control Fc fusion protein was expressed in COS-1 cells, lysed with the radioimmunoprecipitation assay (RIPA) buffer (1% NP-40, 0.1% deoxycholate [DOC], 0.1% SDS, 150 mM NaCl, 10 mM Tris-HCl [pH 7.8], and 1 mM EDTA) containing a proteinase inhibitor cocktail, purified on protein A-Sepharose beads, and quantified as described previously (24). Affinity precipitation was performed as described previously (24) except that purified GST-NMHC-IIA or GST-NMHC-IIB was incubated with purified gB-Fc-, gD-Fc-, or control-Fc-conjugated protein A-Sepharose beads and detected with anti-NMHC-IIA or -NMHC-IIB antibody.
Establishment of cell lines stably expressing shRNA against NMHC-IIB.
COS-1 cells were transfected with pSSSP-NMHC-IIB or pSSSP-Cre and selected with 1 μg puromycin/ml in maintenance medium. Resistant cells transfected with pSSSP-Cre were designated COS-shControl cells and used as a control. Single colonies transfected with pSSSP-NMHC-IIB were isolated and screened by immunoblotting with anti-NMHC-IIB antibody, which led to isolation of COS-shNMHC-IIA cells. COS-shControl and COS-shNMHC-IIB cells in 24-well plates were incubated with YK333 expressing EGFP at an MOI of 1. After viral adsorption for 1 h, the inoculum was removed and the cells were washed twice and refed with the appropriate medium. At 5 h postinfection, cell samples were analyzed using a FACSCalibur cytometer with Cell Quest software.
Cell fusion assay.
COS-1 cells in a 24-well plate were transfected with pPEP98-gB, pPEP99-gD, pPEP101-gL, pPEP100-gH, and pCAGT7, and these transfectants were used as effector cells (23). COS-shControl and COS-shNMHC-IIB cells in 24-well plates were transfected with pT7EMCLuc, and these transfectants were used as target cells (23). As an internal control, pRL-CMV (Promega) containing the Renilla luciferase gene driven by the cytomegalovirus (CMV) promoter was cotransfected into the target cells. At 6 h posttransfection, the effector cells were detached by 0.04% EDTA in phosphate-buffered saline (PBS), washed once with maintenance medium, and cocultured with the target cells for 18 h. Firefly and Renilla luciferase activities were independently assayed by the dual-luciferase reporter assay system (Promega) with a Veritas luminometer (Promega). The relative fusion activity was calculated as (firefly luciferase activity)/(Renilla luciferase activity).
Immunoblotting and immunofluorescence.
Immunoblotting and immunofluorescence were performed as described previously (45).
Biotinylation of cell surface proteins of HSV-1-infected cells.
COS-1 or IC21 cells were incubated with wild-type HSV-1(F), YK711 (MEF-gB), or YK716 (gH-MEF) at an MOI of 50 at 4°C for 2 h in the absence or presence of 20 μM ML-7. In the case of ML-7 treatment, cells were pretreated with 20 μM ML-7 for 30 min at 37°C before virus inoculation. The cells were then transferred to 37°C for 15 min in the absence or presence of 20 μM ML-7, washed four times with ice-cold PBS, and biotinylated twice (15 min each time) with cleavable sulfo–N-hydroxysuccinimide (NHS)–SS-biotin (Pierce). After two washes with Dulbecco modified Eagle medium (DMEM) supplemented with 0.2% bovine serum albumin (BSA), free SH groups were quenched by addition of a solution of 5 mg iodoacetamide/ml in PBS containing 1% BSA and the cells were harvested, solubilized in radioimmunoprecipitation assay (RIPA) buffer containing a proteinase inhibitor cocktail, precipitated with streptavidin beads, and analyzed by immunoblotting with anti-NMHC-IIB, anticadherin, anti-α-tubulin antibody, or streptavidin-horseradish peroxidase.
Infection inhibition assays.
For inhibition with ML-7, COS-1 cells pretreated with various concentrations of ML-7 for 30 min were incubated with YK333 or influenza virus at an MOI of 1 in the same concentrations of ML-7 for 1 h. After removal of unadsorbed virus, the cells were refed with medium containing the same concentrations of ML-7. At 5 h postinfection for YK333 and at 7 h for influenza virus, cell samples were analyzed using a FACSCalibur cytometer with Cell Quest software.
PEG treatment.
COS-1 cells pretreated with or without 20 μM ML7 at 37°C for 5 min were incubated with YK333 at an MOI of 1 in 20 μM ML-7 at 37°C for 30 min. After viral adsorption, unadsorbed virus was removed, and the cells were washed twice and exposed to 40% polyethylene glycol (PEG) as described previously (24, 46). After PEG removal, the cells were refed with medium 199 containing 1% fetal calf serum (FCS). At 5 h postinfection, cell samples were analyzed using a FACSCalibur cytometer with Cell Quest software.
Establishment of IC21 cells stably overexpressing NMHC-IIB, NMHC-IIA, or nectin-1.
IC21 cells were transfected with a mouse NMHC-IIB expression vector (NMHC-IIB in pCAG-Flag-IRES-puro) (44), a human NMHC-IIA expression vector (pMxs-NMHC-IIA) (24), human nectin-1 (pMxs-nectin-1) (22), or pMxs-puro (47) and selected with 0.25 to 1 μg puromycin/ml in maintenance medium. Resistant cells transfected with pMxs-puro, pMxs-NMHC-IIA, pMxs-nectin-1, or NMHC-IIB in pCAG-Flag-IRES-puro were designated IC21/puro, IC21/hNMHC-IIA, IC21/hnectin-1, and IC21/mNMHC-IIB, respectively. IC21/puro, IC21/hNMHC-IIA, IC21/hnectin-1, and IC21/mNMHC-IIB cells in 24-well plates were incubated with YK333 at an MOI of 5. After viral adsorption for 1 h, unadsorbed virus was removed and the cells were washed twice and refed with the appropriate medium. At 10 h postinfection, cell samples were analyzed by fluorescence microscopy (Olympus IX71) or a FACSCalibur cytometer with Cell Quest software.
RESULTS AND DISCUSSION
Association of NMHC-IIB with HSV-1 gB.
It has been reported that COS cells, a simian kidney epithelial cell line, express NMHC-IIB but not NMHC-IIA (48). In agreement with that report, we found that COS-1 cells expressed only NMHC-IIB, whereas Vero cells, another simian kidney epithelial cell line, expressed both NMHC-IIA and NMHC-IIB (Fig. 1A). To examine the specific association of HSV-1 gB with NMHC-IIB upon viral entry, COS-1 cells were mock incubated or incubated with wild-type HSV-1(F) at an MOI of 50 at 4°C for 2 h, after which the cells were subjected to a temperature shift to 37°C for 2 min; next, proteins were immunoprecipitated with anti-gB or anti-gE antibody and analyzed by immunoblotting them with the anti-NMHC-IIB antibody. Whereas the anti-gB antibody coprecipitated gB with NMHC-IIB from a lysate of COS-1 cells incubated with HSV-1(F), the anti-gE antibody did not (Fig. 1B). However, the anti-gB antibody did not immunoprecipitate NMHC-IIB from lysates of mock-incubated COS-1 cells (Fig. 1B), indicating that this antibody did not cross-react with NMHC-IIB. Similarly, an anti-Flag antibody coprecipitated MEF-gB with NMHC-IIB from a lysate of COS-1 cells that had been incubated with YK711 (MEF-gB) at an MOI of 50 at 4°C for 2 h and then shifted to 37°C for 2 min, whereas the antibody did not coprecipitate these proteins from a lysate from COS-1 cells incubated with YK716 (gH-MEF) (Fig. 1C). The anti-Flag antibody appeared to immunoprecipitate MEF-gB as efficiently as it immunoprecipitated gH-MEF from lysates of COS-1 cells incubated with YK711 (MEF-gB) or YK716 (gH-MEF) (Fig. 1C). To investigate whether gB interacted with NMHC-IIB on the cell surface, we biotinylated the cell surface proteins of COS-1 cells exposed to YK711 (MEF-gB) or YK716 (gH-MEF) (as described above) and then detected the immunoprecipitated complex of NMHC-IIB and MEF-gB by using streptavidin. The NMHC-IIB that coprecipitated with MEF-gB was biotinylated, whereas no biotinylated protein with a molecular mass similar to that of NMHC-IIB was detected in cells exposed to YK716 (gH-MEF) (Fig. 1E).
FIG 1.
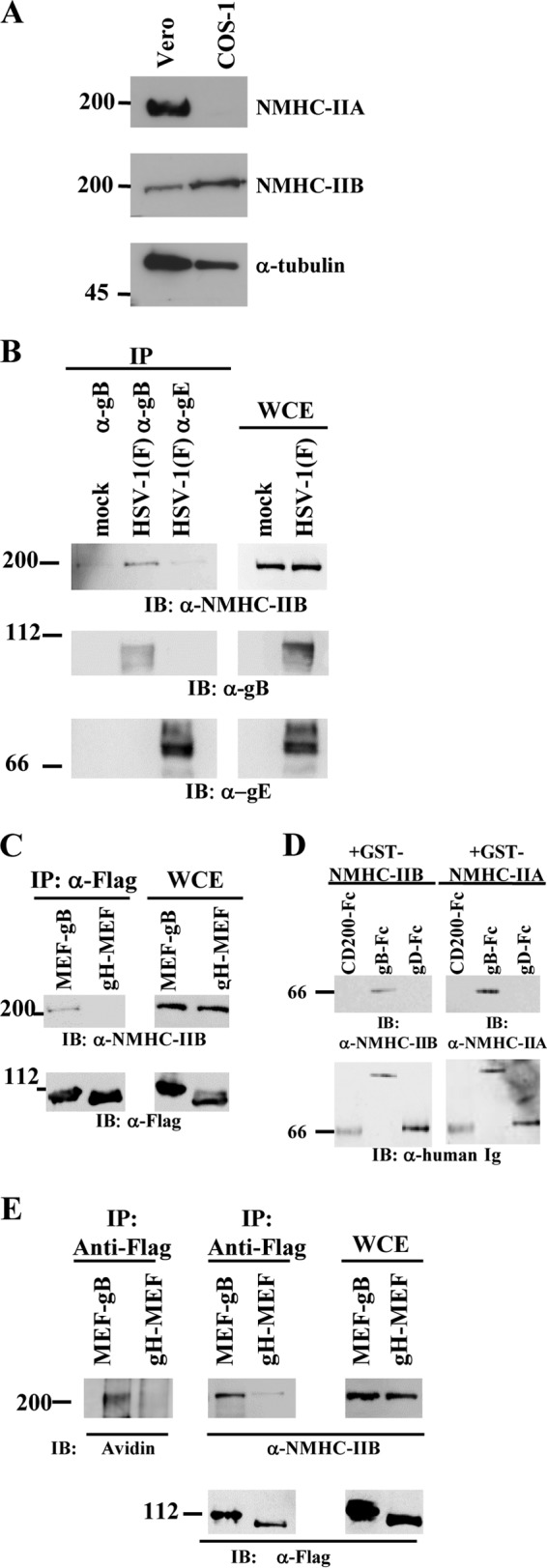
Association of NMHC-IIB with HSV-1 gB. (A) Expression of NMHC-IIA and NMHC-IIB in Vero and COS-1 cells was analyzed by immunoblotting with anti-NMHC-IIA and anti-NMHC-IIB antibody, respectively. (B) COS-1 cells were mock incubated or incubated with wild-type HSV-1(F) at an MOI of 50 at 4°C for 2 h, followed by a temperature shift to 37°C for 2 min, and then immunoprecipitated (IP) with anti-gB or anti-gE antibody and analyzed by immunoblotting (IB) with anti-NMHC-IIB, anti-gB, or anti-gE antibody. WCE, whole-cell extract control. (C) COS-1 cells were incubated with YK711 (MEF-gB) or YK716 (gH-MEF) at an MOI of 50 at 4°C for 2 h, followed by a shift to 37°C for 2 min, and then immunoprecipitated with anti-Flag antibody and analyzed by immunoblotting with anti-NMHC-IIB antibody. WCE, whole-cell extract. (D) Purified GST-NMHC-IIA or GST-NMHC-IIB was reacted with purified gB-Fc, gD-Fc, or control Fc immobilized on protein A-Sepharose beads. The beads were then washed extensively and analyzed by immunoblotting with an anti-NMHC-IIA or -NMHC-IIB antibody. (E) Cell surface proteins of COS-1 cells exposed to YK711 (MEF-gB) or YK716 (gH-MEF) at 4°C for 2 h, followed by a temperature shift to 37°C for 15 min, were biotinylated, immunoprecipitated with an anti-Flag antibody, and analyzed by immunoblotting with streptavidin or anti-Flag or anti-NMHC-IIB antibody. Numbers at left of panels are molecular masses in kilodaltons.
To examine whether both NMHC-IIB and NMHC-IIA directly and specifically bound to gB, purified gB-Fc, gD-Fc, or control-Fc proteins bound to protein A-Sepharose beads were reacted with purified GST-NMHC-IIA or GST-NMHC-IIB, which consisted of GST and the rod domain of human NMHC-IIA or NMHC-IIB, respectively. Purified NMHC-IIA and NMHC-IIB were pulled down by the gB-Fc fusion protein but not by the gD-Fc and control-Fc fusion proteins (Fig. 1D). Collectively, these results suggested that human NMHC-IIB can directly bind to HSV-1 gB in vitro and that the endogenous simian NMHC-IIB in COS-1 cells specifically associated with HSV-1 gB on the cell surface upon HSV-1 entry. Notably, the functions and sequences of mammalian NMHC-IIB proteins are highly conserved, and the identity and similarity between the amino acid sequences of human and mouse NMHC-IIB are 99% and 100%, respectively (49).
Effect of ML-7 on NMHC-IIB cell surface expression upon HSV-1 entry and HSV-1 infection.
We previously reported that ML-7, a specific inhibitor of MLCK, inhibits cell surface expression of NMHC-IIA and HSV-1 infection in cell cultures and in a mouse model (24). MLCK also regulates the function and distribution of NM-IIB (25). To examine the effect of ML-7 on cell surface expression of NMHC-IIB upon HSV-1 entry, cell surface proteins of COS-1 cells infected with wild-type HSV-1(F) at an MOI of 50 at 4°C for 2 h, followed by a temperature shift to 37°C for 15 min, were precipitated with avidin beads and analyzed by immunoblotting with anti-NMHC-IIB, anti-α-tubulin, or anti-pan-cadherin antibody. In agreement with our previous report on NMHC-IIA (24), ML-7 inhibited expression of cell surface NMHC-IIB in COS-1 cells after viral adsorption at 4°C followed by a temperature shift to 37°C, but not that of cadherin (Fig. 2A). To further examine the effect of ML-7 on HSV-1 infection, COS-1 cells infected with YK333 (EGFP) or influenza virus at an MOI of 1 in the presence or absence of ML-7 were analyzed by flow cytometry at 5 or 7 h postinfection, respectively. As shown in Fig. 2B and C, YK333 (EGFP) infection was inhibited by ML-7 in a dose-dependent manner, whereas influenza virus infection was hardly affected by ML-7.
FIG 2.
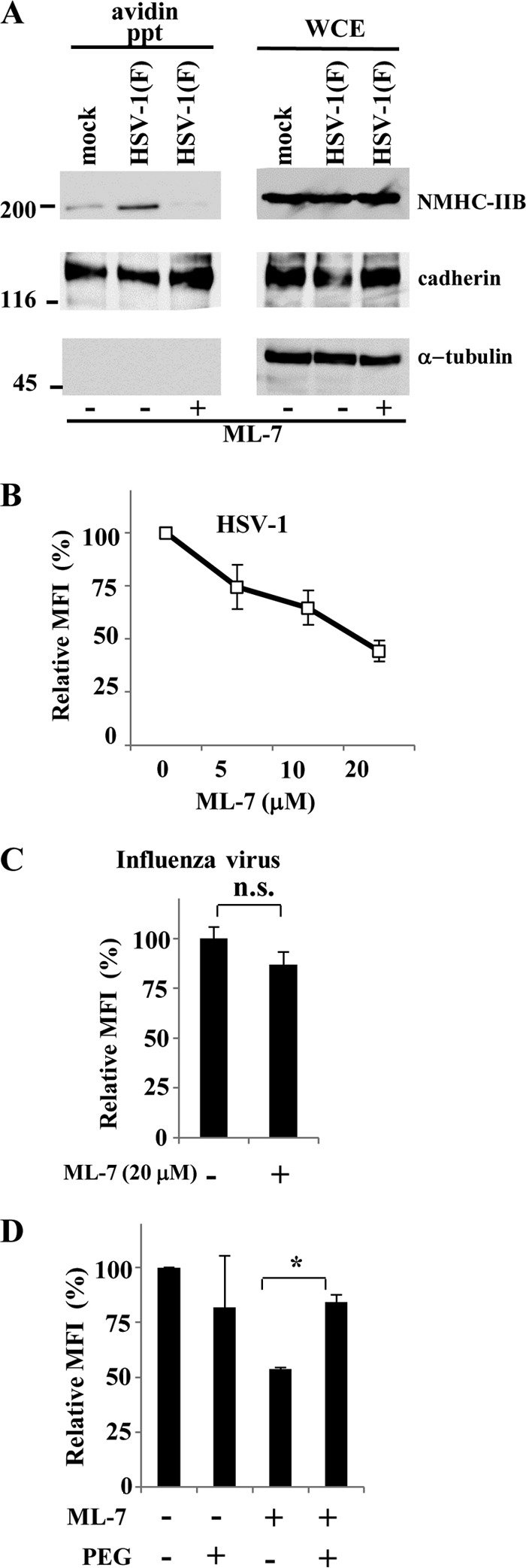
Effect of ML-7 on cell surface expression of NMHC-IIB and HSV-1 infection in COS-1 cells. (A) Cell surface proteins of COS-1 cells incubated with wild-type HSV-1 at an MOI of 50 at 4°C for 2 h, followed by a temperature shift to 37°C for 15 min, in the presence or absence of 20 μM ML-7 were biotinylated, precipitated with avidin beads, and analyzed by immunoblotting with anti-NMHC-IIB or anticadherin or anti-α-tubulin antibody. Numbers at left are molecular masses in kilodaltons. (B and C) COS-1 cells infected with HSV-1 YK333 (EGFP) or influenza virus at an MOI of 1 in the presence of the indicated concentrations of ML-7 (B) or in the presence of 20 μM ML-7 (C) were analyzed by flow cytometry at 5 h and 7 h postinfection, respectively, and mean fluorescence intensities (MFIs) were determined. The mean and standard error of each data set are shown (n = 3; n.s., not significant). The mean value in the absence of ML-7 was normalized to 100% relative MFI. (D) COS-1 cells were infected with YK333 (EGFP) at an MOI of 1 in the absence or presence of 20 μM ML-7 for 1 h and subsequently treated with 40% PEG. Mean fluorescence intensities (MFIs) were determined by flow cytometry at 5 h postinfection. The relative MFI is the MFI of infected cells treated with ML-7 and/or PEG compared with untreated control cells. The mean and standard error of each data set are shown (n = 3; *, P = 0.0077, two-tailed Student t test). The mean value in the absence of ML-7 and PEG was normalized to 100% relative MFI.
To investigate whether ML-7 inhibited HSV-1 infection at the level of viral entry, especially at viral envelope-cell membrane fusion, we tested whether ML-7 inhibition of HSV-1 infection was blocked by treatment with PEG, which can chemically induce fusion of juxtaposed membranes and thereby promote entry of entry-defective or entry-blocked herpesviruses adsorbed on the cell surface (46). As shown in Fig. 2D, in the presence of ML-7, PEG treatment significantly increased YK333 (EGFP) infection compared to cells that were not treated with PEG. These results indicated that inhibition of cell surface expression of NMHC-IIB by ML-7 inhibited HSV-1 infection at the level of viral entry and suggested that NMHC-IIB in COS-1 cells was required for efficient HSV-1 entry.
Effect of NMHC-IIB overexpression on HSV-1 infection.
We next examined whether NMHC-IIB mediated HSV-1 entry. For this purpose, CHO cells, which are resistant to HSV-1 infection, have often been used since overexpression of gD or gB receptors, including nectin-1, nectin-2, HVEM, and PILRα, was shown to make CHO cells become susceptible to HSV-1 infection (11–13, 20). However, we reported previously that HSV-1 entry into CHO cells expressing a gD receptor, nectin-1, was blocked by antisera to NMHC-IIA, suggesting that NMHC-IIA was required for HSV-1 entry into CHO cells expressing the gD receptor (24) and that CHO cells were not suitable for this type of experiment with NMHC-II. Therefore, we used murine macrophage-derived IC21 cells since the cells express NMHC-IIB at a low level and were relatively resistant to HSV infection (data not shown). First, we established IC21 cells that stably overexpressed either the reported gD receptor human nectin-1 (IC21/hnectin-1 cells) or the reported gB receptor human NMHC-IIA (IC21/hNMHC-IIA cells) (Fig. 3A) and examined how the overexpression of these HSV-1 entry receptors affected HSV-1 entry in IC21 cells. Infection of IC21/nectin-1 or IC21/hNMHC-IIA cells with YK333 (EGFP) resulted in a significant increase in the percentage of virus-infected cells compared with the infection of control IC21/puro cells (Fig. 3C and D), which demonstrated that IC21 cells, like CHO cells, can be used as a model for investigating the role of a receptor in HSV-1 entry. As mentioned above, our results also showed that NMHC-IIA and nectin-1 were detectable in IC21/puro cells (Fig. 3A), and this suggested that IC21 cells endogenously express these reported HSV-1 entry receptors. Next, we generated IC21 cells that stably expressed high levels of mouse NMHC-IIB (IC21/mNMHC-IIB cells) (Fig. 3A). The cell surface expression of NMHC-IIB in IC21/mNMHC-IIB cells was increased compared with the expression in control IC21/puro cells (Fig. 3B), and the overexpression of NMHC-IIB significantly enhanced the susceptibility of IC21 cells to HSV-1 infection, similar to what was observed following the overexpression of nectin-1 or NMHC-IIA in these cells (Fig. 3C and D); these results indicated that NMHC-IIB mediated HSV-1 infection. Notably, overexpression of nectin-1 increased the susceptibility of IC21 cells to HSV-1 infection more efficiently than did overexpression of NMHC-IIA or NMHC-IIB (Fig. 3C and D). However, the extent to which the overexpression of each HSV-1 receptor in IC21 cells affected the susceptibility to HSV-1 infection does not necessarily reflect the activity of these receptors in mediating HSV-1 entry; this is because we were not able to evaluate the levels of overexpression of each of the receptors in IC21 cells by using distinct antibodies to detect the HSV-1 receptors.
FIG 3.
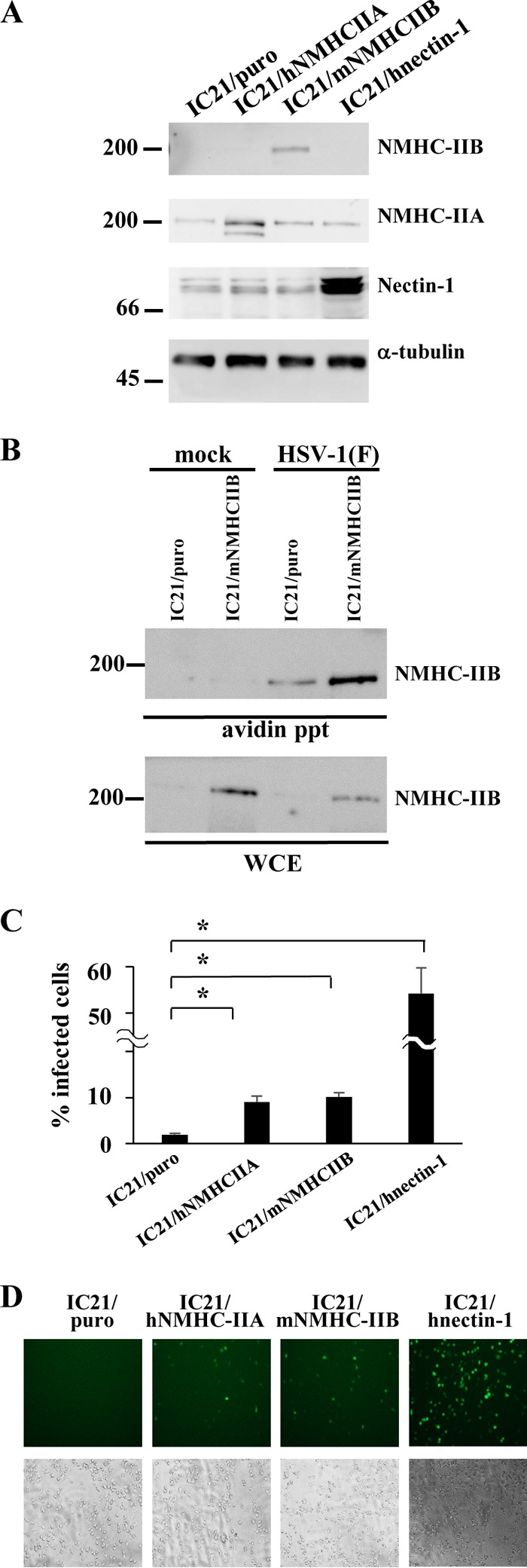
Effect of NMHC-IIB overexpression on HSV-1 infection. (A) Expression of NMHC-IIB, NMHC-IIA, nectin-1, and α-tubulin in IC21/puro, IC21/hNMHC-IIA, IC21/mNMHC-IIB, and IC21/hnectin-1 cells was analyzed by immunoblotting. (B) Cell surface proteins of IC21/puro or IC21/mNMHC-IIB cells incubated with HSV-1 at an MOI of 50 at 4°C for 2 h, followed by a temperature shift to 37°C for 15 min, were biotinylated, precipitated with avidin beads, and analyzed by immunoblotting with anti-NMHC-IIB antibody. WCE, whole-cell extract. Numbers at left of panels A and B are molecular masses in kilodaltons. (C) IC21/puro, IC21/hNMHC-IIA, IC21/mNMHC-IIB, or IC21/hnectin-1 cells were infected with YK333 (EGFP) and analyzed by flow cytometry, and the percentage of infected cells was determined at 10 h postinfection. The mean and standard error of each data set are shown (n = 3; *, P < 0.01, two-tailed Student t test). (D) IC21/puro, IC21/hNMHC-IIA, IC21/mNMHC-IIB, or IC21/hnectin-1 cells were infected with YK333 (EGFP) at an MOI of 1 and analyzed by fluorescence and phase-contrast microscopy at 10 h postinfection.
Effect of NMHC-IIB knockdown in COS-1 cells on HSV-1 infection and cell-cell fusion.
To examine the role of endogenously expressed NMHC-IIB in HSV-1 infection, we generated COS-shNMHC-IIB and COS-shControl cells in which shRNA against NMHC-IIB and control shRNA were expressed, respectively. NMHC-IIB, nectin-1, and α-tubulin expression in COS-shControl and COS-shNMHC-IIB cells were analyzed by immunoblotting to confirm NMHC-IIB knockdown by shRNA against NMHC-IIB (Fig. 4A). Expression of NMHC-IIB, but not of nectin-1 and α-tubulin, was reduced in COS-shNMHC-IIB cells compared to COS-shControl cells. We also confirmed that the cell surface expression of NMHC-IIB in COS/shNMHC-IIB cells was apparently reduced compared to that in control COS-shControl cells (Fig. 4B). COS-shNMHC-IIB and COS-shControl cells were then infected with YK333 (EGFP), and HSV-1 infection was determined by flow cytometry. As shown in Fig. 3C and D, YK333 infection in COS-shNMHC-IIB cells was significantly impaired compared to that in COS-shControl cells, but shRNA against NMHC-IIB had no effect on influenza virus infection.
FIG 4.
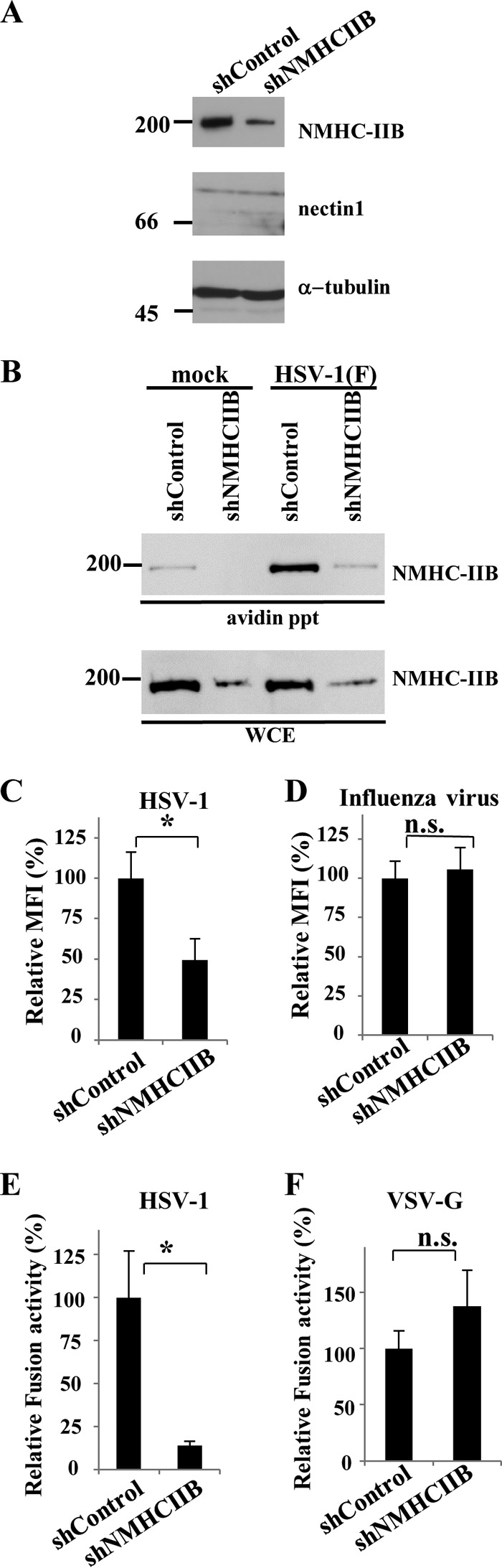
Effects of NMHC-IIB knockdown on HSV-1 infection and cell-cell fusion. (A) NMHC-IIB, nectin-1, and α-tubulin expression levels in COS-shControl and COS-shNMHC-IIB cells were determined by immunoblotting. (B) Cell surface proteins of COS-shControl or COS-shNMHC-IIB cells incubated with HSV-1 at an MOI of 50 at 4°C for 2 h, followed by a temperature shift to 37°C for 15 min, were biotinylated, precipitated with avidin beads, and analyzed by immunoblotting with anti-NMHC-IIB antibody. WCE, whole-cell extract. Numbers at left of panels A and B are molecular masses in kilodaltons. (C and D) COS-shControl and COS-shNMHC-IIB cells infected with YK333 (EGFP) (C) or influenza virus (D) at an MOI of 1 were analyzed by flow cytometry, and mean fluorescence intensities (MFIs) were determined at 5 and 7 h postinfection, respectively. The mean and standard error of each data set are shown (n = 3; *, P = 0.036, n.s., not significant, two-tailed Student t test). The mean value for COS-shControl cells was normalized to 100% relative mean fluorescence intensity (MFI). (E and F) COS-1 cells transfected with expression vectors for T7 polymerase, gB, gD, gH, and gL (E) or expression vectors for T7 polymerase and vesicular stomatitis virus G protein (VSV-G) (F) were cocultured with COS-shControl or COS-shNMHC-IIB cells transfected with a reporter plasmid carrying the luciferase gene driven by the T7 polymerase. The mean and standard error of each data set are shown (n = 3; *, P = 0.034, two-tailed Student t test). The mean value for COS-shControl cells was normalized to 100% relative fusion activity.
To examine the role of NMHC-IIB in HSV-1-mediated membrane fusion, we used a cell-cell fusion assay that allowed measurement of HSV-1-induced membrane fusion based on transient expression of HSV-1 glycoproteins (5). When COS-shNMHC-IIB cells were cocultured with COS-1 cells transiently expressing HSV-1 gB, gD, gH, and gL, the efficiency of cell-cell fusion decreased significantly compared to that of cell-cell fusion when COS-shControl cells were cocultured with COS-1 cells expressing the viral envelope glycoproteins (Fig. 4E). In contrast, NMHC-IIB knockdown had little effect on vesicular stomatitis virus envelope protein G-mediated cell-cell fusion (Fig. 4F).
These results suggested that endogenous NMHC-IIB in COS-1 cells mediated HSV-1 infection and HSV-1 glycoprotein-mediated membrane fusion.
As discussed above, NMHC-II isoforms exhibit a high level of amino acid sequence similarity (25), and therefore, NMHC-II isoforms have been suggested to have overlapping functions. In contrast, accumulating evidence suggests that NMHC-II isoforms have differences in their motor activities, molecular interactions, and cellular and tissue distributions (25). In particular, ablation of the gene encoding NMHC-IIA, -IIB, or IIC in mice showed different phenotypes resulting (50–52). NMHC-IIA ablation resulted in lethality by embryonic day 6.5, with a defect in cell adhesion and visceral endoderm formation (50, 53). NMHC-IIB ablation resulted in survival to embryonic day 14.5, but with marked abnormalities in the heart, including ventricular septal defects and in cardiac myocyte cytokinesis, and brain, including the hydrocephalus and abnormal migration of certain groups of neurons (51, 52). In contrast, NMHC-IIC-ablated mice survive to adulthood and show no obvious defect (53). Because of these diverse functions of NMHC-II isoforms, it was uncertain whether NMHC-IIB, an isoform of NMHC-IIA which is enriched in neuronal tissues that are the most important cellular targets for HSV-1 in vivo (1, 36), can mediate HSV-1 entry by associating with gB like NMHC-IIA. In this study, we have shown a series of data suggesting that NMHC-IIB specifically interacted with HSV-1 gB and mediated HSV-1 entry. Notably, although we have shown that human NMHC-IIB directly bound to gB in vitro, we have not provided data showing that human NMHC-IIB plays a role in HSV-1 entry in human cells. However, as described above, the functions and sequences of mammalian NMHC-IIBs are highly conserved, and no species-specific functional differences of NMHC-IIBs have been reported (49). Therefore, NMHC-IIB in human cells is also likely involved in HSV-1 entry. We also showed that overexpression of NMHC-IIB increased the percentage of cells infected by HSV-1, leading to the conclusion that NMHC-IIB can mediate HSV-1 entry. However, we cannot completely exclude the possibilities that overexpression of NMHC-IIB alters the cell surface expression of nectin-1 and other receptors, affects downstream cellular processes that enhance virus entry, and/or enhances expression of viral glycoproteins resulting in increased cell fusion.
The entry of herpesviruses into a cell is a highly complex process that requires multiple envelope glycoproteins and involves multiple host cellular receptors (54). In HSV, in addition to the gD and gB receptors described above, αvβ6- and αvβ8-integrins were reported to bind to gH/gL and serve as HSV entry receptors (55–57). Similarly, previous studies have reported that (i) members of the integrin family serve as receptors for gH/gL of Epstein-Barr virus (EBV) and human cytomegalovirus (HCMV) and gB of human herpesvirus 8 (HHV-8) and (ii) human leukocyte antigen class II (HLA-II), epidermal growth factor receptor, insulin-degrading enzyme, MAG, and EphrinA2 serve as receptors for EBV gp42, HCMV gB, varicella-zoster virus (VZV) gE, VZV gB, and HHV-8 gH/gL, respectively (23, 58–64). One possible explanation for why herpesviruses use numerous host cellular receptors is that specific interactions between herpesviral envelope glycoproteins and host cellular receptors determine viral cell tropism. In agreement with this hypothesis, previous studies have reported that interactions between EBV gp42 and HLA-II and between EBV gH/gL and integrins are involved in EBV entry into B and epithelial cells, respectively (59, 63). An alternative possibility is that simultaneous interactions between multiple viral envelope glycoproteins and multiple host cellular receptors might enable the viral envelope to stably localize close to the host cellular plasma membrane, which, in turn, might facilitate membrane fusion between the viral envelope and the host cellular membrane. The HSV-1 gD receptors nectin-1 and HVEM, the gB receptors NMHC-IIA and NMHC-IIB, and the gH/gL receptors αvβ6- and αvβ8-integrins are expressed in a variety of cell types (11, 12, 25, 65); therefore, HSV-1 gD, gB, and gH/gL might concurrently interact with these host cellular receptors and thereby contribute to efficient membrane fusion during HSV-1 entry into a cell.
Here, we have demonstrated that cell surface expression of NMHC-IIB was upregulated after adsorption at 4°C followed by a temperature shift to 37°C for 2 to 15 min, as observed with NMHC-IIA in our previous study (24). Currently, the precise mechanism by which the cell surface expression of NMHC-IIA and NMHC-IIB is upregulated upon HSV-1 entry remains to be elucidated. However, HSV-1 entry was reported to rapidly trigger a release of Ca2+ (within 16 s after a shift to 37°C), and this Ca2+ signaling played a critical role in HSV-1 entry (66). Ca2+ is an essential cofactor for the activation of MLCK, which controls the functions and localization of NM through phosphorylation (67). The data presented here and in our previous study have shown that ML-7, a specific inhibitor of MLCK, and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), a Ca2+ chelator, prevented the upregulation of the cell surface expression of NMHC-IIA and/or NMHC-IIB upon viral entry; these results suggested that the upregulation of NMHC-II cell surface expression was regulated by a signaling event(s) during the initiation step of viral entry. In this and previous studies, small amounts of NMHC-IIA and NMHC-IIB were detectable on the surface of mock-infected cells, but whether the NMHC-IIA and NMHC-IIB expressed on the surface of these mock-infected cells are sufficient for HSV-1 entry remains unknown. Moreover, there is a possibility that the 2-h incubation at 4°C followed by a shift up to 37°C allows for sufficient initial membrane fusion and/or pore formation resulting in exposure of surface-bound remaining viral particles and gB to NMHC-IIA and NMHC-IIB. However, previous studies reported that a time lag of 10 to 15 min occurred between viral adsorption at 4°C and the initiation of viral penetration after a temperature shift to 37°C in the case of HSV-1 (68, 69) but not vaccinia virus (70). These findings agree with our observation that the cell surface expression of NMHC-IIA and NMHC-IIB is upregulated after viral adsorption at 4°C followed by a temperature shift to 37°C for 2 to 15 min, and these results suggest that the upregulation of cell surface expression of NMHC-IIA and NMHC-IIB during the initiation of viral entry might be required for efficient viral entry. It has been reported that HSV-1 protein VP22, a virion tegument protein, interacts with NMHC-IIA (71) and that myosin Va, another member of the myosin family, plays roles in the secretion of HSV-1 virions and cell surface expression of HSV-1 envelope glycoproteins (72). Thus, members of the myosin family appear to be involved in multiple steps of HSV-1 replication. Further clarification of the precise roles of these and other myosins in HSV-1 infection may elucidate the molecular basis of multiple aspects of HSV-1 infection.
ACKNOWLEDGMENTS
We thank Ryuichi Nishinakamura for providing the NMHC-IIB expression plasmid, Masayuki Takahashi for providing pEGFP-BRF305, and Shihoko Koyama and Tomoko Ando for excellent technical assistance.
This study was supported in part by the Funding Program for Next Generation World-Leading Researchers and grants for scientific research from the Japan Society for the Promotion of Science (JSPS); a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) and a grant for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan; and grants from the Takeda Science Foundation, Astellas Foundation for Research on Metabolic Disorders, the Sumitomo Foundation, and the Ichiro Kanehara Foundation.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601 InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Strauss SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Herold BC, Visalli RJ, Susmarski N, Brandt CR, Spear PG. 1994. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J Gen Virol 75:1211–1222. doi: 10.1099/0022-1317-75-6-1211. [DOI] [PubMed] [Google Scholar]
- 3.Herold BC, WuDunn D, Soltys N, Spear PG. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J Virol 65:1090–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol 6:401–410. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 5.Turner A, Bruun B, Minson T, Browne H. 1998. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J Virol 72:873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. doi: 10.1006/viro.2000.0713. [DOI] [PubMed] [Google Scholar]
- 7.Spear PG, Manoj S, Yoon M, Jogger CR, Zago A, Myscofski D. 2006. Different receptors binding to distinct interfaces on herpes simplex virus gD can trigger events leading to cell fusion and viral entry. Virology 344:17–24. doi: 10.1016/j.virol.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 8.Avitabile E, Forghieri C, Campadelli-Fiume G. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J Virol 81:11532–11537. doi: 10.1128/JVI.01343-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc Natl Acad Sci U S A 104:18718–18723. doi: 10.1073/pnas.0707452104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. doi: 10.1016/S0092-8674(00)81363-X. [DOI] [PubMed] [Google Scholar]
- 12.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 13.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- 14.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22. doi: 10.1016/S0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 15.Taylor JM, Lin E, Susmarski N, Yoon M, Zago A, Ware CF, Pfeffer K, Miyoshi J, Takai Y, Spear PG. 2007. Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe 2:19–28. doi: 10.1016/j.chom.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J Virol 79:11588–11597. doi: 10.1128/JVI.79.18.11588-11597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fournier N, Chalus L, Durand I, Garcia E, Pin JJ, Churakova T, Patel S, Zlot C, Gorman D, Zurawski S, Abrams J, Bates EE, Garrone P. 2000. FDF03, a novel inhibitory receptor of the immunoglobulin superfamily, is expressed by human dendritic and myeloid cells. J Immunol 165:1197–1209. doi: 10.4049/jimmunol.165.3.1197. [DOI] [PubMed] [Google Scholar]
- 18.Mousseau DD, Banville D, L'Abbe D, Bouchard P, Shen SH. 2000. PILRalpha, a novel immunoreceptor tyrosine-based inhibitory motif-bearing protein, recruits SHP-1 upon tyrosine phosphorylation and is paired with the truncated counterpart PILRbeta. J Biol Chem 275:4467–4474. doi: 10.1074/jbc.275.6.4467. [DOI] [PubMed] [Google Scholar]
- 19.Shiratori I, Ogasawara K, Saito T, Lanier LL, Arase H. 2004. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J Exp Med 199:525–533. doi: 10.1084/jem.20031885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Fan Q, Satoh T, Arii J, Lanier LL, Spear PG, Kawaguchi Y, Arase H. 2009. Binding of herpes simplex virus glycoprotein B (gB) to paired immunoglobulin-like type 2 receptor alpha depends on specific sialylated O-linked glycans on gB. J Virol 83:13042–13045. doi: 10.1128/JVI.00792-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arii J, Wang J, Morimoto T, Suenaga T, Akashi H, Arase H, Kawaguchi Y. 2010. A single-amino-acid substitution in herpes simplex virus 1 envelope glycoprotein B at a site required for binding to the paired immunoglobulin-like type 2 receptor alpha (PILRalpha) abrogates PILRalpha-dependent viral entry and reduces pathogenesis. J Virol 84:10773–10783. doi: 10.1128/JVI.01166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A 107:866–871. doi: 10.1073/pnas.0913351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 25.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. 2009. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li D, Miller M, Chantler PD. 1994. Association of a cellular myosin II with anionic phospholipids and the neuronal plasma membrane. Proc Natl Acad Sci U S A 91:853–857. doi: 10.1073/pnas.91.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nebl T, Pestonjamasp KN, Leszyk JD, Crowley JL, Oh SW, Luna EJ. 2002. Proteomic analysis of a detergent-resistant membrane skeleton from neutrophil plasma membranes. J Biol Chem 277:43399–43409. doi: 10.1074/jbc.M205386200. [DOI] [PubMed] [Google Scholar]
- 28.Olden K, Willingham M, Pastan I. 1976. Cell surface myosin in cultured fibroblasts. Cell 8:383–390. doi: 10.1016/0092-8674(76)90150-1. [DOI] [PubMed] [Google Scholar]
- 29.Gallagher PJ, Herring BP, Stull JT. 1997. Myosin light chain kinases. J Muscle Res Cell Motil 18:1–16. doi: 10.1023/A:1018616814417. [DOI] [PubMed] [Google Scholar]
- 30.Sun Y, Qi Y, Liu C, Gao W, Chen P, Fu L, Peng B, Wang H, Jing Z, Zhong G, Li W. 2014. Nonmuscle myosin heavy chain IIA is a critical factor contributing to the efficiency of early infection of severe fever with thrombocytopenia syndrome virus. J Virol 88:237–248. doi: 10.1128/JVI.02141-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mercer J, Helenius A. 2008. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 32.Schelhaas M, Ewers H, Rajamaki ML, Day PM, Schiller JT, Helenius A. 2008. Human papillomavirus type 16 entry: retrograde cell surface transport along actin-rich protrusions. PLoS Pathog 4:e1000148. doi: 10.1371/journal.ppat.1000148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. 2005. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol 170:317–325. doi: 10.1083/jcb.200503059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Valiya Veettil M, Sadagopan S, Kerur N, Chakraborty S, Chandran B. 2010. Interaction of c-Cbl with myosin IIA regulates Bleb associated macropinocytosis of Kaposi's sarcoma-associated herpesvirus. PLoS Pathog 6:e1001238. doi: 10.1371/journal.ppat.1001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golomb E, Ma X, Jana SS, Preston YA, Kawamoto S, Shoham NG, Goldin E, Conti MA, Sellers JR, Adelstein RS. 2004. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J Biol Chem 279:2800–2808. doi: 10.1074/jbc.M309981200. [DOI] [PubMed] [Google Scholar]
- 36.Phillips CL, Yamakawa K, Adelstein RS. 1995. Cloning of the cDNA encoding human nonmuscle myosin heavy chain-B and analysis of human tissues with isoform-specific antibodies. J Muscle Res Cell Motil 16:379–389. doi: 10.1007/BF00114503. [DOI] [PubMed] [Google Scholar]
- 37.Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor alpha. J Virol 83:4520–4527. doi: 10.1128/JVI.02601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka M, Kodaira H, Nishiyama Y, Sata T, Kawaguchi Y. 2004. Construction of recombinant herpes simplex virus type I expressing green fluorescent protein without loss of any viral genes. Microbes Infect 6:485–493. doi: 10.1016/j.micinf.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haraguchi T, Mizutani T, Yamamichi N, Ito T, Minoguchi S, Iba H. 2007. SiRNAs do not induce RNA-dependent transcriptional silencing of retrovirus in human cells. FEBS Lett 581:4949–4954. doi: 10.1016/j.febslet.2007.09.028. [DOI] [PubMed] [Google Scholar]
- 42.Okuma K, Nakamura M, Nakano S, Niho Y, Matsuura Y. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235–244. doi: 10.1006/viro.1998.9530. [DOI] [PubMed] [Google Scholar]
- 43.Sato MK, Takahashi M, Yazawa M. 2007. Two regions of the tail are necessary for the isoform-specific functions of nonmuscle myosin IIB. Mol Biol Cell 18:1009–1017. doi: 10.1091/mbc.E06-08-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchiyama Y, Sakaguchi M, Terabayashi T, Inenaga T, Inoue S, Kobayashi C, Oshima N, Kiyonari H, Nakagata N, Sato Y, Sekiguchi K, Miki H, Araki E, Fujimura S, Tanaka SS, Nishinakamura R. 2010. Kif26b, a kinesin family gene, regulates adhesion of the embryonic kidney mesenchyme. Proc Natl Acad Sci U S A 107:9240–9245. doi: 10.1073/pnas.0913748107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanamori M, Watanabe S, Honma R, Kuroda M, Imai S, Takada K, Yamamoto N, Nishiyama Y, Kawaguchi Y. 2004. Epstein-Barr virus nuclear antigen leader protein induces expression of thymus- and activation-regulated chemokine in B cells. J Virol 78:3984–3993. doi: 10.1128/JVI.78.8.3984-3993.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarmiento M, Haffey M, Spear PG. 1979. Membrane proteins specified by herpes simplex viruses. III. Role of glycoprotein VP7(B2) in virion infectivity. J Virol 29:1149–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morita S, Kojima T, Kitamura T. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 48.Bao J, Jana SS, Adelstein RS. 2005. Vertebrate nonmuscle myosin II isoforms rescue small interfering RNA-induced defects in COS-7 cell cytokinesis. J Biol Chem 280:19594–19599. doi: 10.1074/jbc.M501573200. [DOI] [PubMed] [Google Scholar]
- 49.Murakami N, Trenkner E, Elzinga M. 1993. Changes in expression of nonmuscle myosin heavy chain isoforms during muscle and nonmuscle tissue development. Dev Biol 157:19–27. doi: 10.1006/dbio.1993.1108. [DOI] [PubMed] [Google Scholar]
- 50.Conti MA, Even-Ram S, Liu C, Yamada KM, Adelstein RS. 2004. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J Biol Chem 279:41263–41266. doi: 10.1074/jbc.C400352200. [DOI] [PubMed] [Google Scholar]
- 51.Tullio AN, Bridgman PC, Tresser NJ, Chan CC, Conti MA, Adelstein RS, Hara Y. 2001. Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II-B heavy chain. J Comp Neurol 433:62–74. doi: 10.1002/cne.1125. [DOI] [PubMed] [Google Scholar]
- 52.Tullio AN, Accili D, Ferrans VJ, Yu ZX, Takeda K, Grinberg A, Westphal H, Preston YA, Adelstein RS. 1997. Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc Natl Acad Sci U S A 94:12407–12412. doi: 10.1073/pnas.94.23.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma X, Jana SS, Conti MA, Kawamoto S, Claycomb WC, Adelstein RS. 2010. Ablation of nonmuscle myosin II-B and II-C reveals a role for nonmuscle myosin II in cardiac myocyte karyokinesis. Mol Biol Cell 21:3952–3962. doi: 10.1091/mbc.E10-04-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gianni T, Salvioli S, Chesnokova LS, Hutt-Fletcher LM, Campadelli-Fiume G. 2013. alphavbeta6- and alphavbeta8-integrins serve as interchangeable receptors for HSV gH/gL to promote endocytosis and activation of membrane fusion. PLoS Pathog 9:e1003806. doi: 10.1371/journal.ppat.1003806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parry C, Bell S, Minson T, Browne H. 2005. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J Gen Virol 86:7–10. doi: 10.1099/vir.0.80567-0. [DOI] [PubMed] [Google Scholar]
- 57.Cheshenko N, Trepanier JB, Gonzalez PA, Eugenin EA, Jacobs WR Jr, Herold BC. 2014. Herpes simplex virus type 2 glycoprotein H interacts with integrin alphavbeta3 to facilitate viral entry and calcium signaling in human genital tract epithelial cells. J Virol 88:10026–10038. doi: 10.1128/JVI.00725-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Akula SM, Pramod NP, Wang FZ, Chandran B. 2002. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407–419. doi: 10.1016/S0092-8674(02)00628-1. [DOI] [PubMed] [Google Scholar]
- 59.Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc Natl Acad Sci U S A 106:20464–20469. doi: 10.1073/pnas.0907508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Huang DY, Huong SM, Huang ES. 2005. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med 11:515–521. doi: 10.1038/nm1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES. 2003. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 424:456–461. doi: 10.1038/nature01818. [DOI] [PubMed] [Google Scholar]
- 62.Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, Holzer A, Schmidt M, Chen J, Konig S, Ensser A, Myoung J, Brockmeyer NH, Sturzl M, Fleckenstein B, Neipel F. 2012. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi's sarcoma-associated herpesvirus. Nat Med 18:961–966. doi: 10.1038/nm.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haan KM, Kwok WW, Longnecker R, Speck P. 2000. Epstein-Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor. J Virol 74:2451–2454. doi: 10.1128/JVI.74.5.2451-2454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Q, Ali MA, Cohen JI. 2006. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 127:305–316. doi: 10.1016/j.cell.2006.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parkinson H, Kapushesky M, Shojatalab M, Abeygunawardena N, Coulson R, Farne A, Holloway E, Kolesnykov N, Lilja P, Lukk M, Mani R, Rayner T, Sharma A, William E, Sarkans U, Brazma A. 2007. ArrayExpress—a public database of microarray experiments and gene expression profiles. Nucleic Acids Res 35:D747–D750. doi: 10.1093/nar/gkl995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cheshenko N, Del Rosario B, Woda C, Marcellino D, Satlin LM, Herold BC. 2003. Herpes simplex virus triggers activation of calcium-signaling pathways. J Cell Biol 163:283–293. doi: 10.1083/jcb.200301084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watanabe T, Hosoya H, Yonemura S. 2007. Regulation of myosin II dynamics by phosphorylation and dephosphorylation of its light chain in epithelial cells. Mol Biol Cell 18:605–616. doi: 10.1091/mbc.E06-07-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elliott G, Hafezi W, Whiteley A, Bernard E. 2005. Deletion of the herpes simplex virus VP22-encoding gene (UL49) alters the expression, localization, and virion incorporation of ICP0. J Virol 79:9735–9745. doi: 10.1128/JVI.79.15.9735-9745.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Galdiero M, Whiteley A, Bruun B, Bell S, Minson T, Browne H. 1997. Site-directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J Virol 71:2163–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Payne LG, Norrby E. 1978. Adsorption and penetration of enveloped and naked vaccinia virus particles. J Virol 27:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Leeuwen H, Elliott G, O'Hare P. 2002. Evidence of a role for nonmuscle myosin II in herpes simplex virus type 1 egress. J Virol 76:3471–3481. doi: 10.1128/JVI.76.7.3471-3481.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roberts KL, Baines JD. 2010. Myosin Va enhances secretion of herpes simplex virus 1 virions and cell surface expression of viral glycoproteins. J Virol 84:9889–9896. doi: 10.1128/JVI.00732-10. [DOI] [PMC free article] [PubMed] [Google Scholar]