

ABSTRACT
In the extracellular environment, cell-free virions seek out naive host cells over long distances and between organisms. This is the primary mechanism of spread for most viruses. Here we provide evidence for an alternative pathway previously undescribed for orthomyxoviruses, whereby the spread of influenza A virus (IAV) infectious cores to neighboring cells can occur within intercellular connections. The formation of these connections requires actin dynamics and is enhanced by viral infection. Connected cells have contiguous membranes, and the core infectious viral machinery (RNP and polymerase) was present inside the intercellular connections. A live-cell movie of green fluorescent protein (GFP)-tagged NS1 of IAV shows viral protein moving from one cell to another through an intercellular connection. The movement of tagged protein was saltatory but overall traveled only in one direction. Infectious virus cores can move from one cell to another without budding and release of cell-free virions, as evidenced by the finding that whereas a neuraminidase inhibitor alone did not inhibit the development of IAV microplaques, the presence of a neuraminidase inhibitor together with drugs inhibiting actin dynamics or the microtubule stabilizer paclitaxel (originally named taxol) precluded microplaque formation. Similar results were also observed with parainfluenza virus 5 (PIV5), a paramyxovirus, when neutralizing antibody was used to block spread by cell-free virions. Intercellular spread of infectious core particles was unaffected or enhanced in the presence of nocodazole for IAV but inhibited for PIV5. The intercellular connections have a core of filamentous actin, which hints toward transport of virus particles through the use of a myosin motor.
IMPORTANCE Here we describe a new method by which influenza A virus (IAV) spreads from cell to cell: IAV uses intracellular connections. The formation of these connections requires actin dynamics and is enhanced by viral infection and the absence of microtubules. Connected cells appeared to have contiguous membranes, and the core infectious viral machinery (RNP and polymerase) was present inside the intercellular connections. Infectious virus cores can move from one cell to another without budding and release of cell-free virions. Similar results were also observed with parainfluenza virus 5 (PIV5).
INTRODUCTION
Influenza A virus (IAV), a member of the Orthomyxoviridae, is an enveloped virus with a negative-strand segmented RNA genome. In virions, the eight RNA segments are decorated with the nucleocapsid protein (NP) and one copy of a heterotrimer of the RNA-dependent RNA polymerase complex consisting of PB1, PB2, and PA. The ribonucleoprotein (RNP) and polymerase complex are the minimal replicative machinery. The ribonucleoproteins are enclosed within a lipid envelope derived from the host cell plasma membrane. Underlying the bilayer is an internal coat of matrix protein (M1). Inserted through the lipid bilayer are the viral integral membrane proteins hemagglutinin (HA) and neuraminidase (NA) and the M2 ion channel protein (1). HA binds to the viral receptor, sialic acid, and also mediates fusion of the viral envelope with an endosomal membrane during virus entry into cells. NA has neuraminidase activity, which cleaves sialic acid from complex carbohydrate chains and is required during virus egress to preclude HA binding to its receptor on the host cell. The proton-selective ion channel M2 activity is required for acidification of the interior of virions during virus uncoating and also for scission of budding virions (1).
Influenza virus particles gain entry to cells by binding to a cell surface molecule containing sialic acid and are then internalized into clathrin-coated pits and transported to endosomes. There, the low-pH environment activates the M2 ion channel and selectively transports protons and potassium into the virion interior to bring about dissociation of the M1 protein from the RNP (2). Low pH (pH 5.0 to pH 5.8, depending on the HA subtype) also triggers an HA protein refolding event which causes the merger of the viral membrane with the endosomal membrane, releasing the RNPs into the cytoplasm for transport to the nucleus, where viral transcription and replication occur.
For the assembly of virions, RNPs exit the nucleus through nuclear pores and traffic on microtubules to the plasma membrane (3). M1 interacts with the viral RNP (4–6) and together with HA, NA, and M2 assembles into viral particles at the plasma membrane. In polarized cells, budding occurs from lipid rafts on the apical surface (7, 8), which are highly enriched in cholesterol and contain large amounts of glycerophospholipids and sphingolipids (9).
The HA-containing lipid rafts (10, 11) coalesce to form “barges” of rafts (from ∼325 to 500 nm at 4 h postinfection [p.i.] to ∼425 to 600 nm at 6 h p.i.) (10). M2 protein associates mainly with the periphery of lipid rafts and is largely excluded from virions (12). In contrast, M1 cannot target the plasma membrane in the absence of other viral proteins (13). M1 expressed alone in cells does not form virus-like particles (14), and M1 likely targets the plasma membrane proteins by associating with the cytoplasmic tails of the viral integral proteins (13, 15–17).
All eight unique vRNPs must be incorporated into a new virus particle for a productive infection to occur, though how packaging is achieved remains controversial (18–20), and two models propose different mechanisms. The random-packaging model suggests that viral RNP (vRNP) segments are randomly incorporated into budding virions and only virions or particle-forming units containing all eight unique segments are infectious. The selective-packing model, which has been rapidly gaining favor (21–23), proposes that each unique vRNP segment contains a distinct property that enables directed packaging to ensure that exactly eight unique segments are incorporated into each budding particle.
The primary delivery system for many enveloped viruses to a cell, including IAV, is by endocytosis of particles and fusion of the envelope with the endosomal membrane. However, for some enveloped viruses, alternative delivery pathways that do not involve either acid-triggered fusion or receptor-binding triggered fusion have been described (24, 25). One emerging model has proposed the use of long, filamentous intercellular connections described as tunneling nanotubules (TNTs) as a means to transport virus particles to neighboring cells (26–28).
TNTs are long membrane-bound extensions (50 to 200 nm in diameter and spanning as much as several cell diameters) that tether two neighboring cells and can link many cells together in tandem, forming complex cellular networks (29). These structures contain filamentous actin (F-actin) but not microtubules and have been described as a means of cellular communication over long distances, particularly transportation of organelles and transmembrane proteins. Once the TNT has been established, the membranes of each cell appear to be fused, as enhanced green fluorescent protein (EGFP)-labeled membrane proteins can transfer from one cell to another. Interestingly, only membrane-bound protein or organelles are able to pass through TNTs and small molecules freely existing in the cytoplasm are not (29).
Intercellular connections have been presented as a means of transporting newly assembled human immunodeficiency virus type 1 (HIV-1) particles to neighboring cells, either by traveling along the outer membrane of a “virological synapse” (30, 31) or the transfer of virus infectious cores within “bridging conduits” (26, 27). The latter pathway has also been described for prions (28). Though many names have been used to describe these intercellular connections (e.g., nanotubules, virological synapse, bridging conduits), the fundamental feature of these structures is the connection of cytoplasm between two cells via membrane-bound extensions. The extent to which these differently named structures are distinct from one another is unclear. Here we will use the term intracellular connection.
Our data show that IAV uses a similar pathway for intercellular infections. This route of spread requires the establishment of intercellular connections connecting the cytoplasm of two neighboring cells, thereby allowing passage of the critical viral components (i.e., RNA, NP, and polymerase) necessary for infection by passing through the thin, membrane-bound connection and into the cell body of a neighboring cell.
MATERIALS AND METHODS
Cells, antibodies, and reagents.
Madin-Darby canine kidney (MDCK) and human alveolar lung cells (A549) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Normal human bronchial epithelial (NHBE) cells were maintained in bronchial epithelial growth medium (BEGM) as recommended by the manufacturer (Lonza). All cells were maintained in a humidified incubator containing 5% CO2 at 37°C. Antibodies specific for influenza virus included goat anti-H3 HA (influenza A/Aichi/2/68 virus) obtained from the National Institute of Allergy and Infectious Diseases Repository, Bethesda, MD, and rabbit serum raised against expressed and purified influenza virus nucleoprotein (NP) used at 1:400 and 1:200, respectively. Antiserum specific for PA was obtained from GeneTex (GTX118991) and used at 1:50.
Antiserum specific for parainfluenza virus 5 (PIV5) hemagglutinin-neuraminidase (HN) (R471) was obtained by injecting rabbits with purified HN soluble ectodomain expressed in Sf9 cells from a recombinant baculovirus (32). For PIV5 NP, a mouse monoclonal antibody (MAb) was used at 1:200 (33). F1a, a MAb specific for PIV5 fusion (F) protein, was used to neutralize cell-free PIV5 virions at a concentration of approximately 4 μg/ml (33). Cytochalasin D, paclitaxel (originally named taxol), and nocodazole were obtained from Sigma-Aldrich and IPA-3 from Calbiochem and used at concentrations of 20 μM, 100 μM, 30 μM, and 30 μM, respectively. Ammonium chloride (Mallinckrodt Pharmaceuticals) was used at a concentration of 4 mM. Zanamivir (GlaxoSmithKline) was used at a concentration of 10 μM, which has been previously shown to inhibit NA activity (34). Wheat germ agglutinin conjugated to Alexa Fluor 488 (Invitrogen) was used at a concentration of 5 μg/ml for 10 min.
The GFP-NS1-expressing virus in the PR8 background has been described previously (35).
Immunofluorescent and live-cell microscopy.
MDCK or A549 cells were grown on glass coverslips and infected with wild-type (wt) influenza A/Udorn/72 virus (IAV) or PIV5 at a multiplicity of infection (MOI) of three PFU per cell unless otherwise specified. Infection was synchronized by allowing virus to attach at 4°C for 1 h followed by washing cells in phosphate-buffered saline (PBS) and overlaying the monolayer with warmed (37°C) Dulbecco's modified Eagle's medium (DME) containing 1% penicillin plus 1% streptomycin and 1 μg/ml N-acetyl trypsin for IAV infections or DMEM containing 1% penicillin plus 1% streptomycin for PIV5 infections. Infected cells were fixed with 10% formalin (Electron Microscopy Sciences, Hatsfield, PA), and cell surfaces were blocked with 10% donkey serum (Sigma-Aldrich) in PBS followed by immunostaining (without permeabilization) with goat anti-H3 HA serum at a concentration of 1:400 or with rabbit anti-HN antibody at a concentration of 1:50 in 1% bovine serum albumin (BSA)–PBS for 1 h at room temperature, followed by extensive washes in PBS. Alternatively, to stain the surfaces of mock-infected cells, wheat germ agglutinin conjugated to Alexa Fluor 488 (Molecular Probes) was used according to the manufacturer's instructions to visualize the cell surface. To stain influenza virus NP or PA, cells were permeabilized with PBS plus 0.1% Tween (PBST) for 3 min and blocked in 10% donkey serum for 10 min at room temperature. Cells were washed with PBST and stained with rabbit anti-NP serum at a concentration of 1:200. For PIV5 NP or P, cells were stained with mouse anti-NP serum at a concentration of 1:200 in 1% BSA–PBST. To stain filamentous actin and tubulin, cells were permeabilized and blocked as above and stained with Alexa Fluor 594-conjugated phalloidin (Molecular Probes) or with 2.5 μg/ml mouse antitubulin serum (sc-32293; Santa Cruz Biotechnology). The secondary antibodies used were goat anti-rabbit IgG and goat anti-mouse IgG conjugated to Alexa Fluor 594 at a concentration of 1:200 in 1% BSA–PBST. After room temperature incubation for 1 h, coverslips were washed in PBST, dipped in water, mounted onto glass slides using Prolong Gold (Invitrogen), and left to set overnight in the dark. For live-cell movies, MDCK cells were seeded onto 42-mm coverslips and infected with a recombinant GFP-tagged NS1 (35) expressing IAV at an MOI of 1. Infected cells on the coverslip were transferred into an open cultivation system (Zeiss) and maintained in warm DMEM buffered with HEPES. The live-cell chamber was mounted on a heated stage to maintain the culture at 37°C.
Immunostained and live cells were imaged with an LSM 5 Zeiss confocal microscope (Thornwood, NY) or a Zeiss Axiovert 200M fluorescence microscope using a 20×, 40×, or 63× objective as indicated. Collected images were modified using Adobe Photoshop CS3.
RESULTS
Cells infected with IAV form intercellular connections containing viral nucleoprotein and polymerase.
To investigate the formation of intercellular connections during influenza A virus (IAV) infection, MDCK cells were infected with Udorn IAV, and at 17 h p.i. cells were fixed and cell surfaces were immunostained for HA. Subsequently, cells were permeabilized and stained internally for NP. Confocal microscopy images showed that in addition to budding virus, long HA-containing protrusions connected to neighboring cells with what appeared to be fused membranes (Fig. 1). We observed these structures in infected cell monolayers as early as 6 h p.i. and as late as 48 h p.i. (data not shown). Furthermore, NP staining was observed within the intercellular connections, suggesting the possibility that IAV vRNPs use the connections as an intercellular gateway to other cells as an alternative egress/entry pathway.
FIG 1.
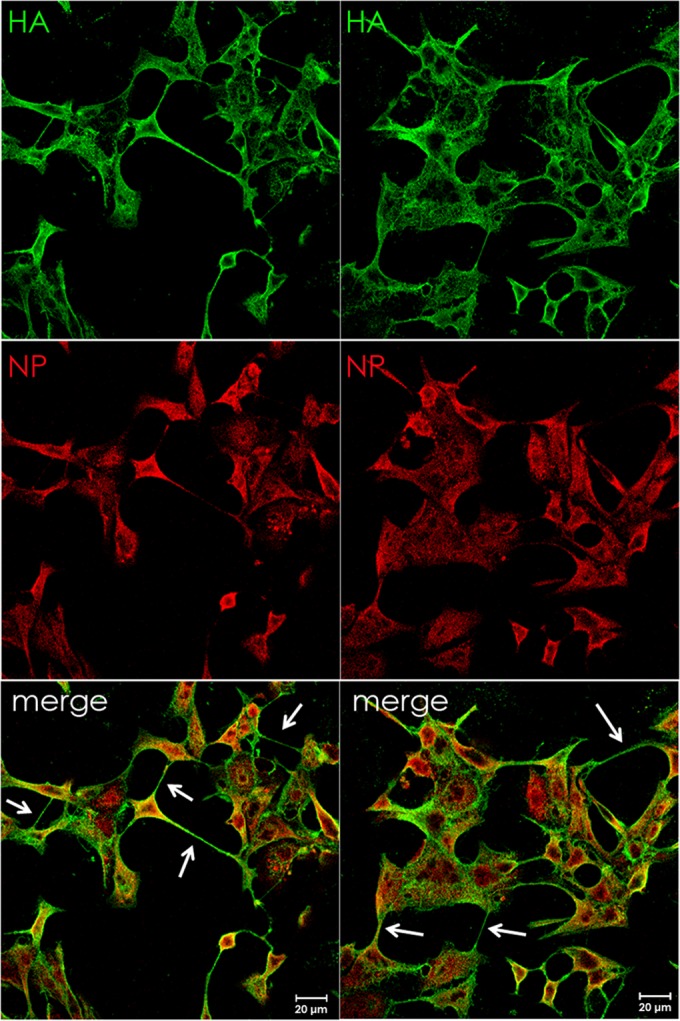
Viral ribonucleoprotein-containing intercellular connections form in IAV-infected cells. MDCK cells were infected with IAV (A/Udorn/72) at an MOI of 3 and fixed at 17 h p.i. Cell surfaces were immunostained for HA (green) and then permeabilized and immunostained for NP (red). Infected cells formed intercellular connections between neighboring cells that appeared to connect the cytoplasm of both cells. Images were photographed on a confocal microscope (Zeiss LSM 5).
To determine if intercellular connections formed in cell types other than MDCK cells, A549 and Vero cells were infected with IAV and stained for HA (Fig. 2). Intercellular connections similar to those seen in MDCK cells were observed. Staining for PA in A549, MDCK, and Vero cells confirmed the presence of RNPs and their associated polymerase complex, as in infected cells RNPs are found with a PA, PB1, and PB2 complex. It has been shown previously that the RNPs with associated polymerase complex is the minimal replicative machinery (36).
FIG 2.

Intercellular connections contain the IAV HA protein and a component (PA) of the polymerase in multiple cell types. A549, MDCK, and Vero cells were infected with IAV at an MOI of 3 and fixed at 17 h p.i. Cell surfaces were immunostained for hemagglutinin protein (HA; green) and then permeabilized and immunostained for viral polymerase (PA; red). Infected cells formed intracellular connections between neighboring cells that appeared to connect the cytoplasm of both cells. Images were photographed on a confocal microscope. Scale bar, 20 μm.
Given the known involvement of F-actin in the TNT structures (29), we were interested in determining whether or not actin dynamics were necessary for the formation of the intercellular connections. To test this, IAV-infected cells were treated with IPA-3, cytochalasin D, or dimethyl sulfoxide (DMSO) (as a solvent control). The microtubule stabilizer paclitaxel and microtubule depolymerizer nocodazole were also included as additional controls. IPA-3 inhibits PAK1 (p21-activated kinase 1) (37), an interacting partner of Rho GTPase family proteins that includes cell division cycle 42 (CDC42) and RAC1. CDC42 is involved in the formation of filopodia, which share many similarities to TNTs, including a bundled F-actin core and a dynamic, probing movement toward neighboring cells. Cytochalasin D binds to F-actin and causes breakage, eventually leading to actin depolymerization (38). The effects of these drugs on the formation of intercellular connections in IAV-infected cells were determined by counting the connected cells under each treatment (Fig. 3).
FIG 3.

Intercellular connections formed during IAV infection require actin dynamics and F-actin but do not require microtubules and occur more frequently during infection. (A) MDCK cells were infected with IAV at an MOI of 3. At 1 h p.i., 30 μM IPA-3, 100 μM paclitaxel (“Taxol”), 20 μM cytochalasin D (CytoD), and 30 μM nocodazole (Noc) were added to infected cells, and the cells were fixed at 18 h p.i. Cell surfaces were immunostained for HA (green) and then permeabilized and immunostained for NP (red). Inset shows a zoomed image of two cells connected by an intercellular connection. Viral NP is clearly visible within the connection (arrows) as well as in the cell body. (B) The bar graph quantifies the percentage of MDCK cell pairs with intercellular connections in drug-treated and control (DMSO) cells infected with IAV. *, P < 0.05. (C) The bar graph quantifies the percentage of MDCK cell pairs connected by intercellular connections in mock, PIV5, or IAV infections. ***, P < 0.001. Images were photographed on a confocal microscope. Scale bar, 20 μm.
Drugs affecting actin dynamics (IPA-3 and cytochalasin D) significantly reduced the number of cells connected by TNTs (Fig. 3). Unexpectedly, the microtubule-affecting drugs also affected the formation of intercellular connections compared to the DMSO control. Addition of the microtubule stabilizer paclitaxel significantly reduced the number of intercellular connections, whereas the microtubule destabilizer nocodazole increased the number of intercellular connections compared to DMSO-treated cells (Fig. 3B). These findings suggest a possible role for the microtubule cytoskeletal network in the regulation of intercellular connection formation. We also quantified the number of intercellular connections in mock- and IAV-infected MDCK cells and found that IAV infection greatly enhanced the formation of intercellular connections (Fig. 3C).
Intercellular connections can be used for spread of infectivity from cell to cell.
The data shown in Fig. 1 to 3 indicate that the intercellular connections that form during IAV infection contain vRNP and that the formation of these connections requires actin dynamics. These findings raise the question as to whether the intercellular connections can mediate cell-to-cell spread of infectivity, as the vRNPs are the minimal replication machinery (36).
To determine if intercellular connections provide a route for viral infection, MDCK cells were infected at a low MOI (0.1) with IAV, and at 2 h p.i. the indicated drugs were added either with or without the NA inhibitor zanamivir. Release of budding virions from the host cell cannot occur efficiently without NA activity, as cell-free virions would be bound at the surface of the host cell due to HA binding sialic acid. Thus, the virus is limited to cell-to-cell spread of infection via transport of vRNP through the intercellular connections. At 48 h p.i., the cells were fixed and immunostained for NP to score the number and size of microplaques. Like a plaque, a microplaque is a clustered grouping of infected cells resulting from cell-to-cell spread of virus. However, instead of measuring large clearings of cells resulting from cytopathic effects, here we score microplaques based on the presence of nucleoprotein within total cells (indicated by nucleoprotein immunostaining and DAPI [4′,6-diamidino-2-phenylindole] staining). Three or more adjacent cells staining positive for nucleoprotein are considered a microplaque. The results are presented in Fig. 4.
FIG 4.

Intercellular connections provide a route for infection of neighboring cells. (A) MDCK cells were infected at an MOI of 0.1 with IAV. At 2 h p.i., 10 mM zanamivir, 30 μM IPA-3, 100 μM paclitaxel (“Taxol”), 20 μM cytochalasin D (CytoD), and 30 μM nocodazole (Noc) were added at 2 h p.i. as indicated, and the cells were incubated for 48 h. Cells were fixed and immunostained for NP (red), and nuclei were stained with DAPI (blue). Images were taken on a fluorescence microscope (Zeiss Axiovert 200M) using a 20× objective. (B) Quantification of the microplaques (NP-positive neighboring cells, white circles) in panel A. ***, P < 0.001. (C) MDCK cells were infected at an MOI of 0.1 with IAV and treated with or without 4 mM ammonium chloride and treated as described for panel A. Cells were fixed and immunostained as described for panel A. Microplaques (NP-positive neighboring cells, white circles) are quantified in a bar graph below the images. *, P < 0.05. Counts for 70 fields of view were included per sample per trial.
Treatment with IPA-3, cytochalasin D, and paclitaxel combined with zanamivir to block cell-free virus spread prevented intercellular spread as indicated by the lack of microplaques formed at 48 h p.i. compared to what was observed with the DMSO solvent control (Fig. 4A and B). This finding is consistent with the data presented in Fig. 3, as treatment with these drugs inhibited the formation of intercellular connections. Notably, the microtubule destabilizer nocodazole resulted in an increase in the number of microplaques (Fig. 4B). Drug concentrations were determined to be nontoxic based on testing with NucGreen Dead 488 (Life Technologies) (data not shown) and based on the ability for cell monolayers to become infected by IAV in the presence of the cytoskeleton-affecting drugs.
As an additional control for IAV cell-to-cell transport, we repeated the experiment shown in Fig. 4A and B using ammonium chloride (AC). AC is a lysosomotropic agent that raises the intraluminal pH of endosomes and inhibits triggering of HA for membrane fusion as well as blocks acidification of the interior of the virion via the M2 ion channel (39). Thus, AC blocks infection by cell-free IAV virions once the particle is internalized and eliminates the possibility that virions are associated with intercellular connections on the extracellular side and “surf” along the connection to bind and enter the neighboring cell, as occurs with HIV (31). The data indicate that cell-to-cell spread of IAV is inhibited in the presence of IPA-3 and cytochalasin D and the microtubule stabilizer paclitaxel but is not inhibited by nocodazole (Fig. 4C). A 4 mM concentration of AC was sufficient to inhibit virus spread, as only 0.2% of the monolayer became infected when cells were pretreated with 4 mM AC and subsequently infected with IAV, whereas posttreatment with 4 mM AC at 2 h p.i. resulted in infection of 14% of the monolayer (data not shown). At 48 h p.i., essentially the entire monolayer of control cells (e.g., with no AC or zanamivir) was infected; therefore, we used 2 days as our endpoint for measuring microplaque formation. Finally, given that microplaques are seen in the presence of nocodazole and DMSO together with AC, we can conclude that spread of IAV infection is occurring in the absence of viral fusion.
Intercellular connections also form in cells infected with another strain of IAV and another RNA virus and contain viral cores.
We examined cells infected with another negative-stranded enveloped RNA virus, parainfluenza virus 5 (PIV5), to test if this virus could also take advantage of intercellular connections as a means of viral cell-to-cell spread. As shown in Fig. 5, when A549, MDCK, or Vero cells infected with PIV5 were examined at 24 h p.i., intercellular connections were observed in all three cell types that stained for cell surface-expressed F protein (green) and internal staining for NP (red) (Fig. 5A) or P (red) (Fig. 5B). As NP and P are part of the vRNP and NP, P, and L form the core replicative machinery (40), the data suggest that RNPs are trafficked to neighboring cells through the intercellular connections. Intercellular connections were also seen with another IAV strain, A/WSN/33 (Fig. 6). Supporting our data shown in Fig. 3C, IAV- or PIV5-infected cells, but not mock-infected cells, contained intercellular connections (Fig. 6).
FIG 5.

Intercellular connections form in paramyxovirus-infected cells and contain the RNP components N and P. A549, MDCK, and Vero cells were infected with PIV5 at an MOI of 3 and fixed at 24 h p.i. Cell surfaces were immunostained for the fusion protein (F; green) and then permeabilized and immunostained for either nucleoprotein (N, red) (A) or P protein (red) (B). Infected cells formed intercellular connections between neighboring cells that appeared to connect the cytoplasm of both cells. Images were photographed on a confocal microscope. Scale bar, 20 μm.
FIG 6.
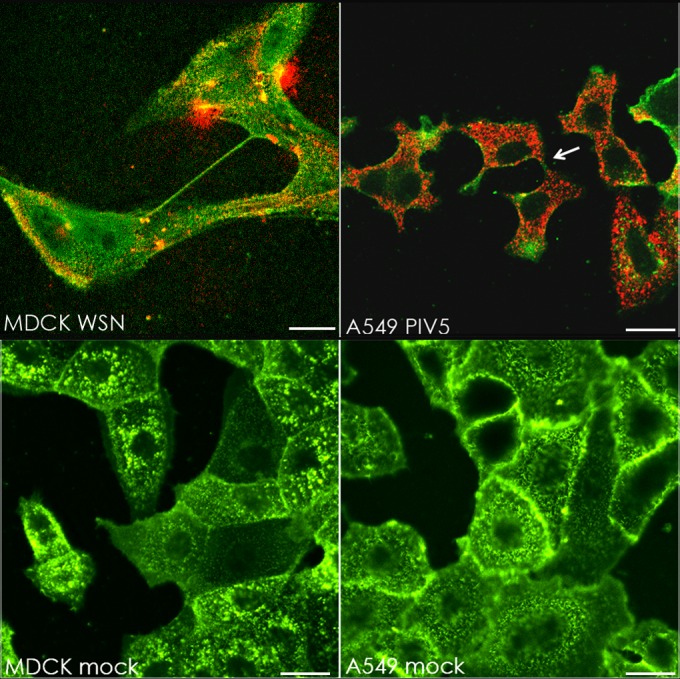
Intercellular connections form in multiple cell types infected with IAV (WSN) or PIV5. (Upper row) MDCK or A549 cells were infected with IAV A/WSN/33 or PIV5, respectively, at an MOI of 3. Cells were fixed at 17 h p.i. and immunostained for surface HA (WSN; green) or HN (PIV5; green) and then immunostained internally for NP (WSN; red) or P (PIV5; red). (Lower row) MDCK and A549 cells were mock infected and fixed 17 h later. Cell surfaces were stained with wheat germ agglutinin conjugated to Alexa Fluor 488 (5 μg/ml for 10 min). The white arrow indicates an example of vRNP-containing cell-cell connections. Images were photographed on a confocal microscope. Scale bar, 20 μm.
Intercellular connections can form in IAV- and PIV5-infected cell lines MDCK, Vero, and A549. Since these are all transformed cell lines, we were interested in whether or not an infected primary cell line would support the formation of intercellular connections. To test this, we infected normal human bronchial epithelial cells (NHBE) with IAV or PIV5 and looked for the presence of intercellular connections as before. Figure 7 shows that IAV-infected, but not mock-infected, NHBE cells formed intercellular connections containing polymerase protein (PA). Interestingly, NHBE cells infected with PIV5 were similar to the mock-infected ones in that we did not see the formation of intercellular connections (data not shown).
FIG 7.
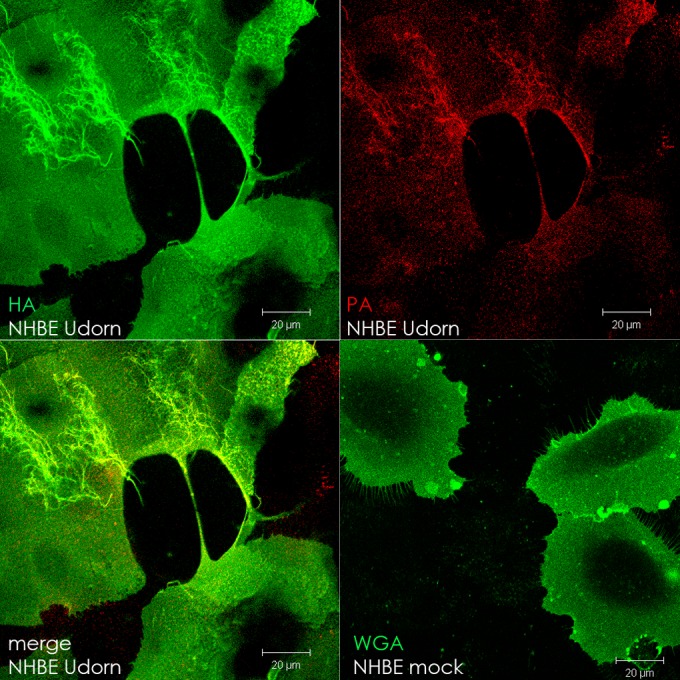
Intercellular connections form in IAV-infected primary cells. Normal human bronchial epithelial cells (NHBE) were infected with IAV (MOI = 1) or mock infected and fixed at 17 h p.i. in 10% formalin. IAV-infected cell surfaces were immunostained for hemagglutinin (HA; green), and then the cells were permeabilized and immunostained for polymerase (PA; red). Mock-infected cells were stained with Alexa Fluor 488-conjugated wheat germ agglutinin (WGA) to visualize the cell surface. Images were photographed on a confocal microscope. Scale bar, 20 μm.
To determine if intercellular connections observed in PIV5-infected MDCK cells could serve as a route to transfer infectious cores intercellularly to a neighboring cell, cells were infected with PIV5 at an MOI of 0.1 and at 2 h p.i. IPA-3, paclitaxel (“Taxol”), cytochalasin D, nocodazole, and DMSO were added to infected cells. To block infection of naive MDCK cells by cell-free PIV5 virions, a neutralizing MAb specific for the fusion (F) protein of PIV5 was added at 2 h p.i. as indicated. After 48 h, the cells were stained for PIV5 NP and microplaques were scored as for IAV. The data show that PIV5 is able to form microplaques in the presence of neutralizing antibody and DMSO, but cell-to-cell spread is significantly reduced in the presence of the cytoskeleton-affecting drugs, including nocodazole (Fig. 8). Pretreatment of cells with the MAb resulted in infection of only 0.03% of the monolayer, whereas posttreatment with the antibody at 2 h p.i. resulted in infection of 5% of the monolayer at 48 h p.i. (data not shown).
FIG 8.
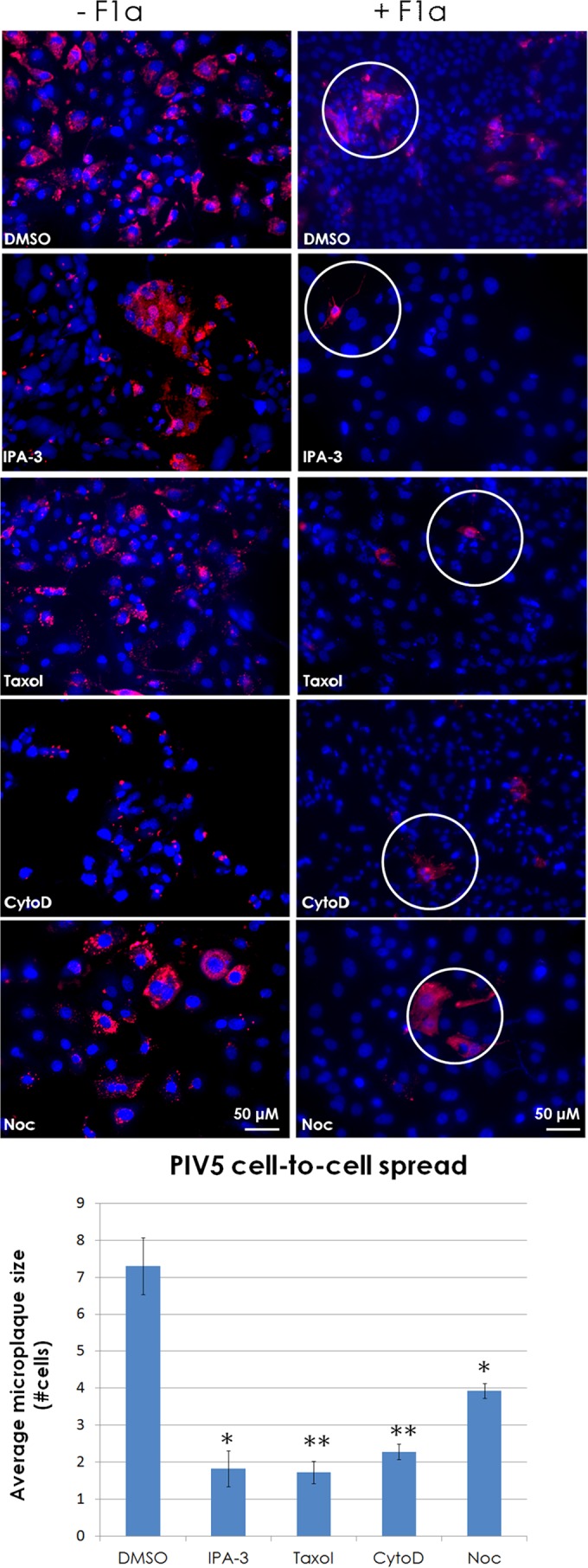
Intercellular connections are utilized by the paramyxovirus parainfluenza virus 5 as a means of cell-to-cell spread. (A) MDCK cells were infected at an MOI of 0.1 with PIV5. At 2 h p.i., cells were treated with neutralizing MAb F1a or left untreated. Cells were then treated with or without IPA-3 (30 μM), paclitaxel (“Taxol”; 100 μM), cytochalasin D (CytoD; 20 μM), or nocodazole (Noc; 30 μM) and incubated for 48 h. Cells were fixed and immunostained for N protein (red), and nuclei were stained with DAPI (blue). Images were taken on a fluorescence microscope with a 20× objective. Microplaque (N-positive neighboring cells, white circles) quantification is shown in the bar graph. *, P < 0.05; **, P < 0.01. Counts for 70 fields of view were included per sample per trial.
Intercellular connections contain filamentous actin.
TNTs are a method of cell-to-cell communication that enables passage of cellular cargo from the cytoplasm of one cell to another (29). These long, thin structures contain filamentous actin (F-actin) and enlist a myosin motor to drive the movement of organelles or other cargo into neighboring cells. The intercellular connections observed during influenza virus infection had a structure similar to that of TNTs and were sensitive to actin-depolymerizing agents but not microtubule-depolymerizing agents (Fig. 3B). Thus, we wanted to determine whether or not filamentous actin (F-actin) was present within the intercellular connections.
To test if the connections seen during IAV infection contained F-actin, MDCK cells infected with IAV were fixed and surface stained for HA and then permeabilized and stained with Alexa Fluor 594-conjugated phalloidin to visualize F-actin. Confocal microscopy showed HA (green) on the surface of the intercellular connections and F-actin (red) within the boundaries of the membrane (Fig. 9A).
FIG 9.
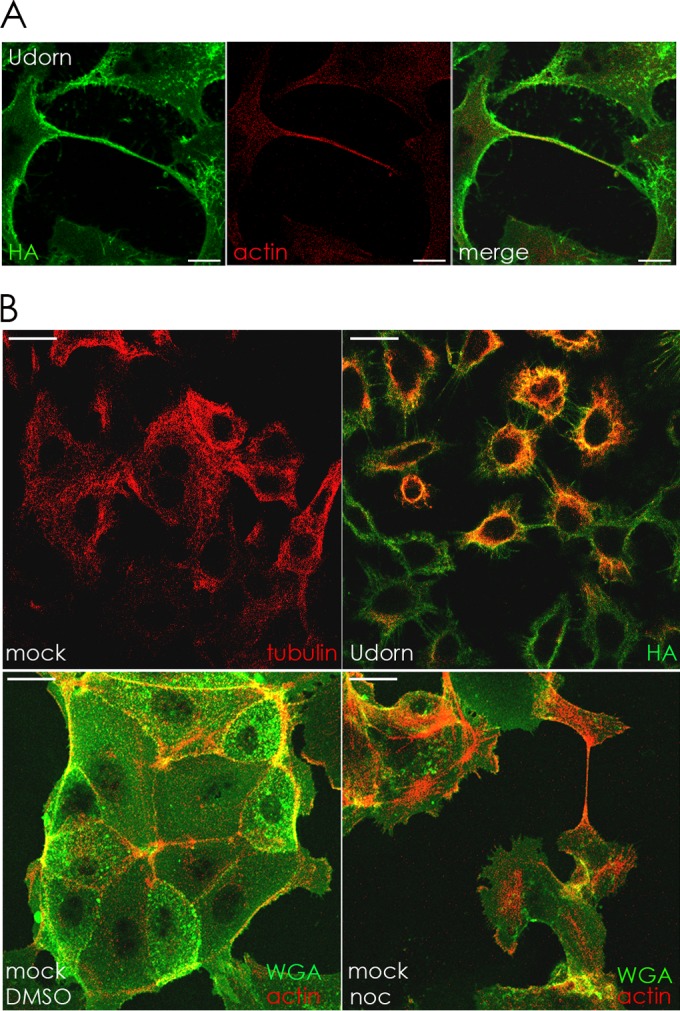
(A) Intercellular connections contain filamentous actin. MDCK cells were infected with IAV at an MOI of 1 and fixed at 12 h p.i. in 10% formalin. Surface HA (green) was immunostained, and then the cells were permeabilized in 0.1% Tween–PBS and stained with phalloidin conjugated to Alexa Fluor 594 to visualize F-actin (red). Images were photographed on a confocal microscope. Scale bar, 20 μm. (B) IAV infection of cells affects microtubule formation, and depolymerization of microtubules in mock-infected cells does not inhibit formation of intercellular connections. Upper row, MDCK cells were mock infected or infected with IAV (MOI = 3) and fixed at 17 h p.i. Cell surfaces were immunostained with antibody to HA (green) and then permeabilized and immunostained with antibody to tubulin (red). Lower row, mock-infected MDCK cells were treated with DMSO or nocodazole (noc) and fixed 17 h later. Cells were stained with Alexa Fluor 488-conjugated WGA (green) to visualize cell surfaces and then permeabilized and stained with Alexa Fluor 594-conjugated phalloidin to visualize F-actin (red). Images were photographed on a confocal microscope. Scale bar, 20 μm.
Examination of mock-infected cells indicated that intercellular connections occurred infrequently compared to the number observed in infected cells (approximately 1.5% of mock cells have intercellular connections compared to 5% for PIV5 and 12% for IAV [Fig. 3C]), suggesting that viral infection increases the frequency of such cellular connections. Although IAV-infected cells exhibit cytopathic effects (CPE) that reduce cell density compared to mock-infected cells, we have examined mock-infected cells at various levels of confluence and do not see a change in the frequency of intercellular connections (data not shown). PIV5 causes virtually no CPE. Thus, the increase in intercellular connection frequency could be the result of a stress response induced by viral infection. As shown in Fig. 4B, nocodazole treatment enhanced the number of intercellular connections in IAV-infected cells compared to DMSO-treated cell monolayers. Therefore, we hypothesized that microtubules are depolymerized during IAV infection.
To test this hypothesis, infected and mock-infected A549 cells were immunostained for tubulin. Examination by confocal microscopy revealed that mock-infected cells contained a visible microtubular network whereas IAV-infected cells contained what appeared to be tubulin monomers or very short microtubules mostly in the center of the cell (Fig. 9B, upper row). Uninfected cells treated with nocodazole had a ruffled plasma membrane compared to DMSO-treated cells and contained intercellular connections containing F-actin (Fig. 9B, lower row).
GFP-tagged NS1 of IAV moves within intercellular connections and enters into a connected cell.
While the immunofluorescent data shown in Fig. 1 to 3 and 5 to 7 coupled with the cell-cell spread data in Fig. 4 and 8 provide evidence for the use of intercellular connections to transport viral cores to neighboring cells, we wanted to show the transport of viral protein through the intercellular connection in live infected cells. To do this live-cell experiment, we used a previously constructed IAV with GFP-tagged NS1 (35). NS1 is a multifunctional protein involved in RNA transport, splicing, and translation and represses the host immune system by preventing activation of the NF-κB pathway (41). While this recombinant virus is slightly attenuated, it is still lethal to mice, and given that it is constructed in a PR8 background, it offers another example of an IAV strain that can induce the formation of intercellular connections.
To make the live-cell movie, MDCK cells were seeded onto a 42-mm coverslip and infected with the GFP-NS1 recombinant virus at an MOI of 1. At 35 h p.i., the coverslip was mounted onto an open cultivation system and supported on a heated stage (37°C) of a confocal microscope. The live-cell movie shows 30 min of active infection with a frame taken every 30 s and can be seen in Movie S1 in the supplemental material. Selected frames from this movie are shown in Fig. 10.
FIG 10.
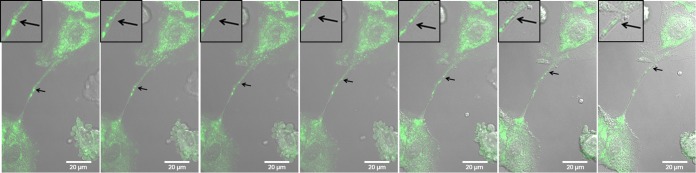
Live-cell images show that GFP-tagged NS1 can be transported from one cell to another through an intercellular connection. MDCK cells were infected with IAV (MOI = 1) that expresses GFP-tagged NS1. Frames from a live-cell movie (see Movie S1 in the supplemental material) show GFP-tagged viral protein (NS1, arrows) moving through an intercellular connection and into the cytoplasm of a neighboring cell. Insets show a zoomed image of the area of interest for enhanced visualization of GFP-tagged protein and membrane border. Images were photographed on a confocal microscope. Scale bar, 20 μm.
Over the course of 30 min, GFP-NS1 can be seen moving within the intercellular connection and into the cell body of a neighboring cell. The cell membrane is clearly visible on either side of the protein as depicted by phase-contrast microscopy (Fig. 10; see Movie S1 in the supplemental material). Black arrows show the location of GFP-NS1 as it progresses away from the lower cell and into the upper cell. Insets magnify the region of interest (Fig. 10). We noted that when particles traveled within the intercellular connections, motion was saltatory but overall had unidirectional movement (see Movie S1 in the supplemental material). This is consistent with active (motor-dependent) movement such as that seen with myosin motors.
DISCUSSION
Assembly, budding, attachment, and uncoating are all critical steps in the primary pathway for cell-to-cell spread of influenza A virus (IAV). While this is an efficient way to travel between human hosts, it also requires a great deal of cellular resources along with inherent risks from exposure to the extracellular environment. During assembly, all the structural viral proteins accumulate together at lipid rafts as localized foci on the plasma membrane and the vRNP is incorporated into these budding virions.
The RNA genomes of IAV and parainfluenza virus 5 (PIV5) are encased in nucleoprotein (NP) and bound by the viral polymerase subunits, which together make up the viral ribonucleoprotein (vRNP) complex (42, 43). The eight unique IAV vRNP segments or the single vRNP of PIV5 constitutes the minimum core components required to elicit a productive infection. All the remaining viral machinery (viral transmembrane and matrix proteins) is needed only for host cell escape (budding and scission) and entry into naive cells from the extracellular environment (attachment and fusion). Utilizing intercellular connections such as those described in this paper as a means to directly access the cytoplasm of a new host cell allows the virus to bypass some of the otherwise critical steps during egress as well as entry. The model presented in Fig. 11 summarizes this secondary route of infection.
FIG 11.

Model for IAV spread via intercellular connections. (A) An IAV-infected cell neighboring an uninfected cell in the beginning stages of establishing an intercellular connection. Filamentous actin (purple) extends through the interior of the membrane-bound extension. (B) The cellular extension connects and fuses to the plasma membrane of the uninfected cell, allowing import of vRNP and establishment of infection. (C) Drugs inhibiting actin dynamics, F-actin, and a microtubule stabilizer (cytochalasin D [cyto D], IPA-3, and paclitaxel [Taxol], respectively) inhibit the formation of intercellular connections and therefore inhibit this form of cell-to-cell spread.
In the current study, we show that loss of microtubules, either during infection by IAV or by treatment with nocodazole, increased the number of intercellular connections as well as intercellular IAV spread (Fig. 3B and C and 4B). This was an unexpected finding, as previously it had been reported that microtubules (together with Rab11) are necessary for transport of vRNP to the plasma membrane as evidenced in a study using fluorescence in situ hybridization (FISH) experiments (3). In that study, in IAV-infected MDCK cells treated with nocodazole or paclitaxel, the NP and vRNP failed to reach the apical surface. However, treatment of MDCK cells with nocodazole caused only a modest decrease in IAV virion production (3). Similar results demonstrating the limited effect of nocodazole treatment on IAV virion production in MDCK cells are shown in Fig. 4A and have also been reported previously (44). Given the lack of requirement for microtubules and sensitivity to paclitaxel, the formation of these connections may be enhanced as a downstream signaling response to a loss of microtubules during IAV infection. Interestingly, nocodazole was slightly inhibitory for PIV5 intercellular spread (Fig. 8), which suggests different requirements in the cellular environment for the PIV5 replication cycle. This may provide an explanation as to why IAV-infected monolayers have more intercellular connections than PIV5-infected cells.
We also used live-cell microscopy to show that a GFP-tagged IAV protein (NS1) can move through the intercellular connections and into a neighboring cell (Fig. 10; see also Movie S1 in the supplemental material). The movement of GFP-NS1 within intercellular connections was saltatory and consistent with movement of cargo by a motor protein. Given that tunneling nanotubules (TNTs) move only vesicular cargo (29), we hypothesize that the GFP-NS1 moving through the interior of the intercellular connection is associated with vesicles possibly during NS1-mediated anti-innate immunity and/or antiapoptotic events occurring in the cytoplasm. Additionally, our data show that these intercellular connections require actin dynamics (Fig. 3) and contain F-actin (Fig. 9A), which suggests the possibility of a myosin motor driving the transport of viral cores through the intercellular connections.
Myosin Va is an actin motor protein known for transporting membranous cargo (45), and this motor was found to be present within TNTs (29). Additionally, myosin Va has been shown to transport herpes simplex type I virions within trans-Golgi vesicles to the plasma membrane (46) and has been shown to bind RNA and RNA-binding proteins (47). However, the enlistment of other actin motors, such as myosin X, which is known to move toward the tips of pseudopodia, cannot be excluded. Future experiments will address the mechanism by which intercellular transport of the core infectious viral machinery (vRNPs) through these cellular connections occurs.
Finally, Rab11 has been shown to play important roles for vRNP trafficking (48). Rab11-containing vesicles can interact with actin through effectors such as FIP2 and myosin Vb (48). As it was noted that TNTs traffic membranous organelles through their interior (29), we speculate that Rab11-vRNP interactions taking place on vesicular membranes may be required for vRNP transport to connected cells. Future experiments will address these possibilities.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Lamb laboratory for valuable discussions and insight.
This research was supported by grant R01 AI-20201 (R.A.L.) from the National Institute of Allergy and Infectious Diseases.
K.L.R. is an associate and R.A.L. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03306-14.
REFERENCES
- 1.Shaw ML, Palese P. 2013. Orthomyxoviridae, p 1151–1185 Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Stauffer S, Feng Y, Nebioglu F, Heilig R, Picotti P, Helenius A. 2014. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J Virol 88:13029–13046. doi: 10.1128/JVI.01430-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amorim MJ, Bruce EA, Read EKC, Foeglein Á Mahen R, Stuart AD, Digard P. 2011. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J Virol 85:4143–4156. doi: 10.1128/JVI.02606-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fontana J, Cardone G, Heymann JB, Winkler DC, Steven AC. 2012. Structural changes in influenza virus at low pH characterized by cryo-electron tomography. J Virol 86:2919–2929. doi: 10.1128/JVI.06698-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye Z, Liu T, Offringa DP, McInnis J, Levandowski RA. 1999. Association of influenza virus matrix protein with ribonucleoproteins. J Virol 73:7467–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elster C, Larsen K, Gagnon J, Ruigrok RWH, Baudin F. 1997. Influenza virus M1 protein binds to RNA through its nuclear localization signal. J Gen Virol 78:1589–1596. [DOI] [PubMed] [Google Scholar]
- 7.Rossman JS, Lamb RA. 2011. Influenza virus assembly and budding. Virology 411:229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nayak DP, Balogun RA, Yamada H, Zhou ZH. 2009. Influenza virus morphogenesis and budding. Virus Res 143:147–161. doi: 10.1016/j.virusres.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerl MJ, Sampaio JL, Urban S, Kalvodova L, Verbavatz J-M, Binnington B, Lindemann D, Lingwood CA, Shevchenko A, Schroeder C, Simons K. 2012. Quantitative analysis of the lipidomes of the influenza virus envelope and MDCK cell apical membrane. J Cell Biol 196:213–221. doi: 10.1083/jcb.201108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leser GP, Lamb RA. 2005. Influenza virus assembly and budding in raft-derived microdomains: a quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology 342:215–227. doi: 10.1016/j.virol.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 11.Hess ST, Kumar M, Verma A, Farrington J, Kenworthy A, Zimmerberg J. 2005. Quantitative electron microscopy and fluorescence spectroscopy of the membrane distribution of influenza hemagglutinin. J Cell Biol 169:965–976. doi: 10.1083/jcb.200412058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossman JS, Jing X, Leser GP, Lamb RA. 2010. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142:902–913. doi: 10.1016/j.cell.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D, Harmon A, Jin J, Francis DH, Christopher-Hennings J, Nelson E, Montelaro RC, Li F. 2010. The lack of an inherent membrane targeting signal is responsible for the failure of the matrix (M1) protein of influenza A virus to bud into virus-like particles. J Virol 84:4673–4681. doi: 10.1128/JVI.02306-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen BJ, Leser GP, Morita E, Lamb RA. 2007. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol 81:7111–7123. doi: 10.1128/JVI.00361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen BJ, Leser GP, Jackson D, Lamb RA. 2008. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol 82:10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barman S, Ali A, Hui EKW, Adhikary L, Nayak DP. 2001. Transport of viral proteins to the apical membranes and interaction of matrix protein with glycoproteins in the assembly of influenza viruses. Virus Res 77:61–69. doi: 10.1016/S0168-1702(01)00266-0. [DOI] [PubMed] [Google Scholar]
- 17.Jin H, Leser GP, Zhang J, Lamb RA. 1997. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J 16:1236–1247. doi: 10.1093/emboj/16.6.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooke CB, Ince WL, Wrammert J, Ahmed R, Wilson PC, Bennink JR, Yewdell JW. 2013. Most influenza A virions fail to express at least one essential viral protein. J Virol 87:3155–3162. doi: 10.1128/JVI.02284-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noda T, Kawaoka Y. 2012. Packaging of influenza virus genome: robustness of selection. Proc Natl Acad Sci U S A 109:8797–8798. doi: 10.1073/pnas.1206736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. 2010. Genome packaging in influenza A virus. J Gen Virol 91:313–328. doi: 10.1099/vir.0.017608-0. [DOI] [PubMed] [Google Scholar]
- 21.Noda T, Sugita Y, Aoyama K, Hirase A, Kawakami E, Miyazawa A, Sagara H, Kawaoka Y. 2012. Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat Commun 3:639. doi: 10.1038/ncomms1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou Y-y, Vafabakhsh R, Doğanay S, Gao Q, Ha T, Palese P. 2012. One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis. Proc Natl Acad Sci U S A 109:9101–9106. doi: 10.1073/pnas.1206069109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noda T, Sagara H, Yen A, Takada A, Kida H, Cheng RH, Kawaoka Y. 2006. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 439:490–492. doi: 10.1038/nature04378. [DOI] [PubMed] [Google Scholar]
- 24.Zhong P, Agosto LM, Munro JB, Mothes W. 2013. Cell-to-cell transmission of viruses. Curr Opin Virol 3:44–50. doi: 10.1016/j.coviro.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mothes W, Sherer NM, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J Virol 84:8360–8368. doi: 10.1128/JVI.00443-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadiu I, Gendelman H. 2011. Human immunodeficiency virus type 1 endocytic trafficking through macrophage bridging conduits facilitates spread of infection. J Neuroimmune Pharmacol 6:658–675. doi: 10.1007/s11481-011-9298-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Kohler K, Oddos S, Eissmann P, Brodsky FM, Hopkins C, Onfelt B, Sattentau Q, Davis DM. 2008. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol 10:211–219. doi: 10.1038/ncb1682. [DOI] [PubMed] [Google Scholar]
- 28.Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J, Olivo-Marin J-C, Mannel D, Zurzolo C. 2009. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11:328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- 29.Rustom A, Saffrich R, Markovic I, Walther P, Gerdes H-H. 2004. Nanotubular highways for intercellular organelle transport. Science 303:1007–1010. doi: 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- 30.Sewald X, Gonzalez DG, Haberman AM, Mothes W. 2012. In vivo imaging of virological synapses. Nat Commun 3:1320. doi: 10.1038/ncomms2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. 2007. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat Cell Biol 9:310–315. doi: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan P, Swanson KA, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc Natl Acad Sci U S A 108:14920–14925. doi: 10.1073/pnas.1111691108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Randall RE, Young DF, Goswami KKA, Russell WC. 1987. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J Gen Virol 68:2769–2780. doi: 10.1099/0022-1317-68-11-2769. [DOI] [PubMed] [Google Scholar]
- 34.Greengard O, Poltoratskaia N, Leikina E, Zimmerberg J, Moscona A. 2000. The anti-influenza virus agent 4-GU-DANA (zanamivir) inhibits cell fusion mediated by human parainfluenza virus and influenza virus HA. J Virol 74:11108–11114. doi: 10.1128/JVI.74.23.11108-11114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, García-Sastre A. 2010. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A 107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang TS, Palese P, Krystal M. 1990. Determination of influenza virus proteins required for genome replication. J Virol 64:5669–5673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deacon SW, Beeser A, Fukui JA, Rennefahrt UEE, Myers C, Chernoff J, Peterson JR. 2008. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem Biol 15:322–331. doi: 10.1016/j.chembiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schliwa M. 1982. Action of cytochalasin D on cytoskeletal networks. J Cell Biol 92:79–91. doi: 10.1083/jcb.92.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinto LH, Holsinger LJ, Lamb RA. 1992. Influenza virus M2 protein has ion channel activity. Cell 69:517–528. doi: 10.1016/0092-8674(92)90452-I. [DOI] [PubMed] [Google Scholar]
- 40.Lamb RA, Parks GD. 2013. Paramyxoviridae: the viruses and their replication, p 957–995 InKnipe DM, Howley PM (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 41.Wang X, Li M, Zheng H, Muster T, Palese P, Beg AA, Garcia-Sastre A. 2000. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol 74:11566–11573. doi: 10.1128/JVI.74.24.11566-11573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. 2012. Organization of the influenza virus replication machinery. Science 338:1631–1634. doi: 10.1126/science.1227270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arranz R, Coloma R, Chichón FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortín J, Martín-Benito J. 2012. The structure of native influenza virion ribonucleoproteins. Science 338:1634–1637. doi: 10.1126/science.1228172. [DOI] [PubMed] [Google Scholar]
- 44.De Conto F, Di Lonardo E, Arcangeletti MC, Chezzi C, Medici MC, Calderaro A. 2012. Highly dynamic microtubules improve the effectiveness of early stages of human influenza A/NWS/33 virus infection in LLC-MK2 cells. PLoS One 7:e41207. doi: 10.1371/journal.pone.0041207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammer JA, Sellers JR. 2012. Walking to work: roles for class V myosins as cargo transporters. Nat Rev Mol Cell Biol 13:13–26. doi: 10.1038/nrm3248. [DOI] [PubMed] [Google Scholar]
- 46.Roberts KL, Baines JD. 2010. Myosin Va enhances secretion of herpes simplex virus 1 virions and cell surface expression of viral glycoproteins. J Virol 84:9889–9896. doi: 10.1128/JVI.00732-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCaffrey MW, Lindsay AJ. 2012. Roles for myosin Va in RNA transport and turnover. Biochem Soc Trans 40:1416–1420. doi: 10.1042/BST20120172. [DOI] [PubMed] [Google Scholar]
- 48.Bruce EA, Stuart A, McCaffrey MW, Digard P. 2012. Role of the rab11 pathway in negative-strand virus assembly. Biochem Soc Trans 40:1409–1415. doi: 10.1042/BST20120166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.