

ABSTRACT
Previous studies have shown that the cancer-causing high-risk human papillomavirus (HPV) E6 oncoproteins have PDZ binding potential, an activity which is important for their ability to support the viral life cycle and to cooperate in the induction of malignancy. However, PDZ interactions are not constitutive, and they can be negatively regulated by phosphorylation within the E6 PDZ binding motif (PBM). In this study, we have investigated the differential regulation of the HPV E6 PBMs from diverse high-risk HPV types. We show that, depending on the HPV type, PDZ binding activity can be regulated by phosphorylation with protein kinase A (PKA) or AKT, which, in turn, inhibits PDZ recognition. Such regulation is highly conserved between E6 proteins derived from HPV-16, HPV-18, and HPV-58 while being somewhat weaker or absent from other types such as HPV-31, HPV-33, and HPV-51. In the case of HPV31, PKA phosphorylation occurs within the core of the E6 protein and has no effect on PDZ interactions, and this demonstrates a surprising degree of heterogeneity among the different high-risk HPV E6 oncoproteins in how they are regulated by different cellular signaling pathways.
IMPORTANCE This study demonstrated that the cancer-causing HPV E6 oncoproteins are all subject to posttranslational modification of their extreme C-terminal PDZ binding motifs through phosphorylation. However, the identities of the kinase are not the same for all HPV types. This demonstrates a very important divergence between these HPVs, and it suggests that changes in cell signaling pathways have different consequences for different high-risk virus infections and their associated malignancies.
INTRODUCTION
Human papillomaviruses (HPVs) are the causative agents of cervical cancer, which remains a leading cause of death in women throughout the world. Over 120 different HPV types have been identified, 12 of which are defined as cancer causing (1, 2). Of these, HPV-16 and HPV-18 are the most important, accounting for approximately 70% of cervical cancers. The remaining cancers are caused by other high-risk (HR) HPV types, which include HPV-31, -33, -35, -51, -52, -58, -39, -45, -56, and -59 (1, 2). HPV-induced carcinogenesis arises from the combined activity of the two major viral oncoproteins E6 and E7, which, by deregulating multiple cellular pathways, including cell cycle control and apoptosis, ultimately induce cell immortalization and, eventually, malignancy (3, 4).
A unique characteristic of the HR HPV E6 oncoproteins is the presence of a class I PDZ (PSD-95/Dlg/ZO-1) binding motif (PBM) at the extreme carboxy terminus, which is absent in the low-risk (LR) non-cancer-causing HPV E6 proteins (5, 6). This region of E6 allows it to interact with a number of cellular PDZ domain-containing proteins, many of which are involved in the regulation of cell junctional integrity and cell signaling pathways (reviewed in reference 7). The first such targets to be identified were the cell polarity regulators Discs Large (hDlg1) (5, 7, 8) and Scribble (hScrib) (9), which were shown to be degraded by HPV-16 and HPV-18 E6 in a proteasome-dependent manner. However, subtle variations in the PBM sequences between HPV-16 and HPV-18 E6 revealed differences in how diverse HPV E6 oncoproteins interact with their PDZ substrates and indicated that HPV-18 E6 preferentially associates with hDlg1, while HPV-16 E6 preferentially binds hScrib (10).
The biological consequences of HPV E6 interaction with PDZ domain-containing targets are very diverse (11). This interaction has been reported to contribute toward the ability of E6 to immortalize rodent cells (8, 12) and human tonsillar keratinocytes (13), and to promote epithelial-to-mesenchymal transition (EMT) characteristics in human foreskin keratinocytes (14). In mice, this region of E6 contributes to the cooperation with E7 in tumor induction and plays an important role in the generation of malignancy in both the cervix and the skin (15, 16). Mutations in this region of E6 in the context of the whole viral genome result in marked defects to the viral life cycle, with reduced rates of viral DNA replication and a reduction in the expansion of replication-competent cells in the basal layers of the organotypic cultures (17, 18). Furthermore, such viral genomes appear unstable over time, with rapid loss of viral episomes upon continued passaging of the cells (17–21).
Recognition of PDZ substrates by HPV-16 and HPV-18 E6 is not constitutive and is subject to posttranslational regulation of both E6 and the substrate (22–24). Depending on the specific HPV type, the E6 PBM can be phosphorylated by either protein kinase A (PKA) or AKT, resulting in disruption of E6 binding to PDZ-containing targets (23, 25). Moreover, we recently showed that this phosphorylated E6 could instead interact with 14-3-3ζ (23), a phospho-serine/threonine binding protein (26). There are seven different isoforms of 14-3-3, which play diverse roles in the regulation of cellular homeostasis, many of these functions being relevant for cancer development (27–29). This suggests that the E6 PBM is actually bifunctional, mediating interaction with either PDZ domain-containing substrates or 14-3-3 family members, depending upon the E6 phosphorylation status. In this study, we have analyzed the phospho-switch regulation of several E6 proteins derived from cancer-causing HPV types. We have found that phospho-regulation of the E6 PBM is a common theme, but the mechanisms by which it occurs and the subsequent consequences are quite diverse.
MATERIALS AND METHODS
Cell culture and transfections.
HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, penicillin-streptomycin (100 U/ml), and glutamine (300 μg/ml). Transient transfections were performed on approximately 7 × 105 cells seeded in 60-mm dishes by standard calcium precipitation (30).
Plasmids.
To generate glutathione S-transferase (GST) fusion proteins, HPV-16, -18, and -31 E6 proteins were cloned into pGEX2T as described previously (31–33). The HPV-33 E6-GST was subcloned directly from pcDNA-33 E6 into compatible BamHI and EcoRI restriction sites of pGEX2T. The HPV-51 E6-GST was generated by mutational alteration of the HPV-18 E6 PBM to match the PBM of HPV-51, while HPV-58, HPV-31 E6 delPDZ, and HPV-31 E6 T145AdelPDZ fusion proteins were generated by subcloning PCR-amplified HPV-58 and HPV-31 E6 into compatible BamHI and EcoRI restriction sites of pGEX2T. Primer sequences used are as follows: for HPV-51 E6, TTTGGATCCATGGCGCGCTTTGAGGATCCA (forward) and GAATTCTACTTGCGTTTCATTGCG (reverse); for HPV-58 E6, ATGGATCCATGTTCCAGGACGCAGAG (forward) and CGGAATTCTTACACTTGTGTTTGTCTGC (reverse); for HPV-31 delPDZ, CCGGATCATGTTCATCAAAAATC (forward) and CCGAATTCTTAAGTACGAGGT (reverse); and for HPV-31 T145AdelPDZ, CCGGATCATGTTCATCAAAAATC (forward) and CCGAATTCTTATGCACGAGTC (reverse).
Resulting plasmids were all verified by DNA sequencing. These HPV-16, HPV-18, HPV-31, HPV-33, HPV-18:HPV-51, and HPV-58 E6 proteins as well as HPV-31 E6 mutants are all depicted schematically in Fig. 1. The Dlg and MAGI-1 expression plasmids have been described previously (7, 34).
FIG 1.

Consensus motifs in the carboxy-terminal regions of diverse HPV E6 oncoproteins. Shown are the amino acid sequences of the C-terminal PDZ binding motifs (PBMs) of HPV-18, -16, -31, -33, -51, and -58, together with the HPV-31 E6 mutant derivatives delPDZ and T145AdelPDZ. At the top are shown the consensus recognition sequences for phosphorylation by PKA (36) and AKT (37). Also shown is the consensus PDZ binding motif.
In vitro phosphorylation and binding assays.
GST fusion proteins were washed with 1× phosphate-buffered saline (PBS) containing 0.1% Tween 20, followed by two washes twice with the respective kinase buffers. The buffers used are as follows: PKA buffer containing 25 mM Tris HCl (pH 7.5), 10 mM MgCl2, and 70 mM NaCl and AKT buffer containing 25 mM Tris HCl (pH 7.5), 10 mM MgCl2, and 5 mM dithiothreitol (DTT).
In vitro phosphorylation of the fusion proteins was carried out using 20 μl of kinase buffer containing 2.5 μCi of radiolabeled 32γ-ATP and 25 U of cAMP-dependent protein kinase catalytic subunit (Promega) for 20 min at 30°C, or 0.2 μg of AKT1 and 4 μl of reaction buffer A from AKT1 kinase enzyme system (Promega) at 30°C for 45 min.
Purified GST fusion proteins (before and after phosphorylation with cold ATP) were incubated with in vitro-translated and radiolabeled Dlg and MAGI-1 or with purified His-tagged 14-3-3ε isoform (Enzolife) for 1 h at room temperature. After a wash with PBS containing 0.1% Tween 20, the bound Dlg and MAGI-1 were subjected to SDS-PAGE and analyzed by autoradiography, while 14-3-3 proteins were detected by Western blotting using anti-14-3-3-specific antibodies.
Immunoprecipitation.
HEK293 cells (7 × 105) were seeded onto 10-cm2 dishes and transfected with 10 μg of HA-tagged HPV-18 E6 using calcium phosphate precipitation (30). Twenty-four hours posttransfection, the cells were collected and lysed using high-salt E1A buffer (500 mM NaCl, 0.1% NP-40, and 50 mM HEPES [pH 7.0]) in the presence of protease and phosphatase inhibitors, followed by gentle syringing and then incubation on ice for 20 min. Cell lysates were then centrifuged at 14,000 rpm for 10 min. The extracted lysates were either pretreated with 2,000 U of λ phosphatase (NEB) at 30°C for 15 min or incubated directly with 30 μl of monoclonal antihemagglutinin (anti-HA)-conjugated agarose beads (Sigma) at 4°C for 3 h. Samples were washed thrice with E1A buffer and then subjected to Western blot analysis.
Antibodies and Western blotting.
The anti-phospho-E6-specific antibody (custom-made by Eurogentec [23]) was raised against the peptide H2N-RQERLQRRRET(PO3H2)QV-COOH in rabbits. For detection of phospho-E6 in a Western blot using this antibody, blocking was done using 3% bovine serum albumin (BSA) in 1× Tris-buffered saline (TBS) containing 0.1% Tween 20 (1× TBST) with gentle rocking at 4°C for 1 h. The blot was then incubated with anti-phospho-E6-specific antibody diluted in 3% BSA in 1× TBST (1:500) overnight with gentle rocking. The blot was washed thrice with 1× TBST and incubated with anti-rabbit horseradish peroxidase (HRP; Dako)-conjugated antibody and developed using the ECL detection system (Amersham).
The following antibodies were also used: mouse anti-6× His (BD Biosciences); rabbit anti-14-3-3ζ, -14-3-3ε, -14-3-3γ, and -14-3-3θ (all from Santa Cruz); mouse anti-HA; and appropriate secondary antibodies conjugated to horseradish peroxidase (Dako).
RESULTS
Phosphorylation of the E6 PBM is conserved across multiple HPV types.
Previous studies have shown that HPV-18, HPV-16, and HPV-31 E6 can be phosphorylated within the PBM, although the identity of the kinase involved, PKA or AKT, depends upon the composition of the PBM, and this can vary (Fig. 1) between different HPV types (23). We were interested in extending these studies to E6 proteins derived from other HPV types to determine whether phospho-regulation of the PBM was a conserved feature among high-risk HPV E6 proteins. To do this, we used HPV-33, -51, and -58 E6 proteins expressed as GST fusion proteins, with HPV-16, -18, and -31 E6-GST fusion proteins for comparison. The purified proteins were incubated with commercially available purified active PKA or AKT together with radiolabeled ATP. The levels of E6 phosphorylation were then ascertained by SDS-PAGE and autoradiography. The results in Fig. 2A demonstrate that HPV-18 E6 is the most susceptible to phosphorylation by PKA, closely followed by HPV-58 E6. HPV-33, -51, and -31 E6 proteins are all somewhat weaker substrates for PKA. In the case of AKT, HPV-16 E6 and HPV-58 E6 are most readily phosphorylated. HPV-31 E6 then follows, with HPV-33 and HPV-51 E6 being very poor substrates for AKT (Fig. 2B). These results demonstrate that while all the high-risk HPV E6 proteins analyzed can be phosphorylated within the PBM, there are major differences in the efficiency with which this occurs.
FIG 2.
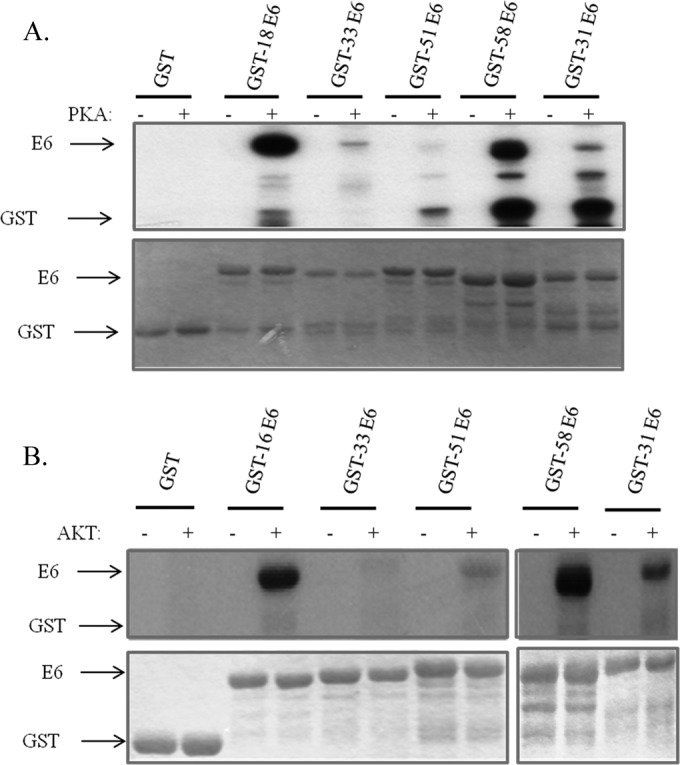
High-risk HPV E6 oncoproteins are phosphorylated by PKA or AKT. (A) Purified GST fusion proteins of HPV-18, -33, -51, -58, and -31 E6, either left untreated (−) or incubated with PKA (+) and 32P-γATP as indicated. Proteins were analyzed by SDS-PAGE and autoradiography. (B) HPV-16, -33, -51, -58, and -31 E6-GST fusion proteins, either left untreated (−) or treated with AKT (+) and 32P-γATP as indicated. In each case the upper portions show the autoradiograms and the lower portions show the Coomassie-stained gels. Arrows indicate the relevant proteins.
Analysis of the HPV-31 E6 carboxy-terminal sequence (Fig. 1) indicates the existence of two potential phospho-acceptor sites (threonine residues) within this region: one within the PBM and one located just upstream. This seemed particularly interesting, since phosphorylation of T145 upstream of the PBM would be unlikely to affect PDZ recognition, while T147 would most likely be inhibitory (23, 35). Therefore, to identify which of these potential sites was phosphorylated, we generated two mutations within the HPV-31 E6 carboxy terminus (see Fig. 1). The purified GST fusion proteins were then subjected to phosphorylation with PKA and AKT; the results obtained are shown in Fig. 3. As can be seen, mutation of both potential phospho-acceptor sites in HPV-31 E6 has no effect on the levels of PKA phosphorylation (Fig. 3A). Although mass spectrometry analysis of PKA-phosphorylated HPV-31 E6 indicated that the potential phospho-acceptor site was S82, the mutant derivative S82A could still nonetheless be phosphorylated by PKA, indicating that HPV-31 E6 is phosphorylated by PKA at more than one site (data not shown). In contrast, the AKT phosphorylation would appear to occur primarily on the carboxy-terminal threonine, lying within the PBM (Fig. 3B).
FIG 3.

HPV-31 E6 is differentially phosphorylated by PKA and AKT. (A) Purified GST fusion proteins of HPV-31 E6 wild-type and PBM mutants, delPDZ and T145AdelPDZ, either left untreated (−) or incubated with PKA (+) and 32P-γATP as indicated. Proteins were analyzed by SDS-PAGE and autoradiography. The upper portion shows the autoradiogram, and the lower portion shows the Coomassie stain of the gel. (B) Experiment similar to that for panel A, but GST fusion proteins were either left untreated (−) or incubated with AKT (+). (C) HPV-18, -31, and -58 E6-GST fusion proteins, either left untreated (−) or incubated with PKA (+) in the presence of cold ATP. These were detected by Western blotting using anti-phospho-E6-specific antibody. The lower portion shows the Ponceau stain of the nitrocellulose membrane confirming equal levels of protein loading. (D) A similar analysis of GST fusion proteins of HPV-16 E6, HPV-31 E6, and the HPV-31 E6 mutants, delPDZ and T145AdelPDZ, either left untreated (−) or incubated with AKT (+) and detected by Western blotting using anti-phospho-E6-specific antibody. Arrows indicate the relevant GST proteins.
To confirm these results, we performed Western blot analysis using antibodies raised against phospho-HPV-18 E6 to determine whether they could also similarly detect HPV-31 and HPV-58 phosphorylated E6 (23). The results in Fig. 3C demonstrate that PKA-phosphorylated HPV-18 and HPV-58 E6 proteins are readily detected by the anti-phospho-E6-specific antibody. In contrast, PKA-phosphorylated HPV-31 E6 is not detected by this antibody, consistent with the HPV-31 E6 PKA phospho-acceptor site lying outside the PBM. In contrast, it is clear that wild-type HPV-31 E6 phosphorylated by AKT is recognized by the anti-phospho-E6 antibody, whereas the two carboxy-terminal mutants of E6 are not (Fig. 3D).
Taken together, these results indicate that while HPV-31 E6 can be phosphorylated by both AKT and PKA, the phospho-acceptor sites are not the same, and only AKT phosphorylates HPV-31 within the PBM. In contrast, HPV-16 (23) and HPV-58 E6 proteins are phosphorylated in the PBM by both AKT and PKA.
Phosphorylation of E6 negatively regulates interaction with Dlg and MAGI-1.
Previous studies showed that PKA phosphorylation of HPV-18 E6 negatively regulates its interaction with Dlg (25) and MAGI-1 (23). While phosphorylation of PBMs is generally assumed to inhibit PDZ recognition (35), we wanted to ascertain whether the other high-risk HPV E6 oncoproteins were subjected to a similar pattern of regulation. To do this, we performed a series of in vitro pulldown assays using phosphorylated GST-E6 and in vitro-translated radiolabeled MAGI-1. The results obtained are shown in Fig. 4 and demonstrate a number of interesting features. Although there are variations in the degree to which the different E6 proteins bind MAGI-1, with HPV-18 and HPV-58 E6 being the strongest, the phosphorylation of HPV-18, -33, -51, and -58 E6 proteins decreased the ability of all to interact with MAGI-1 (Fig. 4A). However, HPV-31 E6 appears to differ: its interaction with MAGI-1 is comparable to that of HPV-18 E6, but PKA phosphorylation has no effect on the interaction (Fig. 4B). This is consistent with the above-described results showing that HPV-31 E6 is phosphorylated by PKA outside the PBM (Fig. 3A and C). Similar results were also seen with Dlg (Fig. 4C). In both cases, the PBM mutation confirmed that HPV-31 E6 PDZ interactions are mediated through classic PBM recognition (Fig. 4B and C). These results demonstrate that although the PDZ recognition of most HPV E6 oncoproteins is negatively regulated by PKA, this does not hold true for HPV-31 E6.
FIG 4.

PKA and AKT phosphorylation negatively regulates interaction of HPV E6 with PDZ domain-containing proteins. (A and B) The indicated GST fusion proteins were either left untreated (−) or subjected to phosphorylation with PKA (+) in the presence of cold ATP. These were then incubated with in vitro-translated radiolabeled MAGI-1. Following extensive washing, the bound MAGI-1 was detected using SDS-PAGE and autoradiography (shown in the upper portion). The lower panels in each case show the Coomassie stain of the gels; arrows indicate the relevant proteins. (C) The indicated GST fusion proteins were either left untreated (−) or subjected to phosphorylation with PKA (+) in the presence of cold ATP. These were then incubated with radiolabeled in vitro-translated Dlg as indicated. Following extensive washing, bound proteins were detected using SDS-PAGE and autoradiography shown in the upper portion. The lower portion shows the Coomassie stain of the gel. The arrows indicate the relevant fusion proteins and in vitro-translated products. (D) Same as in panel C except that the E6 fusion proteins were phosphorylated with AKT (+) in the presence of cold ATP and subsequent interaction with Dlg was then assessed by SDS-PAGE and autoradiography.
We then repeated this analysis following AKT phosphorylation of the different GST-E6 fusion proteins and monitored the effects on interactions with Dlg. The results obtained are shown in Fig. 4D and are again a clear reflection of the capacity of the different E6 proteins to be phosphorylated by AKT within their PBMs. Thus, HPV-18 and HPV-33 E6 are very poor substrates of AKT, and not surprisingly, AKT phosphorylation has no effect on their ability to interact with Dlg. In contrast, HPV-16 and HPV-58 E6 proteins are very good substrates of AKT, and this is reflected in the effects on Dlg recognition, where phosphorylation greatly reduces the interaction with the two E6 proteins. As noted above, HPV-31 E6 is phosphorylated by AKT on the threonine residue embedded within the PBM, and not surprisingly, this also results in a strong inhibition of HPV-31 E6 interaction with Dlg. Taken together, these results demonstrate that the capacities of different HPV E6 oncoproteins to be phosphorylated by different cellular kinases vary considerably among the different high-risk HPV E6 oncoproteins, but the consequences of such phosphorylation events within the E6 PBM are always the same, resulting in an inhibition of E6 PBM PDZ interactions.
Recognition of 14-3-3 family members is conserved among multiple high-risk HPV E6 oncoproteins.
The above-described studies indicate that phosphorylation of the E6 PBM is a common theme in the regulation of E6 function and negatively regulates interaction with PDZ domain-containing substrates. We previously showed that HPV-18 E6 phosphorylated within the PBM could, however, interact with 14-3-3ζ. Since there are seven different isoforms of 14-3-3, we were next interested in determining whether the other high-risk HPV E6 oncoproteins could also associate with 14-3-3 in a similar manner. In order to first ascertain which were the most relevant isoforms to analyze, we performed a series of studies to determine which isoforms could be preferentially bound to HPV-18 E6 in vivo. To do this, HEK293 cells were transfected with a HA-tagged HPV-18 E6 expression plasmid, and after 24 h, the cells were harvested and extracts immunoprecipitated using anti-HA antibody-conjugated agarose beads. Any coimmunoprecipitating 14-3-3 family members were then detected by Western blot analysis using isoform-specific antibodies. The results obtained are shown in Fig. 5A and demonstrate that HPV-18 E6 can strongly interact with 14-3-3ζ, in agreement with our previous observations. Interestingly, additional 14-3-3 isoforms were also identified in complex with HPV-18 E6, and these included 14-3-3ε and 14-3-3θ, with much weaker interaction obtained with 14-3-3γ. However, little or no interaction was detected between HPV-18 E6 and 14-3-3σ or 14-3-3β (data not shown).
FIG 5.
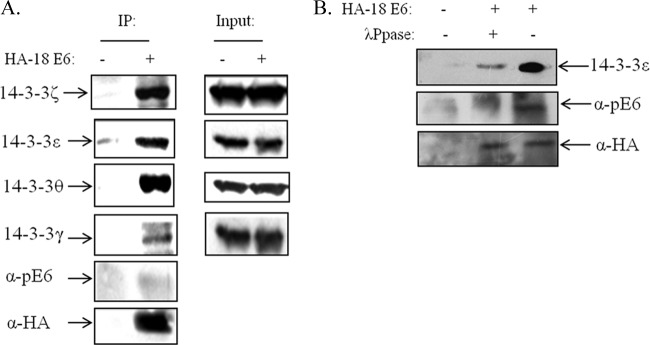
HPV-18 E6 interacts with multiple 14-3-3 isoforms in vivo in a phosphorylation-dependent manner. (A) HEK293 cells were transfected with HA-tagged HPV-18 E6 expression plasmid. After 24 h, the cells were extracted and immunoprecipitated (IP) with anti-HA-conjugated agarose beads, and coprecipitating 14-3-3 isoforms were detected by Western blotting. Also shown is the anti-HA (α-HA) blot for total E6 and anti-phospho-E6 (α-pE6), and the right side shows the protein inputs for the different 14-3-3 isoforms. Note the strong interaction of E6 with 14-3-3ζ, 14-3-3ε, and 14-3-3θ. (B) HEK293 cells were transfected with HA-tagged HPV-18 E6; after 24 h, cell extracts were treated (+) with λ phosphatase (λPpase) or not (−) for 15 min as indicated and then immunoprecipitated with anti-HA-conjugated agarose beads. Bound 14-3-3ε was detected by Western blotting. Also shown is the anti-HA blot for total E6 and anti-phospho-E6.
In order to confirm that the ability of E6 to complex with 14-3-3 isoforms in vivo was dependent upon the phosphorylation of the E6 oncoprotein, we repeated the coimmunoprecipitation assay between HA-tagged HPV-18 E6 and 14-3-3ε, but in this case, we also pretreated the cell extracts with λ phosphatase to remove phosphate groups from HPV-18 E6. The results in Fig. 5B demonstrate that λ phosphatase treatment results in a dramatic reduction in the ability of E6 to interact with 14-3-3ε, and this is reflected in the marked decrease in the levels of phospho-E6, while total E6 levels remain unchanged. Taken together, these results indicate that HPV-18 E6 can interact, in a phosphorylation-dependent manner, with a defined subset of 14-3-3 isoforms in vivo.
We then wanted to ascertain whether the other high-risk HPV E6 oncoproteins could also interact with 14-3-3 in a phosphorylation-dependent manner. In order to do this, the different GST-E6 fusion proteins were purified and phosphorylated using either AKT or PKA. The E6 proteins were then incubated with commercially available, affinity-purified His-tagged 14-3-3ε, and following extensive washing, bound 14-3-3 was detected by Western blotting. The results in Fig. 6A demonstrate that all E6 proteins that are good substrates of PKA are also able to interact strongly with 14-3-3ε postphosphorylation. Likewise, E6 proteins that are good substrates of AKT also interact strongly with 14-3-3ε (Fig. 6B) postphosphorylation. Taken together, these results demonstrate that the E6 PBM of multiple HPV types has dual functions and, depending upon the phosphorylation status, can confer interaction with either PDZ domain-containing proteins or members of the 14-3-3 protein family.
FIG 6.

Diverse HPV E6 oncoproteins interact with 14-3-3 in a phosphorylation-dependent manner. Different HPV E6-GST fusion proteins were purified and phosphorylated with either PKA (A) or AKT (B) in the presence of cold ATP. They were then incubated with purified 14-3-3ε, and after extensive washing, the bound His-tagged 14-3-3ε was detected by Western blotting with anti-6× His antibody. The upper portions show the results of the Western blots, while the lower portions show Ponceau staining of the nitrocellulose membranes confirming equal levels of protein loading. Arrows show the locations of the relevant proteins. (C) HEK293 cells were transfected with HA-tagged HPV-18 and HPV-16 E6 expression plasmids or empty plasmid. After 24 h, the cells were extracted and immunoprecipitated with anti-HA-conjugated agarose beads, and coprecipitating 14-3-3 isoforms were detected by Western blotting. Also shown is the anti-HA blot for total E6 and anti-phospho-E6.
To investigate this further, we then performed a comparative coimmunoprecipitation analysis using HPV-16 and HPV-18 E6 proteins to determine whether there were similar differences in their respective 14-3-3 interaction profiles in vivo. To do this, HEK293 cells were transfected with HA-tagged HPV-16 and HPV-18 E6 expression plasmids, and after 24 h, the cells were harvested and immunoprecipitated with anti-HA-conjugated agarose beads. Western blots were then performed to monitor the levels of E6 phosphorylation using anti-phospho-E6 antibody and 14-3-3ζ- and 14-3-3ε-specific antibodies to detect coimmunoprecipitating proteins. As can be seen from Fig. 6C, HPV-16 E6 shows higher levels of phosphorylation than HPV-18 E6, in agreement with previous studies (23). Interestingly, similar levels of 14-3-3ζ are coimmunoprecipitated with both HPV-18 and HPV-16 E6, while there seems to be weaker association between HPV-16 E6 and 14-3-3ε. These results are a good reflection of the in vitro interaction assays and further support the notion that different E6 oncoproteins exhibit a specific preference for certain combinations of 14-3-3 isoforms.
DISCUSSION
The presence of a PBM on the carboxy termini of cancer-causing E6 oncoproteins is essential for the viral life cycle (17–21) and contributes toward cell transformation (8, 13) and the induction of malignancy (15, 16). While it has been known for some time that this region of E6 could be phosphorylated, which, in turn, would be expected to inhibit E6-PDZ interactions (23, 25), it has only recently become clear that this phosphorylation also confers upon E6 the ability to interact with 14-3-3 family members (23). This indicates that phospho-regulation of E6 is critical in controlling the function of this important region of the E6 oncoprotein. For this reason we have embarked upon a series of studies to compare the relative susceptibility to phosphorylation of the HPV E6 oncoproteins from a panel of different cancer-causing HPV types. We find surprising differences between viruses in terms of the levels of E6 phosphorylation, the identity of the phospho-acceptor site, and the responsible kinase, with corresponding effects on PBM function.
Previous studies had suggested that there were significant differences in how the HPV-16 and HPV-18 E6 oncoproteins were phosphorylated by either PKA or AKT (23). To understand whether phosphorylation of the E6 PBM is a common element in the regulation of cancer-causing HPV E6 functions, we compared the phosphorylation levels of six different HPV E6 oncoproteins. Interestingly, HPV-16, HPV-18, HPV-58, and, to a lesser extent, HPV-31 were all found to be good substrates for phosphorylation by either PKA or AKT, and with the exception of HPV-31 E6 (see below), the phospho-acceptor site was located in the PBM. Lower levels of phosphorylation were found for HPV-33 and HPV-51 E6, and we have not mapped the phospho-acceptor sites on these two E6 proteins. Whether this reflects intrinsically low levels of phosphorylation, or other phospho-acceptor sites, or indicates that phosphorylation may occur through an as-yet-unidentified kinase remains to be determined. However, the consequences of phosphorylation for PDZ recognition were found to be similar between the different E6 oncoproteins, with phosphorylation of the PBM, even if weak, leading to a reduction in PDZ binding potential. An interesting exception to this was found to be HPV-31 E6, which has two potential phospho-acceptor sites within the last six amino acids of its carboxy terminus. This protein is a substrate for both AKT and PKA, but AKT would appear to phosphorylate the downstream T147 within the PBM, while PKA phosphorylates HPV-31 E6 outside the PBM at a residue(s) that remains to be identified. This would tend to suggest that the activity of HPV-31 E6 is regulated differently from those of the other HPV E6 oncoproteins that we have analyzed. Indeed, in the presence of PKA, PDZ recognition by HPV-31 E6 was unaffected, since the PBM was left unphosphorylated. Instead, phosphorylation of HPV-31 E6 occurred at S82, and it will now be of interest to determine how this phosphorylation might affect the other activities of HPV-31 E6, such as E6AP recognition and p53 degradation. In contrast, phosphorylation of HPV-31 E6 by AKT occurred on T147 within the PBM and, as with HPV-16 E6 and HPV-58 E6, this results in an inhibition of PDZ recognition. Whether these differences reflect differences in the pathologies found to be associated with HPV-31 remains to be determined, but comparative studies on the different viral life cycles in vitro should take these aspects into consideration.
Having previously demonstrated that 14-3-3ζ was an interacting partner of HPV-18 E6 in vivo (23), we wanted to determine whether any of the other high-risk HPV E6 proteins shared the capacity to interact with 14-3-3 in a phosphorylation-dependent manner. Since there are seven 14-3-3 isoforms, we first determined which were recognized by HPV-18 E6 in vivo. We found that 3 isoforms are bound strongly: 14-3-3ζ, which is consistent with our previous studies (23), plus 14-3-3ε and 14-3-3θ. These isoforms all have important roles to play in diverse aspects of cellular homeostasis (27–29), and it is now imperative to ascertain how E6 interaction with these isoforms affects E6 activity and 14-3-3 functions. These findings have also facilitated a series of studies to assess the ability of the other high-risk HPV E6 oncoproteins to interact with 14-3-3 in a phosphorylation-dependent manner. We show that phosphorylation of E6 negatively regulates PDZ interactions, and instead confers interaction with 14-3-3ε in a highly conserved manner across multiple HPV E6 types. Interestingly, this is also a direct reflection of the susceptibility of a given E6 protein to be phosphorylated by a particular kinase.
Taken together, these studies have defined a common mechanism of regulation of diverse high-risk HPV E6 oncoproteins with respect to the regulation of their PDZ and 14-3-3 interaction profiles. However, these studies have also revealed surprising differences in the forms in which these regulations take place, including major differences in the kinases involved. These studies also highlight the potential relevance of alterations in AKT and PKA signaling pathways, both under different environmental conditions and in different anatomical locations. Such alterations might have a significant impact on the particular tropisms of different high-risk HPV types, but they might also impact the likelihood of such infections progressing to malignancy. Future studies will investigate these aspects.
ACKNOWLEDGMENTS
This work was supported in part through research grants from the Wellcome Trust (0923450/Z10/Z and 09345/B/10/Z) and the Associazione Italiana per la Ricerca sul Cancro (IG2013N.14025). S.S.B. is supported by an ICGEB Pre-Doctoral Fellowship.
We are grateful to David Pim for valuable comments on the manuscript.
REFERENCES
- 1.Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V. 2009. A review of human carcinogens—part B: biological agents. Lancet Oncol 10:321–322. doi: 10.1016/S1470-2045(09)70096-8. [DOI] [PubMed] [Google Scholar]
- 2.de Villiers E-M, Fauquet C, Broker TR, Bernard H-U, zur Hausen H. 2004. Classification of papillomaviruses. Virology 324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 3.Mantovani F, Banks L. 2001. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene 20:7874–7887. doi: 10.1038/sj.onc.1204869. [DOI] [PubMed] [Google Scholar]
- 4.Münger K, Basile JR, Duensing S, Eichten A, Gonzalez SL, Grace M, Zacny VL. 2001. Biological activities and molecular targets of the human papillomavirus E7 oncoprotein. Oncogene 20:7888–7898. doi: 10.1038/sj.onc.1204860. [DOI] [PubMed] [Google Scholar]
- 5.Lee SS, Weiss RS, Javier RT. 1997. Binding of human virus oncoproteins to hDlg/SAP97, a mammalian homolog of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A 94:6670–6675. doi: 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. 1997. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 7.Gardiol D, Kühne C, Glaunsinger B, Lee SS, Javier R, Banks L. 1999. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 18:5487–5496. doi: 10.1038/sj.onc.1202920. [DOI] [PubMed] [Google Scholar]
- 8.Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. 1997. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc Natl Acad Sci U S A 94:11612–11616. doi: 10.1073/pnas.94.21.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakagawa S, Huibregtse JM. 2000. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol Cell Biol 20:8244–8253. doi: 10.1128/MCB.20.21.8244-8253.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas M, Massimi P, Navarro C, Borg JP, Banks L. 2005. The hScrib/Dlg apico-basal control complex is differentially targeted by HPV-16 and HPV-18 E6 proteins. Oncogene 24:6222–6230. doi: 10.1038/sj.onc.1208757. [DOI] [PubMed] [Google Scholar]
- 11.Banks L, Pim D, Thomas M. 2012. Human tumour viruses and the deregulation of cell polarity in cancer. Nat Rev Cancer 12:877–886. doi: 10.1038/nrc3400. [DOI] [PubMed] [Google Scholar]
- 12.Spanos WC, Hoover A, Harris GF, Wu S, Strand GL, Anderson ME, Klingelhutz AJ, Hendriks W, Bossler AD, Lee JH. 2008. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. J Virol 82:2493–2500. doi: 10.1128/JVI.02188-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spanos WC, Geiger J, Anderson ME, Harris GF, Bossler AD, Smith RB, Klingelhutz AJ, Lee JH. 2008. Deletion of the PDZ motif of HPV16 E6 preventing immortalization and anchorage-independent growth in human tonsil epithelial cells. Head Neck 30:139–147. doi: 10.1002/hed.20673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watson RA, Thomas M, Banks L, Roberts S. 2003. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J Cell Sci 116:4925–4934. doi: 10.1242/jcs.00809. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen ML, Nguyen MM, Lee D, Griep AE, Lambert PF. 2003. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6's induction of epithelial hyperplasia in vivo. J Virol 77:6957–6964. doi: 10.1128/JVI.77.12.6957-6964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shai A, Brake T, Somoza C, Lambert PF. 2007. The human papillomavirus E6 oncogene dysregulates the cell cycle and contributes to cervical carcinogenesis through two independent activities. Cancer Res 67:1626–1635. doi: 10.1158/0008-5472.CAN-06-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee C, Laimins LA. 2004. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J Virol 78:12366–12377. doi: 10.1128/JVI.78.22.12366-12377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delury CP, Marsh EK, James CD, Boon SS, Banks L, Knight GL, Roberts S. 2013. The role of protein kinase A regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. J Virol 87:9463–9472. doi: 10.1128/JVI.01234-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brimer N, Vande Pol SB. 2014. Papillomavirus E6 PDZ interactions can be replaced by repression of p53 to promote episomal HPV genome maintenance. J Virol 88:3027–3030. doi: 10.1128/JVI.02360-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicolaides L, Davy C, Raj K, Kranjec C, Banks L, Doorbar J. 2011. Stabilization of HPV16 E6 protein by PDZ proteins, and potential implications for genome maintenance. Virology 414:137–145. doi: 10.1016/j.virol.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 21.Lorenz LD, Rivera Cardona J, Lambert PF. 2013. Inactivation of p53 rescues the maintenance of high risk HPV DNA genomes deficient in expression of E6. PLoS Pathog 9:e1003717. doi: 10.1371/journal.ppat.1003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narayan N, Massimi P, Banks L. 2009. CDK phosphorylation of the discs large tumour suppressor controls its localisation and stability. J Cell Sci 122:65–74. doi: 10.1242/jcs.024554. [DOI] [PubMed] [Google Scholar]
- 23.Boon SS, Banks L. 2013. High-risk human papillomavirus E6 oncoproteins interact with 14-3-3ζ in a PDZ binding motif-dependent manner. J Virol 87:1586–1595. doi: 10.1128/JVI.02074-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narayan N, Subbaiah VK, Banks L. 2009. The high-risk HPV E6 oncoprotein preferentially targets phosphorylated nuclear forms of hDlg. Virology 387:1–4. doi: 10.1016/j.virol.2009.02.030. [DOI] [PubMed] [Google Scholar]
- 25.Kühne C, Gardiol D, Guarnaccia C, Amenitsch H, Banks L. 2000. Differential regulation of human papillomavirus E6 by protein kinase A: conditional degradation of human discs large protein by oncogenic E6. Oncogene 19:5884–5891. doi: 10.1038/sj.onc.1203988. [DOI] [PubMed] [Google Scholar]
- 26.Muslin AJ, Tanner JW, Allen PM, Shaw AS. 1996. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84:889–897. doi: 10.1016/S0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- 27.Oh S, Shin S, Lightfoot SA, Janknecht R. 2013. 14-3-3 proteins modulate the ETS transcription factor ETV1 in prostate cancer. Cancer Res 73:5110–5119. doi: 10.1158/0008-5472.CAN-13-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reinhardt HC, Yaffe MB. 2013. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol 14:563–580. doi: 10.1038/nrm3640. [DOI] [PubMed] [Google Scholar]
- 29.Aitken A. 2006. 14-3-3 proteins: a historic overview. Semin Cancer Biol 16:162–172. doi: 10.1016/j.semcancer.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 30.Wigler M, Pellicer A, Silverstein S, Axel R, Urlaub G, Chasin L. 1979. DNA-mediated transfer of the adenine phosphoribosyltransferase locus into mammalian cells. Proc Natl Acad Sci U S A 76:1373–1376. doi: 10.1073/pnas.76.3.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagasaka K, Pim D, Massimi P, Thomas M, Tomaić V, Subbaiah VK, Kranjec C, Nakagawa S, Yano T, Taketani Y, Myers M, Banks L. 2010. The cell polarity regulator hScrib controls ERK activation through a KIM site-dependent interaction. Oncogene 29:5311–5321. doi: 10.1038/onc.2010.265. [DOI] [PubMed] [Google Scholar]
- 32.Pim D, Thomas M, Javier R, Gardiol D, Banks L. 2000. HPV E6 targeted degradation of the discs large protein: evidence for the involvement of a novel ubiquitin ligase. Oncogene 19:719–725. doi: 10.1038/sj.onc.1203374. [DOI] [PubMed] [Google Scholar]
- 33.Thomas M, Massimi P, Banks L. 1996. HPV-18 E6 inhibits p53 DNA binding activity regardless of the oligomeric state of p53 or the exact p53 recognition sequence. Oncogene 13:471–480. [PubMed] [Google Scholar]
- 34.Glaunsinger BA, Lee SS, Thomas M, Banks L, Javier R. 2000. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 19:5270–5280. doi: 10.1038/sj.onc.1203906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Dasgupta J, Ma RZ, Banks L, Thomas M, Chen XS. 2007. Structures of a human papillomavirus (HPV) E6 polypeptide bound to MAGUK proteins: mechanisms of targeting tumor suppressors by a high-risk HPV oncoprotein. J Virol 81:3618–3626. doi: 10.1128/JVI.02044-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. 2010. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 37.Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC. 2000. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem 275:36108–36115. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]